

Abstract
T cell receptors (TCRs) are immune proteins that specifically bind to antigenic molecules, which are often foreign peptides presented by major histocompatibility complex proteins (pMHCs), playing a key role in the cellular immune response. To advance our understanding and modeling of this dynamic immunological event, we assembled a protein–protein docking benchmark consisting of 20 structures of crystallized TCR/pMHC complexes for which unbound structures exist for both TCR and pMHC. We used our benchmark to compare predictive performance using several flexible and rigid backbone TCR/pMHC docking protocols. Our flexible TCR docking algorithm, TCRFlexDock, improved predictive success over the fixed backbone protocol, leading to near-native predictions for 80% of the TCR/pMHC cases among the top 10 models, and 100% of the cases in the top 30 models. We then applied TCRFlexDock to predict the two distinct docking modes recently described for a single TCR bound to two different antigens, and tested several protein modeling scoring functions for prediction of TCR/pMHC binding affinities. This algorithm and benchmark should enable future efforts to predict, and design of uncharacterized TCR/pMHC complexes.
Keywords: TCR, MHC, RosettaDock, ZRANK, protein docking, immune recognition
Introduction
Specific recognition of peptides that are presented by major histocompatibility complexes (pMHCs) by T cell receptors (TCRs) is a key event in the cellular immune response. This molecular binding event is mediated by flexible complementarity determining region (CDR) loops on the TCR, which are determined for each T cell via gene rearrangement during development in the thymus. This results in a clonally and structurally diverse set of TCRs in vivo (at least 1 × 109 clones in humans1) that can bind and initiate responses to an immense variety of antigens.
Given their importance in vaccine design,2–4 autoimmune disease,5, 6 and their potential as therapeutics for cancer7–9 and HIV,10 TCRs have been studied extensively to understand their recognition of antigens at the molecular level. This has been facilitated by an increasing number of structurally characterized TCR/pMHC complexes, permitting reviews of docking orientation11 and CDR loop conformational changes during binding.12
Despite these advances, much remains to be understood regarding the dynamic and molecular basis of TCR/pMHC recognition before modeling can accurately recapitulate and predict these interactions. In addition to the challenge of modeling side chains, CDR loops, and peptide flexibility, the docking orientation of TCRs over pMHCs must be determined. While this quaternary structure is conserved in general (with a roughly diagonal orientation of the two TCR chains over the peptide), structures still exhibit notable variability (∼70°) in the TCR/pMHC crossing angle,11 and highly tilted docking modes have been observed for autoimmune TCRs engaging peptides presented by Class II MHCs.6
Here we describe the use of protein–protein docking to accurately predict TCR/pMHC recognition based on the structures of the unbound proteins. Protein–protein docking has advanced greatly over the past two decades, spurred by improvements in scoring functions, computational efficiency, as well as community interaction via the ongoing CAPRI blind docking experiment.13 Improved conformational searching has provided the ability to predict structures of complexes from unbound components, even in the presence of conformational changes.14 In this study, we modified the docking program RosettaDock,15 to predict TCR/pMHC recognition in combination with the program ZRANK.16 Both RosettaDock and ZRANK have been highly successful in the CAPRI protein docking experiment,17–20 and we previously adapted ZRANK specifically to score refined protein–protein docking predictions from RosettaDock.21
To facilitate the testing and development of predictive docking methods, we assembled a benchmark set of 20 TCR/pMHC complexes that have separately solved structures of their unbound components, including 17 Class I MHC-containing complexes and 3 Class II MHC-containing complexes. This benchmark is analogous to our benchmark for protein–protein docking, which is widely used to develop and test protein docking algorithms22 but does not contain TCR/pMHC structures due to their generally conserved binding mode. As with a recently released structure-based binding affinity benchmark,23 we also collected binding affinity data for TCR/pMHC interactions in our benchmark, and we assessed the ability of protein design functions to discriminate high affinity interactions from low and moderate affinity interactions based on structures. This initial success shows that protein–protein docking algorithms are capable of generating accurate structural models of TCR/pMHC binding using unbound structures, and can provide mechanistic insights into this class of dynamic immunological interactions.
Results
Docking benchmark
From an exhaustive search of TCR/pMHC complex structures as well as individually solved TCR and pMHC structures in the PDB,24 we assembled a benchmark of 20 docking test cases (Table I and Supporting Information Table S1). This highly diverse set of structures contains 12 unique TCRs, 9 MHC alleles, and peptides associated with viruses, cancer, and autoimmunity. A subset of cases feature the same TCR bound to multiple antigens featuring distinct peptides and/or MHC alleles (e.g., the 2C TCR in 1MWA, 2CKB, and 2OI9, the LC13 TCR in 1MI5, 3KPR, and 3KPS, and the A6 TCR in 1AO7, 3H9S, and 3PWP) and highlight the multi-specific nature of TCR/pMHC recognition. Among the various MHC alleles, amino acid differences (e.g., 20% of residues vary between HLA-A2 and HLA-B8) do not substantially alter the MHC backbone conformations within each class (seven Class I and two Class I MHCs), but impart changes to the side chains of the peptide binding groove and the nearby residues that interact directly with the TCR. This includes residue changes R65Q and A69T from HLA-A2 to HLA-B3501 in two out of three “restriction triad” residues at the TCR interface,25 or the peptide groove substitution of D77S from H2-Kb to H2-Kbm3, leading in a change in MHC-bound peptide conformation.26
Table I.
The TCR Docking Benchmark
| Complexa | TCRa | TCR name | pMHCa | pMHC name | Peptide sequence | RMSDb | Difficultyc |
|---|---|---|---|---|---|---|---|
| CD8+ TCR; Class I MHC | |||||||
| 1AO7 | 3QH3 | A6 | 1DUZ | Tax/HLA-A2 | LLFGYPVYV | 1.23 | Rigid |
| 1MI5 | 1KGC | LC13 | 1M05 | EBV/HLA-B8 | FLRGRAYGL | 1.25 | Medium |
| 1MWA | 1TCR | 2C | 1LEK | dEV8/H2-Kbm3 | EQYKFYSV | 1.14 | Rigid |
| 1OGA | 2VLM | JM22 | 2VLL | flu/HLA-A2 | GILGFVFTL | 1.36 | Medium |
| 2BNR | 2BNU | 1G4 | 1S9W | NY-ESO9C/HLA-A2 | SLLMWITQC | 0.72 | Rigid |
| 2CKB | 1TCR | 2C | 1LEG | dEV8/H2-Kb | EQYKFYSV | 1.17 | Rigid |
| 2NX5 | 2NW2 | ELS4 | 2NW3 | EBV/HLA-B*3501 | EPLPQGQLTAY | 1.14 | Rigid |
| 2OI9 | 1TCR | 2C | 3ERY | QL9/H2-Ld | QLSPFPFDL | 1.10 | Medium |
| 2PYE | 2PYF | 1G4 c5c1 | 1S9W | NY-ESO9C/HLA-A2 | SLLMWITQC | 0.88 | Rigid |
| 3H9S | 3QH3 | A6 | 3H7B | Tel1p/HLA-A2 | MLWGYLQYV | 1.31 | Medium |
| 3KPR | 1KGC | LC13 | 3KPQ | ABCD3/HLA-B*4405 | EEYLKAWTF | 1.37 | Medium |
| 3KPS | 1KGC | LC13 | 3KPP | ABCD3/HLA-B*4405 | EEYLQAFTY | 1.31 | Medium |
| 3PWP | 3QH3 | A6 | 3PWL | HuD/HLA-A2 | LGYGFVNYI | 1.24 | Rigid |
| 3QDG | 3QEU | DMF5 | 1JF1 | MART1-ELA/HLA-A2 | ELAGIGILTV | 0.96 | Medium |
| 3QDJ | 3QEU | DMF5 | 2GUO | MART1-AAG/HLA-A2 | AAGIGILTV | 0.94 | Medium |
| 3SJV | 3SKN | RL42 | 1M05 | EBV/HLA-B8 | FLRGRAYGL | 0.96 | Rigid |
| 3UTT | 3UTP | 1E6 | 3UTQ | Insulin/HLA-A2 | ALWGPDPAAA | 0.75 | Rigid |
| CD4+ TCR; Class II MHC | |||||||
| 2IAM | 2IAL | E8 | 1KLG | TPImut/HLA-DR1 | GELIGILNAAKVPAD | 0.87 | Rigid |
| 2IAN | 2IAL | E8 | 1KLU | TPI/HLA-DR1 | GELIGTLNAAKVPAD | 0.82 | Rigid |
| 2PXY | 2Z35 | 1934.4 | 1K2D | MBP/H2-IAu | RGGASQYRPSQ | 1.18 | Medium |
PDB code.
