


Abstract
RNA-seq has proven to be a powerful technique for transcriptome profiling based on next-generation sequencing (NGS) technologies. However, due to the short length of NGS reads, it is challenging to accurately map RNA-seq reads to splice junctions (SJs), which is a critically important step in the analysis of alternative splicing (AS) and isoform construction. In this article, we describe a new method, called TrueSight, which for the first time combines RNA-seq read mapping quality and coding potential of genomic sequences into a unified model. The model is further utilized in a machine-learning approach to precisely identify SJs. Both simulations and real data evaluations showed that TrueSight achieved higher sensitivity and specificity than other methods. We applied TrueSight to new high coverage honey bee RNA-seq data to discover novel splice forms. We found that 60.3% of honey bee multi-exon genes are alternatively spliced. By utilizing gene models improved by TrueSight, we characterized AS types in honey bee transcriptome. We believe that TrueSight will be highly useful to comprehensively study the biology of alternative splicing.
INTRODUCTION
RNA-seq is a powerful tool for transcriptome profiling based on ultra high-throughput next-generation sequencing (NGS) technologies. It was shown that RNA-seq is a more accurate method to survey the entire transcriptome in a quantitative and high-throughput fashion than expressed sequence tag (EST) sequencing and microarray technology (1). One of the key advantages of RNA-seq is efficiency in providing information about genome-wide splicing events. Information on splice junctions (SJs), especially those involved in alternative splicing (AS), is critical for isoform identification and quantification (2–4). Although de novo transcriptome assemblers have been developed very recently (5,6), reference-based mapping methods remain most widely used to reliably construct isoforms when the reference genome is available (2–4). The exact mapping of SJ spanning reads serves as a foundation for many RNA-seq-related studies. However, the short length of NGS reads makes the task of mapping SJ spanning reads extremely challenging.
A considerable amount of all RNA-seq reads span SJ sites and cannot be mapped directly to the reference genome as a whole sequence without gaps. Early RNA-seq mapping methods utilized existing gene annotations to narrow down mapping possibilities (7–10). However, even for the human genome and genomes of other well-studied model organisms, gene annotation is still not complete (11). Hence, the approaches relying on gene annotation are not able to fully utilize the power of RNA-seq in finding novel isoforms.
There are two approaches for RNA-seq read mapping without use of gene annotation. The first one is the ‘exon inference’ method implemented in TopHat (12), which utilizes fully aligned reads to ‘re-predict’ exons and constructs potential exon–exon junctions. To identify junction spanning reads, TopHat uses Bowtie (13) to map initially un-mapped (IUM) reads onto new reference sequences created from potential exon–exon junctions. SJs detected by this approach are expected to have high confidence, because they are supported by inferred exons with reasonably high coverage. However, when exons are not correctly predicted, either because a particular gene/isoform has low coverage in the RNA-seq data or exon length is shorter than read length, a substantial number of junctions would be missed.
The second method is the gapped alignment, which adopts the ‘anchor-extension’ strategy used in EST mapping [e.g. BLAT (14)]. This approach, implemented in MapSplice (15) and several others methods (16–19), is powerful in finding SJ spanning reads, regardless of the expression level of the corresponding transcript. Thus, it is particularly useful for detecting minor isoforms that are expressed at low levels and often use unannotated splice sites. Notably, this type of splice form has recently been reported as a prominent source of isoform diversity from a deep survey on human pre-mRNAs (11). To adopt this logic, in the new version of TopHat (version 2) only short reads are mapped using the ‘re-predict’ strategy while the mapping of long reads has also used the gapped alignment strategy.
The ‘anchor-extension’ strategy tends to produce multiple ways in which a candidate RNA-seq read can be split (Figure 1), especially when the read covers just a few bases on one side of the junction. It is reasonable to expect that at least one of the multiple splitting conformations is the true gapped alignment. MapSplice provides a ‘splice junction inference’ module to predict the true alignment by integrating ‘tag mapping significance’ (i.e. the more locations the short sequence on one side of read can be aligned to, the smaller is its tag significance) and RNA-seq distribution entropy (see ‘Mapping entropy’ in ‘Materials and Methods’ section). Although tag significance works for final junction scoring, it does not help for choosing the right candidate. In fact, a read can often be mapped to the reference with different gap size (i.e. the tag on one side might be mapped to several homologous locations). As shown in Figure 1, the orange part of the read (11 bp) is considered as a ‘tag’ in MapSplice that evaluates junction reliability by estimating the overall mapping significance. However, both ‘green’ and ‘red’ junctions have the same 11 bp tag (while the ‘green’ one is correct).
Figure 1.

Ambiguous split read resolved by TrueSight. A 75 bp read (SRR065504.21341241.2) from a human RNA-seq sample (detailed description in ‘Real datasets’ section) has two distinct splitting patterns, labeled in green and red. Mapping length on left and right side of both junctions is 11 and 64 bp, respectively. The same 11 bp sequence (orange) and donor splice site signal (red GT) exist in both gapped alignments. The junction shown in green has a higher TrueSight score (0.171) than the red junction (0.143) and supports a determination of exon skipping for gene KLC2, which is annotated by the UCSC Known Gene model (indicated by the green arrow on the left). MapSplice reported the junction shown in red and made an incorrect alignment for this read, whereas TopHat had not found an alignment for this read. Note that the gene model is for comparison only here, and was not used in TrueSight’s mapping procedure.
To improve sensitivity and specificity of mapping SJ spanning RNA-seq reads, we developed a new method, called TrueSight. The method incorporates information from (i) RNA-seq mapping quality and (ii) coding potentials from the reference genome sequences into a unified model that utilizes adaptive training by iterative logistic regression for de novo identification of SJs and filtering out unreliable SJs. To our knowledge, this is the first method that integrates RNA-seq alignment quality and coding potentials of DNA sequence to achieve more reliable read mapping. Our method also can map RNA-seq reads that span more than one SJ, which happens quite often when reads are longer than 100 bp (note that ∼30% of human exons are shorter than 100 bp). To our knowledge, among current RNA-seq alignment tools, only TopHat (v1.4.1) [We are aware that TopHat has a recent update to v2.0 and it supports Bowtie2. However, based on our evaluation, there were only minor differences in SJ finding between TopHat v1.4.1 and v2.0 when using Bowtie. Also, we observed TopHat performance to significantly drop if Bowtie2 (which is still a beta version) was used as the mapping program. We therefore, decided to use TopHat v1.4.1 in this study], MapSplice (v1.15.2) and PASSion (v1.2.0, specifically designed for paired-end reads) (20) can handle reads spanning more than one junction. In this article, we compare performance of TrueSight with these three methods.
