


Abstract
Multiple conserved structural cis-acting regulatory elements have been recognized both in the coding and untranslated regions (UTRs) of the hepatitis C virus (HCV) genome. For example, the cis-element 5BSL3.2 in the HCV-coding region has been predicted to use both its apical and internal loops to interact with the X RNA in the 3′-UTR, with the IIId domain in the 5′-UTR and with the Alt sequence in the coding region. Additionally, the X RNA region uses a palindromic sequence that overlaps the sequence required for the interaction with 5BSL3.2, to dimerize with another HCV genome. The ability of the 5BSL3.2 and X RNA regions to engage in multi-interactions suggests the existence of one or more molecular RNA switches which may regulate different steps of the HCV life cycle. In this study, we used biophysical methods to characterize the essential interactions of these HCV cis-elements at the molecular level. Our results indicate that X RNA interacts with 5BSL3.2 and another X RNA molecule by adopting two different conformations and that 5BSL3.2 engages simultaneously in kissing interactions using its apical and internal loops. Based on these results, we propose a mode of action for possible molecular switches involving the HCV RNA.
INTRODUCTION
Hepatitis C virus (HCV), a blood-borne infectious pathogen, affects almost 200 million people worldwide (∼3% of human population) (1), being today the leading cause of cirrhosis and liver transplants in the USA (2). The current antiviral therapy of pegylated interferon-α and ribavirin provides sustained virologic response in <50% of patients with chronic genotype 1 HCV and a large number of infected patients are either intolerant to this treatment or fail to achieve a sustained viral response (3–6). Very recently, two new drugs, Teleprevir and Boceprevir (both of which are HCV protease inhibitors) to be given in combination with interferon and ribavirin, were licensed (7). However, these treatments are associated once again with several side effects such as severe anemia, fatigue, dysgeusia, gastrointestinal side effects and skin lesions.
HCV, which belongs to the Flaviviridae family, is the first member of the Hepacivirus genus (8). The ∼9.6-kb HCV genomic RNA contains a single open reading frame (ORF) that encodes for a ∼3000-amino acid polyprotein, which is co- and post-translationally cleaved by several host and viral proteases to yield the non-structural proteins NS2, NS3, NS4A, NS4B, NS5A and NS5B, and the structural proteins E1, E2, p7 and the capsid core. HCV escapes all existing therapeutic agents via its error-prone RNA-dependent RNA polymerase, NS5B, which confers it a high mutational rate (9–11).
The ORF is flanked on both sides by highly structured and conserved 5′- and 3′-untranslated regions (UTRs) (Figure 1A). The highly structured 341-nt 5′-UTR (Figure 1A, green) folds into a four-domain structure that serves as an internal ribosome entry site (IRES) that directs the cap-independent translation of the ORF and contains sequence signals required for viral RNA synthesis (12–14). The 225-nt 3′-UTR has a tri-domain organization, comprised of a non-conserved 40-nt variable region, a poly (U/UC) stretch of 20–200 nt and a 3′-terminal conserved 98-nt sequence named X-RNA (Figure 1A, purple). The tripartite 3′-UTR forms defined stem–loop structures with sequences, some of which proposed, while others established to engage cellular or viral proteins (15–19). Both the 5′- and 3′-UTRs contain cis-acting replication elements crucial for RNA replication and translation (16,19). In addition, phylogenetically conserved RNA secondary structures in the HCV-coding region have been identified (15,19–23). Exceptionally conserved codons in the NS5B-coding region of HCV RNA were proposed to serve as RNA signals for different viral functions (21,24–26), five stem–loop structures (5BSL1, 5BSL2, 5BSL3.1, 5BSL3.3 and 5BSL3.4) being predicted for these conserved sequences (24–26). Subsequently, another highly conserved stem–loop element located between 5BSL3.1 and 5BSL3.3 and spanning nt 9263–9310 was identified in the NS5B-coding region and named 5BSL3.2 (also known as SL9266 or SL-V) (Figure 1B, left) (21,23). Using site-directed mutagenesis, it has been shown that 5BSL3.2 contains RNA elements vital for viral RNA replication (21,23,27). The 5BSL3.2 secondary structure, composed of two short stems separated by an 8-nt internal bulge loop on the 3′-side and capped with a 12-nt apical loop (Figure 1B, left), is supported by enzymatic probing and chemical mapping data (21,23). Both the 5BSL3.2 terminal loop and internal bulge are crucial for HCV RNA replication, while its upper stem serves as an essential scaffold whose disruption leads to loss of HCV replication (21,23,27). Although no RNA–RNA interaction between stem–loops in the NS5B-coding region and 3′-UTR was predicted computationally, the Bartenschlager group showed an interaction between the apical loop of 5BSL3.2 in the coding region (nt 9281–9287) and the X RNA located in the 3′-UTR (nt 9540–9546) indirectly through genetic evidence (Figure 1B). These interactions were postulated to involve the formation of a kissing complex; however, this proposal could not be verified directly by NMR spectroscopy (23), and it has been proposed that a chaperoning protein, most likely NS5B, mediates these kissing interactions (23). Thus, although the existence of such interactions is indirectly supported by mutagenesis data (23,27), prior to this study the existence of a kissing complex between 5BSL3.2 and X RNA has not been directly proved.
Figure 1.


(A) Schematic representation of the full-length HCV RNA. 5′-UTR: green; coding region: red (indicated by the parallel lines is the long stretch of nucleotides that code for the viral proteins); 3′-UTR: purple. The colored lines represent the interactions investigated in this study: IIId apical loop–5BSL3.2 internal loop: mustard; Alt–5BSL3.2 internal loop: brown; 5BSL3.2 apical loop–X55: blue; X55 (genome 1)–X55 (genome 2): black. (B) Kissing interactions between 5BSL3.2 (left) and X55 (right). Encircled on 5BSL3.2 is the adenine replaced with 2-amino purine in the fluorescence spectroscopy assay. Note that the X55 conformation in this figure is as proposed by the Bartenschlager group (23). (C) Kissing interactions between two X55 molecules. (D) Kissing interactions between 5BSL3.2 (left) and Alt (right). (E) Kissing interactions between 5BSL3.2 (left) and IIId (right).
Interestingly, the sequence nt 9540–9546 within the HCV 3′-UTR X RNA, which contains the sequence required for interactions with 5BSL3.2, has been shown to participate in the HCV genome dimerization via a kissing complex intermediate (Figure 1C) (28), suggesting that X RNA might function as a molecular switch by engaging in long-range interactions with different partners at different times in the HCV life cycle.
Stem–loop structured cis-acting regulatory elements (CREs) usually contain internal loops crucial for function, and 5BSL3.2 is no exception, possessing such an internal bulge (nt 9297–9301) (Figure 1B). Mutational studies have shown that both the UCACAGC motif located in the 5BSL3.2 apical loop as well as the GCCCG motif located in the 5BSL3.2 internal bulge are virtually invariant among different genotypes and crucial for viral replication (20,21,23). Deletions in the internal loop of 5BSL3.2, randomization of its sequence or as little as a single-nucleotide substitution were found inhibitory for HCV replication (23). As described above, the 5BSL3.2 apical loop motif UCACAGC interacts with X RNA; however, the function of the 5BSL3.2 internal loop GCCCG motif was unknown until its long-range interaction with an unpaired sequence nt 9106–9123 located ∼200 nt upstream (named Alt) (Figure 1D) has been predicted in silico (20). Mutational analysis showed this interaction to be essential for HCV replication (20). Subsequently, it has been reported that the IRES associates with the 3′-end of the NS5B coding region, through the interactions of the IIId domain with the 5BSL3.2 internal loop (Figure 1E) (22). Although according to thermodynamic predictions the 5BSL3.2–IIId interacting loops might be extended to a duplex due to sequence complementarity, this model was not favored as the structure of domain IIId contains a loop E motif (22). It is interesting that the same motif GCCCG in the 5BSL3.2 internal bulge loop is predicted to interact with two different regions: the Alt sequence, located 200 nt upstream of 5BSL3.2, and the IIId domain, located in 5′-UTR IRES of the HCV genome, respectively.
This intricate network of proposed long-range interactions involving overlapping regions of 100% conservancy within the HCV genome that engage different partners at different times (Figure 1A) suggests a complex mechanism used by the virus to progress through its life cycle, which might rely on the existence of several RNA molecular switches. The present work analyzes these various predicted interactions of the cis-element 5BSL3.2 at the molecular level and proposes a possible model involving each of the interactions within the HCV life cycle.
