


Abstract
Sequence-specific DNA-binding proteins must quickly and reliably localize specific target sites on DNA. This search process has been well characterized for monomeric proteins, but it remains poorly understood for systems that require assembly into dimers or oligomers at the target site. We present a single-molecule study of the target-search mechanism of protelomerase TelK, a recombinase-like protein that is only active as a dimer. We show that TelK undergoes 1D diffusion on non-target DNA as a monomer, and it immobilizes upon dimerization even in the absence of a DNA target site. We further show that dimeric TelK condenses non-target DNA, forming a tightly bound nucleoprotein complex. Together with theoretical calculations and molecular dynamics simulations, we present a novel target-search model for TelK, which may be generalizable to other dimer and oligomer-active proteins.
INTRODUCTION
Many essential cellular processes depend on protein–DNA interactions at specific sequences in the genome. Sequence-specific proteins (SSPs) must quickly and reliably localize target sites that are typically only a few base pairs in length among kilobases of non-target genomic DNA. Several studies of the interaction of SSPs along DNA have revealed some aspects of the mechanism by which target-finding occurs (1–4). According to current models, a protein binds and scans non-target regions of DNA by 1D diffusion facilitated by 3D hopping, until the protein identifies its target sequence (5–7).
Most studies till date have focused on the target search mechanism of a particular class of sequence-specific proteins. Experimental and theoretical studies of target search have mainly considered proteins that are either monomeric [e.g. certain restriction enzymes such as FokI and mismatch repair proteins such as MutH and T4 endonuclease V, DNA repair protein hOgg1 (8–11)], or when oligomeric, pre-assembled in solution [e.g. BbvCl, LacI repressor, EcoRV, EcoRI, Msh2-Msh6, MutS, Mlh1-Pms1, p53 (12–17)]. These examples bind DNA and locate a target site as a single functional unit. Many proteins, however, function exclusively as dimers or oligomers at a target site, but they are monomeric in solution, only assembling into higher-order complexes on DNA. Such proteins are ubiquitous in the cell, and they are involved in a range of cellular functions, including DNA repair, replication, transcription and translation (18–20). Common examples of such proteins include certain recombinases, select type II and III restriction enzymes, transcription factors, integrases, DNA-repair proteins and signal transducers (20–23). Despite the abundance of cellular proteins that must dimerize or oligomerize onto DNA in addition to identifying a target site, little work has been done to understand the complete mechanism by which they localize both their target sequence and protein partner(s). An exception is type III restriction endonucleases (4,24), in which two distinct protein monomers find two separate target sites by a facilitated 1D diffusion and then use sliding to assemble a higher-order complex on the DNA.
Proteins that must assemble higher-order complexes on single target sites must not only locate their DNA target sites but must also find their protein partner(s). This added complexity gives rise to new questions: How do two protein monomers find each other? Is dimerization required for target-site specificity, or do individual monomers hold the capacity to identify a target sequence? Do protein monomers or dimers undergo 1D diffusion? If proteins diffuse in 1D as monomers, what happens if two proteins encounter each other along non-target DNA? Answers to these questions may provide a comprehensive model for the target-search process of dimer-active proteins.
In the prokaryotic organism Klebsiella oxytoca, protelomerase TelK is a protein that is encoded by the lysogenic phage K02 on infection. Unlike the other well-known lysogenic λ phages, whose DNA is integrated into the Escherichia coli chromosome, the phage K02 DNA instead forms a linear plasmid with hairpin ends. Replication of this linear plasmid generates catenated genomes that are resolved by TelK (25). The protein localizes two 56-bp target sequences at opposite poles of the DNA circle replication intermediate, and it generates hairpin-capped ends via site-specific excision, strand exchange and re-ligation. TelK-induced DNA hairpin formation occurs independently of adenosine triphosphate (ATP) or other cofactors such as Mg2+, and TelK is a single-turnover protein (25). TelK shares sequence and structural homology to tyrosine recombinases (Y-recombinases) and type IB topoisomerases (26). The crystal structure of TelK538, a truncation mutant of wild-type TelK, complexed with a 44mer target-DNA substrate shows two tightly bound TelK monomers that form the active dimer oriented head-to-head on the dyad symmetric DNA target site. Dimerization induces a sharp 73° bend in the DNA substrate (26). Although TelK is known to be a monomer in solution, target-sequence activity requires TelK to dimerize at its target site in the correct head-to-head orientation (25), making it an ideal protein for the study of target-search by SSPs that assemble into higher-order complexes on DNA.
To understand the target-site search mechanism for this protein, we studied the interaction of TelK with both DNA lacking and containing the target sequence. Through a combination of single-molecule experiments—including total internal reflection fluorescence microscopy (TIRFM) and optical trapping (27,28)—and theoretical analysis in the form of molecular dynamics (MD) simulations (29) and stochastic simulations, we determined the novel search mechanism TelK uses to identify its DNA target site. As a monomer, TelK undergoes 1D diffusion along non-specific DNA, and it is able to bind to the target site preferentially. There, the target-immobilized monomer waits for a second binding partner to form an active protein complex. Surprisingly, if two monomers coalesce on non-target DNA, they also immobilize and condense DNA. This transient tight-binding is reversible; non-target bound dimers will eventually dissociate into monomers along DNA or into solution. We propose this target-search model for TelK may be applicable to other proteins that are active as dimers or oligomers at DNA target sites.
MATERIALS AND METHODS
DNA synthesis
TIRFM non-target DNA substrate
A 48-kb DNA molecule with biotin at both ends was synthesized by filling in the cos ends of λ-DNA with biotinylated dNTPs. Briefly, 1 µg of λ-DNA (D1501, Promega, Madison, WI, USA) was incubated at room temperature for 10 min with 1 U of DNA Polymerase I Large Klenow Fragment (M0210S, NEB, Ipswich, MA, USA) and 40 µM of dATP, dGTP, dTTP (R0141, R0171 and R0161, respectively; Fermentas, Glen Burnie, MD, USA) and biotinylated dCTP (19518-018, Invitrogen, Carlsbad, CA, USA). The reaction was stopped by heating for 10 min at 75°C and run through a Qiagen polymerase chain reaction (PCR) purification kit (28104, Qiagen, Valencia, CA, USA) with a 50 -μl DNA elution volume.
TIRFM target DNA substrate
The target DNA substrate for use in our TIRFM assays was synthesized based on the 2.9-kb origin pSKN plasmid harbouring the TelK target site, and the 6.9-kb integration vector of Bacillus subtilis, pKSV7 (30), plus an unrelated 4-kb fragment from phage G of Bacillus megaterium (31). These plasmids were built by enlarging the parent pSKN (25) with non-E. coli sequences to 12.9-kb length to span across the TIRFM glass pedestals.
Optical trap non-target DNA substrate
A 3.4-kb DNA molecule with a single biotin and digoxigenin at each end was synthesized by amplifying a segment of the pBR322 DNA plasmid (Fermentas, Glen Burnie, MD, USA). A 5′-biotinylated forward and a 5′-digoxigenated reverse PCR primer (Integrated DNA Technologies, Coralville, IA, USA) was used for amplification along with a high fidelity Phusion PCR kit (F-513S, Finnzymes, Woburn, MA, USA). Subsequent DNA purification was performed with a Qiagen PCR purification kit (28104, Qiagen, Valencia, CA, USA) with a 50-μl DNA elution volume. The purity of the DNA product was confirmed by running an agarose gel.