The honey bee (Apis mellifera) is an excellent model organism to study genes and molecular pathways that are involved in behavioral plasticity. In the past decade, microarray technology has been utilized extensively to identify differentially expressed genes in the brain associated with different behavioral states (21,22), with some recent studies using RNA-seq technology instead (23). However, detailed characterization of AS in honey bee genome has not been done yet despite the fact that AS is an important mechanism for increasing the diversity and complexity of phenotypes. For example, the AS of anaplastic lymphoma kinase gene serves as an important regulator in honey bee larval differentiation (24) and the skipping of one exon in gemini transcription factor leads to honey bee worker sterility (25). Using new high coverage RNA-seq transcriptome profiling and gene models improved by TrueSight, we performed a comprehensive survey of AS in honey bee. We also assessed the accuracy of the TrueSight algorithm and compared it with existing tools (TopHat, MapSplice and PASSion), with previously published RNA-seq datasets of human, Drosophila melanogaster, Arabidopsis thaliana and Caenorhabditis elegans (see ‘Results’ section).
MATERIALS AND METHODS
The mapping procedure of TrueSight can be divided into two parts. The first part includes finding full-length read alignment and initial gapped alignments of IUM reads. The second part applies an expectation maximization algorithm for logistic regression, utilizing information from both DNA sequence and RNA-seq alignments, to find more accurate alignments for IUM reads. Model parameters are not pre-determined; instead, they are estimated iteratively.
Mapping full-length RNA-seq reads
First, TrueSight attempts to map each read onto the reference genome by Bowtie (version 0.12.8). Reads successfully mapped, constitute a set of fully mapped reads. Remaining IUM reads considered as candidate SJ spanning reads are subjected to the new algorithm of gapped alignment. Note that unlike existing gapped alignment methods, which work independently of fully aligned reads, the mapping of full-length reads is incorporated into a classifier in the logistic regression model to aid SJ inference (see ‘Coverage score’ section).
Mapping IUM reads to potential SJs
The IUM reads are mapped to potential SJs using an anchor-extension strategy. Each IUM read is split into N segments and mapped individually using Bowtie. The length of segments can be set to a number between 18 and 25 bp. We expect N–M segments would have a full-length alignment on the reference if the original read spans M SJs (note that we assume the distance between any two SJs in one read is larger than segment size; thus M < N), and we utilize these N–M aligned segments as ‘anchors’ to traverse all possible paths of N–M anchors (Figure 2). For each path, we search gapped alignments for these M unmapped segments from the original read based on their positions within the path. For example, in Figure 2, in order to find mapping of fragment 1 L, we index the reference region [−I, 0] from anchors using a k-mer hash table, where I is the expected maximum intron length (e.g. 200 kb) and k is set to 5. Using the k-mer hash table we can locate tentative alignments for 1 L, with edit distance between 1 L and reference sequence not greater than the number of mismatches allowed.
Figure 2.

An IUM read is split into four segments (N = 4). Segments 2 and 4 can be fully mapped onto the reference (Segment 4 has two potential alignments, labeled as 4_1 and 4_2), while Segments 1 and 3 cannot be fully aligned and are considered as junction spanning segments (M = 2). Segments 1 and 3 are split (shown by red solid lines) into left parts (1 L, 3 L) and right parts (1 R, 3 R). We utilize Segments 2 and 4 as ‘anchors’ and traverse each ‘path’ (2 → 4_1 and 2 → 4_2) by searching gapped alignments for Segments 1 and 3. There are four possible gapped alignments for this IUM read: A → C, A → D, B → C and B → D. In TrueSight, a logistic regression model integrating multiple features scores each candidate and infers the alignment with the highest confidence.
Canonical (GT-AG) SJs (26) have the highest priority in this mapping procedure. Semi-canonical (AT-AC or GC-AG) and non-canonical splice sites are reported only when no canonical junctions exist for that IUM read. Note that TrueSight users can turn off the search for semi/non-canonical junctions if they are only interested in GT-AG canonical SJs. After initial gapped mapping, the whole set of IUM reads is divided into three sets: (i) a set of ‘canonical Uniquely Splitting Reads’ (USRs), in which all reads have unique gapped alignment on canonical SJs; (ii) a set of ‘canonical Multiple Splitting Reads’ (MSRs), where all possible SJs, possibly originated from alternative spliced alignments (as in Figure 1), are retained as undecided junctions for further selection and (iii) a set of ‘non-canonical (including semi-canonical) Uniquely Splitting Reads’ (NUSRs). We only retain NUSRs with no mismatches.
The rationale behind TrueSight is that we believe that mere sequence alignment does not use all information available for RNA-seq read mapping. An IUM read may have several alternative gapped alignments to the reference genome, while only one of these candidate alignments is spanning across real intron. Therefore, to achieve enhanced specificity, it is extremely important to rigorously post-process MSRs produced by the initial gapped alignment that have high sensitivity.
