

Abstract
A variety of antiprion compounds have been reported that are effective in ex vivo and in vivo treatment experiments. However, the molecular mechanisms for most of these compounds remain unknown. Here we classified antiprion mechanisms into four categories: I, specific conformational stabilization; II, nonspecific stabilization; III, aggregation; and IV, interaction with molecules other than PrPC. To characterize antiprion compounds based on this classification, we determined their binding affinities to PrPC using surface plasmon resonance and their binding sites on PrPC using NMR spectroscopy. GN8 and GJP49 bound specifically to the hot spot in PrPC, and acted as “medical chaperones” to stabilize the native conformation. Thus, mechanisms I was predominant. In contrast, quinacrine and epigallocathechin bound to PrPC rather nonspecifically; these may stabilize the PrPC conformation nonspecifically including the interference with the intermolecular interaction following mechanism II. Congo red and pentosan polysulfate bound to PrPC and caused aggregation and precipitation of PrPC, thus reducing the effective concentration of prion protein. Thus, mechanism III was appropriate. Finally, CP-60, an edarabone derivative, did not bind to PrPC. Thus these were classified into mechanism IV. However, their antiprion activities were not confirmed in the GT + FK system, whose details remain to be elucidated. This proposed antiprion mechanisms of diverse antiprion compounds could help to elucidate their antiprion activities and facilitate effective antiprion drug discovery.
Keywords: prion protein, anti-prion compounds, action mechanism, medical chaperones
Introduction
Transmissible spongiform encephalopathies (TSEs) are neurodegenerative diseases that include Creutzfeldt–Jakob disease, chronic wasting disease, scrapie, bovine spongiform encephalopathy, and others. Although the conversion of prion protein (PrP) from the normal cellular form (PrPC) to the misfolded scrapie form (PrPSc), and the accumulation of PrPSc in the central nervous system are the characteristic features of these diseases,1, 2 the detailed structure of PrPSc and the details of the conversion reaction remain unknown. The occurrence of TSEs is associated with specific mutations in PrP, inoculation with infectious material, or spontaneous onset. Because there are currently no established therapies, it is important to identify compounds with therapeutic or prophylactic activity against TSEs. The purpose of this study was to understand the various mechanisms of action of previously reported antiprion compounds and obtain further insights into optimizing their antiprion activities as well as the conversion mechanism of prion proteins.
To date, no purification method for a unique PrPSc strain has been fully established;3 thus, quantitative binding experiments for a single PrPSc conformer is not feasible. Ideal stoichiometric determinations of ligand binding akin to the oxygen–hemoglobin binding scheme4 are rather restricted because of the limitation of quantifying PrPSc.
In contrast, populations of PrPSc are considered to be kinetically regulated.5 If the reaction rate of the PrPC→PrPSc conversion could be successfully reduced to a certain level, the population of available PrPSc would gradually be reduced.6 Following this strategy, we can completely examine the binding properties of ligands with PrPC and evaluate their stabilization effects on PrPC, their interference effects on the interaction between PrPC and PrPSc, and such other effects. These activities are well represented by the notion of chemical chaperones.6
We previously reported a variety of antiprion compounds that were discovered based on the structures and dynamics of prion proteins.6, 7 Among these antiprion compounds, GN86 and its derivatives8 are unique because of their affinities for PrPC, their binding sites at atomic resolution, and their antiprion activities that were clarified in both ex vivo and in vivo experiments.6, 8 Other compounds have also been reported to bind to PrPC and inhibit its pathogenic conversion.7, 9 Several groups have also reported various antiprion compounds based on ex vivo and/or in vivo experiments.10–13 However, the details of their antiprion activities remain unknown. Thus, we attempted to characterize their antiprion mechanisms based on their binding properties to PrPC.
Although previous studies on antiprion compounds primarily emphasized the biological effects of these compounds (IC50), this study examined the various mechanisms of antiprion effects based on their binding properties to PrPC. Thus, this study may offer new insights into the mechanisms of prion diseases. To characterize these antiprion compounds, we determined their binding affinities to PrPC using surface plasmon resonance (SPR) and their binding sites on PrPC using NMR spectroscopy.
We selected 13 typical antiprion compounds, including GJP49, GJP14, quinacrine, Congo red, epigallocatechin gallate (EGCG), pentosan polysulfate (PPS), CP-60, Edaravone derivative 13, D-PEN, and Indole-3-glyoxylamide derivatives (see Table I). We classified these compounds into several classes based on their binding properties to PrPC. GJP49 and GJP14 are antiprion compounds that were discovered through in silico screening.7 Quinacrine, which has been used as an antimalarial drug for over 60 years, has also been shown to be an antiprion compound.10 Congo red, a dye used for staining amyloid fibrils, has been reported to have antiprion activity.14 EGCG is a polyphenolic compound in tea that has been reported to have antiprion activity,15 and PPS, which is used as a therapeutic agent for chronic cystitis, has also been reported to be an antiprion compound.16, 17 We also examined CP-60,11 edaravone derivative 13,18 D-PEN,19 and indole-3-glyoxylamide derivatives.20 In addition, we discuss a novel concept, “medical chaperone,” in the context of further optimization of these previously identified antiprion compounds.
Table I.
Antiprion Compounds Used in This Study
| Compound | Chemical structure | M.W. | Relative PrPSc levela (%) | Antiprion mechanism | References |
|---|---|---|---|---|---|
| GJP49 | 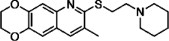 |
344.5 | 47.4 | I | 7 |
| GJP14 | 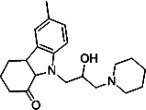 |
342.5 | 51.3 | I | 7 |
| Quinacrine | 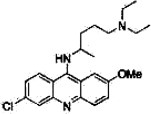 |
400 | Toxicb | II | 10 |
| Congo red | 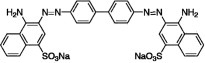 |
696.7 | 39.6 | II + III | 14 |
| Epigallo-catechin gallate (EGCG) |  |
458.4 | 65.6 | II + III | 15 |
| Pentosan polysulfate (PPS) | 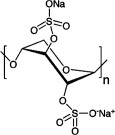 |
366.3n | NDc | II + III | 16, 17 |
| CP-60 | 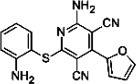 |
333.4 | 94.3 | IVd | 11 |
| Edaravone derivative 13 | 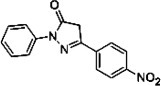 |
281.3 | 93.0 | IVd | 18 |
| D-PEN | 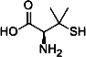 |
149.2 | 95.5 | IVd | 19 |
| Indole-3-glyoxylamide derivative 2 | 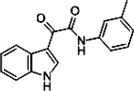 |
278.3 | 81.8 | IVd | 20 |
| Indole-3-glyoxylamide derivative 10 | 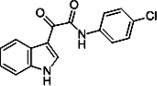 |
298.7 | 110 | IVd | 20 |
| Indole-3-glyoxylamide derivative 12 | 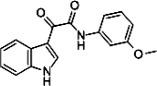 |
294.3 | 103 | IVd | 20 |
| Indole-3-glyoxylamide derivative 13 | 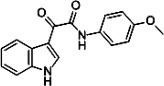 |
294.3 | Toxice | IVd | 20 |
Mean PrPSc level (%) using 10 μM of each compound on Fukuoka-1 strain-infected GT + FK cells.