Root mean square distance (in Å) between backbone atoms of the TCR/pMHC interface and the corresponding atoms in the unbound structures.
Docking difficulty, as defined by interface RMSD and contacting residues.22
Conformational changes between unbound and bound structures are higher on average than for unbound antibody/antigen docking test cases,22 with interface root-mean-square distances (RMSDs; calculated using backbone atoms of all TCR and pMHC residues within 10.0 Å of the binding partner in the bound structure) ranging from 0.72 to 1.37 Å. Many of the larger conformational changes take place on the TCR side of the interface, with the notable exception of 2NX5 which features a flexible “bulged” 11-mer peptide that results in a 1.0 Å binding conformational change in the pMHC (Supporting Information Table S1). Within the pMHC side of the interface, peptides typically accounted for more conformational change than the MHC helices (Supporting Information Table S1), however in the case of 3H9S the HLA-A2 MHC dominated the conformational change (1.02 Å vs. 0.79 Å for the peptide); this is thought to be due to dynamic motions of the HLA-A2 helices induced when bound to the Tel1p peptide.27 Using our previously established criteria to evaluate docking difficulty based on interface conformational changes,22 nearly half (nine cases) are classified as medium difficulty, and the remaining 11 cases are rigid-body (Table I).
To assess the structural diversity within the TCR docking benchmark, we compared rigid-body docking orientations, in addition to backbone conformations, among the TCR/pMHC structures (Fig. 1). The crossing angle, the angle between the inter-domain vector of the TCR, and the antigen binding groove vector of the MHC, varied greatly, ranging between 22° and 69° (Supporting Information Table S1), which is approximately the range noted by Rudolph and Wilson in their review of TCR/pMHC structures.11 The incident angle, corresponding to the tilt of the TCR with respect to the MHC's peptide-binding plane (see Materials and Methods for details), varied less than the crossing angle, but it can be seen in Figure 1(A) that these angular differences represent considerable changes in docking orientation. Superposition of bound TCR CDR loops [Fig. 1(C)] indicates a large degree of structural variability, particularly among the CDR3α and CDR3β loops of the TCR, and to a smaller extent for the germ-line CDR1α and CDR2α loops. In the superposition of pMHCs [Fig. 1(D)], the peptide backbone conformations show substantial diversity, driven by varying peptide sequences and lengths, as well as MHC allele and bound TCR.
Figure 1.
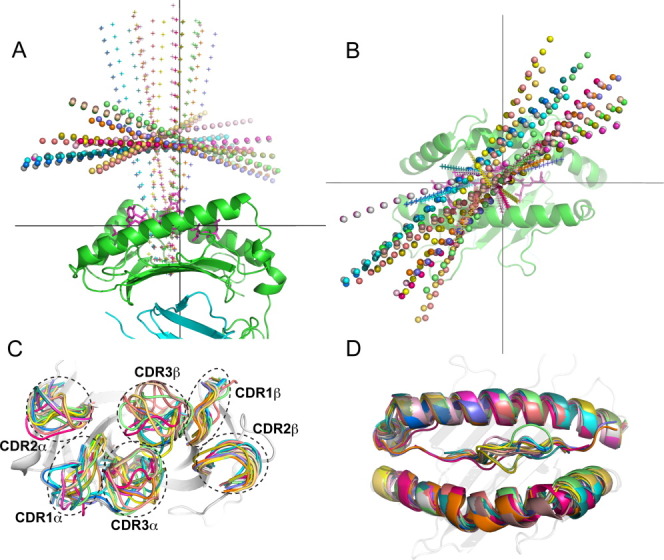
Diversity of docking orientations and conformations for the 20 complexes in the TCR docking benchmark. A: Side and (B) top view of TCR docking orientations, denoted by TCR inter-domain axes (spheres) and pseudo-symmetric axes between TCR variable domains (“+”); the pMHC from 1AO7 is shown for reference (peptide = magenta sticks, MHC = green cartoon, β2m = cyan cartoon). C: CDR loops from superposed TCRs and (D) superposed pMHC structures, with the 1AO7 TCR and MHC shown for reference.
In addition to analyzing the variation among the bound TCR/pMHC complexes in the benchmark, we calculated the binding conformational changes of the TCRs (calculated using the unbound and bound TCR structures from each test case) as a function of position (Fig. 2). We found that the CDR3α loop exhibited the largest average conformational change upon binding, followed by CDR3β and CDR1α, in agreement with previous findings from Armstrong et al. in a study of 12 TCR/pMHC complexes.12 Some less pronounced conformational changes can be seen for the other CDRs and sites of pMHC binding. We also observed conformational changes around residue 40 in the TCR β chains; this occurs in the turn between the C and C′ strands and contains a mobile glycine residue. As this region is distant from the pMHC binding site, we did not investigate this further with respect to implications on pMHC binding.
Figure 2.
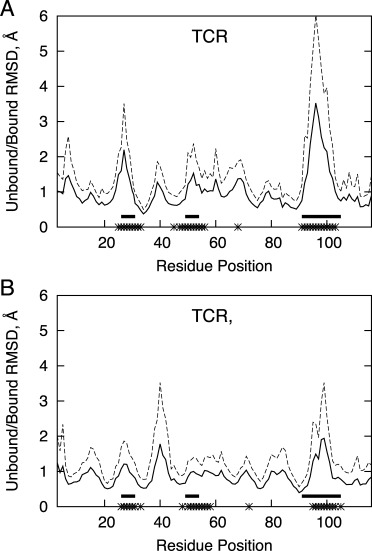
Backbone RMSD between residues of bound and unbound TCR variable domains after structural superposition, for the α chain (A) and β chain (B). Average RMSD is shown as a solid line, and dotted line represents the average plus one standard deviation. Solid horizontal bars represent the positions of CDR loops (based on the IMDB definition28), and asterisks on the x-axis indicate positions at the interface with pMHC (within 6.0 Å) for three or more benchmark complexes.