Initial spliced alignment datasets
Initial Positive Set 
For semi-supervised training of model parameters, we defined a positive set of spliced alignments  by selecting USRs satisfying the following criteria: (i) no mismatches for alignments on either side of SJ and (ii) the SJ is supported by at least five USRs. Empirically, SJs selected from the above criteria have high accuracy and carries features of true positive junctions. We simulated a human RNA-seq dataset consisting of 20 million reads with 100 bp length (see ‘Simulated datasets’ section), 134 794 SJs were selected for
by selecting USRs satisfying the following criteria: (i) no mismatches for alignments on either side of SJ and (ii) the SJ is supported by at least five USRs. Empirically, SJs selected from the above criteria have high accuracy and carries features of true positive junctions. We simulated a human RNA-seq dataset consisting of 20 million reads with 100 bp length (see ‘Simulated datasets’ section), 134 794 SJs were selected for  Based on information from RefSeq, Ensembl, spliced EST and UCSC Known Gene models, 96.39% of all alignments in
Based on information from RefSeq, Ensembl, spliced EST and UCSC Known Gene models, 96.39% of all alignments in  were confirmed to match existing annotation.
were confirmed to match existing annotation.
Initial Negative Set 
A negative set of spliced alignment  was made from MSRs and NUSRs for which either of the following two conditions holds: (i) the MSR was not supported by any USR and (ii) the NUSR was the only read that supports a SJ and its mapping length on one side of the junction is shorter than 10 bp. In the same simulated human RNA-seq dataset mentioned above, 142 308 SJs originated from MSRs were selected as
was made from MSRs and NUSRs for which either of the following two conditions holds: (i) the MSR was not supported by any USR and (ii) the NUSR was the only read that supports a SJ and its mapping length on one side of the junction is shorter than 10 bp. In the same simulated human RNA-seq dataset mentioned above, 142 308 SJs originated from MSRs were selected as 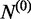 ; 99.71% of these SJs were not annotated; also 61 712 SJs originated from NUSRs were added to
; 99.71% of these SJs were not annotated; also 61 712 SJs originated from NUSRs were added to  ; 99.14% of these SJs were not annotated.
; 99.14% of these SJs were not annotated.
Logistic regression features
Splicing signal features
We designate an SJ of interest as  , where p refers to the donor site position (first base of intron) and q refers to the acceptor site position (first base of downstream exon). For simplicity, chromosome name is omitted in the following discussion (although we do consider it in the TrueSight source code) and in all formulas below, we assume that SJs are on the forward strand.
, where p refers to the donor site position (first base of intron) and q refers to the acceptor site position (first base of downstream exon). For simplicity, chromosome name is omitted in the following discussion (although we do consider it in the TrueSight source code) and in all formulas below, we assume that SJs are on the forward strand.
Exact splice site detection is critical for prediction of eukaryotic multi-exon gene structure and AS. Several ab initio gene prediction tools (27–34) can predict splice sites with high accuracy using just the DNA sequence information. However, all these algorithms have an underlying assumption of absence of AS. Alternative isoforms could be efficiently predicted if EST information is available (35). Still, the amount of EST was limited until the advent of NGS and RNA-seq (1). The success of DNA-based splice site prediction strongly indicates that information on splice sites is embedded in DNA sequence. This observation motivated us to develop a novel approach for SJ detection that integrates RNA-seq mapping with splice site signals and coding potentials defined by DNA sequence.
Starting with a set of highly confident SJs,  , we use a
, we use a  -order [
-order [ , chosen by the size of
, chosen by the size of  ] Markov chain (MC) to model both donor and acceptor sites:
] Markov chain (MC) to model both donor and acceptor sites:
 , and
, and  In order to avoid over-fitting in training,
In order to avoid over-fitting in training,  -order MC model, we require each (
-order MC model, we require each ( -mer has at least 100 instances in
-mer has at least 100 instances in  on average; thus,
on average; thus,  is chosen as the largest integer satisfying:
is chosen as the largest integer satisfying:  .
.
We also define parameters of a background Markov model
using GT-AG containing sequences randomly chosen from the reference genome.
Nucleotides at position  (last three base pairs from upstream exon and first 20 base pairs on intron) and
(last three base pairs from upstream exon and first 20 base pairs on intron) and 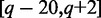 (last 20 base pairs on intron and first three base pairs from downstream exon) were selected to represent donor and acceptor site sequences, respectively. The Markov model defines a score of a SJ:
(last 20 base pairs on intron and first three base pairs from downstream exon) were selected to represent donor and acceptor site sequences, respectively. The Markov model defines a score of a SJ:
 |
We also define position weight matrix (PWM) (36) to score splice sites. In contrast to the MC model, the score assumes that nucleotides in adjacent positions are independent, whereas each position has a specific nucleotide frequency distribution. The PWM score is defined as:
 |
where  refers to the nucleotide frequencies at
refers to the nucleotide frequencies at 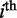 position, obtained from all donor/acceptor sequences in
position, obtained from all donor/acceptor sequences in  , and
, and 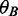 stands for the background nucleotide frequencies obtained from non-splice site sequences (defined above).
stands for the background nucleotide frequencies obtained from non-splice site sequences (defined above).
Coding potential feature
It was shown earlier that algorithms that incorporate protein-coding potential predict splice sites better than algorithms using splicing signals only (37). Protein-coding potential measure provides other advantages. For instance, with uneven distribution of RNA-seq reads on transcripts, some exon regions may not be fully covered RNA-seq reads, specifically exons related to low expression transcripts. Also, exons shorter than RNA-seq read length cannot be aligned with full-length reads. In these cases, RNA-seq alone does not provide enough information for exon delineation, whereas sequence properties of coding regions may help extend the mapping and identify true locations for ambiguously split reads.