TIRF microscopy
TIRFM instrument
We used a total internal reflection fluorescence microscope as previously described (32). A spot-fitting algorithm allowed for nanometre-scale localization of the fluorescent spot position, as previously detailed (33).
Quantum dot labelling of TelK
Full-length TelK640 (referred to as TelK in the text) was purified using a previously established protocol (25). This full-length wild-type TelK was used for all TIRF and optical trap experiments. TelK contained a 6-amino acid N-terminal His tag. To label TelK, we incubated the protein with a 10× excess of Anti-His quantum dots (QDs) (Qdot 565 Antibody Conjugation Kit, Invitrogen, Carlsbad, CA, USA, Q22032MP) on ice for 2 h and re-suspended the solution in a total volume of 20 μl of 1× TelK buffer (20 mM of Tris–HCl, 50 mM of potassium glutamate, 1 mM of Dithiothreitol, 0.1 mM of ethylenediaminetetraacetic acid). We estimated a QD labelling efficiency of ∼84%, measured by an agarose gel-shift assay comparing the mobility of DNA, DNA + TelK and DNA + QD-TelK (Supplementary Figure S1a). The activity of QD-labelled TelK was shown to be unaffected by QD labelling when compared with unlabelled protein, as shown by agarose gel electrophoresis (Supplementary Figure S1b).
TIRF chamber design
DNA bridges were formed on an etched glass surface with glass pedestals measuring 1 µm in height and separated by 7 µm (Institute of Microchemical Technology, Kanagawa, Japan) using a modification of a previous design (34). A circular glass coverslip (26022, Ted Pella, Redding, CA, USA) was placed on two 50-µm thick plastic spacers flanking the glass grating, such that a 50-µm wide channel was formed over the glass surface. The channel was filled with buffer using a pipette from one end, and a kimwipe (Kimberly-Clark, Irving, TX, USA) on the opposite end to create fluid flow through the channel. A solution of 33 µM neutravidin in water (Thermo Scientific, Waltham, MA, USA) was flowed in and was allowed to adsorb onto the glass surface for 5 min. The neutravidin was rinsed with phosphate-buffered saline (PBS). Twenty microlitres of an 80 nM solution of biotinylated λ-DNA (see DNA synthesis, ‘Materials and Methods’ section) was flowed in and, subsequently, rinsed with PBS. A pre-determined concentration of QD-labelled TelK was flowed in and incubated for 5 min with the DNA bridges. The unbound labelled TelK was rinsed from the channel with deoxygenated TelK buffer. Deoxygenation was achieved by adding an oxygen scavenging system (100 nM of glucose oxidase, 1.5 mM of catalase, 56 mM of glucose) to TelK buffer. The 50-µm plastic spacers were removed, leaving behind ∼50 nm of fluid between the glass pedestals and the coverslip surface to allow TIRF illumination. The coverslip was then sealed to the etched glass slide using nail polish before imaging.
Mean square displacement and diffusion coefficient analysis
Continuous image sequences were acquired for up to 50 s at 100 ms per frame. For each trajectory, the mean square displacement (MSD) of the spot was calculated. The diffusion coefficient (D) for each TelK spot was determined by fitting the first 0.6 s of the MSD versus time plot to a line through the following relation: 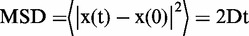 , as in previous studies (15,35–37). Mobile spots showed linear MSD versus time traces (Supplementary Figure S3a) characteristic of Brownian 1D diffusion. An offset at t = 0 because of the small thermally driven longitudinal motion of the DNA bridges was most apparent for stationary spots (Supplementary Figure S3b), which exhibited far smaller MSDs than mobile spots. Stationary spots showed a linear MSD versus time regime but also exhibited deviations from linearity after ∼1 s consistent with confined diffusion (38).
, as in previous studies (15,35–37). Mobile spots showed linear MSD versus time traces (Supplementary Figure S3a) characteristic of Brownian 1D diffusion. An offset at t = 0 because of the small thermally driven longitudinal motion of the DNA bridges was most apparent for stationary spots (Supplementary Figure S3b), which exhibited far smaller MSDs than mobile spots. Stationary spots showed a linear MSD versus time regime but also exhibited deviations from linearity after ∼1 s consistent with confined diffusion (38).
Optical trap
The dual-trap optical tweezers set-up has been described in detail previously (39). Briefly, the instrument consisted of two optical traps generated by two orthogonally polarized beams from a single 5-W, 1064-nm fibre coupled laser (YLR-5-1064-LP; IPG Photonics, Oxford, MA, USA). The position of one trap relative to the other was controlled by a piezoactuated mirror stage (Nano-MTA-2; Mad City Labs, Madison, WI, USA). A custom flow cell served as the experimental trap chamber, and it could be displaced relative to the two traps in all directions by a three-axis translational stage (ESP300; Newport, Irvine, CA, USA). Optical trap microspheres were prepared according to previously published protocols (40). Deoxygenated TelK buffer, prepared as described in the TIRFM ‘Materials and Methods’ section, was used for all optical trap experiments.
Optical trap experiments were performed in a laminar flow chamber, made by cutting flow channels into parafilm, which was then melted between two glass coverslips. This chamber is designed to enable control of DNA tether exposure to a set concentration of protein. Two streams, one containing buffer only, the other containing a pre-determined concentration of TelK, were flowed side-by-side at 100 µm/s using a syringe pump (702000; Harvard Apparatus, Holliston, MA, USA). A ∼200-µm boundary between the two streams was thus created. Tethers were formed in the protein-free buffer stream and, subsequently, moved to the TelK stream within 2 s.
Calculation of expected TelK-induced DNA condensation step size
We calculated the expected condensation step size of the DNA substrate in our optical trap assays based on a previously described model (41). This model takes into account three contributions to the change in free energy when DNA-binding proteins bend DNA: (i) the increase in distance between adjacent base pairs because of applied tension F; (ii) DNA kinking by angle θ induced by the protein; and (iii) DNA bending and end-to-end shortening beyond the protein-binding site:
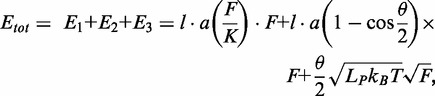 |
where (l·a) = 56 × 0.34 nm = 19.04 nm represents the length of DNA contacted by TelK, θ = 73° is the kink angle, Lp = 50 nm and K = 1100 pN are the persistence length and the stretch modulus of DNA under optical trap conditions, respectively (deoxygenated TelK buffer; ‘Materials and Methods’ section), kB is Boltzmann’s constant and T is the absolute temperature. The expected change in DNA extension as a function of tension was determined from the derivative of the free energy with respect to force. For the average tension at which tethers are held in the optical trap of 5.2 ± 1.3 pN, the expected DNA condensation of 7.5 ± 0.4 nm, is in excellent agreement with our average observed large step size of 7.2 nm.
Molecular dynamics simulations
An active but C-terminally truncated TelK mutant—TelK538, the same protein used in crystal structures—was used for all MD simulations, complexed to a 44-bp DNA substrate containing the TelK target sequence. Five atomic models of TelK–DNA were built based on a crystal structure of a TelK538 dimer complexed with double-stranded DNA (Protein Data Bank entry code 2V6E): (i) a TelK538 dimer bound to target DNA; (ii) a TelK538 dimer bound to non-target DNA; (iii) a TelK538 monomer bound to target DNA; (iv) a TelK538 monomer bound to non-target DNA; and (v) a TelK538 dimer without DNA. Non-target DNA was simulated by mutating nucleotides that make contacts with TelK538 in the crystal structure; the mutated sites are shown in Supplementary Figure S2. The topology file of DNA and protein along with the missing hydrogen atoms was generated using the psfgen plug-in of VMD (42). Each complex was placed in a water box with 0.15 mol/l of NaCl. The total size of the simulated systems lies in the 270 000–350 000 atom range.