TCRFlexDock performance
After assembly of our benchmark set of structures, we developed a flexible backbone docking protocol using RosettaDock15and ZRANK21 to perform TCR/pMHC docking (Fig. 3), which we refer to as TCRFlexDock. Our approach employs iterative Monte Carlo moves of rigid-body positions and side chain packing combined with refinement of CDR loop (and optionally peptide) backbone conformations, analogous to the SnugDock protocol developed for antibody/antigen docking.29 Unlike SnugDock, we utilized the recently developed kinetic closure algorithm (KIC)30 for flexible CDR loop modeling, which shows higher performance than the standard Rosetta protocol (cyclic coordinate descent or CCD) in producing loop structural models30 and in sequence design predictions at an antibody/antigen interface.31 All unbound TCR and pMHC structures were initially set to the same starting position with a diagonal crossing angle (45°) and 25 Å separation between the unbound TCR and pMHC centers (details in the Methods), which provided space between binding partners for initial conformational sampling. The CDR3 loops were then given an initial KIC perturbation and refinement, followed by a random rigid-body perturbation of the pMHC (a 3 Å and 8° Gaussian movement), and a low resolution docking search. Then two steps of Monte Carlo minimization (rigid body and side chain movements) and CDR (and optionally peptide) loop refinement were performed. This docking protocol was run 1000 times per complex to generate 1000 docked models, which were re-scored using the docking scoring function ZRANK with refinement weights21 to select the top predictions. We authored the docking portion of this protocol using the Rosetta Scripts language;32 the code is available as Supporting Information.
Figure 3.
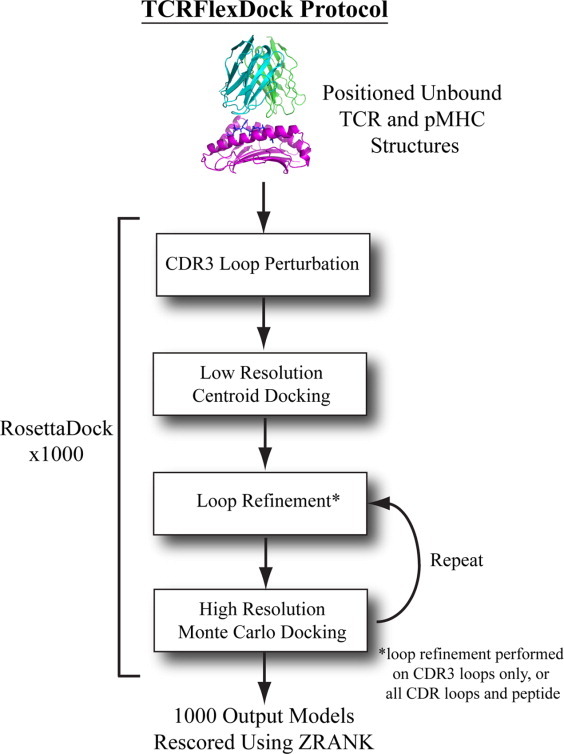
The multi-stage TCR/pMHC docking protocol, TCRFlexDock. After the unbound TCR and pMHC oriented to a starting position, the CDR3 loops are perturbed and minimized, and a coarse-grained centroid docking search is followed by two iterations of CDR (and optionally peptide) loop refinement and 6D rigid body movements plus side chain repacking. This is repeated 1000 times, resulting in 1000 predictions per test case, which are then scored using ZRANK to select docking models. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
We compared two versions of TCRFlexDock, moving the backbones of CDR3s (“CDR3”) and moving the backbones of all CDRs and the peptide (“CDRPep”), with the fixed backbone docking protocol of RosettaDock (“Fixedbb”) for predictive performance on the TCR docking benchmark (Fig. 4). We employed the criteria for docking model evaluation used in the CAPRI experiment,33 facilitating comparison with results from other studies, and considered medium and high accuracy CAPRI predictions to be near-native hits. While the fixed backbone protocol performed well, it was outperformed by the TCRFlexDock protocols, particularly when considering the top 10 and top 30 predictions for each test case. Success rates were 80% and 100% for the CDRPep protocol for the top 10 and top 30 predictions, respectively (CDRPep results for the top 10 predictions are in Supporting Information Table S2). Interface RMSDs of predictions [backbone distance from the bound interface; Fig. 4(B)] were also improved for both the CDR3 and CDRPep TCRFlexDock protocols versus the Fixedbb protocol, and all three docking protocols had dramatically improved RMSDs versus the input structures.
Figure 4.
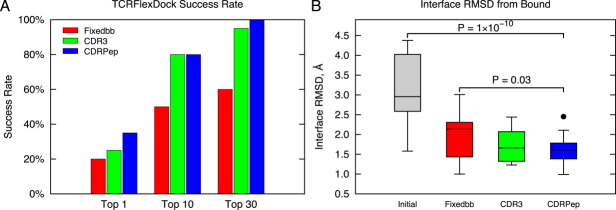
TCRFlexDock success rates on the TCR docking benchmark. A: Success using a fixed backbone (Fixedbb) docking protocol is compared to TCRFlexDock with flexible CDR3 loops (CDR3) and flexible CDR loops and peptide (CDRPep). Success was evaluated for the top 1, top 10, and top 30 predictions (selected by ZRANK from 1000 RosettaDock models per test case). B: Distribution of interface backbone RMSDs of the best model among the top 30 predictions for the three docking protocols. “Initial” corresponds to the initial RMSDs of the complexes that were input to the docking search. P-values were calculated using the Wilcoxon rank sum test. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
To explore changes in binding energy landscape due to sampling during docking simulations, we compared scores versus RMSDs from bound interface for all 1000 models from each docking simulation, which is shown for two test cases in Figure 5. In addition to the Fixedbb and CDRPep protocols, we tested fixed backbone docking with the bound structures (Boundbb) to gauge how much correct backbone conformations aid the docking search (prior to docking, bound side chains were removed and modeled, and the TCR and pMHC structures were set to the same starting position as the unbound structures). While for the 1AO7 test case there was some improvement in the binding funnel between the Fixedbb and the flexible CDRPep protocols, the improvement was more pronounced for the test case 2PYE, which features a high affinity TCR. For both cases, docking with the bound backbone led to very clear binding funnels, underscoring the importance of accurate backbone conformation in high accuracy docking results.
Figure 5.
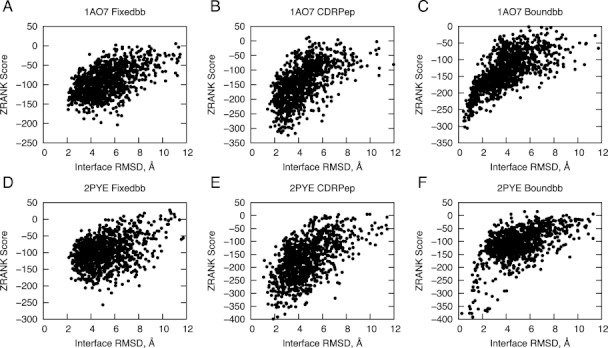
Binding funnels for two benchmark test cases. ZRANK scores versus interface RMSD for 1000 docking models are shown for 1AO7 (A, B, and C) and 2PYE (D, E, and F). The Fixedbb protocol (with flexible side chains only) (A and D) yielded less success than the CDRPep protocol (with flexible backbones for CDR loops and peptide and flexible side chains for all residues) (B and E). For comparison, the Boundbb results are also shown (C and F), which used the backbone conformations from the bound structures as input to fixed backbone docking (with side chains removed and rebuilt), yielding the most distinct energy funnels and most accurate models.