Relative PrPSc level is 37.1 at 2 μM for quinacrine.
Though PrPSc level at 10 μM cannot determined because the compound concentration could not be determined, relative PrPSc level at 10 μg/mL is 24.4 for PPS.
Binding to PrPC nor antiprion activities in GT + FK system were not confirmed.
Relative PrPSc level is 144 at 5 μM for indole-3-glyoxylamide derivative 13.
Results
GJP49 and GJP14 specifically bind to PrPC
We previously reported the binding affinity of GJP49 for PrPC as determined by SPR.7 The SPR data were fit using a 1:1 binding model of equilibrium analysis, and the dissociation constant (KD) was estimated to be 50.8 μM. To identify the interaction sites on PrPC for GJP49, we measured the 1H-15N HSQC spectra with or without GJP49 and superimposed them, as shown in Figure 1(A). We also calculated the chemical shift perturbations (Δδ) of 1H and 15N nuclei in PrPC upon binding with this compound [Fig. 1(B)], and significantly perturbed PrPC residues were mapped onto the three dimensional structure of mouse PrP (PDB ID: 1AG2). Figure 1(E) shows the specific binding of GJP49 to the C-terminal region of helix B and at part of the B-C loop of PrPC.
Figure 1.
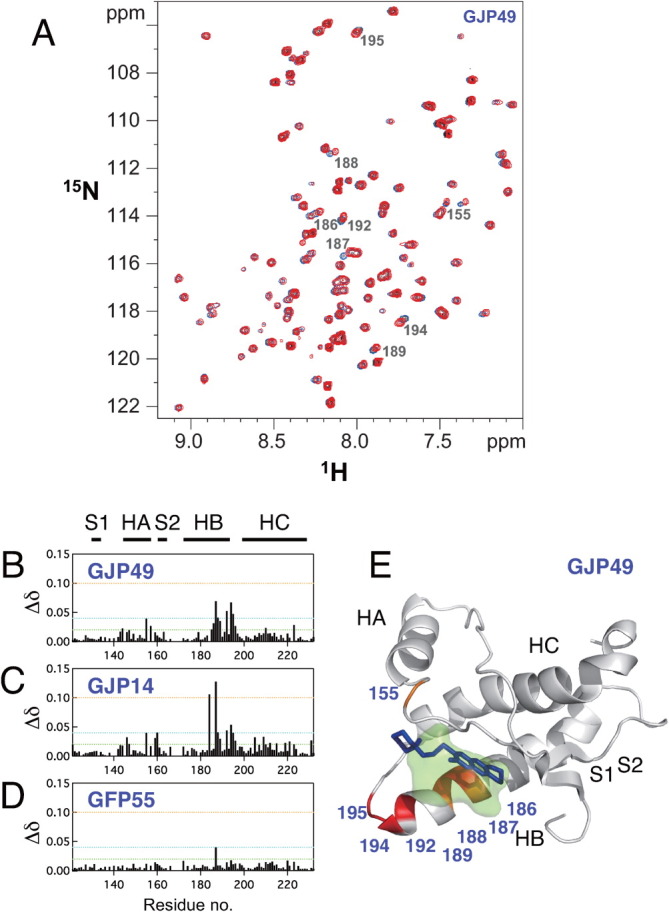
Characteristics of the binding sites for the antiprion compound GJP49. GJP49 was discovered by in silico screening using Autodock.7 (A) 1H-15N HSQC spectra of 15N-labeled recombinant prion protein, mouse PrP(121–231) (33 μM), with (red) or without (blue) GJP49 (500 μM). (B–D) Plots of chemical shift perturbations (Δδ = [(Δδ1H)2 + 0.17(Δδ15N)2]1/2) as a function of the residue number. (E) Mapping of the significantly perturbed residues on the three-dimensional structure of the prion (1AG2). The perturbed residues with Δδ values of >0.04 ppm are shown in red, and those with 0.4 < Δδ < 0.3 ppm are shown in orange. The binding pocket is overlaid in green. S1, HA, S2, HB, and HC indicate S1 strand, helix A, S2 strand, helix B, and helix C, respectively. The image was created using PyMol.
We also examined the interaction sites for GJP14, another effective antiprion compound that was found by in silico screening.7 This compound also bound to the same region of PrPC [Fig. 1(C)]. The region of PrPC that binds GJP49 and GJP14 corresponds to that of GN8, another antiprion compound that we examined in a previous article.6 This region of PrPC undergoes a global fluctuation on a time scale of micro- to milliseconds.21
As a negative control for these binding experiments, we also examined the interaction sites of GFP55, which belongs to a low-binding, ineffective group of compounds.7 In contrast to GJP49 and GJP14, the addition of GFP55 did not result in a significant chemical shift change [Fig. 1(D)]. Therefore, we considered that GJP49 and GJP14 specifically bound to a flexible pocket of PrPC and inhibited the pathogenic conversion of PrPC to PrPSc.
Quinacrine binds to PrPC in a nonspecific manner
We analyzed SPR responses to examine the specificity of the interaction between quinacrine and PrPC [Fig. 2(A)]. The SPR response increased as the concentration of test compound increased, without any trend toward saturation [inset in Fig. 2(A)]. The SPR response for quinacrine could not be fit using a simple binding model, which suggested nonspecific adhesion possibly due to strong hydrophobic interactions.
Figure 2.

Surface plasmon resonance sensorgrams for various antiprion compounds. (A) Quinacrine; the concentrations from bottom to top are: 0.781, 1.56, 3.13, 6.25, 12.5, 25, 50, and 100 μM. (B) Epigallocathechin gallate; the concentrations from bottom to top are: 3.91 7.81, 15.6, 31.3, 62.5, and 125 μM. (C) Congo red; the concentrations from bottom to top are: 1.56, 3.13, 6.25, 12.5, 25, 50, and 100 μM. (D) Pentosan polysulfate; the concentrations is: 1.95 μg/mL. (E–H) CP60, Edarabone derivative 13, D-PEN, and indole-3-glyoxylamide derivative 10; the concentrations from bottom to top are: 0.75, 1.25, 2.5, 5.0, 10, 20, and 40 μM. The symbol * indicates spike noise. Insets show the compound concentration dependence of the Biacore response. For quinacrine, Congo red, and pentosan polysulfate binding experiments, recombinant mouse PrP(121–231) was fixed on the surface of the sensor chip. For epigallocatechin gallate, CP60, edarabone derivative 13, D-PEN, and indole-3-glyoxylamide derivative 10 binding experiments, recombinant mouse PrP(23–231) was fixed on the surface of the sensor chip.