Simulations were carried out using the program NAMD 2.8 (43) with the CHARMM27 force field for DNA (44), the CHARMM22 force field for proteins with CMAP corrections (45) and the TIP3P water model (46). Periodic boundary conditions were applied, and the Particle Mesh Ewald method (47) was used to calculate full electrostatic interactions. The van der Waals (vdW) energy was calculated using a smooth cut-off of 12 Å. The system temperature was maintained at 295 K using a Langevin thermostat that was applied only to the oxygen atoms of water with a damping coefficient of 0.1 ps−1 (48).
All systems were energy minimized for 8000 steps and heated to 295 K in 4 ps. After that, systems were subjected to a 500 ps isothermal-isobaric (NPT) equilibration with the protein backbone constrained and a 2 ns canonical ensemble (NVT) equilibration without constraint before production runs. Finally, an 80 ns production run in an NVT ensemble was performed for each system (Supplementary Table S1 lists all simulations). Data analysis of MD trajectories and snapshots of the molecular structures were realized with VMD (42).
Stochastic simulations
Stochastic simulations were performed to determine the first passage times for TelK dimerization and target-finding as a function of occupancy of TelK on DNA. Custom MATLAB code was used to implement the simulations. We assumed an L = 3.4-kb-long (1.156-μm) molecule of DNA—the same length as that used in the optical trap measurements—which contained the target sequence at its centre. Reflecting boundary conditions were applied. TelK monomers were modelled as point particles and randomly assigned uniformly along the DNA. Particles were allowed to diffuse in 1D with coefficient D = 1.8 µm2/s and dissociate with rate constant koff = 0.24 s−1, average values corresponding to those determined from the TIRFM experiments for TelK monomers. New particles could also bind to the DNA with a preset rate constant kon. Binding and dissociation established a steady-state occupancy of TelK on DNA determined by
During the simulations, particles were advanced incrementally until two particles came into contact anywhere along the DNA and dimerized (defining the first-passage time tdimer) or until a protein monomer came into contact with the target site (defining ttarget). Dimers were assumed to be immobile and not to dissociate. Each simulation was run 100 times for 100 000× 0.1 s time steps, for each of several DNA occupancies, which was achieved by varying kon over the range 0.0015–4 s−1 to cover experimentally observed occupancies.
RESULTS
TelK exhibits two distinct modes of interaction to non-target DNA
We first investigated the interaction between TelK and non-target DNA. We used TIRF microscopy to image individual QD-labelled proteins on linearly extended DNA that lacked the TelK target sequence. This was achieved by depositing 48.5-kb end-biotinylated λ-DNA across neutravidin-coated pedestals etched in a glass surface (‘Materials and Methods’ section; Figure 1a). These formed extended DNA ‘bridges’ with which QD-TelK interacted. The etched glass chamber was then sealed with a glass coverslip, and TIRF imaging was achieved by creating an evanescent light field through this coverslip. TelK640 (the full-length wild-type 640 amino acid protein henceforth referred to as TelK) was labelled with anti-histidine conjugated QDs with an ∼84% labelling efficiency (‘Materials and Methods’ section; Supplementary Figure S1a and b). Control assays showed that QD-labelled TelK displayed comparable activity to unlabelled TelK in bulk (Supplementary Figure S1c). Before a typical measurement, QD-labelled TelK was flowed into the chamber and left to incubate with the DNA bridges for 5 min to allow TelK to bind to the DNA bridges. Unbound TelK and free QD were, subsequently, rinsed away with a small volume (50 µl) of imaging buffer (20 mM of Tris–HCl, 50 mM of potassium glutamate, 1 mM of Dithiothreitol, 0.1 mM of ethylenediaminetetraacetic acid, 100 nM of glucose oxidase; 1.5 mM of catalase; 56 mM of glucose; ‘Materials and Methods’ section) to reduce the fluorescence background. A low background concentration of TelK remained in solution throughout the experiment because of unbound protein not removed during rinsing and from dissociation of bound protein from DNA.
Figure 1.

TelK monomers diffuse along non-target DNA, whereas dimers immobilize. (a) Schematic of TIRFM experimental set-up and representative fluorescence image. An etched glass slide with 1 × 1 µm pedestals separated by 7-µm etches was coated with neutravidin. Dual-biotinylated λ-DNA was flowed in to form DNA bridges, and the chamber was, subsequently, incubated with QD-labelled TelK monomers (here, 340 nM). QDs stuck to the glass surface were used as reference spots to ensure that drift and background motion were minimal. TelK concentrations used in all TIRF experiments ranged from 70 to 1350 nM. (b) Kymograph of QD-labelled TelK on λ-DNA showing mobile (1–31 s) and stationary (32–50 s) states after analysis and background subtraction with Gaussian fitting of spots. (c) Fluorescence intensity corresponding to the kymograph. The intensity of the mobile TelK doubles as it becomes immobile along the λ-DNA bridge. QD-blinking events (arrows), known to occur for single QDs, are observed only before TelK immobilization. (d) Diffusion coefficient and lifetime for blinking fluorescence spots (n = 114, red) and non-blinking spots (n = 81, blue). The shaded area represents the limit of sensitivity of our assay. The distribution of diffusion coefficients (right panel) is bimodal, with blinkers diffusing approximately four orders of magnitude faster than non-blinkers. As shown in the lifetime distributions (bottom panel), blinking spots also remained DNA-bound for shorter times (4.2 s) than non-blinkers (>50 s).
The positions of individual protein units on DNA were tracked by Gaussian-fitting the fluorescent intensity (33). In Figure 1b, a kymograph of TelK’s motion along DNA shows Brownian 1D diffusional motion along the DNA backbone as TelK searches for a target site. However, we also observed a stationary mode of TelK interaction with non-target DNA. The same fluorescent spot is observed to switch abruptly from a ‘mobile’ mode undergoing rapid 1D diffusion to a clearly distinguishable ‘stationary’ mode. Intensity analysis of the mobile and stationary part of the trace reveals that the mobile part of the trace is roughly half as bright as the stationary part of the trace (Figure 1c). Also, QDs are known to exhibit ‘blinking’ behaviour, where the QD fluorescence intensity stochastically switches on and off (49). In our image analysis, we defined blinking spots as those whose intensity dropped to zero for at least one frame (0.1 s). In Figure 1b, the mobile part of the trace shows on/off QD behaviour, whereas the stationary part of the trace does not blink off. Based on their intensity and on/off behaviour, we deduced that the mobile part of the trace represents the motion of a single labelled TelK monomer, whereas the stationary and non-blinking part of the trace represents two TelK proteins within a diffraction limited spot, presumably from binding of a second TelK monomer in solution.