Prediction of binding modes of the 43F3 TCR bound to different pMHCs
Having demonstrated that TCRFlexDock can predict bound TCR/pMHC docking orientations from unbound structures, we applied it to a new system where no unbound TCR structure was available, hence not included in our benchmark. The 42F3 TCR recognizes the QL9 peptide bound to the H2-Ld MHC (the same antigen as test case 2OI9 in our benchmark), and in a recent study this TCR was found to bind to an unrelated peptide (p3A1; sequence SPLDSLWWI; also presented by H2-Ld) with a dramatically different orientation.34 As this shift in docking orientation would not be predicted by homology modeling using 42F3/QL9/H2-Ld as the template, or the notion of conserved contacts between this TCR and the H2-Ld MHC, we considered this to be an ideal case to test our docking protocol. We generated two test cases (named, as with the benchmark cases, after the bound structures): 3TF7 (42F3/QL9/H2-Ld) and 3TJH (42F3/3A1/H2-Ld) (Table II). The input TCR for each case was taken from the other case's bound structure, which in light of the considerable CDR3α conformational change34 provides a useful test of flexible CDR modeling. For 3TJH, the p3A1 peptide was modeled using fixed backbone mutagenesis in Rosetta35 onto an unbound peptide/H2-Ld structure (PDB code 1LD9), for which the peptide (sequence YPNVNIHNF) shares only the key anchor residue Pro2 with p3A1.
Table II.
The 42F3 TCR Test Cases
| Complexa | TCRa | pMHCa | pMHC name | Peptide sequence |
|---|---|---|---|---|
| 3TF7 | 3TJH | 3ERY | QL9/H2-Ld | QLSPFPFDL |
| 3TJH | 3TF7 | 1LD9b | 3A1/H2-Ld | SPLDSLWWIb |
PDB code.
The peptide in the unbound 1LD9 pMHC structure was mutated in silico to match the sequence of the peptide targeted by 42F3 (mutant sequence positions shown in bold).
We employed TCRFlexDock (with CDRPep flexibility) to predict the structures of these two complexes, generating 1000 predictions per case (as with the benchmark); predicted structures are shown in Figure 6. For the 3TF7 complex, a near-native prediction was ranked number 4, with an interface RMSD of 2.02 Å and two CAPRI stars (medium accuracy). The prediction for 3TJH was slightly less accurate (3.01 Å from the bound interface), but still was within the CAPRI criteria for one star (acceptable prediction), which is notable given that the pMHC structure was modeled for this case. Among the correctly predicted contacts for this interface was the hydrogen bond between the side chains of residues Lys95 of the TCR α chain and Asp4 of the peptide [Fig. 6(B)], one of the three CDR3-peptide contacts observed in the crystal structure34 and the only one between two side chains.
Figure 6.
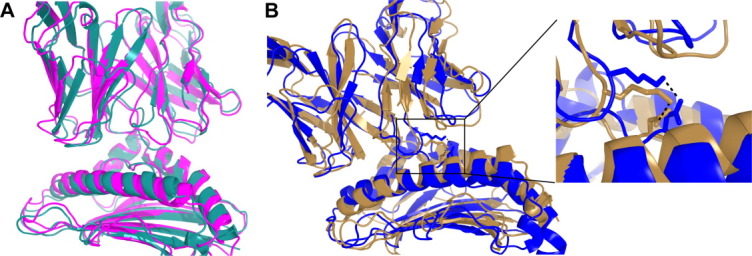
Flexible docking predictions for the 42F3 TCR bound to two different peptides presented by the same MHC. A: The 42F3/QL9/H-2Ld complex (magenta; PDB code 3TF7) and TCRFlexDock model 4 (cyan). This prediction has interface RMSD of 2.02 Å with the bound complex and two CAPRI stars. B: The 42F3/p3A1/H-2Ld complex (gold; PDB code 3TJH) and TCRFlexDock model 4 (blue). This prediction has an interface RMSD of 3.01 Å from the bound complex and one CAPRI star. Shown to the right is the hydrogen bond (from the modeled complex and the crystal structure) between Lys95 of the 42F3 TCR α chain and Asp4 of p3A1. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Prediction of TCR binding affinities
Several recent studies that have demonstrated the potential of protein docking and design scoring functions to predict binding affinities based on structures of complexes,23, 36, 37 yet such energetic predictions are not always accurate, as observed when scoring the complexes between a TCR and several super antigens featuring a mobile loop.38 We tested the performance of predictive scoring algorithms in the context of TCR/pMHC affinity prediction (Fig. 7 and Supporting Information Table S3). In addition to the complexes in our TCR docking benchmark with characterized binding affinities, we included five additional high affinity TCRs with characterized antigen binding affinities and solved TCR/pMHC structures, to examine whether such functions can discriminate between normal and high affinity complexes. For both Rosetta35 (using “ddg” weights) and ZAFFI,39 there were strong correlations between predicted and measured binding affinities (R = 0.79 for both). We evaluated the performance of two additional functions (Supporting Information Table S3): ZRANK's refinement function21 (R = 0.75), which was used to select TCRFlexDock predictions, and the atomic contact energy (ACE) statistical potential (R = 0.73), which was derived over 10 years ago from contact frequencies of atoms in monomeric crystal structures.40 Given that Rosetta, ZAFFI, and ZRANK utilize complex weighted energy functions that include statistical energy terms (ZAFFI and ZRANK include ACE in their functions), the strong performance of ACE alone is impressive.
Figure 7.
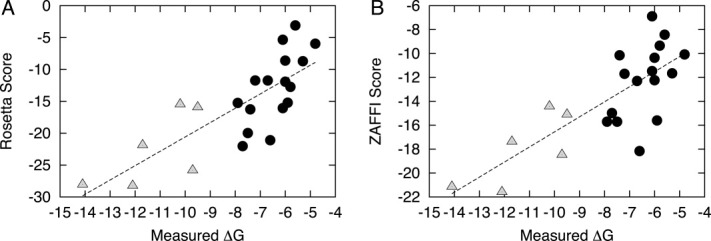
Scores versus measured binding affinities for 22 TCR/pMHC complexes, using (A) Rosetta and (B) ZAFFI. The complexes include 17 structures from the TCR docking benchmark and five additional complexes of high affinity TCR mutants with antigens. Low and moderate affinity complexes (black circles) are distinguished from high affinity complexes (gray triangles). Best-fit lines are shown for each function; the correlation between scores and experimental measurements is 0.79 for both.