It has been reported that quinacrine accumulates in the brain after long-term administration.22 In addition to PrPC binding, quinacrine may also bind to other proteins without being degraded, which would cause the observed accumulation in the brain and significant side effects. To characterize the interaction sites between quinacrine and PrPC, we obtained the chemical shift perturbations (Δδ) from the 1H-15N HSQC spectra [Fig. 3(C)] and mapped these onto the three dimensional structure [Fig. 3(D)]. When quinacrine was added to a 30.6 μM solution of PrPC at a final concentration of 500 μM, no significant change in chemical shift was observed. When we increased the concentration of quinacrine (to 5.3 mM), the chemical shift perturbations increased to >0.04 for residues covering the entire molecular surface, particularly around parts of helices A, B, and C, including residues Y225, Y226, and D227. This result strongly suggested a nonspecific interaction.
Figure 3.

Chemical shift perturbations (Δδ) as a function of residue number and the mapping of Δδ onto the three dimensional structure of recombinant mouse PrP(121–231) (PDB ID: 1AG2). (A, B) GJP49, (C, D) quinacrine, and (E, F) epigallocatechin gallate (EGCG). Final concentrations of the protein and compounds were, respectively, 31 μM and 500 μM for GJP49 and EGCG binding experiments and 32 μM and 5.3 mM for quinacrine binding experiment.
Based on the results of an ex vivo assay using the GT + FK cell line,23 the IC50 values for GN8 and quinacrine were 1.40 μM and 1.11 μM, respectively.8 The mean Δδ values with GN8 at 1.0 mM and PrPC at 26 μM roughly corresponded to those for quinacrine at 5.3 mM and PrPC at 32.4 μM, respectively. Although their IC50 values were close, quinacrine required an approximately fourfold greater concentration than GN8 to reach the same Δδ level in its NMR spectrum.
This discrepancy can be explained by the differences in the specificities of the ligand–protein interaction (i.e., the binding regions for quinacrine are broadly distributed over the entire protein surface). In addition, the local structural stability of the ligand–protein complex may be important because structural fluctuations may reduce the Δδ values through an averaging effect. Quinacrine has also been reported to be transported into the intracellular space by endocytosis; lysosomes have a 10,000 times greater concentration of quinacrine than what is found in the extracellular space.24 Thus, locally concentrated quinacrine may have been able to interact with PrPC in the ex vivo experiment.
EGCG binds to PrPC in a nonspecific manner and partially aggregates PrPC
Although we measured the SPR responses of PrPC upon binding with EGCG [Fig. 2(B)], the interaction was too strong to reverse the sensorgram to its original level, even after an exhaustive washing and subsequent recovery procedure. Before NMR determinations, we found that mixing PrPC and EGCG at a molar ratio of 1:10 resulted in precipitating 80% of the PrPC (data not shown). When we measured the 1H-15N HSQC spectrum for PrPC and EGCG at a molar ratio of 1:8.2, we observed large chemical shift perturbations (Δδ) for the regions that diffusely covered the entire molecule [Fig. 3(F)], which indicated nonspecific binding. Although EGCG binds to PrPC, which forms aggregates and causes the precipitation of PrPC, a significant fraction of this complex remains in the soluble phase.
Congo red promotes aggregation and reduces the concentration of PrPC
Affinity analysis showed that increased concentrations of Congo red resulted in significantly increased SPR responses [Fig. 2(C)] without any significant saturation effect [inset in Fig. 2(C)]. Thus, Congo red nonspecifically adheres to PrPC. In addition, PrPC was precipitated as the concentration of Congo red was increased in the NMR tube [Fig. 4(A)]. To characterize this interaction at the atomic level, we measured the NMR spectrum for a sample of PrPC and Congo red at a molar ratio of 1:2, but we could not observe any significant chemical shift perturbation (Δδ; data not shown). When we increased the molar ratio of PrPC and Congo red to 1:16.3, PrPC was precipitated [Fig. 4(A)] and the signal heights were shifted by 20% [Fig. 4(B)].
Figure 4.
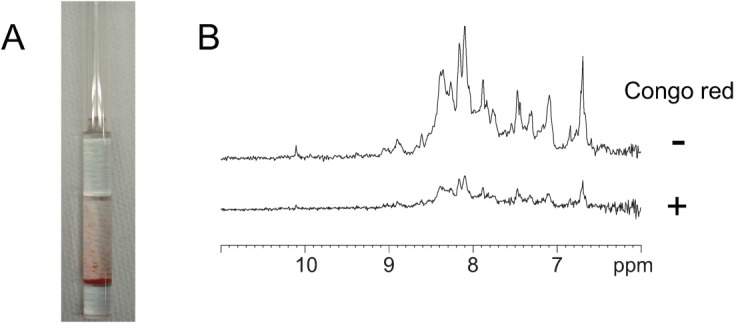
Aggregation and precipitation of mouse PrP(121–231) by Congo red. (A) Precipitation of mouse PrP(121–231) by Congo red in a Shigemi tube. (B) 1D 1H NMR spectra with (lower trace) and without (upper trace) Congo red.
Consequently, we could not obtain a high quality two-dimensional spectrum for the chemical shift perturbation. Thus, under in vivo conditions, Congo red binds nonspecifically to PrPC and promotes its aggregation. In addition, even at a lower PrPC concentration, this would reduce the amount of available PrPC required for the conversion reaction. Congo red also induced aggregation of hen lysozyme and reduced the protein concentration (Supporting Information Fig. 1), indicating that the aggregation by Congo red is not specific to PrPC.
PPS promotes aggregation and reduces the concentration of PrPC
Affinity analysis showed that increased concentrations of PPS resulted in significantly increased SPR responses, and incomplete dissociation [Fig. 2(D)]. The amount of PPS that was bound was greater for full-length PrP, but PPS did significantly bind to the C-terminal domain of this protein (data not shown). Thus, PPS adheres strongly and nonspecifically to PrPC. In addition, PrPC was precipitated as the concentration of PPS was increased (data not shown), as was observed with PrPC and Congo red. Thus, under in vivo conditions, PPS nonspecifically binds to PrPC and promotes its aggregation. PPS induced aggregation of hen lysozyme and reduced the protein concentration (Supporting Information Table I), indicating that the aggregation by PPS is not specific to PrPC.