We performed this TIRFM assay over a range of TelK concentrations from 70 to 1350 nM. Analysis over many DNA bridges revealed a clear bimodal distribution in the population of fluorescently labelled protein, showing species of mobile and immobile spots (Figure 1d). Mobile spots diffused rapidly along DNA with an average diffusion coefficient D = 0.74 ± 0.1 µm2/s [mean ± standard error of the mean (SEM)], whereas stationary spots had a diffusion coefficient of (3.0 ± 0.4) × 10−4 µm2/s (mean ± SEM). Diffusion coefficients were calculated by fitting the linear part of the fluorescent protein’s MSD dependence on time (‘Materials and Methods’ section and Supplementary Figure S3). Based on the bimodal distribution of diffusion coefficients (Figure 1d), a cut-off of D ∼ 0.01 µm2/s was used to classify spots as mobile or stationary. In a subset of movies, we observed stationary fluorescent spots stuck to the glass pedestals. When available, these reference spots were also tracked. The mean diffusion coefficient from reference spots was D ∼ 1 × 10−4 µm2/s, providing an estimate of the instrument noise and of the sensitivity limit of our measurements. Subtracting the reference spot motion from the DNA-bound TelK positions had only a negligible effect on our estimates of the diffusion coefficients. Beyond differences in mobility on DNA, mobile spots dissociated from the DNA at a rate of koff = 0.24 ± 0.07 s−1 (mean ± SEM), whereas most (88%) stationary spots remained stably bound for the duration of the TIRFM experiments (typically 50 s; i.e. koff < 0.02 s−1) (Figure 1d). On occasions, stationary spots were observed to dissociate from DNA (12%) or separate into two mobile spots (2%), and a small fraction of mobile spots were observed to coalesce into an immobile spot (2%) (Supplementary Figure S4).
As in Figure 1b, overall protein mobilities displayed a distinct correlation between TelK diffusion coefficient and QD fluorescence behaviour. All mobile diffraction-limited fluorescent spots showed blinking behaviour, compared with only ∼14% stationary spots (Figure 1d). This analysis strongly suggests that TelK monomers are able to exhibit the characteristic 1D diffusion of SSPs with non-target DNA, but that dimerization (or oligomerization) between TelK monomers causes 1D diffusion to cease. We attribute the small 14% fraction of blinking spots belonging to the stationary population to the ∼16% unlabelled ‘dark’ protein from our QD labelling (Supplementary Figure S1a and b).
Trajectories such as that shown in Figure 1b demonstrate that a minimum of two TelK proteins are sufficient to cause the protein immobilization on the λ-DNA bridges. In most cases, we were not able to observe directly a switch from the mobile to stationary state. However, most of the stationary spots observed under low-TelK concentration (0–200 nM) exhibited the same characteristic fluctuating pattern in fluorescence intensity as the dimer in Figure 1b and c (81%). As TelK concentration was increased (>400 nM), we observed an increasing number of trajectories (∼85%) exhibiting multiple plateaus in fluorescence intensity, which we attributed to higher-order (>2) TelK assemblies within a diffraction-limited spot. We believe these results are consistent with monomeric TelK being mobile and dimers being stationary, although larger aggregates are possible and may also lead to immobilization. Because TelK is functional as a dimer, our results strongly suggest that the active state of the protein is not competent to search for the target site.
Dimer-induced condensation of non-target DNA by TelK causes protein immobilization
What is the mechanism for the observed dimer immobilization? Crystal structures of the TelK dimer in complex with target DNA indicate a conformation in which monomer–monomer contacts and a tight interaction to the target sequence kink the DNA by an angle of 73° (26). Dimer immobilization observed in our TIRF measurements suggests that similar monomer–monomer contacts may also be responsible for dimer immobilization on non-target DNA. To test this hypothesis, we next used an optical trap to probe the TelK–DNA conformation on an extended DNA molecule. Using a previously established protocol (‘Materials and Methods’ section), a 3.4-kb DNA molecule lacking the target sequence was tethered between two microspheres held in two optical traps. The DNA tether was held at a low force [5.2 ± 1.3 pN, mean ± standard deviation (SD)] comparable with that experienced by the extended DNA bridges in the TIRF assay (3.1 ± 2.7 pN, mean ± SD.; ‘Materials and Methods’ section; Supplementary Figure S5). For our optical trap assay, we designed a sample chamber that allowed us to control when a DNA tether was exposed to TelK (Figure 2a). Chambers were designed with two main parallel channels, one containing protein-free buffer, the other with a fixed concentration of TelK (‘Materials and Methods’ section). A laminar flow of 100 µm/s created a ∼200-µm boundary between the two channels. This design allowed us to move a DNA tether from the flow channel containing protein-free buffer through the buffer boundary into the second channel containing TelK within 2 s.
Figure 2.

TelK dimers condense non-target DNA. (a) Schematic of optical trap experimental set-up. A DNA tether was formed in a stream containing buffer only and, subsequently, moved into a stream with buffer containing TelK. TelK binding to the DNA tether causes DNA condensation. (b) Time traces showing single DNA condensation events for tethers exposed to 100 nM TelK (blue) fit with a step-finder algorithm (red), and protein-free control trace (grey). Three characteristic times are highlighted in a sample trace: the time to first condensation, the condensed state lifetime and uncondensed state time. Traces offset for clarity. (c) The time for first TelK-induced condensation (green) is strongly dependent on (TelK), whereas the dwell time of the condensed state (blue) and the time between condensation events (red) do not depend on (TelK). (d) Normalized histogram of step size (Δx) for all observed condensation events (n = 93) because of DNA-induced TelK bending, with Gaussian fit to the condensation step size expected from the TelK dimer-DNA crystal structure.
Despite the absence of the TelK target site on the DNA, we repeatedly observed the DNA end-to-end extension to decrease by a few nanometres on exposure to TelK (blue traces, Figure 2b). These condensation events were transient with an average dwell time of 22 ± 6 s (mean ± SEM) and occurred repeatedly at TelK concentrations ranging from 40 to 270 nM. In contrast, control experiments performed in buffer lacking TelK showed no DNA condensation (gray trace, Figure 2b). Our ability to control when DNA was first exposed to TelK allowed us to measure accurately the time to the first condensation event. As expected, this first condensation time was strongly dependent on TelK concentration (green data points, Figure 2c), indicating binding of one or multiple TelK to the DNA. In contrast, the average condensed state lifetime and the time between subsequent condensation events were independent of TelK concentration (blue, red data points, Figure 2c), indicating that the same individual TelK complex was responsible for those condensation events. At high (>200 nM) concentrations of TelK, we mostly observed multiple simultaneous condensation events that were indistinguishable from each other. Therefore, we restricted our analysis to tethers showing individual separate condensation steps.
The size of these individual condensation events was determined with a step-finding algorithm (50). The step-size distribution for DNA condensation by TelK is displayed in Figure 2d. Condensation behaviour is expected as a result of TelK dimer formation at the DNA target site, as the 73° bend of the target DNA observed in crystal structures condenses the DNA, reducing its end-to-end extension. However, DNA condensation in the absence of the target site is not expected. The observed condensation in the optical trap measurements suggests that the same dimer-DNA conformation may be adopted on non-target DNA.
To test this suggestion, we performed a series of MD simulations to study the interaction of (i) a TelK dimer with target DNA, (ii) a TelK dimer with non-target DNA and (iii) a TelK monomer with non-target DNA. The simulations reveal that dimers induce near identical bend angles (∼70°) onto DNA with and without the target sequence (Supplementary Figure S2). We used the bend angle observed in these MD simulations to estimate the change in DNA end-to-end extension as a result of TelK dimerization on the non-target DNA substrate used in our optical trap assays. We compared the extension of the condensed DNA to that of linearly extended DNA taking into account the bend angle observed in the MD simulation, the DNA elastic properties and the force at which the tether was held in the optical trap (41) (‘Materials and Methods’ section), as shown in Figure 3a. Based on this calculation, we expect a condensation step size of 7.5 nm at a tension of 5.2 pN. As shown in Figure 2d, the observed step-size distribution is bimodal, with one peak at 7.2 nm, in excellent agreement with the prediction. This result indicates that a significant fraction of condensation events observed correspond to formation of DNA–TelK dimer complexes similar to those seen in crystal structures. Simulations of a TelK monomer binding to non-target DNA further show that the monomeric form of TelK is unable to bend DNA as a dimer does. Instead, monomeric TelK interaction with DNA results in a large decrease in the bend angle (Figure 3b). A histogram of the end-to-end distance of the 44-bp DNA substrate shows significant condensation in the case of the TelK dimer, but on average no condensation for a TelK monomer on non-target DNA (Figure 3c). MD results provide strong support for the claim that the condensation events observed in the optical trap measurements correspond to TelK dimerization.