Discussion
We have developed and benchmarked predictive docking protocols by assembling a set of diverse test cases for an immunologically critical system, TCR/pMHC recognition. Our flexible docking protocol, TCRFlexDock, gave near-native docking models in the top 10 predictions for the vast majority of the benchmark structures (16 out of 20 cases). The docking protocols we describe here are to our knowledge the first protocols for predictive TCR/pMHC docking that were tested against a set of TCR/pMHC complexes. Recent studies have performed docking of autoimmune TCRs to Class II antigens41, 42 using established homology modeling and docking approaches, but given their focus on predicting single uncharacterized complexes, it is difficult to determine their predictive performance or general applicability. Another study used steered molecular dynamics to simulate TCR/pMHC binding for three complexes, but components for each simulation were taken from the respective bound complex, greatly simplifying the modeling of the binding process.43
While TCRFlexDock produced relatively accurate docking models (average interface RMSDs less than 2 Å from bound) for all TCR/pMHC test cases, with recapitulation of key interface contacts, further improvements in docking accuracy can improve models closer to crystallographic resolution for analysis requiring fine structural details, such as computational mutagenesis. The performance of our TCR docking approaches on the benchmark, as well as recent observations of TCR/pMHC recognition, provides possible means to improve in this regard. Simulating flexibility of the mobile CDR3 loops provided a notable improvement in docking accuracy over fixed backbone docking, yet sampling loop conformations closer to the bound structure would likely provide greater success. This could potentially be achieved by using conformational propensities derived from a study of TCR CDR loop conformations, as performed by Al-Lazikani et al.,44 but updated to include CDR3 loops as performed recently for antibody CDRs.45 Along with improved simulation of flexible CDR loops, other areas of the binding interface with less pronounced conformational changes could also be addressed, such as the MHC helices, which have been observed to undergo dynamic changes as in the test case 3H9S.46Another means to improve docking performance would be to guide the docking search using likely contacts between TCRs and MHCs, as with the docking program HADDOCK, which uses “ambiguous interaction restraints” as key input parameters.47 The concept of conserved contacts between TCRs and MHCs,48 as well as recently described distinct “codons” of germline TCR/pMHC recognition49 could be used in this context; however recent structural studies have indicated that such simplifications could be misleading.34, 50 Finally, some improvement could be achieved by modeling (or constraining) TCR/pMHC interactions in the context of the immunological synapse. The quaternary structure of a TCR, pMHC, and CD4 protein has recently been determined by X-ray crystallography;51 such a structure, along with an analogous structure (or model) including the CD8 homodimer for Class I MHCs could potentially help to guide or filter models to identify putative TCR/pMHC complexes from a docking search.
The predictive performance for TCR/pMHC binding affinities provides evidence that scoring functions currently in use for scoring protein–protein interfaces can be applied to predict affinities of this system. This would be immensely useful as a potential means to evaluate binding affinities of a range of antigens to a single TCR, or to discriminate models of high affinity TCRs generated using in silico methods. However, given that our study used the ideal case where the bound structure was known rather than relying on a docking model or set of docking models, it is likely that scoring would need to be adapted to accommodate potential noise within a set of docking predictions or models. Recent use of distributions of docking scores from unbound rigid-body docking to discriminate binding from non-binding proteins demonstrates that even in the absence of a single high-resolution structure, predictions of binding partners can be made.52
Predictions of docked TCR structures as well as pMHC binding affinities are particularly attractive in light of recent progress in next generation sequencing technologies, which have made it possible to characterize the vast repertoire of TCRs in vivo,53, 54 and have helped to characterize many “public” TCRs with antigen-specific sequences shared by many individuals in a population.55 By combining the flexible docking approaches described here with modeling of TCR structures from sequence, it should be possible to predict the structures of these TCRs in complex with cognate pMHCs, particularly when their antigenic targets are known. The use of modeled TCRs instead of unbound structures would likely yield more challenging cases; the improvements to the docking procedure noted above would likely help in this scenario. Docking with modeled immune proteins has been demonstrated with antibodies using RosettaDock29 and similar methods could be developed to model TCR CDRs from sequence. Another recent study modeled the structures of six TCR clones bound to the NY-ESO-1 antigen and HLA-A2, but structures were pre-docked to the antigen based on a homologous complex structure (1G4/NY-ESO9C/HLA-A2; test case 2BNR) prior to CDR modeling.56 Our approach is applicable to the more general and complex scenario of unbound TCRs binding to pMHCs without pre-orientation to a known structure, and can reproduce a substantial variety of docking orientations as seen in the benchmark cases and the 42F3 TCR.
By building an extensible framework to perform these simulations, our flexible TCR docking algorithm could be readily combined with methods for modeling the structures of peptides bound to MHCs,57 which would be useful in cases for which the pMHC structure has not been characterized. Given that even point mutations in a peptide can impact TCR specificity, avidity, and composition of T cell repertoires,58 accurately representing peptide conformations would be essential in such a modeling pipeline. With the rapid expansion of immunoinformatics tools and data, predictive tools to describe TCR/pMHC recognition as described here have great potential to transform immunology and therapeutics research.
Materials and Methods
Docking benchmark assembly
To ensure sufficient structural quality, we only selected crystallographic structures with resolution better than 3.25 Å, as with our protein–protein docking benchmark.22 We performed a search for test cases using all structures in the Protein Data Bank24 (PDB) in June 2012. In the event that multiple structures were available for a particular protein or complex, we selected the structure with the highest resolution. We omitted structures that included other proteins (e.g., superantigens) bound at the TCR/pMHC interface, or peptides covalently attached to the MHC or TCR, due to the concern that indirect effects from these interactions may alter the TCR/pMHC interface and thereby bias the docking results. For the test cases 2IAM and 2IAN, the superantigen bound to the pMHC structures (PDB codes 1KLG and 1KLU) was not significantly contacting the TCR binding site or interacting with the peptide, thus the cases were retained.
Though unbound structures with missing interface residues were avoided, some were nonetheless included in the benchmark as judged by the extent of the missing atoms. This included the unbound ELS4 TCR (PDB code 2NW2), which lacks coordinates for one residue (and portions of two adjacent residues) in the CDR3α loop, yet the core of the binding interface was intact. Residue mismatches between bound and unbound structures at the binding interface were not permitted. The 2C TCR bound to QL9/H2-Ld had several mismatching residues between unbound and bound TCRs (1TCR vs. 2O19), but these were away from the interface and the mutations were introduced to improve solubility and not pMHC binding.59
Calculation of crossing and incident angles
The relative orientation between a TCR and a pMHC can be described using two angles: crossing angle and incident angle. The crossing angle is the angle between the inter-domain TCR vector and the vector along the MHC helices; its calculation is described in detail by Rudolph et al.11 Briefly, the inter-domain TCR vector was calculated using the centroids of the disulfide bonds in the two TCR variable domains. The vector along the MHC helices was calculated using singular value decomposition of the alpha carbon atoms for the helical residues delineated by Rudolph and Wilson. This produces three (orthogonal) eigenvectors, of which one is the vector along the MHC helices (corresponding to the largest eigenvalue), and one is the normal vector to the MHC helix plane (corresponding to the smallest eigenvalue). The incident angle is the angle between the pseudo two-fold symmetry axis that relates the TCR variable domains and the normal to the MHC helix plane. The axis of symmetry was calculated using a modified version of the FAST structural alignment program,60 and the normal vector to the MHC plane was taken from the singular value decomposition described above.