CP-60, Edaravone derivative 13, D-PEN, and indole-3-glyoxylamide derivatives do not bind to PrPC
The responses on the SPR sensorgrams using mouse PrP(23–231) and CP-60 [Fig. 2(E)], edaravone derivative 13 [Fig. 2(F)], D-PEN [Fig. 2(G)], indole-3-glyoxylamide derivative 10 [Fig. 2(H)], and indole-3-glyoxylamide derivatives 2, 12, and 13 (data not shown) were all minimal. Thus, these data suggested that these compounds did not directly interact with either the C- or N-terminal domain of PrPC. As shown in Table I, CP-60, edaravone derivative 13, D-PEN, and indole-3-glyoxylamide derivatives did not inhibit PrPSc formation. The mean PrPSc levels using 10 μM of each of these compounds were 94.3, 93.0, 95.5, 81.8, 110, and 103% for CP-60, edaravone derivative 13, D-PEN, and indole-3-glyoxylamide derivatives 2, 10, and 12, respectively. Indole-3-glyoxylamide derivative 13 was toxic at a concentration of 10 μM. Thus we could not confirm the antiprion activities for these compounds. This could depend on the strain used for the ex vivo assay.
In this study, we used GT + FK cells that produce significant amounts of PrPres as well as PrPC. Hence the assay system using GT + FK cells is reliable, reproducible, and less sensitive to minor perturbations such as those due to the culture conditions.9 However, it has been reported that administering these compounds to prion-infected mice days after the onset of their symptoms extended their life spans.19 Although these compounds are reported to have antiprion activities, we could not confirm them. In other systems, they may possibly interact with the intermediate prion protein (PrP*), PrPSc, membrane proteins, or other relevant proteins,25 and may regulate a prion's toxicity.
Discussion
Classifying antiprion mechanisms
Although there have been many reports on antiprion compounds, their mechanisms of action have not been clearly defined. In particular, comparisons of the mechanisms between various antiprion compounds are not available. Antiprion compounds can be classified as shown in Table II and Figure 5 based on their binding properties to PrPC. We classified antiprion activities into four categories: I, specific conformational stabilization of PrPC;6 II, nonspecific stabilization including the interference with the interaction between PrPC and PrPSc as well as the contribution of binding to the hot spots; III, aggregation and precipitation to reduce the amount of available PrPC; and IV, interactions with molecules other than PrPC. Here, we examined the activities of representative antiprion compounds (GJP49, GJP14, quinacrine, EGCG, Congo red, PPS, CP-60, edaravone derivative 13, D-PEN, and indole-3-glyoxylamide derivatives) and characterized their antiprion activities based on their binding properties with PrPC.
Table II.
Classification of the Antiprion Mechanisms
| Class | Mechanism | Biacore | NMR |
|---|---|---|---|
| I | Specific conformational stabilization of PrPC | Specific binding | Specific binding |
| II | Nonspecific stabilization of PrPC | Nonspecific binding | Nonspecific binding |
| III | Aggregation and precipitation to reduce PrPC population | Nonspecific binding | Not detectable |
| IV | Interaction with molecules other than PrPC | No binding | No interaction |
Figure 5.
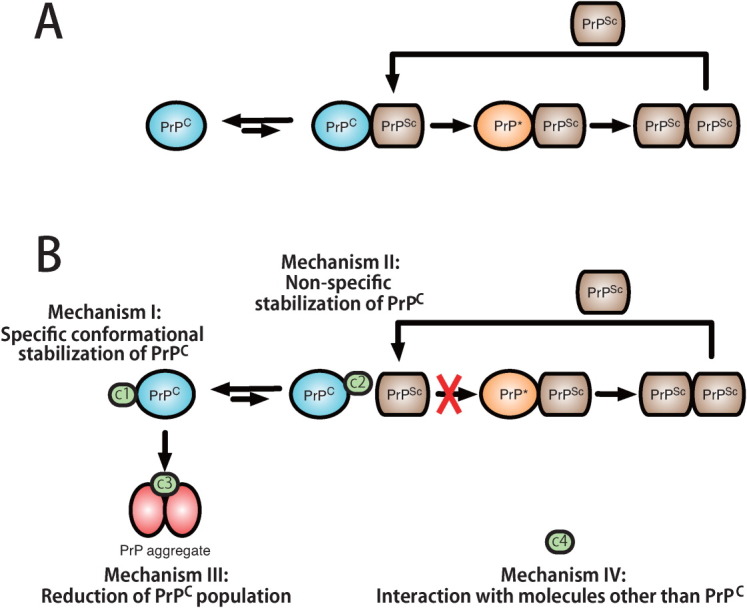
Illustration of the pathogenic conversion process from PrPC to PrPSc in the absence (A) and presence (B) of antiprion compounds and the classification of antiprion mechanisms based on interactions with PrPC. Mechanism I molecules (c1), designated “medical chaperones,” stabilize the PrPC conformation. Mechanism II molecules (c2) bind to PrPC nonspecifically to stabilize the PrPC and interfere with the interaction between PrPC and PrPSc. Mechanism III molecules (c3) induce prion protein aggregation and cause precipitation, which reduces the amount of PrPC. Mechanism IV molecules (c4) interact with molecules other than PrPC, such as PrPSc, PrP*, or membrane proteins.
Characterizing antiprion compounds
GN8 and GJP49 directly bound to PrPC at specific binding sites on PrPC and inhibited its pathogenic conversion. These binding regions are shown in Figure 1(B,E), and are essentially the same as those for GN8 (i.e., hot spot).6 Specific binding to this region is associated with mechanisms I.
In contrast, quinacrine and EGCG bound nonspecifically to PrPC as shown in Figure 3(C,E). To check correlation between the Δδ values for each compound, we compared Δδ values for each residue between each compound (Supporting Information Fig. 2). However, except GJP14-GJP49, clear correlation could not be observed. This also indicates the nonspecific binding property of quinacrine and EGCG. This nonspecific binding may be attributed to mechanism II. For conformational stabilization, binding to the specific hot spot that is responsible for conformational instability is required, while for interference between PrPC and PrPSc, nonspecific binding to PrPC would be allowed to some extent because they mutually interact with large contact areas that form oligomers,26 amyloid fibrils,27 or nonspecific aggregates28 depending on the characteristics of the relevant strains. Hence, small compounds can interfere with these interactions at various nonspecific sites. However, these binding regions of these compounds also included the hot spots, therefore, there may be partially attributed to mechanism I.