MATERIALS AND METHODS
RNA samples
A series of RNA oligonucleotides [wild-type X55 RNA (55 nt), IIId RNA (37 nt) and Alt (31 nt), truncated 5BSL3.2_TRUNC (38 nt) and 5BSL3.2_short (24 nt)] were transcribed from synthetic DNA templates using T7 RNA polymerase in vitro transcription reactions. The RNAs were purified by 10% polyacrylamide, 8 M urea gel electrophoresis, recovered by electrophoretic elution and dialyzed against 1 mM cacodylic acid, pH 6.5. The wild-type 48-nt 5BSL3.2 RNA was chemically synthesized by Dharmacon, Inc. since the in vitro transcription of 5BSL3.2 required insertion of extra Gs on the 5′-end of 5BSL3.2 RNA and this altered its folding. X55_mut, an X55 mutant containing mutations in the region proposed to interact with 5BSL3.2, has also been chemically synthesized by Dharmacon, Inc. Unless otherwise specified, all RNA samples were annealed prior to use by heating at 95°C for 5 min, followed by snap-cooling on ice. Supplementary Table S1 describes the sequences of all RNAs used in this study as well as their position with respect to the entire HCV genome.
HCV core peptide
The first two basic domains of the HCV core protein, comprising amino acid residues 2–23 and 38–74, were combined to form a 58-amino acid sequence core peptide, which was named the ‘2BD core’ peptide (28,29). The core 2BD peptide was chemically synthesized and purified by the Peptide Synthesis Unit at the University of Pittsburgh, Center for Biotechnology and Bioengineering, and was reconstituted in 1 mM cacodylic acid, pH 6.5. The sequence of the 2BD peptide is PRRGPRLGVRATRKTSERSQPRGRRQPIPKVRHQTGRRGSRPNWGPNDPRRRSRNLGK.
Native gel electrophoresis
The RNA samples were diluted from stock solutions to 0.05–20 µM concentrations, and following boiling and snap-cooling, they were incubated in the presence of 0.2–10 mM MgCl2 either at 22°C (temperature at which a kissing complex conformation is maintained) or at 55°C (temperature which promotes the formation of the duplex conformation) for 2.5 h. The samples were electrophoresed on 10–15% native gels in either Tris–Borate EDTA (TBE) buffer or Tris–Borate Magnesium (TBM) buffer at 4°C.
To study the effect of the 2BD core peptide on the magnesium-treated IIId–5BSL3.2 complex samples, the peptide was directly added to the RNA, followed by incubation for 30 min at room temperature. These samples were subsequently treated with proteinase K for peptide digestion (concentration of 190 µg/ml) for 1 h prior to being run on native gels. The native gels were run at 4°C, 40–100 V for 1.5–10 h and were visualized by UV shadowing at 254 nm using an AlphaImager HP (AlphaInnotech, Inc.) or by using SYBr Gold stain for gels containing nanomolar concentrations of RNA.
Fluorescence spectroscopy
Experiments were performed on a J.Y. Horiba Fluoromax-3 fluorimeter equipped with variable temperature control. The excitation wavelength was set at 310 nm, and the emission spectra were recorded from 330 to 450 nm. All experiments were performed at 22°C.
X55–5BSL3.2 RNA heterodimerization
All RNA constructs used in the fluorescence assays were numbered serially from 5′ to 3′ beginning with 1; however, the true position in reference to the full-length HCV genome is given in parentheses. For the fluorescence spectroscopy experiments, 2-aminopurine (2-AP)—an adenine fluorophore analog—was used to monitor the interactions between X55 RNA and 5BSL3.2 RNA. 5BSL3.2 RNA was 2-AP labeled at position 21 (nt 9283 in full-length genome) (circled in Figure 1B), forming 5BSL3.2_21AP that was chemically synthesized and purified by Dharmacon, Inc. The dissociation constant of the kissing complex formed by 5BSL3.2_21AP and wild-type X55 RNA was determined by titrating a fixed concentration of the fluorescent 5BSL3.2_21AP (76 nM) in 1 mM cacodylic acid pH 6.5 containing 5 mM MgCl2 with increasing concentrations of wild-type X55. The kissing complex dissociation constant was determined by fitting the binding curve to Equation (1):
 |
(1) |
where IB and IF are the steady-state fluorescence intensities of the free and bound 5BSL3.2_21AP, [5BSL3.2_21AP]t is the total concentration of 5BSL3.2_21AP RNA and [X55]t is the total concentration of X55 RNA.
5BSL3.2–IIId or 5BSL3.2–Alt heterodimerization
The interactions of 5BSL3.2 RNA with IIId or Alt were monitored by fluorescence spectroscopy by using a 5BSL3.2 RNA labeled at position 35 (nt 9297 in full-length genome) by 6-methyl isoxanthopterin (6MI), a guanine fluorophore analog (Figure 1B). The labeled 5BSL3.2_35_6MI was chemically synthesized by Fidelity Systems, Inc. The dissociation constants of the kissing complexes formed by 5BSL3.2_35_6MI with wild-type IIId or with Alt RNA were determined by titrating a fixed concentration of the fluorescent 5BSL3.2_35_6MI (100 nM) in 1 mM cacodylic acid pH 6.5 containing 5 mM MgCl2 with increasing concentrations of either wild-type IIId or Alt. The kissing complex dissociation constant was determined by fitting the binding curves to an equation similar to Equation (1), where [5BSL3.2_21AP]t is replaced by [5BSL3.2_35_6MI]t, which is the total concentration of 5BSL3.2_35_6MI RNA, and [X55]t is replaced by either [IIId]t or [Alt]t, which are the total concentrations of IIId or Alt RNA, respectively.
Effects of Alt RNA on the X55–5BSL3.2 RNA heterodimerization
The effects of Alt RNA on the X55–5BSL3.2 interactions were evaluated by fluorescence spectroscopy. 76 nM of 2-AP-labeled 5BSL3.2 (5BSL3.2_2-AP) was treated with Alt (1:1 ratio) in the presence of 5 mM MgCl2, after which wild-type X55 was titrated into the mixture in nanomolar increments. The dissociation constant of the kissing complex formed by 5BSL3.2_21AP and wild-type X55 RNA, in the presence of equimolar ratio of Alt RNA, was determined by fitting the binding curve to Equation (1).
RESULTS AND DISCUSSION
X RNA is involved both in homodimerization with another genome as well as in long-range interactions with 5BSL3.2 RNA
We have previously shown that the first 55 nt of X RNA—henceforth named X55—have similar interactions when isolated, or in the context of the entire 98 nt X RNA (28). Thus, in all further studies we have used X55 RNA as a model system for X RNA. X55 RNA harbors a 16-nt palindromic sequence (nt 9531–9546 in the HCV genome, highlighted in red in Figure 2) responsible for the in vitro HCV genomic RNA dimerization (28,29). We have shown that this dimerization occurs through the formation of a kissing complex intermediate that is converted to a more stable extended duplex conformation by the viral core protein (28). Interestingly, the X55 dimer linkage sequence required for the genome homodimerization (nt 9531–9546) overlaps with the X55 sequence proposed to be involved in long-range interactions with 5BSL3.2 (nt 9540–9546). This overlap makes the existence of a tri-complex formed by two X55 and one 5BSL3.2 molecule impossible. To be able to engage in both types of interactions, it is likely that X RNA exists in more than one monomeric conformation, and this is indeed predicted when this RNA is folded with the M-fold secondary structure prediction software (30) (Figure 2). To investigate the possible conformations adopted by X55 RNA, we analyzed it in higher percentage gels containing varying MgCl2 concentrations in the range 1–10 mM. As seen in Figure 3A, the monomeric form of X55 is resolved into two bands at lower concentrations of 1 and 5 mM MgCl2, which collapse into a single wider band at 10 mM MgCl2. Present also on these gels is the dimer band corresponding to the X55–X55 kissing complex intermediate (110 nt) (28). It is interesting to note that based on the different intensities of the two monomeric bands, one of the two X55 monomer conformations (conformation 2) is preferred over the other (conformation 1) at low MgCl2 concentrations. In Figure 3A (extreme right panel), it is also shown that in the absence of MgCl2 in a native TBE gel, X55 exists in a single monomeric conformation. Note that the kissing dimer band is absent in the TBE gel (Figure 3A, extreme right panel), as magnesium ions stabilize the kissing complex structure, and these ions are chelated in the TBE gel (31–33). As far as we know, this is the first time when the existence of two monomeric conformations of X55 RNA has been demonstrated experimentally. Based on these experiments, we cannot determine unambiguously which of the two predicted conformations of the first 55 nt of X RNA (labeled X RNA-1 and X RNA-2 in Figure 2) corresponds to the two monomer bands labeled as conformation 1 and conformation 2 in Figure 3A. Nonetheless, as the MgCl2 concentration increases, the intensity of the band corresponding to X55 conformation 2 decreases, with the concomitant increase in the intensity of the band corresponding to the X55–X55 homodimer (Figure 3A). This suggests that the X55 monomer in conformation 2 is engaging in the X55–X55 homodimerization, corresponding most likely to the X RNA-2 structure depicted in Figure 2, right.