RosettaDock
We used RosettaDock15 (Rosetta version 3.2) to perform local docking searches using two basic protocols: fixed backbone and flexible loops. Fixed backbone docking was performed using the “docking_protocol” executable, restricted to local searching with Gaussian perturbations of 3 Å and 8° (“-docking:dock_pert 3 8”). The default rotamer library was appended with extra chi1 and chi2 aromatic rotamers (“-ex1 −ex2aro”), unbound rotamers (“-unboundrot”), and off-rotamer minimization (“-dock_rtmin”). About 1000 models were produced by each docking run (“-nstruct 1000”), and each model was scored using the ZRANK program with weights developed for docking models refined using RosettaDock.21 We also tested the use of the RosettaDock score for each model, which was from the interface score term (“I_sc”) of the RosettaDock output, as employed by Sircar and Gray for antibody/antigen docking model evaluation,29 but we found the ZRANK score to be superior (Supporting Information Table S2).
Implementation of the flexible TCR docking protocol, TCRFlexDock, was performed using RosettaScripts;32 a sample script is provided in the Supporting Information text. In addition to the perturbation and side chain sampling flags specified in the Methods for rigid-body docking, we used the flags (“-loops:outer_cycles 1 -loops:max_inner_cycles 100”) to limit the number of KIC loop refinement iterations during the docking run. Additionally, we made two modifications to the Rosetta source code related to RosettaScripts.30 One modification allowed the use of the docking scoring function in the high resolution docking search, while using the standard “score12” weights for side chain packing, consistent with the scoring in the default fixed backbone docking protocol in Rosetta 3.2 and the original RosettaDock implementation.15 The other modification allowed multiple loops to be specified in RosettaScripts within a single LoopRemodel construct, permitting the simultaneous refinement of multiple CDR loops (and optionally peptide). Details are given below:
In the file DockAndRetrieveSidechains.cc (used by RosettaScripts for docking), the default scoring function for the high-resolution docking search (“scorehi”) was set to “score12” (Rosetta's standard high resolution scoring function). In order to maintain consistency with the fixed backbone docking used here and the original implementation of RosettaDock,15 we modified the default “scorehi” to NULL so that the DockingMover would apply its default settings, using the high-resolution docking scoring function for scoring docking predictions and “score12” for side chain packing during the docking search.
In order to support definition of multiple loops in a single RosettaScripts entity, we modified the “loop_start_pdb_num” and “loop_end_pdb_num” tags in LoopRemodel.cc to “loop_start_pdb_nums” and “loop_end_pdb_nums”. Iterators were then used to parse the specified start and end indices for each loop, and (as with the specification of single loops) the add_loop() function was used to add each successive loop to the loop protocol.
For input to RosettaDock, the TCR and pMHC of all test cases were placed in the same initial orientation, described by the following three steps. The TCR variable domain pseudo two-fold symmetry axis was aligned with the normal vector to the MHC helix plane (resulting in an incident angle of 0°). The TCR was then rotated so that the inter-domain axis was 45° from the MHC helix vector in the MHC helix normal plane (resulting in a crossing angle of 45°). Finally, the pMHC was moved along the MHC helix normal axis so that there was a 25 Å distance between the center of mass of the MHC helix (using α-carbon atoms) and the center of the TCR inter-domain vector (calculated using each domain's disulfide bonds). This operation was implemented in a C++ program, which is available from the authors upon request.
For the test case 2NX5, we used the Modeller program,61 release 9v8, to add the missing atoms (six backbone atoms from three residues) to the unbound CDR3α loop prior to docking in Rosetta. Ten models were produced using the automodel class with the unbound structure and the sequence including the missing residues, and the top structural model was selected based on Modeller's DOPE score.
Docking model evaluation
We used the CAPRI criteria33, 62 to evaluate docking predictions, using inter-residue contacts in addition to interface and ligand RMSD to categorize predictions as incorrect, acceptable (*), medium accuracy (**), or high accuracy (***). For success rate calculation, near-native hits were defined as predictions with medium or high CAPRI accuracy.
Binding affinity prediction
We used several programs to score TCR/pMHC complexes for binding affinity prediction. For Rosetta,35 we used the “score_jd2” executable in release 3.2 with “DDG” weights “-score:weights ddg”. The pMHC, TCR, and complex were scored separately, and the ΔG prediction was calculated by subtracting the TCR and pMHC scores from that of the complex. The ZAFFI score was calculated using weighted terms from Rosetta and ZRANK as previously described,39 and the ACE40 score was calculated by ZRANK.
Figures
Molecular structures were visualized using PyMOL (http://www.pymol.org), and data analysis figures were produced using gnuplot (http://www.gnuplot.info). Superposition of molecular structures for visualization purposes was performed using FAST.60
Acknowledgments
The athors thank Brian M. Baker (University of Notre Dame) for providing coordinates of the unbound A6 TCR and the c134/Tax/HLA-A2 complex prior to their official release, and for helpful comments. They also thank Mary Ellen Fitzpatrick (Boston University) for computing support, Howook Hwang and Thom Vreven for valuable discussions, and the Scientific Computing Facilities at Boston University for computing resources. Additionally, David Cole (Cardiff University), Malkit Sami (Immunocore Ltd), and Pierre Rizkallah (Cardiff University) kindly provided details regarding affinity measurement conditions for several high affinity TCRs.