Congo red and PPS induced PrPC aggregation and reduced its effective concentration. In general, many small compounds inhibit amyloid formation by binding with amyloidogenic proteins via π–π interactions and form aggregates.29, 30 Thus, their mechanisms of action are basically described by mechanism III. However, the nonspecific binding properties of these compounds indicate that they may also act by mechanism II, especially at low concentrations of these compounds and protein.
Finally, CP-60, Edaravone derivative 13, D-PEN, and indole-3-glyoxylamide derivatives do not bind to PrPC, and we could not confirm their antiprion activities. Though in other system, they may possibly interact with other proteins or membranes and thus, indirectly reduce the amount of PrPSc. Thus, mechanism IV could be assigned.
Compounds associated with mechanism I share a common binding site
We compared the binding sites for the compounds that bound directly to PrPC and found that GJP49, GJP14, quinacrine, and EGCG bound to a common site, the C-terminal region of helix B and the B-C loop, which is shown in Figure 3(A,C,E) (blue boxes) and Figure 3(B,D,F) (blue oval shapes). In a previous study, we demonstrated that GN8 also bound to this region.6, 8 This region is known to undergo global fluctuations on a time scale of micro- to milliseconds.6 Each of the compounds examined here with antiprion activity via direct binding to PrPC had affinity for this region. Thus, these compounds can be referred to as “medical chaperones” that stabilize the native conformation of the target protein and inhibit its transition to the abnormal conformation, PrPSc.
Medical chaperones can be considered to suppress the seismic fluctuations of a protein (i.e., protein quakes),31, 32 which results in destroying its native structure. Among the compounds examined in this study, GJP49 specifically bound to the common binding site. Thus, GJP49 may not induce any side effects and could be a candidate therapeutic agent for prion diseases.
It has been assumed that factor X is required for the pathogenic conversion of PrP,25 although factor X has not yet been identified. It is reported that quinacrine binds to Tyr225, Tyr226, and Gln227 (Asp227 for mouse)24 which is located near the factor X” binding sites.33 As shown in Figure 3(D), Δδ values in the presence of 2.67 mM quinacrine indicated that Tyr225, Tyr226, and Gln227 were also involved in the binding with quinacrine. Thus, quinacrine may inhibit the pathogenic conversion of PrP by competitive binding with factor X. However, it must be noted that even the maximum Δδ values for Tyr226 at this site were comparable to that at other quinacrine binding sites [Fig. 3(C)], and that this site was not a common binding site for the other compounds (Fig. 3).
Medical chaperones
It is considered that a high-energy barrier exists between PrPC and PrPSc,34, 35 and that prion proteins in the PrPC state rarely overcome this barrier; they only achieve the PrPSc state when triggered by some unknown causes. Compounds in the first and second classes (GN8, GJP49, GJP14, quinacrine, and EGCG) bound to the residues surrounding the major binding pocket, which may be the cause of the seismic fluctuations that lead to pathogenic conversion. Medical chaperones prevent these protein quakes and suppress these fluctuations. Thus, these would stabilize the PrPC conformation (i.e., reduce the free energy level of PrPC), which would result in an increase in the energy barrier between PrPC and PrPSc.36 Therefore, these would suppress the pathogenic conversion of PrPC to PrPSc.
Although the PrPC conformations are nearly identical between species, PrPSc conformations are quite heterogeneous and are referred to as different “strains.” Strain conformation is conserved after transmission between individuals.37, 38 Antiprion compounds that vary significantly in their efficiency, depending on the strain,39 may directly affect the PrPSc conformation rather than the PrPC conformation. A great advantage of a “medical chaperone” is that its effect is strain-independent6, 40 because it acts on PrPC which has a common structure in nearly all mammals.
Medical chaperones also act by mechanism II, interference with the interaction between PrPC and PrPSc, as described in the following section.
Interference with the interaction between PrPC and PrPSc during the template-dependent self-replication process
PrPC interacts with PrPSc during the process of pathogenic conversion. Compounds that bind to PrPC nonspecifically can interfere with the interaction between PrPC and PrPSc. Although there have been no evidence for the direct interference of the interaction between PrPC and PrPSc, this mechanism constructs the critical difference from the originally proposed chemical chaperone for the misfolding diseases.41 Specific binding of a medical chaperone with this function could be advantageous in clinical use by reducing side effects.
The possibility of administering a cocktail of antiprion compounds
We classified 13 different antiprion compounds into four classes based on their antiprion mechanisms of action. Compounds associated with different mechanisms of action can be administered together (e.g., GJP49 and indole-3-glyoxylamide derivative 13). GJP49 binds specifically to PrPC and suppresses the available population in the intermediate state (PrP*), while indole-3-glyoxylamide derivative 13 does not bind to PrPC, which indicates that this compound interacts with molecules other than PrPC. Indole-3-glyoxylamide derivative 13 reduced PrPSc formation at the nanomolar concentration range when it was screened using a scrapie infected mouse cell line (SMB).42 Cocktail administration of antiprion compounds might possibly reduce the concentration of each compound, which could decrease their associated side effects. Cocktail administration has also been reported for HIV43, 44 and yeast prions,45 and could be useful for mammalian prion diseases.
Toward further optimization of chemical structures
GJP49, GJP14, quinacrine, and EGCG bind to the major pocket of PrPC and contribute to its structural stabilization. We need to quantitatively evaluate the degree of stabilization using relaxation measurements for PrPC, particularly around the major pocket in the presence of each compound. We also need to determine the structures of complexes using X-ray crystallography. This dynamical and structural information will be important for the rational optimization of antiprion compounds.
A specific interaction at the common binding site is critical for antiprion efficiency according to mechanism I. Specific interactions may also be advantageous for reducing side effects. Therefore, selecting specifically interacting derivatives may be important for compound optimization. For this purpose, we may able to use the Δδ values from NMR as shown in Figure 3. For example, quinacrine binds to PrPC in a nonspecific manner [Figs. 2(A) and 3(C,D)] and has been reported to be toxic22 (Table I). By selecting specifically interacting derivatives of quinacrine, we may be able to find compounds that have fewer side effects than quinacrine.
It should be noted that the above discussion was focused entirely on the interactions between compounds and PrPC. When the interactions of compounds with PrPSc or the cell surface can be monitored, we will obtain additional insights into the detailed mechanisms of prion diseases, which will aid in the development of therapeutics.
Conclusion
Elucidating the molecular mechanisms of action of previously identified antiprion compounds is one of the most important steps for developing novel antiprion drugs and optimizing the most promising compounds. When this strategy (i.e., “medical chaperones”) has been established for prion diseases, it could also be applied to the rational design of drugs for other neurodegenerative diseases, such as Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis.