In our algorithm, both coding and non-coding regions are modeled using fifth-order Markov models trained on sequences associated with the  set. For a junction
set. For a junction  in
in 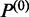 , fragments
, fragments  and
and  are selected into a training set of protein coding regions to define parameters of the exon Markov model:
are selected into a training set of protein coding regions to define parameters of the exon Markov model:  . Sequences in fragments
. Sequences in fragments  and
and  are used for training an intron Markov model:
are used for training an intron Markov model: 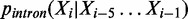 . To define a coding potential score for
. To define a coding potential score for  , 80 bp long fragments are selected. Notably, for exons and introns shorter than 80 bp, the 80 bp fragment may contain mislabeled sequences. Still, as such events are observed with low frequency, they are expected to have negligible effect on the Markov model parameters (the average exon and intron sizes in human are 327 bp and 7215 bp, respectively). We define the coding potential score as follows:
, 80 bp long fragments are selected. Notably, for exons and introns shorter than 80 bp, the 80 bp fragment may contain mislabeled sequences. Still, as such events are observed with low frequency, they are expected to have negligible effect on the Markov model parameters (the average exon and intron sizes in human are 327 bp and 7215 bp, respectively). We define the coding potential score as follows:
 |
RNA-seq mapping derived features
Coverage score
Fully aligned RNA-seq reads are used to compute a ‘coverage score’. Intuitively, for positions close to exon boundaries, one would expect mapping coverage (by reads that have gapless alignments) to be lower than in the rest of the region. Let i be a genomic position of the ‘first’ base of fully aligned read,  be the total number of reads mapped to position i, and l be the read length. Coverage for interval (a, b) is defined as:
be the total number of reads mapped to position i, and l be the read length. Coverage for interval (a, b) is defined as:  . The coverage score for a donor site is then:
. The coverage score for a donor site is then:  . If p corresponds to a real donor site,
. If p corresponds to a real donor site,  would be the exon region enriched by full-length read alignments, whereas fewer full alignments would be found in region
would be the exon region enriched by full-length read alignments, whereas fewer full alignments would be found in region  (reads with their first base aligned within this region would span across the donor splice site). Similarly, a coverage score for an acceptor site is:
(reads with their first base aligned within this region would span across the donor splice site). Similarly, a coverage score for an acceptor site is:  . Sum of the donor and acceptor coverage scores is the coverage score for the junction:
. Sum of the donor and acceptor coverage scores is the coverage score for the junction: 
Intron size
A set of introns in  provides data to compute the distribution of intron size. Empirically, a candidate SJ with an excessively long genomic span is likely to be incorrect, though our gapped alignment algorithm can accept large introns (with default 200 kb). We use percentile rank on introns and define a critical intron size,
provides data to compute the distribution of intron size. Empirically, a candidate SJ with an excessively long genomic span is likely to be incorrect, though our gapped alignment algorithm can accept large introns (with default 200 kb). We use percentile rank on introns and define a critical intron size, 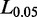 as one longer than length of 95% of introns. If candidate intron size
as one longer than length of 95% of introns. If candidate intron size  , we set
, we set  ; otherwise
; otherwise 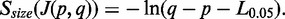
Junction mapping number
This score is equal to the number of USRs mapped onto
is equal to the number of USRs mapped onto  .
.
Length of the shorter side of the alignment
This feature is defined as the maximum length  of the shorter side of gapped alignment spanning
of the shorter side of gapped alignment spanning  among all reads mapped onto this junction. The smaller is the value of
among all reads mapped onto this junction. The smaller is the value of  , the greater is the chance that
, the greater is the chance that  is a false positive.
is a false positive.
Mapping entropy
Let  be the fraction of USRs that span
be the fraction of USRs that span  at position i of the read. The Shannon entropy is then (15):
at position i of the read. The Shannon entropy is then (15):  . Given sufficient sequencing depth, the position of a SJ on RNA-seq read is assumed to have a uniform distribution (9). Therefore, the values of
. Given sufficient sequencing depth, the position of a SJ on RNA-seq read is assumed to have a uniform distribution (9). Therefore, the values of 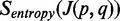 for true SJs with high coverage are expected to be larger than the values for false-positive junctions.
for true SJs with high coverage are expected to be larger than the values for false-positive junctions.
Multiple mapping score
 , where
, where  is number of reads mapped onto
is number of reads mapped onto  and
and  is number of multiple splitting patterns for ith read mapped onto
is number of multiple splitting patterns for ith read mapped onto  ;
;  for a USR. The score reflects mapping ambiguity. Small
for a USR. The score reflects mapping ambiguity. Small  implies that reads mapped onto
implies that reads mapped onto 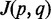 have many other spliced alignments to the genome, thus the mapping support for the particular
have many other spliced alignments to the genome, thus the mapping support for the particular  is weak.
is weak.
Number of mismatches
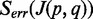 is defined as the mean number of alignment mismatches of all reads mapped onto
is defined as the mean number of alignment mismatches of all reads mapped onto  .
.
Summary
For each SJ, the 10 score values form a vector of 10 features. To discriminate positive (correct) and negative (incorrect) sets of candidate gapped alignments, we propose an iterative algorithm that finds parameters of a logistic regression function simultaneously with using the function for classification of the alignment.
Expectation-maximization with logistic regression
All junctions inferred from USRs, MSRs and NUSRs (n of them) constitute the data set for analysis. Let
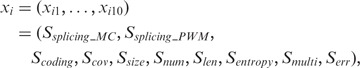 |
where  stands for value of jth feature for SJ
stands for value of jth feature for SJ  . Note that
. Note that  values are scaled to interval (0:1).
values are scaled to interval (0:1).
Initial sets  and
and  were selected by empirical criteria described above. We consider
were selected by empirical criteria described above. We consider and
and  junctions as ‘labeled’ [denoted as
junctions as ‘labeled’ [denoted as  ], while junctions initially not selected are considered as ‘unlabeled’ [denoted as
], while junctions initially not selected are considered as ‘unlabeled’ [denoted as  ]. Semi-supervised training methods working with both labeled and unlabeled data can be applied (38).
]. Semi-supervised training methods working with both labeled and unlabeled data can be applied (38).
We use a general classification expectation–maximization algorithm (CEM) (39) with logistic classifiers (40) to estimate probabilities (SJ scores, or SJS; see Supplementary Methods for details) for initially ‘unlabeled’ junctions to be true junctions. Similar to the EM algorithm (except an additional classification step between E-step and M-step), the CEM algorithm can be considered as a k-means clustering method and can efficiently optimize classification maximum likelihood (39). A detailed description of the algorithm is provided in Supplementary Methods.