Figure 3.

MD simulations confirm that Telk dimers bend non-target DNA, whereas monomers cannot. (a) Schematic representation of DNA condensation as would be measured in optical trap experiments (brown line) and MD simulations (boxed region). ΔL is defined as the difference between the contour length and the end-to-end distance of the 44-bp DNA substrate. MD snapshots of a TelK dimer-DNA complex and a TelK monomer-DNA complex are shown. DNA adopts a bent conformation in interaction with the TelK dimer, but not with the TelK monomer. (b) DNA bend angle induced by a TelK dimer (red) and a TelK monomer (blue). (c) Distribution of the change in end-to-end distance in the 44-bp MD DNA substrate (ΔL) induced by a TelK dimer and monomer in MD simulations.
As shown in Figure 2d, the condensation step-size distribution displays a second peak at 4.5 nm, corresponding to ∼50% of all observed condensation events. This suggests that TelK condensation may have two distinct tight-binding conformations on non-target DNA. Unfortunately, MD simulations do not reproduce this feature within the simulation time frame, instead producing only a single condensed conformation. We speculate that the smaller step may result from misoriented dimers that meet along the DNA (Supplementary Discussion). Such a conformation would not be observable with MD simulations, which start from the correctly oriented DNA–TelK crystal structure.
MD simulations also show that the TelK centre of mass (CoM) dislocation along the axis of non-target DNA is greater for monomeric TelK than for dimeric TelK (Figure 4a). A similar mobility discrepancy is observed for the rotational motion of TelK around the DNA axis; the TelK monomer is able to rotate around DNA more readily, whereas a TelK dimer does not rotate (Figure 4b). Together, these simulations suggest that TelK dimers exhibit tight-binding behaviour on DNA, resulting in limited translocation along the DNA backbone. In contrast, TelK monomers exhibit higher mobility along DNA because of weaker binding interactions.
Figure 4.

TelK monomers (blue) are more mobile on non-target DNA compared to dimers (red). (a) Dislocation of TelK dimer and monomer along non-target DNA axis based on MD simulations. (b) Rotation of TelK linker domain (SER208-ASN229). The snapshots show the starting and final positions of the linker domain (magenta) in TelK monomer and dimer after 80 ns. DNA is shown in yellow, and TelK is shown in cyan.
Monomeric TelK bind preferentially to the target DNA sequence
The experiments and simulations above establish that TelK dimers become immobilized on non-target DNA, yet likely form complexes that are close to those depicted in crystal structures of TelK on target DNA. If dimers form tightly bound and immobile complexes on non-target DNA, how is target search accomplished? To answer this question, we performed TIRF assays with 12.8-kb DNA bridges containing a single TelK target site located near the centre of the molecule (‘Materials and Methods’ section). The concentration of TelK was varied from 70 to 1350 nM as for the TIRF assay with λ-DNA bridges [despite the presence of a target sequence, we did not observe cutting of the DNA bridges by TelK. We attribute this to an inhibitory effect because of the tension applied on the DNA molecule when stretched over the TIRF chamber glass pedestals (Supplementary Discussion)]. Across all assayed TelK concentrations, we observed a preference for TelK binding to the centre of the target DNA substrate, compared with random TelK binding along non-target λ-DNA (Figure 5a and b). We also observed mostly dimers (non-blinking spots) at the target site (78%). To determine whether dimers or monomers confer target-site specificity, we considered a subset of our data by comparing only DNA tethers that showed one single blinking fluorescent spot per molecule of extended target or non-target DNA. For all singly occupied DNA bridges containing the target sequence, all observed blinking fluorescent spots were located at the target site, indicating that monomeric TelK has a binding preference for the target site. Based on the TelK–QD labelling efficiency, we are confident that at least 84% of these spots are monomers. Importantly, none of these blinking spots at the target site underwent 1D diffusion [D = (3.8 ± 0.4) × 10−5 µm2/s; mean ± SEM]. In contrast, blinking singly occupied λ-DNA bridges lacking the target sequence did not display a binding site-preference along the DNA, and the single QD–TelK underwent 1D diffusion (0.88 ± 0.10 µm2/s; mean ± SEM, comparable with that obtained for all mobile spots on non-target DNA).
Figure 5.

TelK binds preferentially to target DNA. (a) Average TelK fluorescent spot positions along target (green, n = 182) and non-target (red, n = 253) DNA, obtained by aligning and overlapping fluorescence images. TelK concentration range used in all TIRF experiments ranged from 70 to 1350 nM. (b) Position probability distribution of TelK along target (green) and non-target (red) DNA.
Our TIRF results are supported by MD simulations of a single TelK monomer on target and non-target DNA substrates. Monomers along target DNA exhibited greater stability than monomers along non-target DNA. As shown in Supplementary Figure S7a and b, the contact area and number of hydrogen bonds between a TelK monomer and target DNA remain constant during simulations, whereas both decrease in simulations of TelK monomer-non-target DNA complexes. MD simulation results indicate that TelK monomers have an increased affinity for the target sequence.
Repeating the optical trap experiments with DNA containing the TelK target site, we again observed repeated condensation of the DNA. The average condensation dwell time was 21 ± 2 s (mean ± SEM), with an average step-size of 7.7 ± 0.2 nm (mean ± SEM; Supplementary Figure S6 and Supplementary Discussion), in good agreement with the value measured with non-target DNA. Interestingly, we observed less of the 4.5-nm condensation events, which we attributed to misoriented dimers. We speculate that the presence of the target site may bias dimer formation into the more ‘proper’ configuration consistent with crystal structures.
DISCUSSION
The process by which site-specific proteins find their DNA target sites has remained poorly characterized for proteins that are functional on DNA as dimers or higher-order protein complexes but are monomeric in solution. Our TIRF studies demonstrate that TelK diffuses in 1D as a monomer along non-target DNA, yet becomes immobile as it encounters its target sequence. Therefore, monomer target-site immobilization confers specificity for TelK dimerization at the target site. MD simulations confirm experimental observations of monomer target-site specificity and provide molecular details of TelK’s interactions with target versus non-target DNA (Supplementary Figure S7). We also showed that TelK dimers become stationary and condense non-target DNA in our TIRF and trap measurements. However, protein immobilization on non-target DNA is intuitively counter-productive for the localization of the DNA target site, as it truncates the search process prematurely. Similarly, non-target DNA condensation is unexpected for a protein requiring sequence specificity for activity because condensation is usually predicted to occur at the target site before catalytic activity (51).