Materials and Methods
Compounds
Compounds GJP49, GJP14, GJP55, and Indole-3-glyoxylamide derivatives were purchased from ASINEX (Moscow, Russia). Quinacrine, Congo red, and EGCG were purchased from Sigma-Aldrich (Japan, Tokyo). PPS, CP-60, Edaravone derivative 13, and D-PEN were obtained from bene-Arzneimittel GmbH (Munich, Germany), Ambinter (Orléans, France), Labotest (Niederschöna, Germany), and Wako Pure Chemical Industries (Osaka, Japan), respectively.
Recombinant mouse PrP
An expression plasmid for recombinant mouse PrP residues 23–231 [PrP(23–231)] was prepared according to a previously described protocol.6 An expression plasmid for mouse PrP residues 121–231 [PrP(121–231)] was a kind gift from Professor Kurt Wüthrich and Dr. Simone Hornemann.46 Recombinant PrP was prepared as previously described.6 The concentrations of mouse PrP(23–231) and PrP(121–231) were estimated by absorbance at 280 nm using specific absorbances (ε280) = 2.68 and 1.49 (mg/mL)−1 cm−1, respectively.
SPR measurements
Interactions between prion proteins and the different compounds were analyzed using the Biacore T200 system (GE Healthcare, Buckinghamshire, UK). Recombinant mouse PrP was immobilized on a sensor chip (CM5) according to the manufacturer's instructions. Varying concentrations of each compound were injected into the running buffer (10 mM HEPES buffer, pH 7.4, containing 0.15 M NaCl, 0.1% Surfactant P20, and 5% DMSO) for 1 to 2 min at a flow rate of 30 mL/min. Running buffer without compounds was then injected for 10 min at the same flow rate. Data were corrected by subtracting the response of blank sensor chip from that of protein-bound sensor chip, so the contribution of nonspecific binding of the compounds to SPR chip surface was removed.
Nuclear magnetic resonance (NMR) measurements and data analysis
For NMR measurements, mouse PrP(121–231) uniformly labeled with 15N was prepared in 30 mM acetate-d3 buffer (pH 4.5) containing 1 mM NaN3, 4.5 μM AEBSF, 20 μM EDTA, 0.4 μM Bestatin, 0.06 μM pepstatin, 0.06 μM E-64, and 1 nM DSS dissolved in 90% H2O/10% D2O. NMR spectra were recorded at 20.0°C on a Bruker Avance600 spectrometer (Bruker BioSpin, Rheinstetten, Germany) at Gifu University. The spectrometer operated at a 1H frequency of 600.13 MHz and a 15N frequency of 60.81 MHz. A 5-mm 1H inverse detection probe with triple-axis gradient coils was used for all measurements. 1H-15N HSQC spectra were acquired with 2048 complex points covering 9600 Hz for 1H and 256 complex points covering 1200 Hz for 15N. NMR data were processed using the TOPSPIN software package (Bruker BioSpin, Rheinstetten, Germany) and NMR assignment and integration software Sparky.47 Resonance frequencies in these spectra were identified using the chemical shift lists for mouse PrP(121–231).48 The backbone 1H and 15N chemical shifts for a compound-bound protein were assigned by tracing the corresponding peaks in 1H-15N HSQC spectra determined with varying concentrations of the compounds. The protein structures were generated using PyMOL Molecular Graphics System Version 0.99rc9 (Schrödinger, LLC, NY).
Ex vivo assay for antiprion activity
An ex vivo assay was performed as described previously.7 Briefly, we used an immortalized neuronal mouse cell line that was persistently infected with a human TSE agent (Fukuoka-1 strain),23 which was designated GT + FK. This cell line was grown and maintained in 5% CO2 at 37°C in D-MEM (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (Equitech-bio, Kerrville, TX), 50 U/mL penicillin G sodium, and 50 μg/mL streptomycin sulfate (Invitrogen, Carlsbad, CA). Stock solutions of the test compounds were prepared fresh in 100% DMSO at a concentration of 10 mM and stored at 4°C. Before use, the test compounds were diluted with medium to a concentration of 10 μM. Control cells were treated with medium containing solvent alone (0.1%). Approximately 3.0 × 105 cells were plated in each well of a six-well plate, and treatment with a test compound was started 15 h later. After 72 h of treatment, cells were lysed in 150 μL of 1× Triton X-100/DOC lysis buffer.49 Western blotting for PrPSc was done as described previously.7
Acknowledgments
The authors thank Professor Kurt Wüthrich and Dr. Simone Hornemann for kindly providing them with the expression plasmid for mouse PrP(121–231). Pentosan polysulfate was kindly provided by Professor Masami Niwa and Professor Noriyuki Nishida. The authors thank Dr. Tomoharu Matsumoto, Ms. Junko Matsubara, and Ms. Miki Horii for their assistance with protein sample preparation. The authors thank Ms. Tomomi Saeki for technical help with evaluating antiprion activity.