Figure 2.

Alternate conformations of HCV X RNA region as predicted by the Mfold software (30). Shown in red is the 16-nt palindromic sequence named the dimer linkage sequence (DLS) (29).
Figure 3.

(A) Left: 12% TBM gels of X55 RNA in the presence of 1, 5 and 10 mM MgCl2. Extreme right: 12% TBE gel of X55 RNA. (B) 12% TBM gel in the presence of 1, 5 and 10 mM MgCl2. Lane 1: 5BSL3.2 RNA (48 nt); lane 2: X55 RNA (55 nt); lane 3: 5BSL3.2 and X55 RNAs in a 1:1 ratio. The gels were visualized by UV shadowing at 254 nm.
Next we inquired if X RNA interacts with 5BSL3.2 via the formation of a kissing complex, as proposed based on genetic data (21,23), by performing TBM native gel electrophoresis experiments in which we used the full-length 5BSL3.2 (48 nt, spanning the sequence 9263–9310 in the HCV genome) and X55 RNA. As shown in Figure 3B, at all MgCl2 concentrations investigated there is a single band for 5BSL3.2 (lane 1), whereas X55 (lane 2) shows two lower monomeric bands and a top band corresponding to the homodimeric X55–X55 kissing complex (110 nt) (28). When 5BSL3.2 and X55 are mixed in equimolar concentrations (Figure 3B, lane 3), in addition to the X55–X55 homodimer band (110 nt), there is a new upper band that we assign to the X55–5BSL3.2 complex (103 nt). It is interesting to note that upon the formation of the X55–5BSL3.2 complex, the lower band corresponding to the monomeric X55 conformation 1 disappeared in as low as 1 mM MgCl2, whereas the band corresponding to the monomeric X55 conformation 2 disappeared only in 10 mM MgCl2. This result suggests that the X55 monomer in conformation 1 engages in interactions with 5BSL3.2, in agreement with our previous observation that the X55 monomer in conformation 2 engages in interactions with another X55 RNA. Although X55 was mixed in an equimolar ratio with 5BSL3.2, there is a 5BSL3.2 monomer band also present in lane 3 (48 nt; compare lanes 1 and 3), as not all of the X55 RNA is available for binding 5BSL3.2, being involved in the formation of the X55–X55 homodimer.
Since the monomeric conformations of X55 RNA can be distinctly observed only in MgCl2 concentrations <10 mM, all further experiments analyzing the 5BSL3.2–X55 interactions were carried out in the presence of 5 mM MgCl2. To confirm that the X55–5BSL3.2 complex is formed by the interactions between the 5BSL3.2 terminal loop and the X55 sequence spanning the nt 9540–9546 (Figure 1B), we introduced four non-complementary mutations in the X55 sequence, creating X55_mut RNA (Supplementary Figure S1A), and showed by TBM gel electrophoresis that X55_mut RNA is no longer able to form a kissing complex with 5BSL3.2 (Supplementary Figure S1B).
Taken together, these results indicate that the X55–X55 homodimer coexists with the X55–5BSL3.2 complex, and moreover, that the formation of the X55–5BSL3.2 heterodimer does not require the presence of a chaperoning protein, as previously proposed (23). Additionally, in the absence of the core protein, the X55–5BSL3.2 complex is preferred over the X55–X55 complex, as indicated by the intensities of the bands corresponding to the respective complex (Supplementary Figure S1C).
Next, we investigated the dependence of the X55–5BSL3.2 complex upon the presence of magnesium by TBE/TBM native gel electrophoresis (31–34). Two identical sets of RNA samples were prepared in the presence of 5 mM MgCl2, one being analyzed on a native TBM gel containing 5 mM MgCl2 (Figure 4B) and the second on a native TBE gel containing EDTA that will chelate the magnesium ions (Figure 4A). 5BSL3.2 RNA is monomeric in both the TBE and TBM gel, respectively (Figure 4A and B, lane 1). Three bands are present for X55 RNA in the TBM gel (monomeric conformations 1 and 2 and homodimeric kissing complex) (Figure 4B, lane 2), all of which collapse into a single monomeric band in the TBE gel (Figure 4A, lane 2). The equimolar mixture of 5BSL3.2 and X55 shows both the X55–X55 homodimer and the X55–5BSL3.2 heterodimer in the TBM gel (Figure 4B, lane 3), whereas in a TBE gel only the bands corresponding to the monomeric 5BSL3.2 monomeric X55 are present (Figure 4A, lane 3). This result strongly supports the hypothesis that both the X55–X55 homodimer and the X55–5BSL3.2 heterodimer are magnesium-dependent kissing complexes.
Figure 4.

(A) 12% TBE gel and (B) 12% TBM gel. The RNA samples (15 µM) were prepared similarly for both gels, being pre-incubated in the presence of 5 mM MgCl2 at 22°C unless otherwise specified. The gels were visualized by UV shadowing at 254 nm. Lane 1: 5BSL3.2 RNA (48 nt); lane 2: X55 RNA (55 nt); lane 3: 5BSL3.2 and X55 RNAs in a 1:1 ratio; lane 4: 5BSL3.2 and X55 RNAs in a 1:1 which following incubation were treated with core 2BD and proteinase K; lane 5: 5BSL3.2 and X55 RNAs were pre-incubated in the presence of 5 mM MgCl2 at 55°C for 2.5 h. (C) Fluorescence spectroscopy: a solution of 76 nM 5BSL3.2_21AP in 1 mM cacodylic acid pH 6.5 containing 5 mM MgCl2 was titrated with increasing concentrations of wild-type X55 (black squares). A kissing complex dissociation constant of (42 ± 6 nM) was determined by fitting the binding curve to Equation (1). In control experiments, 5BSL3.2_21AP (76 nM) was titrated with IIId, which does not bind the apical loop of 5BSL3.2 (black triangles), as well as with X55 in the absence of MgCl2 (black circles).
Our earlier results indicate that the metastable magnesium-dependent kissing X55 homodimer complex gets converted to the more stable magnesium-independent X55 duplex in the presence of the core peptide 2BD (28). The core 2BD peptide, which was produced by fusing the first two basic domains of the core protein (amino acids 2–23 and 38–74), has been previously shown to have nucleic acid chaperoning activity similar to the full-length core protein (29). Given the ability of X55 RNA to also form a kissing complex with 5BSL3.2, we inquired if these interactions are affected by the presence of the core protein. As seen in Figure 4A and B (lane 4), in both TBM and TBE gels in the presence of the core 2BD peptide, the X55–5BSL3.2 kissing complex band is absent, X55 existing only in homodimeric form (X55–X55), whereas all 5BSL3.2 is monomeric (compare lanes 3 and 4 in Figure 4A and B). Thus, the equilibrium between the X55–X55 and 5BSL3.2–X55 kissing complexes is completely shifted toward the more stable X55–X55 homodimer duplex in the presence of the core 2BD peptide, resulting in the release of free monomeric 5BSL3.2. These results strongly suggest that X55 could function as a molecular switch in the HCV life cycle, wherein in the absence of the core protein the replication favorable interaction between 5BSL3.2–X55 is preferred, whereas in the presence of the core protein, X55 forms the more stable X55–X55 duplex disrupting the replication favorable X55–5BSL3.2 heterodimer.
To obtain quantitative information about the X55–5BSL3.2 kissing complex, we used a fluorescence spectroscopy binding assay, in which the highly fluorescent purine analog 2-AP replaced the adenine at position 21 in the apical loop of the 5BSL3.2 sequence UCACAGC (position 9283 in the full-length HCV genome) (circled in Figure 1B). The 2-AP steady-state fluorescence is very sensitive to its microenvironment, being quenched by stacking interactions (35–37). Thus, when a kissing complex is formed by X55 RNA with the apical loop of 5BSL3.2 in the presence of MgCl2, the steady-state fluorescence of the 2-AP reporter will be quenched due to the change in its microenvironment from a single-stranded loop nucleotide to a base-paired stacked nucleotide in the kissing complex. Nanomolar increments of wild-type X55 RNA in the range 0–250 nM were titrated into a fixed concentration of the 2-AP-labeled 5BSL3.2. However, since X55 RNA could also form homodimers in the presence of MgCl2, we employed TBM native gel electrophoresis to assess the X55–X55 dimer formation at the low nanomolar concentrations used in the fluorescence spectroscopy assay. As seen in Supplementary Figure S2, no X55–X55 dimer was present when X55 and 5BSL3.2 were mixed in an equimolar ratio at the concentrations of 50, 100 and 300 nM, which span the range of concentrations used in the fluorescence spectroscopy assay (Supplementary Figure S2, lanes 3, 6 and 9).