We suspect condensation and immobilization may be essential target-search features for proteins that must assemble into oligomeric complexes on DNA and that require more complex mechanisms for target-site identification than monomeric or pre-assembled SSPs. DNA bending has been thought to occur exclusively at target sites for sequence-specific protein–DNA systems. Only at the target-site do protein–DNA electrostatic interactions become maximized, and catalysis occurs hand-in-hand with DNA topological distortions (41). However, as has been previously suggested for certain SSPs (52), a protein’s ability to bend DNA indiscriminately may aid target-site identification. Erie et al. suggest that dimer-active proteins may bend DNA to ‘test’ their compatibility with that particular DNA sequence via protein–DNA and protein–protein interactions (53). Transcription factor HoxD9 has also been shown by Clore et al. (54,55) to bind non-target DNA with the same affinity and in the same binding mode as target-DNA as a means of enhancing the rate of target identification. Dimer-active proteins such as TelK may use this mechanism to maximize their electrostatic contacts with the DNA during their ‘sampling’ state, not only during site-specific catalysis.
Our optical trap data may provide some support for a ‘sampling’ mechanism in TelK. The mean dwell time of the condensation events (22 ± 6 s; mean ± SEM) in the trap measurements is noticeably shorter than the immobilized-state lifetime observed in our TIRF experiments, which spanned minutes (Figures 1d and 2c). However, the time between condensation events was independent of TelK concentration, suggesting that a single intra-molecular dimerization event was responsible for multiple sequential condensation and de-condensation events. We suspect this indicates that a TelK dimer may repeatedly attempt to bend its DNA substrate.
If non-target DNA condensation occurs too readily, it may significantly slow or inhibit target-finding and substrate formation. At first glance, it may seem that two monomers will find each other more quickly on DNA abundantly coated with protein, when in direct competition with the process of finding a target site. However, through stochastic simulations (‘Materials and Methods’ section), we show there is a wide range of conditions over which target localization occurs more rapidly than dimerization. Our simulations use experimentally measured kinetic parameters for TelK monomer unbinding (koff = 0.24 s−1) and diffusion (D = 0.74 µm2/s) (for simplicity, dimers were assumed to be immobile and to remain stably bound). Varying the average protein occupancy (the mean number of TelK monomers per DNA molecule), we calculated the rate of dimerization kdimer on non-target DNA versus the target-finding rate ktarget.
In comparing the two competing processes of protein dimerization versus protein target-finding, two regimes were observed as shown in Figure 6b. At low-TelK occupancies, the rate for target-site identification was significantly higher than for dimerization. However, at high occupancies, dimerization occurred faster than target-finding. This can be conceptualized by considering two competing processes: a monomer finding a target site versus a monomer finding another monomer. For a DNA molecule coated with N monomers, on average, the minimum inter-monomer distance scales as 1/N2 (Supplementary Discussion). Conversely, on average, the distance between a target site and the closest monomer scales as 1/N. As a result, the rates at which TelK diffuses over those distances depend differently on the number of proteins on DNA. When diffusion is the rate-limiting step to dimerization or target finding, kdimer scales as N4, whereas ktarget scales as N2, as seen in Figure 6b (at low TelK levels, the rate-limiting step is binding of TelK rather than diffusion, and kdimer scales instead as N2). The net result is that, at low-protein concentrations, the rate for a protein to diffuse to a target site is consistently higher than a protein diffusing into another protein. At high-protein concentrations, where the occupancy of protein along DNA is large, dimerization is faster than target localization.
Figure 6.

Kinetics of TelK-induced dimerization and target search. (a) Model of TelK target search. TelK monomers in solution bind DNA with rate kon and scan rapidly along non-target DNA with mean diffusion coefficient Dmonomer = 1.8 µm2/s. Monomers localize the target site with rate ktarget and bind tightly or dissociate from non-target DNA with average rate koff = 0.24 s−1. Preferential and stable binding of a TelK monomer allows a second monomer to dimerize at the target site and form a kinked DNA–TelK complex primed for catalysis. Occasionally, mobile monomers encounter each other on non-target DNA with rate kdimer and form stable, immobile dimers (Ddimer < 1 × 10−4 µm2/s) that ‘test’ their substrate by condensing the DNA transiently. Eventually, dimers on non-target DNA dissociate or separate into mobile monomers again (rate < 0.01 s−1). (b) Implementation of kinetic model in (a) via stochastic simulations using experimentally derived kinetic parameters. Average rates for first dimerization kdimer (blue, black), and first target localization ktarget (red) as a function of average protein occupancy, <N>, on DNA. For mean occupancies >1, target-finding and dimerization rates obey simple scaling laws of N2 and N4, respectively. For mean occupancies <1, dimerization follows of N2 scaling. Provided TelK occupancy is reasonably small, target localization consistently occurs faster than dimerization. Simulated rates are in good agreement with experimentally derived rates of first dimerization.
TelK’s host organism, Klebsiella oxytoca, is believed to maintain a cellular TelK copy number <20 for a ∼52-kb genome, or less than one dimer per 5 kb of DNA (data not published). These estimates are firmly placed in the low-occupancy regime predicted in our simulations. Therefore, we expect that target-site localization occurs faster than dimerization at biologically relevant TelK concentrations, allowing non-target DNA condensation to provide site-specificity in dimer-active proteins without hindering the kinetics of target sequence localization. Our simulations would also predict that the activity of the protein should decrease at high concentrations because of inhibition of target finding by immobile dimers. This is consistent with the observation of TelK inhibition at high TelK concentrations (>400 nM) in bulk (Supplementary Figure S8). A target-search mechanism in which protein dimers immobilize along non-target DNA is thus viable provided the DNA is sparsely coated. Based on our results, we propose a mechanism for dimer-active TelK in which target site identification occurs primarily by protein monomers diffusing to the target and preferentially binding to the target sequence, followed by dimerization at the target site.
The results of our kinetic simulations in principle can be compared with our optical trap data. As DNA is exposed to TelK at a well-defined time in our optical trap assay, we can measure directly the time for the first DNA condensation event (the first passage time) as a function of TelK concentration. Using our TIRF assay to calibrate occupancy on DNA against TelK incubation concentration (‘Materials and Methods’ section; Supplementary Figure S9), we compared our theoretical and experimental dimerization rates. The measured first passage times in our optical trap yield average rates (0.02–0.21 s−1) in good agreement with dimerization rates obtained in our simulations (0.02–0.11 s−1) for comparable mean occupancies (<N> = 0.4 − 0.8). This agreement further supports our dimer-active protein model for target search.
The literature to-date suggests an all-encompassing model for protein target-search in which proteins scan non-target DNA in their fully functional form (5,12). However, our studies of protelomerase TelK, a protein that is functional only as a dimer, reveal that additional variables must be taken into consideration when describing target-search mechanisms for proteins that assemble into dimer-active complexes on DNA. Although 1D scanning is a fundamental characteristic of target search by SSPs, we show that, for the dimer-active protein TelK, scanning only occurs as a monomer, and immobilization occurs when it forms a tightly-bound dimer on DNA or localizes the target sequence as a monomer. We speculate that the target-search mechanism proposed as a result of this work is extendable to several different protein families that share characteristics with TelK, such as those that assemble higher-order complexes onto their DNA target sites. Future studies of other members of the recombinase protein family will further our understanding of this novel target search mechanism, and its applicability to dimer and oligomer SSPs.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Table 1, Supplementary Figures 1–9, Supplementary Methods, Supplementary Discussion and Supplementary References [56–59].