Glossary
Abbreviations
- AEBSF
4-(2-Aminoethyl) benzenesulfonyl fluoride hydrochloride
- DMSO
dimethyl sulfoxide
- DSS
4,4-dimethyl-4-silapentane-1-sulfonic acid
- EDTA
ethylenediaminetetraacetic acid
- EGCG
epigallocatechin gallate
- HSQC
Heteronuclear Single Quantum Coherence
- IC50
inhibitory concentration for 50% reduction of PrPSc
- KD
dissociation constant
- PPS
pentosan polysulfate
- PrP
prion protein
- PrP(23–231)
PrP residues 23–231
- PrP(121–231)
PrP residues 121–231
- PrP*
the intermediate state between PrPC and PrPSc
- PrPC
the cellular form of PrP
- PrPSc
the scrapie form of PrP
- SPR
surface plasmon resonance
- TSEs
transmissible spongiform encephalopathies
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Prusiner SB. Prions. Proc Natl Acad Sci USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chesebro B. Prion protein and the transmissible spongiform encephalopathy diseases. Neuron. 1999;24:503–506. doi: 10.1016/s0896-6273(00)81105-8. [DOI] [PubMed] [Google Scholar]
- 3.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 4.O'Riordan JF, Goldstick TK, Ditzel J, Ernest JT. Characterization of oxygen-hemoglobin equilibrium curves using nonlinear regression of the Hill equation: parameter values for normal human adults. Adv Exp Med Biol. 1983;159:435–444. doi: 10.1007/978-1-4684-7790-0_37. [DOI] [PubMed] [Google Scholar]
- 5.Harrison PM, Bamborough P, Daggett V, Prusiner SB, Cohen FE. The prion folding problem. Curr Opin Struct Biol. 1997;7:53–59. doi: 10.1016/s0959-440x(97)80007-3. [DOI] [PubMed] [Google Scholar]
- 6.Kuwata K, Nishida N, Matsumoto T, Kamatari YO, Hosokawa-Muto J, Kodama K, Nakamura HK, Kimura K, Kawasaki M, Takakura Y, Shirabe S, Takata J, Kataoka Y, Katamine S. Hot spots in prion protein for pathogenic conversion. Proc Natl Acad Sci USA. 2007;104:11921–11926. doi: 10.1073/pnas.0702671104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hosokawa-Muto J, Kamatari YO, Nakamura HK, Kuwata K. Variety of antiprion compounds discovered through an in silico screen based on cellular-form prion protein structure: Correlation between antiprion activity and binding affinity. Antimicrob Agents Chemother. 2009;53:765–771. doi: 10.1128/AAC.01112-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kimura T, Hosokawa-Muto J, Kamatari YO, Kuwata K. Synthesis of GN8 derivatives and evaluation of their antiprion activity in TSE-infected cells. Bioorg Med Chem Lett. 2011;21:1502–1507. doi: 10.1016/j.bmcl.2010.12.132. [DOI] [PubMed] [Google Scholar]
- 9.Kimura T, Hosokawa-Muto J, Asami K, Murai T, Kuwata K. Synthesis of 9-substituted 2,3,4,9-tetrahydro-1H-carbazole derivatives and evaluation of their anti-prion activity in TSE-infected cells. Eur J Med Chem. 2011;46:5675–5679. doi: 10.1016/j.ejmech.2011.08.039. [DOI] [PubMed] [Google Scholar]
- 10.Doh-Ura K, Iwaki T, Caughey B. Lysosomotropic agents and cysteine protease inhibitors inhibit scrapie-associated prion protein accumulation. J Virol. 2000;74:4894–4897. doi: 10.1128/jvi.74.10.4894-4897.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perrier V, Wallace AC, Kaneko K, Safar J, Prusiner SB, Cohen FE. Mimicking dominant negative inhibition of prion replication through structure-based drug design. Proc Natl Acad Sci USA. 2000;97:6073–6078. doi: 10.1073/pnas.97.11.6073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kocisko DA, Baron GS, Rubenstein R, Chen J, Kuizon S, Caughey B. New inhibitors of scrapie-associated prion protein formation in a library of 2000 drugs and natural products. J Virol. 2003;77:10288–10294. doi: 10.1128/JVI.77.19.10288-10294.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Korth C, May BC, Cohen FE, Prusiner SB. Acridine and phenothiazine derivatives as pharmacotherapeutics for prion disease. Proc Natl Acad Sci USA. 2001;98:9836–9841. doi: 10.1073/pnas.161274798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caughey B, Ernst D, Race RE. Congo red inhibition of scrapie agent replication. J Virol. 1993;67:6270–6272. doi: 10.1128/jvi.67.10.6270-6272.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rambold AS, Miesbauer M, Olschewski D, Seidel R, Riemer C, Smale L, Brumm L, Levy M, Gazit E, Oesterhelt D, Baier M, Becker CF, Engelhard M, Winklhofer KF, Tatzelt J. Green tea extracts interfere with the stress-protective activity of PrP and the formation of PrP. J Neurochem. 2008;107:218–229. doi: 10.1111/j.1471-4159.2008.05611.x. [DOI] [PubMed] [Google Scholar]
- 16.Doh-ura K, Ishikawa K, Murakami-Kubo I, Sasaki K, Mohri S, Race R, Iwaki T. Treatment of transmissible spongiform encephalopathy by intraventricular drug infusion in animal models. J Virol. 2004;78:4999–5006. doi: 10.1128/JVI.78.10.4999-5006.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsuboi Y, Doh-Ura K, Yamada T. Continuous intraventricular infusion of pentosan polysulfate: clinical trial against prion diseases. Neuropathology. 2009;29:632–636. doi: 10.1111/j.1440-1789.2009.01058.x. [DOI] [PubMed] [Google Scholar]
- 18.Kimata A, Nakagawa H, Ohyama R, Fukuuchi T, Ohta S, Doh-ura K, Suzuki T, Miyata N. New series of antiprion compounds: pyrazolone derivatives have the potent activity of inhibiting protease-resistant prion protein accumulation. J Med Chem. 2007;50:5053–5056. doi: 10.1021/jm070688r. [DOI] [PubMed] [Google Scholar]
- 19.Sigurdsson EM, Brown DR, Alim MA, Scholtzova H, Carp R, Meeker HC, Prelli F, Frangione B, Wisniewski T. Copper chelation delays the onset of prion disease. J Biol Chem. 2003;278:46199–46202. doi: 10.1074/jbc.C300303200. [DOI] [PubMed] [Google Scholar]
- 20.Thompson MJ, Borsenberger V, Louth JC, Judd KE, Chen B. Design, synthesis, and structure-activity relationship of indole-3-glyoxylamide libraries possessing highly potent activity in a cell line model of prion disease. J Med Chem. 2009;52:7503–7511. doi: 10.1021/jm900920x. [DOI] [PubMed] [Google Scholar]
- 21.Kuwata K, Kamatari YO, Akasaka K, James TL. Slow conformational dynamics in the hamster prion protein. Biochemistry. 2004;43:4439–4446. doi: 10.1021/bi036123o. [DOI] [PubMed] [Google Scholar]
- 22.Nakajima M, Yamada T, Kusuhara T, Furukawa H, Takahashi M, Yamauchi A, Kataoka Y. Results of quinacrine administration to patients with Creutzfeldt-Jakob disease. Dement Geriatr Cogn Disord. 2004;17:158–163. doi: 10.1159/000076350. [DOI] [PubMed] [Google Scholar]