The titration of the 2-AP-labeled 5BSL3.2 with nanomolar increments of the complementary unlabeled X55 RNA in the presence of 5 mM MgCl2 resulted in a decrease in the steady-state 2-AP fluorescence emission at 371 nm (Figure 4C, black squares), due to the base-stacking interactions arising when the 2-AP in 5BSL3.2 (position 9283) base pairs with the uracil at position 9544 in the unlabeled X55 RNA. A dissociation constant of 42 ± 6 nM was determined for the kissing complex formed by X55 and 5BSL3.2 by fitting the binding curve (black squares) illustrated in Figure 4C with Equation (1) (see ‘Materials and Methods’ section). In a control experiment, we showed that when the 2-AP-labeled 5BSL3.2 is titrated with X55 RNA in the absence of MgCl2, no 2-AP steady-state fluorescence quenching is observed (Figure 4C, black circles). The X55–5BSL3.2 kissing complex has a much lower dissociation constant (Kd = 42 ± 6 nM) than that of the X55–X55 homodimeric kissing complex (Kd = 318 ± 58) reported in our earlier study (28). This result is consistent with our native gel electrophoresis results, which indicated that in the absence of the core protein, the X55–5BSL3.2 kissing complex is formed in higher percentage than the X55–X55 kissing complex (Supplementary Figure S1C). To confirm the specificity of the fluorescence spectroscopy assay, in a second control experiment we titrated the 2-AP-labeled 5BSL3.2 with the 37-nt RNA IIId derived from the HCV IRES (Figure 4C, black triangles). IIId has complementarity for the internal loop of 5BSL3.2, but not for its apical loop where the 2-AP reporter is located. As expected, no significant quenching was observed for the 2-AP steady-state fluorescence.
Taken together, these results indicate that X RNA exists in more than one conformation and can engage in both X55–X55 homodimer interactions as well as in X55–5BSL3.2 kissing complex interactions. The Bartenschlager group had proposed that the interaction of X55 with 5BSL3.2 may be mediated by the NS5B protein binding (23); however, we demonstrate in this study that the initiation of this X55–5BSL3.2 complex is independent of chaperoning proteins. Nevertheless, it could be possible that the NS5B protein plays a role in stabilizing the already initiated 5BSL3.2–X55 kissing complex and this in turn could serve as a template for negative strand synthesis. Cheng and co-workers had earlier reported strong binding of the NS5B protein to the NS5B-coding RNA (38) and later Lee et al. (39) demonstrated that 5BSL3.2 was the primary determinant of this interaction. Our assumption that X55–5BSL3.2 serves as a template for negative RNA synthesis is further supported by the findings that the 3′-UTR by itself was a weak template in in vitro transcription, but when 44 nt of 3′-UTR were combined with 430 nt from the 3′-end of NS5B coding region (which includes the 5BSL3.2 sequence), a highly efficient template was obtained which yielded product equivalent to the size of input RNA (40).
5BSL3.2 engages in long-range kissing interactions with the IIId domain located in the 5′-UTR IRES and with the upstream coding region Alt
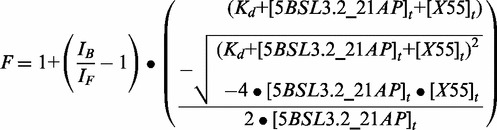
Next we performed native gel electrophoresis experiments to analyze the proposed interactions of the 5BSL3.2 highly conserved GCCCG internal loop motif with the Alt sequence and the IIId domain, respectively (Figure 1D and E). Similarly as described above, we have performed TBM gels in the presence of as low as 0.5 mM Mg2+ ions and TBE gels in which these ions are chelated by EDTA. Figure 5A and B shows the interactions between full-length 5BSL3.2 and IIId in a TBE and TBM gel, respectively. As expected, both IIId (37 nt) and 5BSL3.2 (48 nt) migrate as monomers (Figure 5A and B, lanes 1 and 2); however, the IIId domain does not migrate according to its size in a native gel, most likely due to the presence of an E-loop motif in its structure (41). The formation of the E-loop motif is independent on the presence of Mg2+ ions (22,41), hence the same migration pattern is observed for IIId in the TBE gel. However, in a control denaturing gel, IIId migrates according to its size (Supplementary Figure S3). A higher molecular weight species is present in the TBM gel (Figure 5B, lane 3), corresponding to the 5BSL3.2–IIId complex; however, this complex dissociates in the TBE gel (Figure 5A, lane 3), indicating that magnesium is required for its stabilization. This results strongly supports the proposal that 5BSL3.2 engages in kissing interactions with the IIId domain (22).
Figure 5.
(A) 12% TBE gel and (B) 12% TBM gel. The RNA samples (15 µM) were prepared similarly for both gels, being pre-incubated in the presence of 0.5 mM MgCl2. Lane 1: IIId RNA (37 nt); Lane 2: 5BSL3.2 RNA (48 nt); Lane 3: 5BSL3.2 and IIId RNAs in a 1:1 ratio; lane 4: 5BSL3.2 and IIId RNAs in a 1:1 ratio, which following incubation were treated with core 2BD and proteinase K. The gels were visualized by UV shadowing at 254 nm.
To test whether the metastable magnesium-dependent 5BSL3.2–IIId kissing complex gets converted to a stable duplex in the presence of a chaperoning protein, we treated the 5BSL3.2–IIId kissing complex (in the presence of magnesium) with the HCV chaperoning core 2BD peptide and analyzed it in TBE and TBM gels (Figure 5A and B, lane 4). Since they have an identical size, the kissing complex conformation cannot be distinguished from the duplex conformation in a TBM gel. However, in a TBE gel, the 5BSL3.2–IIId kissing complex whose formation is dependent on Mg2+ will dissociate into monomers, whereas the duplex which is Mg2+ independent will remain intact. As seen in Figure 5A, lane 4, the core treated 5BSL3.2–IIId kissing complex dissociates into monomers in the absence of Mg2+, indicating that the core peptide does not facilitate its conversion to the stable duplex conformation.
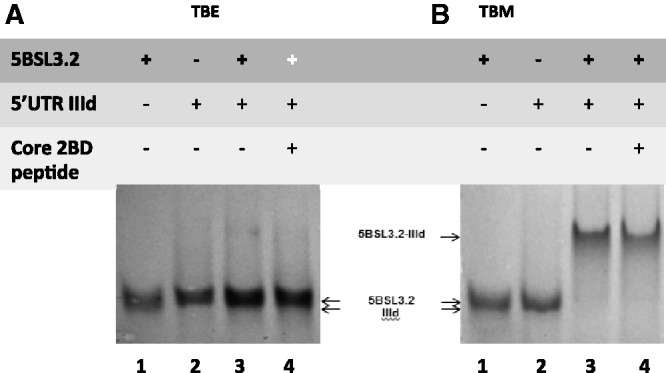
In a similar manner using TBM and TBE native gel electrophoresis, we analyzed the proposed interactions between 5BSL3.2 and Alt, the sequence located 200 nt upstream from it (20). The Alt sequence used in this study is 31 nt long and contains the invariant sequence CGGGC, proposed to bind to the GCCCG internal loop of 5BSL3.2 (20). In a TBM gel (Figure 6A, lanes 1 and 2), both free Alt and 5BSL3.2 RNAs migrate as monomers, whereas when mixed in equimolar ratios they form the 5BSL3.2–Alt complex (Figure 6A, lane 3). However, this complex dissociates in the TBE gel (Figure 6B, lane 3), suggesting that the 5BSL3.2–Alt complex is mediated by magnesium-dependent kissing interactions.
Figure 6.
(A) 12% TBM gel and (B) 12% TBE gel. The RNA samples (15 µM) were prepared the same for both gels, being pre-incubated in the presence of 0.5 mM MgCl2. Lane 1: Alt RNA (31 nt); lane 2: 5BSL3.2 RNA (48 nt); lane 3: 5BSL3.2 and Alt RNAs in a 1:1 ratio. The gels were visualized by UV shadowing at 254 nm.
To confirm that 5BSL3.2 participates in the kissing interactions with IIId and Alt by using its internal bulge, control experiments were performed in which we used either a 24-nt 5BSL3.2 containing only the apical stem–loop (5BSL3.2_sh, Supplementary Figure S4A) or a 38-nt 5BSL3.2 (5BSL3.2_TRUNC, Supplementary Figure S4B), which contains both the apical loop and the internal loop sequences. Although the GCCCG sequence is present in 5BSL3.2_TRUNC, it does not adopt an internal loop structure due to alternative base pairing caused by the absence of the last 5 bp in the lower stem region.