Sorting out MSRs and predicting splice junctions from RNA-seq data
There are two reasons to use SJSs. First, SJSs are utilized for identifying true junctions from MSRs data. As it is reasonable to expect one of the multiple split alignments to be the true gapped alignment, the SJ with the highest score is retained as predicted SJ. To assess the contributions of each of the 10 features in CEM algorithm to the MSRs classification, we ran TrueSight on simulated dataset (see below) and plotted area under curve (AUC) values (calculated from ROC curves based on 10 000 data points) of the full model, as well as each individual feature (Supplementary Methods and Supplementary Table. S1). It is shown in Figure 3 that the CEM algorithm using the model with all features achieves the best performance in selecting true positive splice junctions from all the MSRs.
Figure 3.

Comparison of AUC values for each feature in inferring true MSRs. The full model (black column), utilizing features derived from DNA sequence (light gray columns) and RNA-seq features (dark gray columns), has the best overall performance.
Second, after sorting out all MSRs, all splice junctions in USRs, NUSRs and MSRs are binned together as candidate SJs (even with low SJS). With SJS assigned, several selection criteria (e.g. to suppress low score non-canonical junctions) are applied to select the best candidate junctions and only reads covering these selected junctions will be reported in the final output (in the Binary Alignment/Map (BAM) format). For reads spanning more than one SJ, we can use three options to combine the SJS for the covered SJs: ‘minimum’, ‘mean’ and ‘product’. We choose to use ‘minimum’ because it achieves highest AUC values in differentiating true and false multiple gapped alignments in our simulated datasets (described in ‘Results’ section). In case of multiple SJ per read (n) the read alignment is presented in the BAM file with a tag ‘AS’ and the read’s junction total score:
where  is SJS for ith junction that the read spans across.
is SJS for ith junction that the read spans across.
RESULTS
Performance evaluation
Real dataset
To assess the accuracy of the TrueSight algorithm and compare with existing tools (TopHat, MapSplice and PASSion), we selected RNA-seq datasets of human, D. melanogaster, A. thaliana and C. elegans. For each genome, we built a combined annotation of introns from several sources, to achieve a more comprehensive evaluation reference (Supplementary Table S3). Introns predicted as SJs were divided into four classes (Supplementary Table S4): (i) introns matching annotated known introns; (ii) introns not annotated while both donor and acceptor splice sites were annotated as parts of other introns; (iii) introns with only one annotated splice site and (iv) introns where both splice sites are novel.
Even though the current annotation of transcriptomes, including those from human are still incomplete (10,11), several conclusions can be reasonably drawn (Figure 4). Introns with both ends annotated (column ‘known introns’ in Supplementary Table S4) are likely to be true introns (SJs). For this type of SJ, TrueSight and MapSplice are more sensitive than TopHat and PASSion. We expect SJs with both novel splice sites (column ‘both novel’ in Supplementary Table S4) to have a high probability to be incorrect; MapSplice makes the largest number of predictions in this category of SJs.
Figure 4.

Performance of four SJ detection tools on four real RNA-seq datasets. We label ‘known introns’ as true junctions (gray bars) and ‘both novel’ in Supplementary Table S4 as false junctions (gray lines).
Simulated datasets
We used Cufflinks (3) to estimate expression levels from a human RNA-seq dataset (Supplementary Table S3) based on isoforms defined by UCSC Known Gene models. To build test datasets similar to real transcriptome sequencing data, we used Maq (41) to generate simulated Illumina reads with an error rate of 0.02, and with abundance proportional to the human dataset based on UCSC Known Gene models. Three paired-end datasets of 20 million reads were generated with 50, 75 and 100 bp read lengths, respectively.
All four programs were tested with default settings (the number of mismatches was set as two). As shown in Figure 5 (for overall performance, Table 1) for all three datasets, TrueSight shows higher sensitivity among the four tools, which is even more pronounced for low coverage SJs. In terms of specificity, TrueSight, TopHat and PASSion performed substantially better than MapSplice. TrueSight also performed better than the other three tools for aligning reads that span more than one SJ (Supplementary Table S2).
Figure 5.

Evaluation of TrueSight, TopHat, MapSplice and PASSion on simulated datasets. The sensitivity and specificity is plotted as function of cumulative junction coverage. The sensitivity is the ratio of detected positive junctions over all junctions covered by simulated reads; specificity is the ratio of positive junctions over all reported ones. Overall SN and SP are summarized in Table 1.
Table 1.
Overall accuracy performance of the four methods (TrueSight, TopHat, MapSplice and PASSion) on simulated RNA-seq datasets
| Dataset | Tools | Total | True | False | SNa (%) | SPb (%) |
|---|---|---|---|---|---|---|
| 50 bp | TrueSight | 151 565 | 148 372 | 3193 | 93.55 | 97.92 |
| TopHat | 139 426 | 136 335 | 3091 | 87.45 | 97.81 | |
| MapSplice | 171 550 | 135 130 | 36 420 | 87.85 | 78.79 | |
| PASSion | 135 823 | 130 525 | 5298 | 88.08 | 96.13 | |
| 75 bp | TrueSight | 156 558 | 154 245 | 2313 | 95.51 | 98.55 |
| TopHat | 150 723 | 147 481 | 3242 | 92.43 | 97.88 | |
| MapSplice | 161 043 | 143 834 | 17 209 | 91.03 | 89.34 | |
| PASSion | 140 037 | 135 481 | 4556 | 89.30 | 96.78 | |
| 100 bp | TrueSight | 159 403 | 157 430 | 1973 | 96.53 | 98.79 |
| TopHat | 156 506 | 152 739 | 3767 | 94.60 | 97.62 | |
| MapSplice | 164 456 | 155 984 | 8472 | 96.28 | 94.88 | |
| PASSion | 141 344 | 137 035 | 4309 | 89.30 | 96.98 |
aSensitivity is the fraction of simulated junctions correctly detected by TrueSight; bSpecificity is the fraction of true junctions (comparing with RefSeq, Ensembl, spliced EST and UCSC Known Gene) among all predicted junctions. Best sensitivity and specificity are highlighted.
SN, sensitivity; SP, specificity.