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Warren RL, Freeman JD, Zeng T, Choe G, Munro S, Moore R, Webb JR, Holt RA. Exhaustive T-cell repertoire sequencing of human peripheral blood samples reveals signatures of antigen selection and a directly measured repertoire size of at least 1 million clonotypes. Genome Res. 2011;21:790–797. doi: 10.1101/gr.115428.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sette A, Fikes J. Epitope-based vaccines: an update on epitope identification, vaccine design and delivery. Curr Opin Immunol. 2003;15:461–470. doi: 10.1016/s0952-7915(03)00083-9. [DOI] [PubMed] [Google Scholar]
- 3.Slansky JE, Jordan KR. The Goldilocks model for TCR-too much attraction might not be best for vaccine design. PLoS Biol. 2010;8 doi: 10.1371/journal.pbio.1000482. 8:e1000482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koup RA, Douek DC. Vaccine design for CD8 T lymphocyte responses. Cold Spring Harb Perspect Med. 2011;1:a007252. doi: 10.1101/cshperspect.a007252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ely LK, Burrows SR, Purcell AW, Rossjohn J, McCluskey J. T-cells behaving badly: structural insights into alloreactivity and autoimmunity. Curr Opin Immunol. 2008;20:575–580. doi: 10.1016/j.coi.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 6.Wucherpfennig KW, Call MJ, Deng L, Mariuzza R. Structural alterations in peptide-MHC recognition by self-reactive T cell receptors. Curr Opin Immunol. 2009;21:590–595. doi: 10.1016/j.coi.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, Zheng Z, Nahvi A, de Vries CR, Rogers-Freezer LJ, Mavroukakis SA, Rosenberg SA. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8:299–308. doi: 10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, Wunderlich JR, Nahvi AV, Helman LJ, Mackall CL, Kammula US, Hughes MS, Restifo NP, Raffeld M, Lee CC, Levy CL, Li YF, El-Gamil M, Schwarz SL, Laurencot C, Rosenberg SA. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29:917–924. doi: 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Varela-Rohena A, Molloy PE, Dunn SM, Li Y, Suhoski MM, Carroll RG, Milicic A, Mahon T, Sutton DH, Laugel B, Moysey R, Cameron BJ, Vuidepot A, Purbhoo MA, Cole DK, Phillips RE, June CH, Jakobsen BK, Sewell AK, Riley JL. Control of HIV-1 immune escape by CD8 T cells expressing enhanced T-cell receptor. Nat Med. 2008;14:1390–1395. doi: 10.1038/nm.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rudolph MG, Stanfield RL, Wilson IA. How TCRs bind MHCs, peptides, and coreceptors. Annu Rev Immunol. 2006;24:419–466. doi: 10.1146/annurev.immunol.23.021704.115658. [DOI] [PubMed] [Google Scholar]
- 12.Armstrong KM, Piepenbrink KH, Baker BM. Conformational changes and flexibility in T-cell receptor recognition of peptide-MHC complexes. Biochem J. 2008;415:183–196. doi: 10.1042/BJ20080850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Janin J. Protein-protein docking tested in blind predictions: the CAPRI experiment. Mol Biosyst. 2010;6:2351–2362. doi: 10.1039/c005060c. [DOI] [PubMed] [Google Scholar]
- 14.Zacharias M. Accounting for conformational changes during protein–protein docking. Curr Opin Struct Biol. 2010;20:180–186. doi: 10.1016/j.sbi.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 15.Gray JJ, Moughon S, Wang C, Schueler-Furman O, Kuhlman B, Rohl CA, Baker D. Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J Mol Biol. 2003;331:281–299. doi: 10.1016/s0022-2836(03)00670-3. [DOI] [PubMed] [Google Scholar]
- 16.Pierce B, Weng Z. ZRANK: reranking protein docking predictions with an optimized energy function. Proteins. 2007;67:1078–1086. doi: 10.1002/prot.21373. [DOI] [PubMed] [Google Scholar]
- 17.Wiehe K, Pierce B, Tong WW, Hwang H, Mintseris J, Weng Z. The performance of ZDOCK and ZRANK in rounds 6–11 of CAPRI. Proteins. 2007;69:719–725. doi: 10.1002/prot.21747. [DOI] [PubMed] [Google Scholar]
- 18.Hwang H, Vreven T, Pierce BG, Hung JH, Weng Z. Performance of ZDOCK and ZRANK in CAPRI rounds 13–19. Proteins. 2010;78:3104–3110. doi: 10.1002/prot.22764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chaudhury S, Sircar A, Sivasubramanian A, Berrondo M, Gray JJ. Incorporating biochemical information and backbone flexibility in RosettaDock for CAPRI rounds 6–12. Proteins. 2007;69:793–800. doi: 10.1002/prot.21731. [DOI] [PubMed] [Google Scholar]
- 20.Sircar A, Chaudhury S, Kilambi KP, Berrondo M, Gray JJ. A generalized approach to sampling backbone conformations with RosettaDock for CAPRI rounds 13–19. Proteins. 2010;78:3115–3123. doi: 10.1002/prot.22765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pierce B, Weng Z. A combination of rescoring and refinement significantly improves protein docking performance. Proteins. 2008;72:270–279. doi: 10.1002/prot.21920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hwang H, Vreven T, Janin J, Weng Z. Protein–protein docking benchmark version 4.0. Proteins. 2010;78:3111–3114. doi: 10.1002/prot.22830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moal IH, Agius R, Bates PA. Protein-protein binding affinity prediction on a diverse set of structures. Bioinformatics. 2011;27:3002–3009. doi: 10.1093/bioinformatics/btr513. [DOI] [PubMed] [Google Scholar]
- 24.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burrows SR, Chen Z, Archbold JK, Tynan FE, Beddoe T, Kjer-Nielsen L, Miles JJ, Khanna R, Moss DJ, Liu YC, Gras S, Kostenko L, Brennan RM, Clements CS, Brooks AG, Purcell AW, McCluskey J, Rossjohn J. Hard wiring of T cell receptor specificity for the major histocompatibility complex is underpinned by TCR adaptability. Proc Natl Acad Sci USA. 2010;107:10608–10613. doi: 10.1073/pnas.1004926107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luz JG, Huang M, Garcia KC, Rudolph MG, Apostolopoulos V, Teyton L, Wilson IA. Structural comparison of allogeneic and syngeneic T cell receptor-peptide-major histocompatibility complex complexes: a buried alloreactive mutation subtly alters peptide presentation substantially increasing V(beta) interactions. J Exp Med. 2002;195:1175–1186. doi: 10.1084/jem.20011644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Borbulevych OY, Insaidoo FK, Baxter TK, Powell DJ, Jr, Johnson LA, Restifo NP, Baker BM. Structures of MART-126/27–35 Peptide/HLA-A2 complexes reveal a remarkable disconnect between antigen structural homology and T cell recognition. J Mol Biol. 2007;372:1123–1136. doi: 10.1016/j.jmb.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ehrenmann F, Kaas Q, Lefranc MP. IMGT/3Dstructure-DB and IMGT/DomainGapAlign: a database and a tool for immunoglobulins or antibodies, T cell receptors, MHC, IgSF and MhcSF. Nucleic Acids Res. 2010;38:D301–D307. doi: 10.1093/nar/gkp946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sircar A, Gray JJ. SnugDock: paratope structural optimization during antibody-antigen docking compensates for errors in antibody homology models. PLoS Comp Biol. 2010;6:e1000644. doi: 10.1371/journal.pcbi.1000644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mandell DJ, Coutsias EA, Kortemme T. Sub-angstrom accuracy in protein loop reconstruction by robotics-inspired conformational sampling. Nat Methods. 2009;6:551–552. doi: 10.1038/nmeth0809-551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Babor M, Mandell DJ, Kortemme T. Assessment of flexible backbone protein design methods for sequence library prediction in the therapeutic antibody Herceptin-HER2 interface. Protein Sci. 2011;20:1082–1089. doi: 10.1002/pro.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fleishman SJ, Leaver-Fay A, Corn JE, Strauch EM, Khare SD, Koga N, Ashworth J, Murphy P, Richter F, Lemmon G, Meiler J, Baker D. RosettaScripts: a scripting language interface to the Rosetta macromolecular modeling suite. PLoS One. 2011;6:e20161. doi: 10.1371/journal.pone.0020161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mendez R, Leplae R, De Maria L, Wodak SJ. Assessment of blind predictions of protein–protein interactions: current status of docking methods. Proteins. 2003;52:51–67. doi: 10.1002/prot.10393. [DOI] [PubMed] [Google Scholar]