As seen in Supplementary Figure S4C left, in a TBM gel IIId and 5BSL3.2_sh migrate as monomers (lanes 1 and 2). When an equimolar ratio of 5BSL3.2_sh and IIId is mixed (Supplementary Figure S4C left, lane 3), no higher molecular weight complex is observed, indicating that IIId is unable to interact with 5BSL3.2_sh since it lacks the complementary internal bulge motif GCCCG. Similarly, IIId and 5BSL3.2_TRUNC migrate as monomers in a TBM gel (Supplementary Figure S4C right, lanes 1 and 2) and no complex species are present when they are mixed in a 1:1 ratio (Supplementary Figure S4C right, lane 3), indicating that in the absence of the 5BSL3.2_TRUNC internal loop structure, IIId cannot interact with it. These results are in agreement with earlier studies demonstrating that the lower duplex of 5BSL3.2 is essential to maintain the structural integrity of this CRE (21,23).
In a similar manner, to evaluate whether 5BSL3.2_sh and 5BSL3.2_TRUNC can interact with Alt, their mixtures were examined in a TBM gel (Supplementary Figure S4D, left and right). No complex species are observed when Alt is mixed in a 1:1 ratio with either 5BSL3.2_sh (Supplementary Figure S4D; left, lane 3) or with 5BSL3.2_TRUNC (Supplementary Figure 4D; right, lane 3), whereas as expected, free Alt and 5BSL3.2_sh and 5BSL3.2_TRUNC migrate as monomers (lanes 1 and 2). Taken together, these results clearly indicate that 5BSL3.2 requires its internal bulge loop to engage in kissing interactions with either the IIId domain, or with Alt.
These experiments, which used the wild-type sequences of the proposing interacting regions, provide direct in vitro evidence of kissing complex formation between 5BSL3.2–IIId and 5BSL3.2–Alt. Although very little information is available on the Alt sequence (20), there are many studies on IIId and its interactions. NMR spectroscopy studies of the IIId structure showed that its folding is independent of magnesium ions (41). The loop E motif of IIId has been predicted to recruit several cellular proteins (42) and to be responsible for positioning the other domains of the IRES (43). Most importantly, IIId is part of the larger structural motif that is required for binding of the ribosomal subunits (44). Antisense oligos and aptamers synthesized against IIId strongly inhibited translation in several studies (45), and one of them competed with 40S ribosomal subunit for IIId binding (46). Thus, at least one of the roles of IIId is to regulate the translational machinery. Our results confirm that IIId participates in long-range interactions with the highly conserved internal loop of 5BSL3.2 in the coding region and show direct evidence that these are mediated via a kissing complex that cannot be converted to an extended duplex by the core protein, despite the base pair potential due to sequence complementarity.
IIId and Alt compete for kissing interactions with the internal loop of 5BSL3.2
Next, we obtained quantitative data on the 5BSL3.2–IIId and 5BSL3.2–Alt interactions by fluorescence spectroscopy, by using a 5BSL3.2 construct in which the guanine at position 35 located in the 5BSL3.2 internal bulge has been replaced with the fluorophore 6MI. 6MI is a guanosine analog, whose steady-state fluorescence is very sensitive to its microenvironment, being quenched upon its involvement in base pairing (47). The titration of the 6MI-labeled 5BSL3.2 with nanomolar increments of the complementary unlabeled construct, either IIId or Alt, resulted in a decrease in the steady-state 6-MI fluorescence emission at 430 nm due to the base-stacking interactions arising when the 6MI in the labeled RNA (5BSL3.2_35_6MI) and the cytosine or uracil in the unlabeled complementary RNA (IIId or Alt) base pair. Figure 7A (black squares) shows the binding curve of IIId to 5BSL3.2_35_6MI that was fitted with Equation (1) (see ‘Materials and Methods’ section) to determine a dissociation constant of 46 ± 2 nM. The Berzal–Herranz group reported a dissociation constant of 49.5 nM for the 5BSL3.2–IIId complex using a larger fragment of the HCV 5′-UTR. The Kd value we determined for the complex formed by the isolated 5BSL3.2 and IIId stem–loop is in close agreement, suggesting that IIId interacts with 5BSL3.2 in a similar manner, when it is isolated or in the context of the larger 5′-UTR.
Figure 7.

Fluorescence spectroscopy. (A) 5BSL3.2_6MI in 1 mM cacodylic acid pH 6.5 was titrated with increasing concentrations of IIId RNA in the presence (black squares) or absence (black circles) of 5 mM MgCl2 as well as with X55 RNA in the presence of 5 mM MgCl2 (black triangles). (B) Similarly, 5BSL3.2_6MI in 1 mM cacodylic acid pH 6.5 was titrated with increasing concentrations of Alt RNA in the presence (black squares) or absence (black circles) of 5 mM MgCl2. The dissociation constants of the 5BSL3.2–IIId (46 ± 2 nM) and of the 5BSL3.2–Alt (34 ± 5 nM) kissing complexes were determined by fitting their corresponding binding curve (black squares) to Equation (1) in which 5BSL3.1_21AP was replaced by 5BSL3.2_6MI and X55 was replaced by IIId or Alt RNA, respectively. (C) 5BSL3.2_21AP in 1 mM cacodylic acid pH 6.5 containing 5 mM MgCl2 was first pre-incubated in a 1:1 ratio with Alt RNA, after which it was titrated with increasing concentrations of X55 RNA. The dissociation constant of the 5BSL3.2–X55 RNA kissing complex in the presence of Alt RNA (80 ± 3 nM) was determined by fitting the binding curve to Equation (1).
Similarly, a value of 34 ± 5 nM was obtained for the dissociation constant of the 5BSL3.2–Alt kissing complex, by fitting their binding curve illustrated in Figure 7B (black squares) with Equation (1). In control experiments we show that in the absence of magnesium ions, neither IIId (Figure 7A, black circles) nor Alt (Figure 7B, black circles) interact with 5BSL3.2, consistent with the results of the TBE native gel electrophoresis experiments (Figures 5A and 6B, respectively). In a second control experiment, to confirm the specificity of the fluorescence spectroscopy assay, we have titrated the 6MI-labeled 5BSL3.2 with X55 RNA, in the presence on 5 mM MgCl2. X55 RNA is not predicted to bind the 5BSL3.2 internal loop, and, as expected, no significant quenching was observed for the 6MI steady-state fluorescence (Figure 7A, black triangles). The 5BSL3.2–IIId interaction is predicted to be important in regulating translation (22,48), whereas the 5BSL3.2–Alt interaction is essential for replication (20). Since the isolated IIId and Alt sequences have very similar affinities for the 5BSL3.2 internal loop, it is possible that the deciding factor as to which interaction will occur first would depend on the accessibility of this internal loop to either IIId or Alt. Both the interactions of the 5BSL3.2 terminal loop with X RNA and its internal loop interactions with Alt have been identified as essential for replication (20,22). Thus, we inquired if the 5BSL3.2 internal bulge loop interactions with Alt have any impact on the 5BSL3.2 terminal loop interactions with X55. We conducted fluorescence spectroscopy studies, in which Alt was first incubated with the fluorescent 5BSL3.2_21AP, in an equimolar ratio in the presence of 5 mM MgCl2 to allow the pre-formation of the kissing complex between the internal loop of 5BSL3.2_2-AP (which is not fluorescently labeled in the internal loop) and Alt. Subsequently, nanomolar increments of X55 were titrated into the 5BSL3.2_21AP-Alt mixture and the change in the steady-state fluorescence of 2-AP, which reports on the formation of the kissing complex involving the 5BSL3.2 apical loop and X55, was monitored. As shown in Figure 7C in the presence of Alt, a Kd value of 80 ± 3 nM was determined for the 5BSL3.2–X55 kissing complex, which is almost double the Kd value for 5BSL3.2–X55 interaction measured in the absence of Alt, indicating some local steric hindrance posed by Alt. Since these studies were performed with truncated RNAs, it is possible that such steric hindrance effects might be amplified in the context of the entire HCV genome.
Possible involvement of RNA–RNA molecular switches in the HCV life cycle
Several examples of long-range RNA–RNA interactions either mediated directly by base pairing or indirectly by protein chaperones exist among flaviviruses (49,50). In the case of HCV, we provide biophysical evidence for the existence of three essential kissing interactions mediated by base pairing in the absence of any chaperoning protein, all of which involve the highly conserved 5BSL3.2 element: X55–5BSL3.2, 5BSL3.2–IIId and 5BSL3.2–Alt. The dissociation constant for the homodimer X55–X55 kissing complex was determined to be 318 ± 58 nM in our earlier study (28). In this study, we determined the dissociation constants of the X55–5BSL3.2 complex, 42 ± 6 nM (Figure 4C), of the 5BSL3.2–IIId complex, 46 ± 2 nM (Figure 7A) and of the 5BSL3.2–Alt complex, 34 ± 5 nM (Figure 7B). Based on this information, we envision a model according to which the 5BSL3.2 CRE could swap partners to participate in several molecular switches that might control the progression through the HCV life cycle (Figure 8).