FUNDING
National Institutes of Health [9P41GM104601 to X.Z. and K.S.]; Cell mechanics [R01 GM073655 to K.S.]; National Science Foundation [082265, Physics Frontier Center: Center for the Physics of Living Cells]; Burroughs-Wellcome Fund: Career Awards at the Scientific Interface (to Y.R.C.); National Science Foundation Graduate Research Fellowship [0913128 to M.P.L.]; East Asia and Pacific Summer Institutes fellowship [ID OISE-0913128 to M.P.L.]; Texas Advanced Computing Center and the National Center for Supercomputing Applications via XSEDE Resource Allocation Committee [MCA93S028]. Funding for open access charge: Burroughs-Wellcome Fund.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Prof. Toshio Yanagida, Dr Komori Tomotaka, Thomas Martin and all the members of the Ha, Selvin, Yanagida and Chemla laboratories for helpful discussions and for their generous advice.
REFERENCES
- 1.Koslover EF, Diaz de la Rosa MA, Spakowitz AJ. Theoretical and computational modeling of target-site search kinetics in vitro and in vivo. Biophys. J. 2011;101:856–865. doi: 10.1016/j.bpj.2011.06.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van den Broek B, Lomholt MA, Kalisch SMJ, Metzler R, Wuite GJL. How DNA coiling enhances target localization by proteins. Proc. Natl Acad. Sci. USA. 2008;105:15738–15742. doi: 10.1073/pnas.0804248105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gowers DM, Wilson GG, Halford SE. Measurement of the contributions of 1D and 3D pathways to the translocation of a protein along DNA. Proc. Natl Acad. Sci. USA. 2005;102:15883–15888. doi: 10.1073/pnas.0505378102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ramanathan SP, van Aelst K, Sears A, Peakman LJ, Diffin FM, Szczelkun MD, Seidel R. Type III restriction enzymes communicate in 1D without looping between their target sites. Proc. Natl Acad. Sci. USA. 2009;106:1748–1753. doi: 10.1073/pnas.0807193106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Halford SE, Marko JF. How do site-specific DNA-binding proteins find their targets? Nucleic Acids Res. 2004;32:3040–3052. doi: 10.1093/nar/gkh624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kolomeisky AB. Physics of protein-DNA interactions: mechanisms of facilitated target search. Phys. Chem. Chem. Phys. 2011;13:2088–2095. doi: 10.1039/c0cp01966f. [DOI] [PubMed] [Google Scholar]
- 7.Sudhanshu B, Mihardja S, Koslover EF, Mehraeen S, Bustamante C, Spakowitz AJ. Tension-dependent structural deformation alters single-molecule transition kinetics. Proc. Natl Acad. Sci. USA. 2011;108:1885–1890. doi: 10.1073/pnas.1010047108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bitinaite J, Wah DA, Aggarwal AK, Schildkraut I. FokI dimerization is required for DNA cleavage. Proc. Natl Acad. Sci. USA. 1998;95:10570–10575. doi: 10.1073/pnas.95.18.10570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ban C, Yang W. Structural basis for MutH activation in E.coli mismatch repair and relationship of MutH to restriction endonucleases. EMBO J. 1998;17:1526–1534. doi: 10.1093/emboj/17.5.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dowd DR, Lloyd RS. Biological significance of facilitated diffusion in protein-DNA interactions. Applications to T4 endonuclease V-initiated DNA repair. J. Biol. Chem. 1990;265:3424–3431. [PubMed] [Google Scholar]
- 11.Blainey PC, van Oijent AM, Banerjee A, Verdine GL, Xie XS. A base-excision DNA-repair protein finds intrahelical lesion bases by fast sliding in contact with DNA. Proc. Natl Acad. Sci. USA. 2006;103:5752–5757. doi: 10.1073/pnas.0509723103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Widom J. Target site localization by site-specific, DNA-binding proteins. Proc. Natl Acad. Sci. USA. 2005;102:16909–16910. doi: 10.1073/pnas.0508686102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van den Broek B, Noom MC, Wuite GJ. DNA-tension dependence of restriction enzyme activity reveals mechanochemical properties of the reaction pathway. Nucleic Acids Res. 2005;33:2676–2684. doi: 10.1093/nar/gki565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Langowski J, Alves J, Pingoud A, Maass G. Does the specific recognition of DNA by the restriction endonuclease Ecori involve a linear diffusion step - investigation of the processivity of the Ecori endonuclease. Nucleic Acids Res. 1983;11:501–513. doi: 10.1093/nar/11.2.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gorman J, Chowdhury A, Surtees JA, Shimada J, Reichman DR, Alani E, Greene EC. Dynamic basis for one-dimensional DNA scanning by the mismatch repair complex Msh2-Msh6. Mol. Cell. 2007;28:359–370. doi: 10.1016/j.molcel.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mendillo ML, Mazur DJ, Kolodner RD. Analysis of the interaction between the Saccharomyces cerevisiae MSH2-MSH6 and MLH1-PMS1 complexes with DNA using a reversible DNA end-blocking system. J. Biol. Chem. 2005;280:22245–22257. doi: 10.1074/jbc.M407545200. [DOI] [PubMed] [Google Scholar]
- 17.Gorman J, Greene EC. Visualizing one-dimensional diffusion of proteins along DNA. Nat. Struct. Mol. Biol. 2008;15:768–774. doi: 10.1038/nsmb.1441. [DOI] [PubMed] [Google Scholar]
- 18.Singh V, Ekka MK, Kumaran S. Second Monomer Binding Is the Rate-Limiting Step in the Formation of the Dimeric PhoP-DNA Complex. Biochemistry. 2012;51:1346–1356. doi: 10.1021/bi201257d. [DOI] [PubMed] [Google Scholar]
- 19.Erie DA, Yang G, Schultz HC, Bustamante C. DNA bending by Cro protein in specific and nonspecific complexes: implications for protein site recognition and specificity. Science. 1994;266:1562–1566. doi: 10.1126/science.7985026. [DOI] [PubMed] [Google Scholar]
- 20.Marianayagam NJ, Sunde M, Matthews JM. The power of two: protein dimerization in biology. Trends Biochem. Sci. 2004;29:618–625. doi: 10.1016/j.tibs.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 21.Vangent DC, Vink C, Groeneger AAMO, Plasterk RHA. Complementation between HIV integrase proteins mutated in different domains. EMBO J. 1993;12:3261–3267. doi: 10.1002/j.1460-2075.1993.tb05995.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hai T, Curran T. Cross-family dimerization of transcription factors Fos Jun and Atf creb alters DNA-binding specificity. Proc. Natl Acad. Sci. USA. 1991;88:3720–3724. doi: 10.1073/pnas.88.9.3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shuai K, Horvath CM, Huang LH, Qureshi SA, Cowburn D, Darnell JE., Jr Interferon activation of the transcription factor Stat91 involves dimerization through SH2-phosphotyrosyl peptide interactions. Cell. 1994;76:821–828. doi: 10.1016/0092-8674(94)90357-3. [DOI] [PubMed] [Google Scholar]