By plotting the TrueSight SJS distribution for both true and false junctions from the three simulated datasets (Supplementary Figure S1), we observed distinct SJS patterns: 95% of true junctions have SJS >0.5, whereas only 60% of false junctions had SJS >0.5. Comparing the SJS distribution across the three datasets with different read lengths, we found that the power of TrueSight to separate true and false SJ is higher in samples with longer reads, which is consistent with the trend in sensitivity and specificity in Figure 5. The performance in prediction of non-/semi-canonical junctions is shown in Supplementary Table S5. TopHat does not appear to be the best tool for finding non-canonical junctions in the three datasets [consistent with earlier observations (15)]. Although TopHat recovered the largest portion of semi-canonical junctions among the four tools, it also had the largest number of false predictions. TrueSight has almost the same sensitivity but higher specificity in prediction of non-/semi-canonical junctions than MapSplice.
We also used Cufflinks (3) to assess an impact of SJ mapping on transcript construction. Since the output format of PASSion is not suitable for Cufflinks, we only assessed Cufflinks performance based on RNA-seq mapping results obtained by TrueSight, TopHat and MapSplice. By comparing with the UCSC Known Gene models, we showed that the sensitivity and specificity of assembled intron-chains inferred from the TrueSight mapping were higher than those obtained from other tools for majority of datasets (Supplementary Figure S2). These results indicate that more accurate RNA-seq read mapping to SJs would lead, as expected, to better construction of transcripts.
Implementation and running time
All computationally intensive parts of TrueSight, including RNA-seq gapped alignment and EM semi-supervised training, were written in C++ and were then wrapped up by Perl scripts as a pipeline. Tested on a simulated dataset with 20 million read pairs (read length is 100 bp), TrueSight took 35 CPU hours (TopHat took 26 CPU hours, MapSplice took 19 CPU hours and PASSion took 26 CPU hours). Users can utilize multi-cores to accelerate the running time of TrueSight.
Application to honey bee transcriptomes
RNA-seq has been shown to be very effective in revealing AS (42,43). Still, a detailed analysis of AS for a number of species has not been reported yet. Having a particular interest in honey bee, we generated 380 million, 100 bp paired-end reads (i.e. 190 million pairs) through RNA sequencing using Illumina HiSeq 2000 based on 10 dissected honey bee fat body tissues (Supplementary Methods and Supplementary Table S6). The TrueSight program was run with default parameters and mapped all the RNA-seq reads from each sample onto honey bee genome assembly version 4 (44).
Improving GLEAN honey bee gene models
The honey bee GLEAN consensus gene set (45) was created by integrating the output of multiple gene prediction algorithms with a goal to balance sensitivity and specificity. Notably, the GLEAN models have not captured AS isoforms in an extensive manner due to the limited amount of transcriptome information previously available for the honey bee genome sequencing project (44). Having new deep RNA-seq data, we applied TrueSight to find SJs essential for AS identifications and to improve the GLEAN gene models (Supplementary Methods). The improved gene models were used to survey of AS patterns in the honey bee genome (see below; improved gene models available in Supplementary Table S8).
In comparison with the original GLEAN set of gene models, 5873 new exons were added, 1059 of them were Cassette Exons. A total of 4122 of the newly added exons were novel terminal exons. After this refinement of GLEAN models, the number of SJs increased from 53 884 to 70 022. The newly added junctions are likely to be involved in various types of AS. Also, we have identified 2803 novel multi-exon transcripts in inter-genic regions annotated with respect to the GLEAN models, an indication that the GLEAN annotation of 10 098 genes has been incomplete. These improved gene models will be made publicly available on the BeeBase browser for the community.
Alternative splicing in the honey bee transcriptomes
Based on the deep coverage honey bee RNA-seq dataset and the gene models improved by TrueSight, we conducted a survey of AS variants in honey bee. There are four principal types of AS (46): (i) intron retention (IR), in which an intron may be retained as part of a mature transcript or spliced out; (ii) exon skipping, in which a cassette exon (CE) may be included or not in transcripts; (iii) alternative use of splice sites (donor/acceptor), leading to alternative exon boundaries (AEB) and (iv) alternative terminal exons (ATE), in which alternative first exons or alternative last exons are used. Overall, 81% of the AS genes were found in at least eight samples (out of 10) (Figure 6; Supplementary Table S7 has the list of AS genes). We also observed that different AS types showed variations in frequencies among the 10 individual samples (Figure 6). Almost 75% of CE and 73% of IR were shared by at least eight samples (out of 10), whereas ∼50% of AEB and ATE events were shared by at least eight samples, indicating a higher level of variation for AEB and ATE. The criteria used in detecting IR are summarized in Supplementary Methods. Distributions of various AS types in the honey bee transcriptome are characterized in Table 2. We found that 2596 (out of 3645) honey bee AS genes have Drosophila orthologs and were shared by all 10 RNA-seq samples used in this study, with 41.1% of them (1068) categorized as AS genes in the Drosophila gene models (flybase version r5.42). We leave further analysis of AS in honey bee for a future study.
Figure 6.

Variation of AS and different subtypes (including IR, CE, AEB and ATE) among 10 honey bee samples used in this study. Different colors are referring to different total number of samples, where a given feature is shared. Particularly, red color indicates percent of the AS type shared in all 10 samples, magenta indicates presence in 9 out of 10, and so on.
Table 2.
Counts of different types of alternative splicing events in honey bee transcriptome
| AS event | Number | Exons involveda | Genes involved (%) |
|---|---|---|---|
| Intron retention | 5258 | 9047 | 2848 (48.0) |
| Cassette exon | 1731 | 1731 | 1336 (14.3) |
| AEB | |||
| Alternative donor site | 2684 | 2441 | 1972 (21.1) |
| Alternative acceptor site | 4461 | 3959 | 2806 (30.0) |
| ATE | |||
| Alternative first exon | 1382 | 1382 | 1061 (11.3) |
| Alternative last exon | 507 | 507 | 456 (4.87) |
aFor retained introns, two flanking exons are counted as ‘involved’ exons.)