Figure 8.

HCV model. (I) Co-existing conformation 1 and conformation 2 of X RNA. (II) Binding of PCBP2 (orange circle) to the 5′-UTR; binding of PTB (purple circle) to the 3′-UTR, which might stabilize conformation 1 of X RNA. (III) IIId interactions with the 5BSL3.2 internal loop bring the 5′-UTR in the closer proximity of the 3′-UTR. (IV) PTB and PCBP2 oligomerization and the binding of the La protein might promote the formation of a protein bridge that will stabilize the circularization of the HCV genome. (V) Upon the formation of this protein bridge, IIId could be released from its interactions with the internal loop of 5BSL3.2 and can be bound by the ribosome allowing for the translation initiation. (VI) Upon the accumulation of the viral proteins, both PTB and PCBP2 could be cleaved by a viral protease, leading to their loss of interaction with the La protein and disruption of the protein bridge, which in turn results in the linearization of the HCV genome. (VII) The X RNA in conformation 1 could interact now with the terminal loop of 5BSL3.2, and Alt with the internal loop of 5BSL3.2, which will set the platform for replication initiation. Upon accumulation of sufficient HCV genomes X RNA could homodimerize through a kissing complex intermediate that is converted to a stable duplex conformation by the core protein. Upon the duplex formation, X RNA can no longer engage in interactions with the terminal loop of 5BSL3.2, the replication might be inhibited, switching to the packaging mode.
The magnesium concentration within the HCV virion is not known; however, we speculate that once the viral genome is exposed to the intracellular magnesium concentration of 1–5 mM, its 3′-UTR X RNA will fold into two conformations that co-exist in equilibrium: conformation 1, which favors the intramolecular interactions with 5BSL3.2, and conformation 2, which favors the intermolecular interactions with another genome (Figure 8I). Although in the TBM native gel electrophoresis (Figure 3A) conformation 2 is predominant, it is unlikely that the HCV genome will dimerize upon cell entry, due to the low copy number of genomes. It is also possible that the viral RNA genome will engage in interactions with other cellular components, which might affect the equilibrium between the X RNA conformations. Several cellular proteins have been shown to interact with the HCV RNA, having binding sites either within the 5′-UTR, 3′-UTR or on both (51,52). Among these, the polypyrimidine tract-binding protein (PTB) has been shown to have a high-affinity binding site within the HCV 3′-UTR (Figure 8II) and a weaker binding site within the 5′-UTR (53–55). Similarly, the poly C-binding protein 2 (PCBP2), a member of the cellular heterogeneous nuclear ribonucleoprotein (hnRNP) family, binds to the stem–loop 1 within the 5′-UTR (Figure 8, II) and also has a weaker affinity for the 3′-UTR, being implicated in the translation of the viral proteins, and in HCV replication (56). The La protein (Lupus antigen) has also been shown to bind to the HCV 5′-UTR (57). These proteins might play a role in the stabilization of various RNA secondary and tertiary structures that would facilitate the viral translation, replication and/or packaging. PTB has been shown to bind to the stem–loops SL2 and SL3 within the 3′-UTR X RNA (55), and by stabilizing conformation 1, could shift the equilibrium between the two X RNA conformations in favor of conformation 1. When in conformation 1, X RNA is able to interact with the terminal loop of 5BSL3.2; however, it is not clear if these interactions are still possible in the presence of PTB.
The IIId domain located within the HCV IRES could interact with the internal loop of 5BSL3.2 located at the end of the coding region, bringing the 5′-UTR close to the 3′-UTR (Figure 8III), and if interactions between X RNA and the terminal loop of 5BSL3.2 are still possible in the presence of PTB, 5BSL3.2, would in essence act as a scaffold that brings together the 5′- and 3′-UTRs. As mentioned above, PTB, PCBP2 and La proteins have binding sites within the 5′-UTR and 3′-UTR; thus, when the two ends of the genome are brought in close proximity by long-range RNA–RNA interactions involving 5BSL3.2, a protein bridge could promote and stabilize the genomic RNA circularization. Both PTB and PCBP2 can oligomerize, and recently it has been shown that a peptide comprising the La protein amino acids 11–28 interacts with both PCBP2 and PTB (58). Thus, it is plausible that such a protein bridge could form, and upon its formation, the 5BSL3.2 could be released from its interactions with IIId and possibly X RNA (Figure 8, IV), allowing the ribosome 40S subunit to engage in interactions with the IIId terminal loop for which it has a strong affinity (59). This would set the stage for the initiation of synthesis of viral proteins (Figure 8V), whose efficiency could be greatly enhanced by the circularization of the HCV genome (49). If possible in the presence of PTB, the X55–5BSL3.2 interactions would be disrupted by the translation machinery. Upon the excessive accumulation of the viral proteins, we speculate that the viral protease NS3 could cleave PCBP2 and/or PTB, similarly to the polio virus case where it has been reported that upon its accumulation, the viral protein 3CDpro cleaves PCBP2 as well as PTB (56,58,60) resulting in the polio genome linearization. Interestingly, only the intact PTB protein containing all of four RNA recognition motifs is able to interact with the 28-amino acid La peptide, but not the two fragments obtained upon PTB cleavage (58). Thus, it is plausible that the cleavage of the PTB and/or PCBP2 proteins by a viral protease could disrupt their interactions with the La protein, altering the postulated 5′–3′ UTR protein bridge and resulting in the linearization of the HCV genome (Figure 8VI). In the presence of an excess of the core protein, the IIId interactions with the ribosomes could be inhibited as the HCV core protein has affinity for the IIId terminal loop (61), resulting in the translation inhibition. X RNA could now form a stable kissing complex with the terminal loop of 5BSL3.2 (Figure 8VII). Additionally, in the absence of the competition by IIId, the Alt sequence located 200 nt upstream of 5BSL3.2 could bind to the 5BSL3.2 internal loop (Figure 8VII). We speculate that this complex arrangement of interactions between the 5BSL3.2 terminal loop and X RNA, 5BSL3.2 internal loop and Alt provides a suitable platform for the HCV RNA-dependent RNA polymerase NS5B, to recognize and bind the viral RNA. Supporting this hypothesis is the finding that among the aptamers tested for binding to NS5B, GC-rich motifs and specifically a CGGG motif aptamer exhibited maximal binding (62). This motif has been identified in the context of the lower stem of 5BSL3.2 (nt 4–7 in Figure 1B, nt 9266–9269 in the HCV genome, Figure 1A), which has been shown to bind to NS5B (62). Moreover, a single mutation in the motif, together with the compensatory mutation that maintains the integrity of the lower stem, has been shown to greatly diminish the affinity of NS5B for the 5BSL3.2 CRE (62). The interactions of 5BSL3.2 with Alt involve the formation of several GC base pairs (specifically the CGGG motif) and thus, it is plausible that the formation of an additional CGGG stem could increase the binding affinity of NS5B. The platform thus set with 5BSL3.2–XRNA and 5BSL3.2–Alt interactions is suitable for replication and upon being stabilized by HCV RNA-dependent RNA polymerase NS5B, replication can be initiated. It has been shown that the HCV replication takes place in ‘membranous webs’ which form in the presence of the NS4B viral protein. When the viral RNA accumulates in sufficient high copy numbers, it could undergo homodimerization via the formation of a kissing complex that is converted to a more stable duplex by the core protein. As a result, X RNA will no longer be available for interactions with 5BSL3.2, greatly reducing the affinity of NS5B for the viral RNA and contributing to replication inhibition. Thus, depending on its interacting partners, X RNA might be part of another molecular switch: promoting replication when it interacts with 5BSL3.2, or inhibiting replication and possibly switching to packaging mode when it interacts with the X RNA of another genome. The exact role of the HCV genomic dimerization is not known; our model proposes that it would function to inhibit the viral replication and possibly promote packaging. However, since according to the current understanding in the field there is a single genome packaged within the virion, further investigations might reveal how the two RNA genomes become monomeric again prior to their packaging into new virions.
In summary, we have provided direct biophysical evidence for the existence of several long-range RNA–RNA interactions between highly conserved regions within the HCV genome, which are mediated via the formation of kissing complexes. We have also formulated a model which proposes the existence of two molecular RNA switches which might orchestrate the progression of the HCV life cycle, one involving the 5BSL3.2 element and the second involving the X RNA region. Although largely supported by our data, this model makes many assumptions that will have to be validated by further studies.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Table 1 and Supplementary Figures 1–3.
FUNDING
Pennsylvania State Health Formula Research (to M.R.M.). Funding for open access charge: Research Grant.