- 24.van Aelst K, Toth J, Ramanathan SP, Schwarz FW, Seidel R, Szczelkun MD. Type III restriction enzymes cleave DNA by long-range interaction between sites in both head-to-head and tail-to-tail inverted repeat. Proc. Natl Acad. Sci. USA. 2010;107:9123–9128. doi: 10.1073/pnas.1001637107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang WM, Joss L, Hsieh TT, Casjens S. Protelomerase uses a topoisomerase IB/Y-recombinase type mechanism to generate DNA hairpin ends. J. Mol. Biol. 2004;337:77–92. doi: 10.1016/j.jmb.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 26.Aihara H, Huang WM, Ellenberger T. An interlocked dimer of the protelomerase TelK distorts DNA structure for the formation of hairpin telomeres. Mol. Cell. 2007;27:901–913. doi: 10.1016/j.molcel.2007.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moffitt JR, Chemla YR, Smith SB, Bustamante C. Recent advances in optical tweezers. Annu. Rev. Biochem. 2008;77:205–228. doi: 10.1146/annurev.biochem.77.043007.090225. [DOI] [PubMed] [Google Scholar]
- 28.Selvin PR, Ha T. Single-Molecule Techniques: A Laboratory Manual. Cold Spring Harbor, NY: John Inglis; 2008. [Google Scholar]
- 29.Klepeis JL, Lindorff-Larsen K, Dror RO, Shaw DE. Long-timescale molecular dynamics simulations of protein structure and function. Curr. Opin. Struct. Biol. 2009;19:120–127. doi: 10.1016/j.sbi.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 30.Smith K, Youngman P. Use of a new integrational vector to investigate compartment-specific expression of the Bacillus-subtilis spoiim gene. Biochimie. 1992;74:705–711. doi: 10.1016/0300-9084(92)90143-3. [DOI] [PubMed] [Google Scholar]
- 31.Sun M, Serwer P. The conformation of DNA packaged in bacteriophage G. Biophys. J. 1997;72:958–963. doi: 10.1016/s0006-3495(97)78730-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Selvin PR, Lougheed T, Tonks Hoffman M, Park H, Balci H, Blehm B, Toprak E. Single-Molecule Techniques: A Laboratory Manual. In: Ha T, editor. In Vitro and In Vivo FIONA and Other Acronyms for Watching Molecular Motors Walk. Cold Spring Harbor, NY: Cold Spring Harbor Books; 2008. [Google Scholar]
- 33.Yildiz A, Forkey JN, McKinney SA, Ha T, Goldman YE, Selvin PR. Myosin V walks hand-over-hand: single fluorophore imaging with 1.5-nm localization. Science. 2003;300:2061–2065. doi: 10.1126/science.1084398. [DOI] [PubMed] [Google Scholar]
- 34.Iwaki M, Iwane AH, Shimokawa T, Cooke R, Yanagida T. Brownian search-and-catch mechanism for myosin-VI steps. Nat. Chem. Biol. 2009;5:403–405. doi: 10.1038/nchembio.171. [DOI] [PubMed] [Google Scholar]
- 35.Dahan M, Levi S, Luccardini C, Rostaing P, Riveau B, Triller A. Diffusion dynamics of glycine receptors revealed by single-quantum dot tracking. Science. 2003;302:442–445. doi: 10.1126/science.1088525. [DOI] [PubMed] [Google Scholar]
- 36.Bouzigues C, Morel M, Triller A, Dahan M. Asymmetric redistribution of GABA receptors during GABA gradient sensing by nerve growth cones analyzed by single quantum dot imaging. Proc. Natl Acad. Sci. USA. 2007;104:11251–11256. doi: 10.1073/pnas.0702536104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Courty S, Luccardini C, Bellaiche Y, Cappello G, Dahan M. Tracking individual kinesin motors in living cells using single quantum-dot imaging. Nano Lett. 2006;6:1491–1495. doi: 10.1021/nl060921t. [DOI] [PubMed] [Google Scholar]
- 38.Bannai H, Levi S, Schweizer C, Dahan M, Triller A. Imaging the lateral diffusion of membrane molecules with quantum dots. Nat. Protoc. 2007;1:2628–2634. doi: 10.1038/nprot.2006.429. [DOI] [PubMed] [Google Scholar]
- 39.Bustamante C, Chemla YR, Moffitt JR. In: Single-Molecule Techniques, A Laboratory Manual. Ha T, editor. 2008. High-Resolution Dual-Trap Optical Tweezers with Differential Detection. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 40.Landry MP, McCall PM, Qi Z, Chemla YR. Characterization of photoactivated singlet oxygen damage in single-molecule optical trap experiments. Biophys. J. 2009;97:2128–2136. doi: 10.1016/j.bpj.2009.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van den Broek B, Noom MC, Wuite GJ. DNA-tension dependence of restriction enzyme activity reveals mechanochemical properties of the reaction pathway. Nucleic Acids Res. 2005;33:2676–2684. doi: 10.1093/nar/gki565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33-38–27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 43.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Foloppe N, MacKerell AD. All-atom empirical force field for nucleic acids: I. Parameter optimization based on small molecule and condensed phase macromolecular target data. J. Comput. Chem. 2000;21:86–104. [Google Scholar]
- 45.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 46.Mackerell AD, Feig M, Brooks CL. Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 47.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 48.Martyna GJ, Tobias DJ, Klein ML. Constant-pressure molecular-dynamics algorithms. J. Chem. Phys. 1994;101:4177–4189. [Google Scholar]
- 49.Nirmal M, Dabbousi BO, Bawendi MG, Macklin JJ, Trautman JK, Harris TD, Brus LE. Fluorescence intermittency in single cadmium selenide nanocrystals. Nature. 1996;383:802–804. [Google Scholar]
- 50.Dogterom M, Kerssemakers JWJ, Munteanu EL, Laan L, Noetzel TL, Janson ME. Assembly dynamics of microtubules at molecular resolution. Nature. 2006;442:709–712. doi: 10.1038/nature04928. [DOI] [PubMed] [Google Scholar]
- 51.Slutsky M, Mirny LA. Kinetics of protein-DNA interaction: facilitated target location in sequence-dependent potential. Biophys. J. 2004;87:4021–4035. doi: 10.1529/biophysj.104.050765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grove A, Galeone A, Mayol L, Geiduschek EP. Localized DNA flexibility contributes to target site selection by DNA-bending proteins. J. Mol. Biol. 1996;260:120–125. doi: 10.1006/jmbi.1996.0386. [DOI] [PubMed] [Google Scholar]
- 53.Erie DA, Yang GL, Schultz HC, Bustamante C. DNA bending by Cro protein in specific and nonspecific complexes - implications for protein site recognition and specificity. Science. 1994;266:1562–1566. doi: 10.1126/science.7985026. [DOI] [PubMed] [Google Scholar]
- 54.Iwahara J, Clore GM. Detecting transient intermediates in macromolecular binding by paramagnetic NMR. Nature. 2006;440:1227–1230. doi: 10.1038/nature04673. [DOI] [PubMed] [Google Scholar]
- 55.Iwahara J, Zweckstetter M, Clore GM. NMR structural and kinetic characterization of a homeodomain diffusing and hopping on nonspecific DNA. Proc. Natl Acad. Sci. USA. 2006;103:15062–15067. doi: 10.1073/pnas.0605868103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Biebricher A, Wende W, Escude C, Pingoud A, Desbiolles P. Tracking of single quantum dot labeled ecorv sliding along DNA manipulated by double optical tweezers. Biophys. J. 2009;96:L50–L52. doi: 10.1016/j.bpj.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Grindley ND, Whiteson KL, Rice PA. Mechanisms of site-specific recombination. Annu. Rev. Biochem. 2006;75:567–605. doi: 10.1146/annurev.biochem.73.011303.073908. [DOI] [PubMed] [Google Scholar]
- 58.Grainge I, Jayaram M. The integrase family of recombinase: organization and function of the active site. Mol. Microbiol. 1999;33:449–456. doi: 10.1046/j.1365-2958.1999.01493.x. [DOI] [PubMed] [Google Scholar]
- 59.Lee L, Sadowski PD. Strand selection by the tyrosine recombinases. Prog. Nucleic Acid Res. Mol. Biol. 2005;80:1–42. doi: 10.1016/S0079-6603(05)80001-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.