DISCUSSION
To our knowledge, TrueSight is the first method with the ability to combine RNA-seq mapping with genome-wide splicing signal and coding potential computation from the DNA sequence. In testing on both real and simulated data, TrueSight has shown a better overall performance than existing tools in terms of sensitivity and specificity of detecting SJs, especially in SJs having low coverage by RNA-seq reads. As many AS isoforms are of low coverage, we expect TrueSight will be extremely useful in AS detection. Mapping RNA-seq reads to SJs is a pivotal point in an algorithm of isoform construction utilizing a reference genome. For example, in IsoLasso (4), a recently developed isoform construction algorithm using the TopHat output, inferred SJs are explicitly used to significantly reduce the total number of possible isoforms subjected to the LASSO procedure. We have shown that the sensitivity and specificity of assembled transcript structures (using Cufflinks) from the TrueSight read mapping are better than the ones utilizing other SJ detection tools. We expect that TrueSight will be useful in improving isoform construction and, consequently, in improving the accuracy of estimation of isoform expression levels.
There are several other features that we could incorporate in order to further improve the algorithm. First, we could add an explicit modeling of SJs in untranslated region (UTR). Second, we could use the three-periodic model of a coding region to trace exon reading frames; this addition will enhance modeling of SJs in coding regions and will reduce the number of pairs of candidate splice sites to those that do not disrupt the reading frame. Further making these models local GC content-dependent is an additional option to increase the accuracy.
We used TrueSight and deep RNA-seq data to perform AS analysis for the honey bee, an important model organism whose genome is still lacking a comprehensive gene annotation. We have identified 16 023 instances of AS for 5644 genes, suggesting that 60.3% multi-exon honey bee genes can produce multiple transcripts. The honey bee is a key model organism for studying brain and behavior (22,47). Therefore, our contribution to annotation of honey bee transcriptome based on RNA-seq will facilitate future studies aimed at understanding genetic variations (in particular, AS) and important regulatory networks underlying different behavioral phenotypes (23).
Recent advances in NGS technologies have made it possible to sequence large number of genomes from the tree of life. The G10K project (sequencing 10 000 vertebrate genomes) (48) and the i5k project (sequencing 5000 insect genomes) (49) have been recently initiated and many of these new genomes will also have RNA-seq data available. The TrueSight program can greatly accelerate the annotation of these new genomes and help elucidate the origins of complex traits of different species.
AVAILABILITY
Source code of the TrueSight program is available on our supplementary website:
ACCESSION NUMBERS
RNA-seq data generated in this study have been submitted to the NCBI Sequence Read Archive (SRA) (http://www.ncbi.nlm.nih.gov/Traces/sra/) under accession no. SRA053010.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–8, Supplementary Figures 1, 2 and Supplementary Methods.
FUNDING
National Science Foundation [1054309 to J.M.]; National Institutes of Health [1R21HG006464 to J.M., 1DP1OD006416 to G.E.R. and 5R01HG00783 to M.B.]. Funding for open access charge: National Institutes of Health [1R21HG006464].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank A. Hernandez and the W. M. Keck Center for Comparative and Functional Genomics at the University of Illinois for library preparation and RNA-seq; T. Newman for assistance in the laboratory; J. Kim and J. Hou for useful discussions.
REFERENCES
- 1.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genetics. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guttman M, Garber M, Levin JZ, Donaghey J, Robinson J, Adiconis X, Fan L, Koziol MJ, Gnirke A, Nusbaum C, et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat. Biotechnol. 2010;28:503–510. doi: 10.1038/nbt.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li W, Feng J, Jiang T. IsoLasso: a LASSO regression approach to RNA-Seq based transcriptome assembly. J. Comput. Biol. 2011;18:1693–1707. doi: 10.1089/cmb.2011.0171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robertson G, Schein J, Chiu R, Corbett R, Field M, Jackman SD, Mungall K, Lee S, Okada HM, Qian JQ, et al. De novo assembly and analysis of RNA-seq data. Nat. Methods. 2010;7:909–912. doi: 10.1038/nmeth.1517. [DOI] [PubMed] [Google Scholar]
- 6.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cloonan N, Forrest AR, Kolle G, Gardiner BB, Faulkner GJ, Brown MK, Taylor DF, Steptoe AL, Wani S, Bethel G, et al. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat. Methods. 2008;5:613–619. doi: 10.1038/nmeth.1223. [DOI] [PubMed] [Google Scholar]
- 8.Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008;18:1509–1517. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 10.Sultan M, Schulz MH, Richard H, Magen A, Klingenhoff A, Scherf M, Seifert M, Borodina T, Soldatov A, Parkhomchuk D, et al. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science. 2008;321:956–960. doi: 10.1126/science.1160342. [DOI] [PubMed] [Google Scholar]
- 11.Pickrell JK, Pai AA, Gilad Y, Pritchard JK. Noisy splicing drives mRNA isoform diversity in human cells. PLoS Genet. 2010;6:e1001236. doi: 10.1371/journal.pgen.1001236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kent WJ. BLAT–the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang K, Singh D, Zeng Z, Coleman SJ, Huang Y, Savich GL, He X, Mieczkowski P, Grimm SA, Perou CM, et al. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 2010;38:e178. doi: 10.1093/nar/gkq622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bryant DW, Jr, Shen R, Priest HD, Wong WK, Mockler TC. Supersplat–spliced RNA-seq alignment. Bioinformatics. 