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Shepard CW, Finelli L, Alter MJ. Global epidemiology of hepatitis C virus infection. Lancet Infect. Dis. 2005;5:558–567. doi: 10.1016/S1473-3099(05)70216-4. [DOI] [PubMed] [Google Scholar]
- 2.McCashland T, Watt K, Lyden E, Adams L, Charlton M, Smith AD, McGuire BM, Biggins SW, Neff G, Burton JR, Jr, et al. Retransplantation for hepatitis C: results of a U.S. multicenter retransplant study. Liver Transpl. 2007;13:1246–1253. doi: 10.1002/lt.21322. [DOI] [PubMed] [Google Scholar]
- 3.Afdhal NH. The natural history of hepatitis C. Semin. Liver Dis. 2004;24:3–8. doi: 10.1055/s-2004-832922. [DOI] [PubMed] [Google Scholar]
- 4.Fried MW, Hadziyannis SJ. Treatment of chronic hepatitis C infection with peginterferons plus ribavirin. Semin. Liver Dis. 2004;24:47–54. doi: 10.1055/s-2004-832928. [DOI] [PubMed] [Google Scholar]
- 5.Heathcote J, Main J. Treatment of hepatitis C. J. Viral Hepat. 2005;12:223–235. doi: 10.1111/j.1365-2893.2005.00600.x. [DOI] [PubMed] [Google Scholar]
- 6.Manns MP, Wedemeyer H, Cornberg M. Treating viral hepatitis C: efficacy, side effects, and complications. Gut. 2006;55:1350–1359. doi: 10.1136/gut.2005.076646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Asselah T. Realize the advance in HCV treatment, but remain cautious. J. Hepatol. 2011;55:1457–1460. doi: 10.1016/j.jhep.2011.07.020. [DOI] [PubMed] [Google Scholar]
- 8.Miller RH, Purcell RH. Hepatitis C virus shares amino acid sequence similarity with pestiviruses and flaviviruses, as well as members of two plant virus supergroups. Proc. Natl Acad. Sci. USA. 1990;87:2057–2061. doi: 10.1073/pnas.87.6.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Behrens SE, Tomei L, De Francesco R. Identification and properties of the RNA-dependent RNA polymerase of hepatitis C virus. EMBO J. 1996;15:12–22. [PMC free article] [PubMed] [Google Scholar]
- 10.Holland J, Spindler K, Horodyski F, Grabau E, Nichol S, VandePol S. Rapid evolution of RNA genomes. Science. 1982;215:1577–1585. doi: 10.1126/science.7041255. [DOI] [PubMed] [Google Scholar]
- 11.Simmonds P, Holmes EC, Cha TA, Chan SW, McOmish F, Irvine B, Beall E, Yap PL, Kolberg J, Urdea MS. Classification of hepatitis C virus into six major genotypes and a series of subtypes by phylogenetic analysis of the NS-5 region. J. Gen. Virol. 1993;74:2391–2399. doi: 10.1099/0022-1317-74-11-2391. [DOI] [PubMed] [Google Scholar]
- 12.Shi ST, Lai MM. Hepatitis C viral RNA: challenges and promises. Cell Mol. Life Sci. 2001;58:1276–1295. doi: 10.1007/PL00000939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cohen J. The scientific challenge of hepatitis C. Science. 1999;285:26–30. doi: 10.1126/science.285.5424.26. [DOI] [PubMed] [Google Scholar]
- 14.Tsukiyama-Kohara K, Iizuka N, Kohara M, Nomoto A. Internal ribosome entry site within hepatitis C virus RNA. J. Virol. 1992;66:1476–1483. doi: 10.1128/jvi.66.3.1476-1483.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blight KJ, Rice CM. Secondary structure determination of the conserved 98-base sequence at the 3′ terminus of hepatitis C virus genome RNA. J. Virol. 1997;71:7345–7352. doi: 10.1128/jvi.71.10.7345-7352.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Friebe P, Bartenschlager R. Genetic analysis of sequences in the 3′ nontranslated region of hepatitis C virus that are important for RNA replication. J. Virol. 2002;76:5326–5338. doi: 10.1128/JVI.76.11.5326-5338.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kolykhalov AA, Feinstone SM, Rice CM. Identification of a highly conserved sequence element at the 3′ terminus of hepatitis C virus genome RNA. J. Virol. 1996;70:3363–3371. doi: 10.1128/jvi.70.6.3363-3371.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tanaka T, Kato N, Cho MJ, Shimotohno K. A novel sequence found at the 3′ terminus of hepatitis C virus genome. Biochem. Biophys. Res. Comm. 1995;215:744–749. doi: 10.1006/bbrc.1995.2526. [DOI] [PubMed] [Google Scholar]
- 19.Yi M, Lemon SM. 3′ nontranslated RNA signals required for replication of hepatitis C virus RNA. J. Virol. 2003;77:3557–3568. doi: 10.1128/JVI.77.6.3557-3568.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diviney S, Tuplin A, Struthers M, Armstrong V, Elliott RM, Simmonds P, Evans DJ. A hepatitis C virus cis-acting replication element forms a long-range RNA-RNA interaction with upstream RNA sequences in NS5B. J. Virol. 2008;82:9008–9022. doi: 10.1128/JVI.02326-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.You S, Stump DD, Branch AD, Rice CM. A cis-acting replication element in the sequence encoding the NS5B RNA-dependent RNA polymerase is required for hepatitis C virus RNA replication. J. Virol. 2004;78:1352–1366. doi: 10.1128/JVI.78.3.1352-1366.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Romero-Lopez C, Berzal-Herranz A. A long-range RNA-RNA interaction between the 5' and 3′ ends of the HCV genome. RNA. 2009;15:1740–1752. doi: 10.1261/rna.1680809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Friebe P, Boudet J, Simorre JP, Bartenschlager R. Kissing-loop interaction in the 3′ end of the hepatitis C virus genome essential for RNA replication. J. Virol. 2005;79:380–392. doi: 10.1128/JVI.79.1.380-392.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith DB, Simmonds P. Characteristics of nucleotide substitution in the hepatitis C virus genome: constraints on sequence change in coding regions at both ends of the genome. J. Molec. Evol. 1997;45:238–246. doi: 10.1007/pl00006226. [DOI] [PubMed] [Google Scholar]
- 25.Tuplin A, Wood J, Evans DJ, Patel AH, Simmonds P. Thermodynamic and phylogenetic prediction of RNA secondary structures in the coding region of hepatitis C virus. RNA. 2002;8:824–841. doi: 10.1017/s1355838202554066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hofacker IL, Fekete M, Flamm C, Huynen MA, Rauscher S, Stolorz PE, Stadler PF. Automatic detection of conserved RNA structure elements in complete RNA virus genomes. Nucleic Acids Res. 1998;26:3825–3836. doi: 10.1093/nar/26.16.3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.You S, Rice CM. 3′ RNA elements in hepatitis C virus replication: kissing partners and long poly(U) J. Virol. 2008;82:184–195. doi: 10.1128/JVI.01796-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shetty S, Kim S, Shimakami T, Lemon SM, Mihailescu MR. Hepatitis C virus genomic RNA dimerization is mediated via a kissing complex intermediate. RNA. 2010;16:913–925. doi: 10.1261/rna.1960410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ivanyi-Nagy R, Kanevsky I, Gabus C, Lavergne JP, Ficheux D, Penin F, Fosse P, Darlix JL. Analysis of hepatitis C virus RNA dimerization and core-RNA interactions. Nucleic Acids Res. 2006;34:2618–2633. doi: 10.1093/nar/gkl240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laughrea M, Jette L. Kissing-loop model of HIV-1 genome dimerization: HIV-1 RNAs can assume alternative dimeric forms, and all sequences upstream or downstream of hairpin 248-271 are dispensable for dimer formation. Biochemistry. 1996;35:1589–1598. doi: 10.1021/bi951838f. [DOI] [PubMed] [Google Scholar]
- 32.Takahashi KI, Baba S, Chattopadhyay P, Koyanagi Y, Yamamoto N, Takaku H, Kawai G. Structural requirement for the two-step dimerization of human immunodeficiency virus type 1 genome. RNA. 2000;6:96–102. doi: 10.1017/s1355838200991635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baba S, Takahashi K, Nomura Y, Noguchi S, Koyanagi Y, Yamamoto N, Takaku H, Kawai G. Conformational change of dimerization initiation site of HIV-1 genomic RNA by NCp7 or heat treatment. Nucleic Acids Res. 2001;1:155–156. doi: 10.1093/nass/1.1.155. [DOI] [PubMed] [Google Scholar]