- 23.Milhavet O, McMahon HE, Rachidi W, Nishida N, Katamine S, Mange A, Arlotto M, Casanova D, Riondel J, Favier A, Lehmann S. Prion infection impairs the cellular response to oxidative stress. Proc Natl Acad Sci USA. 2000;97:13937–13942. doi: 10.1073/pnas.250289197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vogtherr M, Grimme S, Elshorst B, Jacobs DM, Fiebig K, Griesinger C, Zahn R. Antimalarial drug quinacrine binds to C-terminal helix of cellular prion protein. J Med Chem. 2003;46:3563–3564. doi: 10.1021/jm034093h. [DOI] [PubMed] [Google Scholar]
- 25.Telling GC, Scott M, Mastrianni J, Gabizon R, Torchia M, Cohen FE, DeArmond SJ, Prusiner SB. Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell. 1995;83:79–90. doi: 10.1016/0092-8674(95)90236-8. [DOI] [PubMed] [Google Scholar]
- 26.Masel J, Genoud N, Aguzzi A. Efficient inhibition of prion replication by PrP-Fc(2) suggests that the prion is a PrP(Sc) oligomer. J Mol Biol. 2005;345:1243–1251. doi: 10.1016/j.jmb.2004.10.088. [DOI] [PubMed] [Google Scholar]
- 27.Wille H, Zhang GF, Baldwin MA, Cohen FE, Prusiner SB. Separation of scrapie prion infectivity from PrP amyloid polymers. J Mol Biol. 1996;259:608–621. doi: 10.1006/jmbi.1996.0343. [DOI] [PubMed] [Google Scholar]
- 28.Pham N, Yin S, Yu S, Wong P, Kang SC, Li C, Sy MS. Normal cellular prion protein with a methionine at position 129 has a more exposed helix 1 and is more prone to aggregate. Biochem Biophys Res Commun. 2008;368:875–881. doi: 10.1016/j.bbrc.2008.01.172. [DOI] [PubMed] [Google Scholar]
- 29.Lamberto GR, Binolfi A, Orcellet ML, Bertoncini CW, Zweckstetter M, Griesinger C, Fernandez CO. Structural and mechanistic basis behind the inhibitory interaction of PcTS on alpha-synuclein amyloid fibril formation. Proc Natl Acad Sci USA. 2009;106:21057–21062. doi: 10.1073/pnas.0902603106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nicoll AJ, Trevitt CR, Tattum MH, Risse E, Quarterman E, Ibarra AA, Wright C, Jackson GS, Sessions RB, Farrow M, Waltho JP, Clarke AR, Collinge J. Pharmacological chaperone for the structured domain of human prion protein. Proc Natl Acad Sci USA. 2010;107:17610–17615. doi: 10.1073/pnas.1009062107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ansari A, Berendzen J, Bowne SF, Frauenfelder H, Iben IE, Sauke TB, Shyamsunder E, Young RD. Protein states and proteinquakes. Proc Natl Acad Sci USA. 1985;82:5000–5004. doi: 10.1073/pnas.82.15.5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Itoh K, Sasai M. Dynamical transition and proteinquake in photoactive yellow protein. Proc Natl Acad Sci USA. 2004;101:14736–14741. doi: 10.1073/pnas.0402978101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cohen FE, Prusiner SB. Pathologic conformations of prion proteins. Annu Rev Biochem. 1998;67:793–819. doi: 10.1146/annurev.biochem.67.1.793. [DOI] [PubMed] [Google Scholar]
- 34.Cohen FE, Pan KM, Huang Z, Baldwin M, Fletterick RJ, Prusiner SB. Structural clues to prion replication. Science. 1994;264:530–531. doi: 10.1126/science.7909169. [DOI] [PubMed] [Google Scholar]
- 35.Cohen FE, Prusiner SB. Structural studies of prion proteins. In: Prusiner SB, editor. Prion biology and diseases. New York: Cold Spring Harbor; 1999. pp. 191–228. [Google Scholar]
- 36.Yamamoto N, Kuwata K. Regulating the conformation of prion protein through ligand binding. J Phys Chem B. 2009;113:12853–12856. doi: 10.1021/jp905572w. [DOI] [PubMed] [Google Scholar]
- 37.Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R, Mastrianni J, Lugaresi E, Gambetti P, Prusiner SB. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science. 1996;274:2079–2082. doi: 10.1126/science.274.5295.2079. [DOI] [PubMed] [Google Scholar]
- 38.Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, Cohen FE, Prusiner SB. Eight prion strains have PrP(Sc) molecules with different conformations. Nat Med. 1998;4:1157–1165. doi: 10.1038/2654. [DOI] [PubMed] [Google Scholar]
- 39.Kawasaki Y, Kawagoe K, Chen CJ, Teruya K, Sakasegawa Y, Doh-ura K. Orally administered amyloidophilic compound is effective in prolonging the incubation periods of animals cerebrally infected with prion diseases in a prion strain-dependent manner. J Virol. 2007;81:12889–12898. doi: 10.1128/JVI.01563-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li J, Browning S, Mahal SP, Oelschlegel AM, Weissmann C. Darwinian evolution of prions in cell culture. Science. 2010;327:869–872. doi: 10.1126/science.1183218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sawkar AR, Cheng WC, Beutler E, Wong CH, Balch WE, Kelly JW. Chemical chaperones increase the cellular activity of N370S beta -glucosidase: a therapeutic strategy for Gaucher disease. Proc Natl Acad Sci USA. 2002;99:15428–15433. doi: 10.1073/pnas.192582899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kanu N, Imokawa Y, Drechsel DN, Williamson RA, Birkett CR, Bostock CJ, Brockes JP. Transfer of scrapie prion infectivity by cell contact in culture. Curr Biol. 2002;12:523–530. doi: 10.1016/s0960-9822(02)00722-4. [DOI] [PubMed] [Google Scholar]
- 43.Montagnier L. 25 years after HIV discovery: prospects for cure and vaccine. Virology. 2010;397:248–254. doi: 10.1016/j.virol.2009.10.045. [DOI] [PubMed] [Google Scholar]
- 44.Shafer RW, Vuitton DA. Highly active antiretroviral therapy (HAART) for the treatment of infection with human immunodeficiency virus type 1. Biomed Pharmacother. 1999;53:73–86. doi: 10.1016/s0753-3322(99)80063-8. [DOI] [PubMed] [Google Scholar]
- 45.Roberts BE, Duennwald ML, Wang H, Chung C, Lopreiato NP, Sweeny EA, Knight MN, Shorter J. A synergistic small-molecule combination directly eradicates diverse prion strain structures. Nat Chem Biol. 2009;5:936–946. doi: 10.1038/nchembio.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hornemann S, Korth C, Oesch B, Riek R, Wider G, Wuthrich K, Glockshuber R. Recombinant full-length murine prion protein, mPrP(23-231): purification and spectroscopic characterization. FEBS Lett. 1997;413:277–281. doi: 10.1016/s0014-5793(97)00921-6. [DOI] [PubMed] [Google Scholar]
- 47.Goddard TD, Kneller DG. SPARKY, version 3. San Francisco: University of California; 2001. [Google Scholar]
- 48.Riek R, Hornemann S, Wider G, Billeter M, Glockshuber R, Wuthrich K. NMR structure of the mouse prion protein domain PrP(121-231) Nature. 1996;382:180–182. doi: 10.1038/382180a0. [DOI] [PubMed] [Google Scholar]
- 49.Nishida N, Harris DA, Vilette D, Laude H, Frobert Y, Grassi J, Casanova D, Milhavet O, Lehmann S. Successful transmission of three mouse-adapted scrapie strains to murine neuroblastoma cell lines overexpressing wild-type mouse prion protein. J Virol. 2000;74:320–325. doi: 10.1128/jvi.74.1.320-325.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.