2010;26:1500–1505. doi: 10.1093/bioinformatics/btq206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Au KF, Jiang H, Lin L, Xing Y, Wong WH. Detection of splice junctions from paired-end RNA-seq data by SpliceMap. Nucleic Acids Res. 2010;38:4570–4578. doi: 10.1093/nar/gkq211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dimon MT, Sorber K, DeRisi JL. HMMSplicer: a tool for efficient and sensitive discovery of known and novel splice junctions in RNA-Seq data. PLoS One. 2010;5:e13875. doi: 10.1371/journal.pone.0013875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang L, Wang X, Wang X, Liang Y, Zhang X. Observations on novel splice junctions from RNA sequencing data. Biochem. Biophys. Res. Commun. 2011;409:299–303. doi: 10.1016/j.bbrc.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y, Lameijer EW, t Hoen PA, Ning Z, Slagboom PE, Ye K. PASSion: a pattern growth algorithm-based pipeline for splice junction detection in paired-end RNA-Seq data. Bioinformatics. 2012;28:479–486. doi: 10.1093/bioinformatics/btr712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Whitfield CW, Cziko AM, Robinson GE. Gene expression profiles in the brain predict behavior in individual honey bees. Science. 2003;302:296–299. doi: 10.1126/science.1086807. [DOI] [PubMed] [Google Scholar]
- 22.Liang ZS, Nguyen T, Mattila HR, Rodriguez-Zas SL, Seeley TD, Robinson GE. Molecular determinants of scouting behavior in honey bees. Science. 2012;335:1225–1228. doi: 10.1126/science.1213962. [DOI] [PubMed] [Google Scholar]
- 23.Ament SA, Wang Y, Chen CC, Blatti CA, Hong F, Liang ZS, Negre N, White KP, Rodriguez-Zas SL, Mizzen CA, et al. The transcription factor ultraspiracle influences honey bee social behavior and behavior-related gene expression. PLoS Genet. 2012;8:e1002596. doi: 10.1371/journal.pgen.1002596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Foret S, Kucharski R, Pellegrini M, Feng S, Jacobsen SE, Robinson GE, Maleszka R. DNA methylation dynamics, metabolic fluxes, gene splicing, and alternative phenotypes in honey bees. Proc. Natl Acad. Sci. USA. 2012;109:4968–4973. doi: 10.1073/pnas.1202392109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jarosch A, Stolle E, Crewe RM, Moritz RF. Alternative splicing of a single transcription factor drives selfish reproductive behavior in honeybee workers (Apis mellifera) Proc. Natl Acad. Sci. USA. 2011;108:15282–15287. doi: 10.1073/pnas.1109343108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burset M, Seledtsov IA, Solovyev VV. Analysis of canonical and non-canonical splice sites in mammalian genomes. Nucleic Acids Res. 2000;28:4364–4375. doi: 10.1093/nar/28.21.4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pertea M, Lin X, Salzberg SL. GeneSplicer: a new computational method for splice site prediction. Nucleic Acids Res. 2001;29:1185–1190. doi: 10.1093/nar/29.5.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie. J. Comput. Biol. 1997;4:311–323. doi: 10.1089/cmb.1997.4.311. [DOI] [PubMed] [Google Scholar]
- 29.Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- 30.Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004;11:377–394. doi: 10.1089/1066527041410418. [DOI] [PubMed] [Google Scholar]
- 31.Ter-Hovhannisyan V, Lomsadze A, Chernoff YO, Borodovsky M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 2008;18:1979–1990. doi: 10.1101/gr.081612.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lomsadze A, Ter-Hovhannisyan V, Chernoff YO, Borodovsky M. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 2005;33:6494–6506. doi: 10.1093/nar/gki937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parra G, Blanco E, Guigo R. GeneID in Drosophila. Genome Res. 2000;10:511–515. doi: 10.1101/gr.10.4.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stanke M, Waack S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics. 2003;19(Suppl. 2):ii215–ii225. doi: 10.1093/bioinformatics/btg1080. [DOI] [PubMed] [Google Scholar]
- 35.Kan Z, Rouchka EC, Gish WR, States DJ. Gene structure prediction and alternative splicing analysis using genomically aligned ESTs. Genome Res. 2001;11:889–900. doi: 10.1101/gr.155001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Staden R. Methods to define and locate patterns of motifs in sequences. Comput. Appl. Biosci. 1988;4:53–60. doi: 10.1093/bioinformatics/4.1.53. [DOI] [PubMed] [Google Scholar]
- 37.Thanaraj TA. Positional characterisation of false positives from computational prediction of human splice sites. Nucleic Acids Res. 2000;28:744–754. doi: 10.1093/nar/28.3.744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu X, Goldberg AB. Introduction to semi-supervised learning. Synth. Lect. Artif. Intell. Mach. Learn. 2009;3:1–130. [Google Scholar]
- 39.Celeux G, Govaert G. A classification EM algorithm for clustering and two stochastic versions. Comput. Stat. Data An. 1992;14:315–332. [Google Scholar]
- 40.Amini MR, Gallinari P. In 15th European Conference on Artificial Intelligence. IOS Press; 2002. Semi-supervised logistic regression; pp. 390–394. [Google Scholar]
- 41.Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 43.Gonzalez-Porta M, Calvo M, Sammeth M, Guigo R. Estimation of alternative splicing variability in human populations. Genome Res. 2012;22:528–538. doi: 10.1101/gr.121947.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weinstock GM, Robinson GE, Gibbs RA, Worley KC, Evans JD, Maleszka R, Robertson HM, Weaver DB, Beye M, Bork P, et al. Insights into social insects from the genome of the honeybee Apis mellifera. Nature. 2006;443:931–949. doi: 10.1038/nature05260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Elsik CG, Mackey AJ, Reese JT, Milshina NV, Roos DS, Weinstock GM. Creating a honey bee consensus gene set. Genome Biol. 2007;8:R13. doi: 10.1186/gb-2007-8-1-r13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463: 457–463. doi: 10.1038/nature08909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chandrasekaran S, Ament SA, Eddy JA, Rodriguez-Zas SL, Schatz BR, Price ND, Robinson GE. Behavior-specific changes in transcriptional modules lead to distinct and predictable neurogenomic states. Proc. Natl Acad. Sci. USA. 2011;108:18020–18025. doi: 10.1073/pnas.1114093108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. (2009) Genome 10 K: a proposal to obtain whole-genome sequence for 10,000 vertebrate species. J. Hered, 100, 659–674. [DOI] [PMC free article] [PubMed]
- 49.Robinson GE, Hackett KJ, Purcell-Miramontes M, Brown SJ, Evans JD, Goldsmith MR, Lawson D, Okamuro J, Robertson HM, Schneider DJ. Creating a buzz about insect genomes. Science. 2011;331:1386. doi: 10.1126/science.331.6023.1386. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.