- 34.Adams JJ, Narayanan S, Liu B, Birnbaum ME, Kruse AC, Bowerman NA, Chen W, Levin AM, Connolly JM, Zhu C, Kranz DM, Garcia KC. T cell receptor signaling is limited by docking geometry to peptide-major histocompatibility complex. Immunity. 2011;35:681–693. doi: 10.1016/j.immuni.2011.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kortemme T, Baker D. A simple physical model for binding energy hot spots in protein-protein complexes. Proc Natl Acad Sci USA. 2002;99:14116–14121. doi: 10.1073/pnas.202485799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kastritis PL, Bonvin AM. Are scoring functions in protein–protein docking ready to predict interactomes? Clues from a novel binding affinity benchmark. J Proteome Res. 2010;9:2216–2225. doi: 10.1021/pr9009854. [DOI] [PubMed] [Google Scholar]
- 37.Vreven T, Hwang H, Pierce BG, Weng Z. Prediction of protein-protein binding free energies. Protein Sci. 21:396–404. doi: 10.1002/pro.2027. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cho S, Swaminathan CP, Bonsor DA, Kerzic MC, Guan R, Yang J, Kieke MC, Andersen PS, Kranz DM, Mariuzza RA, Sundberg EJ. Assessing energetic contributions to binding from a disordered region in a protein-protein interaction. Biochemistry. 2010;49:9256–9268. doi: 10.1021/bi1008968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haidar JN, Pierce B, Yu Y, Tong W, Li M, Weng Z. Structure-based design of a T-cell receptor leads to nearly 100-fold improvement in binding affinity for pepMHC. Proteins. 2009;74:948–960. doi: 10.1002/prot.22203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang C, Vasmatzis G, Cornette JL, DeLisi C. Determination of atomic desolvation energies from the structures of crystallized proteins. J Mol Biol. 1997;267:707–726. doi: 10.1006/jmbi.1996.0859. [DOI] [PubMed] [Google Scholar]
- 41.De Rosa MC, Giardina B, Bianchi C, Carelli Alinovi C, Pirolli D, Ferraccioli G, De Santis M, Di Sante G, Ria F. Modeling the ternary complex TCR-Vbeta/CollagenII(261–273)/HLA-DR4 associated with rheumatoid arthritis. PLoS One. 2010;5:e11550. doi: 10.1371/journal.pone.0011550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kato Z, Stern JN, Nakamura HK, Miyashita N, Kuwata K, Kondo N, Strominger JL. The autoimmune TCR-Ob.2F3 can bind to MBP85–99/HLA-DR2 having an unconventional mode as in TCR-Ob.1A12. Mol Immunol. 2010;48:314–320. doi: 10.1016/j.molimm.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 43.Cuendet MA, Zoete V, Michielin O. How T cell receptors interact with peptide-MHCs: a multiple steered molecular dynamics study. Proteins. 2011;79:3007–3024. doi: 10.1002/prot.23104. [DOI] [PubMed] [Google Scholar]
- 44.Al-Lazikani B, Lesk AM, Chothia C. Canonical structures for the hypervariable regions of T cell alphabeta receptors. J Mol Biol. 2000;295:979–995. doi: 10.1006/jmbi.1999.3358. [DOI] [PubMed] [Google Scholar]
- 45.North B, Lehmann A, Dunbrack RL., Jr A new clustering of antibody CDR loop conformations. J Mol Biol. 2011;406:228–256. doi: 10.1016/j.jmb.2010.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Borbulevych OY, Piepenbrink KH, Gloor BE, Scott DR, Sommese RF, Cole DK, Sewell AK, Baker BM. T cell receptor cross-reactivity directed by antigen-dependent tuning of peptide-MHC molecular flexibility. Immunity. 2009;31:885–896. doi: 10.1016/j.immuni.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dominguez C, Boelens R, Bonvin AM. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J Am Chem Soc. 2003;125:1731–1737. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- 48.Marrack P, Scott-Browne JP, Dai S, Gapin L, Kappler JW. Evolutionarily conserved amino acids that control TCR-MHC interaction. Annu Rev Immunol. 2008;26:171–203. doi: 10.1146/annurev.immunol.26.021607.090421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Garcia KC, Adams JJ, Feng D, Ely LK. The molecular basis of TCR germline bias for MHC is surprisingly simple. Nat Immunol. 2009;10:143–147. doi: 10.1038/ni.f.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stadinski BD, Trenh P, Smith RL, Bautista B, Huseby PG, Li G, Stern LJ, Huseby ES. A role for differential variable gene pairing in creating T cell receptors specific for unique major histocompatibility ligands. Immunity. 2011;35:694–704. doi: 10.1016/j.immuni.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yin Y, Wang XX, Mariuzza RA. Crystal structure of a complete ternary complex of T-cell receptor, peptide-MHC, and CD4. Proc Natl Acad Sci USA. 2012;109:5405–5410. doi: 10.1073/pnas.1118801109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wass MN, Fuentes G, Pons C, Pazos F, Valencia A. Towards the prediction of protein interaction partners using physical docking. Mol Syst Biol. 2011;7:469. doi: 10.1038/msb.2011.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Robins HS, Srivastava SK, Campregher PV, Turtle CJ, Andriesen J, Riddell SR, Carlson CS, Warren EH. Overlap and effective size of the human CD8+ T cell receptor repertoire. Sci Transl Med. 2010;2:47ra64. doi: 10.1126/scitranslmed.3001442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miconnet I. Probing the T-cell receptor repertoire with deep sequencing. Curr Opin HIV AIDS. 2012;7:64–70. doi: 10.1097/COH.0b013e32834ddcae. [DOI] [PubMed] [Google Scholar]
- 55.Li H, Ye C, Ji G, Han J. Determinants of public T cell responses. Cell Res. 2012;22:33–42. doi: 10.1038/cr.2012.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leimgruber A, Ferber M, Irving M, Hussain-Kahn H, Wieckowski S, Derre L, Rufer N, Zoete V, Michielin O. TCRep 3D: an automated in silico approach to study the structural properties of TCR repertoires. PLoS One. 2011;6:e26301. doi: 10.1371/journal.pone.0026301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yanover C, Bradley P. Large-scale characterization of peptide-MHC binding landscapes with structural simulations. Proc Natl Acad Sci USA. 2011;108:6981–6986. doi: 10.1073/pnas.1018165108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kedzierska K, Guillonneau C, Gras S, Hatton LA, Webby R, Purcell AW, Rossjohn J, Doherty PC, Turner SJ. Complete modification of TCR specificity and repertoire selection does not perturb a CD8+ T cell immunodominance hierarchy. Proc Natl Acad Sci USA. 2008;105:19408–19413. doi: 10.1073/pnas.0810274105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Colf LA, Bankovich AJ, Hanick NA, Bowerman NA, Jones LL, Kranz DM, Garcia KC. How a single T cell receptor recognizes both self and foreign MHC. Cell. 2007;129:135–146. doi: 10.1016/j.cell.2007.01.048. [DOI] [PubMed] [Google Scholar]
- 60.Zhu J, Weng Z. FAST: a novel protein structure alignment algorithm. Proteins. 2005;58:618–627. doi: 10.1002/prot.20331. [DOI] [PubMed] [Google Scholar]
- 61.Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A. Comparative protein structure modeling using Modeller. Curr Protoc Bioinformatics. 2006 doi: 10.1002/0471250953.bi0506s15. Chapter 5:Unit 5 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lensink MF, Mendez R, Wodak SJ. Docking and scoring protein complexes: CAPRI 3rd Edition. Proteins. 2007;69:704–718. doi: 10.1002/prot.21804. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.