- 34.Mujeeb A, Ulyanov NB, Georgantis S, Smirnov I, Chung J, Parslow TG, James TL. Nucleocapsid protein-mediated maturation of dimer initiation complex of full-length SL1 stem loop of HIV-1: sequence effects and mechanism of RNA refolding. Nucleic Acids Res. 2007;35:2026–2034. doi: 10.1093/nar/gkm097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tanpure AA, Srivatsan SG. A microenvironment-sensitive fluorescent pyrimidine ribonucleoside analogue: synthesis, enzymatic incorporation, and fluorescence detection of a DNA abasic site. Chemistry. 2011;17:12820–12827. doi: 10.1002/chem.201101194. [DOI] [PubMed] [Google Scholar]
- 36.Zhao C, Marino JP. Synthesis of HIV-1 Psi-site RNA sequences with site specific incorporation of the fluorescent base analog 2-aminopurine. Tetrahedron. 2007;63:3575–3584. doi: 10.1016/j.tet.2006.08.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee HW, Briggs KT, Marino JP. Dissecting structural transitions in the HIV-1 dimerization initiation site RNA using 2-aminopurine fluorescence. Methods. 2009;49:118–127. doi: 10.1016/j.ymeth.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheng JC, Chang MF, Chang SC. Specific interaction between the hepatitis C virus NS5B RNA polymerase and the 3′ end of the viral RNA. J. Virol. 1999;73:7044–7049. doi: 10.1128/jvi.73.8.7044-7049.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee H, Shin H, Wimmer E, Paul AV. Cis-acting RNA signals in the NS5B C-terminal coding sequence of the hepatitis C virus genome. J. Virol. 2004;78:10865–10877. doi: 10.1128/JVI.78.20.10865-10877.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yamashita T, Kaneko S, Shirota Y, Qin W, Nomura T, Kobayashi K, Murakami S. RNA-dependent RNA polymerase activity of the soluble recombinant hepatitis C virus NS5B protein truncated at the C-terminal region. J. Biol. Chem. 1998;273:15479–15486. doi: 10.1074/jbc.273.25.15479. [DOI] [PubMed] [Google Scholar]
- 41.Klinck R, Westhof E, Walker S, Afshar M, Collier A, Aboul-Ela F. A potential RNA drug target in the hepatitis C virus internal ribosomal entry site. RNA. 2000;6:1423–1431. doi: 10.1017/s1355838200000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yen JH, Chang SC, Hu CR, Chu SC, Lin SS, Hsieh YS, Chang MF. Cellular proteins specifically bind to the 5'-noncoding region of hepatitis C virus RNA. Virology. 1995;208:723–732. doi: 10.1006/viro.1995.1204. [DOI] [PubMed] [Google Scholar]
- 43.Jubin R, Vantuno NE, Kieft JS, Murray MG, Doudna JA, Lau JY, Baroudy BM. Hepatitis C virus internal ribosome entry site (IRES) stem loop IIId contains a phylogenetically conserved GGG triplet essential for translation and IRES folding. J. Virol. 2000;74:10430–10437. doi: 10.1128/jvi.74.22.10430-10437.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Odreman-Macchioli FE, Tisminetzky SG, Zotti Mm, Baralle FE, Buratti E. Influence of correct secondary and tertiary RNA folding on the binding of cellular factors to the HCV IRES. Nucleic Acids Res. 2000;28:875–885. doi: 10.1093/nar/28.4.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tallet-Lopez B, Aldaz-Carroll L, Chabas S, Dausse E, Staedel C, Toulme JJ. Antisense oligonucleotides targeted to the domain IIId of the hepatitis C virus IRES compete with 40S ribosomal subunit binding and prevent in vitro translation. Nucleic Acids Res. 2003;31:734–742. doi: 10.1093/nar/gkg139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kikuchi K, Umehara T, Fukuda K, Kuno A, Hasegawa T, Nishikawa S. A hepatitis C virus (HCV) internal ribosome entry site (IRES) domain III-IV-targeted aptamer inhibits translation by binding to an apical loop of domain IIId. Nucleic Acids Res. 2005;33:683–692. doi: 10.1093/nar/gki215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Narayanan M, Kodali G, Singh V, Xing Y, Hawkins ME, Stanley RJ. Differential fluorescence quenching of fluorescent nucleic acid base analogues by native nucleic acid monophosphates. J. Phys. Chem. 2010;114:5953–5963. doi: 10.1021/jp1011507. [DOI] [PubMed] [Google Scholar]
- 48.Romero-Lopez C, Berzal-Herranz A. The functional RNA domain 5BSL3.2 within the NS5B coding sequence influences hepatitis C virus IRES-mediated translation. Cell. Mol. Life Sci. 2011;69:103–113. doi: 10.1007/s00018-011-0729-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alvarez DE, Lodeiro MF, Luduena SJ, Pietrasanta LI, Gamarnik AV. Long-range RNA-RNA interactions circularize the dengue virus genome. J. Virol. 2005;79:6631–6643. doi: 10.1128/JVI.79.11.6631-6643.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thurner C, Witwer C, Hofacker IL, Stadler PF. Conserved RNA secondary structures in Flaviviridae genomes. J. Gen. Virol. 2004;85:1113–1124. doi: 10.1099/vir.0.19462-0. [DOI] [PubMed] [Google Scholar]
- 51.Hwang SB, Lo SY, Ou JH, Lai MM. Detection of cellular proteins and viral core protein interacting with the 5' untranslated region of hepatitis C virus RNA. J. Biomed. Sci. 1995;2:227–236. doi: 10.1007/BF02253382. [DOI] [PubMed] [Google Scholar]
- 52.Fields BN, Knipe DM, Howley PM. Hepatitis C viruses. Fields Virol. 1996:1035–1058. [Google Scholar]
- 53.Ali N, Siddiqui A. Interaction of polypyrimidine tract-binding protein with the 5' noncoding region of the hepatitis C virus RNA genome and its functional requirement in internal initiation of translation. J. Virol. 1995;69:6367–6375. doi: 10.1128/jvi.69.10.6367-6375.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ito T, Lai MM. An internal polypyrimidine-tract-binding protein-binding site in the hepatitis C virus RNA attenuates translation, which is relieved by the 3′-untranslated sequence. Virology. 1999;254:288–296. doi: 10.1006/viro.1998.9541. [DOI] [PubMed] [Google Scholar]
- 55.Tsuchihara K, Tanaka T, Hijikata M, Kuge S, Toyoda H, Nomoto A, Yamamoto N, Shimotohno K. Specific interaction of polypyrimidine tract-binding protein with the extreme 3′-terminal structure of the hepatitis C virus genome, the 3′X. J. Virol. 1997;71:6720–6726. doi: 10.1128/jvi.71.9.6720-6726.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang L, Jeng KS, Lai MM. PolyC-binding protein 2 interacts with sequences required for viral replication in the hepatitis C virus (HCV) 5′-untranslated region and directs HCV RNA replication through circularizing the viral genome. J. Virol. 2011;85:7954–7964. doi: 10.1128/JVI.00339-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ali N, Pruijn GJ, Kenan DJ, Keene JD, Siddiqui A. Human La antigen is required for the hepatitis C virus internal ribosome entry site-mediated translation. J. Biol. Chem. 2000;275:27531–27540. doi: 10.1074/jbc.M001487200. [DOI] [PubMed] [Google Scholar]
- 58.Fontanes V, Raychaudhuri S, Dasgupta A. A cell-permeable peptide inhibits hepatitis C virus replication by sequestering IRES transacting factors. Virology. 2009;394:82–90. doi: 10.1016/j.virol.2009.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Babaylova E, Graifer D, Malygin A, Stahl J, Shatsky I, Karpova G. Positioning of subdomain IIId and apical loop of domain II of the hepatitis C IRES on the human 40S ribosome. Nucleic Acids Res. 2009;37:1141–1151. doi: 10.1093/nar/gkn1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Perera R, Daijogo S, Walter BL, Nguyen JH, Semler BL. Cellular protein modification by poliovirus: the two faces of poly(rC)-binding protein. J. Virol. 2007;81:8919–8932. doi: 10.1128/JVI.01013-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shimoike T, Koyama C, Murakami K, Suzuki R, Matsuura Y, Miyamura T, Suzuki T. Down-regulation of the internal ribosome entry site (IRES)-mediated translation of the hepatitis C virus: critical role of binding of the stem-loop IIId domain of IRES and the viral core protein. Virology. 2006;345:434–445. doi: 10.1016/j.virol.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 62.Kanamori H, Yuhashi K, Uchiyama Y, Kodama T, Ohnishi S. In vitro selection of RNA aptamers that bind the RNA-dependent RNA polymerase of hepatitis C virus: a possible role of GC-rich RNA motifs in NS5B binding. Virology. 2009;388:91–102. doi: 10.1016/j.virol.2009.02.032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.