

Abstract
Molecular dynamics simulations have been used to characterise the binding of the fatty acid ligand palmitate in the barley lipid transfer protein 1 (LTP) internal cavity. Two different palmitate binding modes (1 and 2), with similar protein–ligand interaction energies, have been identified using a variety of simulation strategies. These strategies include applying experimental protein–ligand atom–atom distance restraints during the simulation, or protonating the palmitate ligand, or using the vacuum GROMOS 54B7 force-field parameter set for the ligand during the initial stages of the simulations. In both the binding modes identified the palmitate carboxylate head group hydrogen bonds with main chain amide groups in helix A, residues 4 to 19, of the protein. In binding mode 1 the hydrogen bonds are to Lys 11, Cys 13, and Leu 14 and in binding mode 2 to Thr 15, Tyr 16, Val 17, Ser 24 and also to the OH of Thr 15. In both cases palmitate binding exploits irregularity of the intrahelical hydrogen-bonding pattern in helix A of barley LTP due to the presence of Pro 12. Simulations of two variants of barley LTP, namely the single mutant Pro12Val and the double mutant Pro12Val Pro70Val, show that Pro 12 is required for persistent palmitate binding in the LTP cavity. Overall, the work identifies key MD simulation approaches for characterizing the details of protein–ligand interactions in complexes where NMR data provide insufficient restraints.
Keywords: lipid transfer protein, ligand binding, lipid binding, palmitic acid, molecular dynamics simulation, GROMOS, NMR spectroscopy, Proline, internal cavity
Introduction
The binding of small molecules to proteins is a key step in many biological processes including enzyme catalysis, the immune response and cell signaling.1 Understanding the details of protein–ligand interactions is therefore an important goal in biophysical research.2, 3 It is also essential for successful structure-based drug design.4, 5 In some cases, proteins have binding sites that display a high degree of specificity for binding one type of molecule. In other cases there is much more promiscuity, with a variety of ligands being accommodated, for example, in a hydrophobic cavity within a protein.6–8 Both X-ray crystallography and NMR techniques have the potential to determine in detail the structures of protein–ligand complexes. However, there can be limitations to these structure determination methods, particularly if the ligand retains a degree of mobility when it is bound or if multiple binding modes are adopted by a given ligand.9–13 In this article we explore how MD simulations can help in the characterization of ligand binding in situations like this, using as a test case the binding of the fatty acid palmitate to barley nonspecific lipid transfer protein 1.
Nonspecific lipid transfer proteins 1 (LTP) are found in a wide range of plant species including cereals and fruit.14 The functions of these proteins are not fully established, although it is clear that they have a role in plant defense mechanisms against bacteria and fungi15, 16 and probably also in plant growth and development.17 In addition many LTPs are allergens in plant foods and pollens.18 Plant LTPs adopt a four helix fold that is stabilized by four disulphide bridges and contains a long tunnel-like internal cavity.18, 19 Ligands including fatty acids, acyl coenzyme A, phospholipids, glycolipids, hydroxylated fatty acids, and prostaglandins have been shown to bind in this cavity and a range of structures of plant LTPs bound to lipid and related molecules have been determined.9, 20–25 These demonstrate that the LTP cavity has a plasticity and can adapt and increase its volume on binding larger ligands.9, 20, 26
One particularly interesting feature is that not all ligands show the same binding mode in different species of plant LTPs. In particular, the ligand palmitate binds in an inverted manner in barley LTP compared to maize LTP.21 In maize LTP the palmitate carboxylate head group is exposed, located on the surface of the protein interacting with the side chain of Tyr 81 while the hydrophobic tail of the ligand is buried in the internal cavity.9, 25 In contrast, in barley LTP the palmitate ligand is totally enclosed with the head group at the opposite end of the cavity, positioned close to Leu 14, Val 17, and Ile 69.21 In this binding mode the carboxylate head group is held in a largely hydrophobic environment. Lerche and Poulsen21 proposed that the palmitate head group may make a hydrogen bond or salt bridge to the side chain of Lys 9 when it is bound in the barley LTP cavity. This residue is changed to alanine in the maize LTP sequence and it is suggested that this sequence variation could explain the different binding modes seen for palmitate in barley and maize LTP. However, in the NMR structure of barley LTP and palmitate21 the distance between the palmitate head group and the Lys 9 side chain is quite large (≥0.56 nm).
The position of the palmitate in the LTP cavity was defined in the NMR structure determination by 25 protein–ligand NOE restraints.21 These NOEs are from aliphatic protons on the carbon chain of the palmitate but the NMR data does not identify any interactions involving the carboxylate head group. The barley LTP-palmitate complex provided an ideal system for exploring how MD simulations can enhance structure determinations in which hydrogen bonds or other electrostatic interactions play an important role. Interestingly, the MD simulations have located two different binding modes for palmitate. Both of these involve hydrogen bonds from the ligand to main chain NH groups in the helix A region, residues 4–19, of the protein. For barley LTP the ligand binding appears to exploit irregularities in the intraprotein backbone hydrogen bonds in helix A due to the presence of Pro 12. Comparisons of MD simulations of wild type barley LTP with simulations of two proline mutants suggest that Pro 12 may be essential for the observed ligand binding mode.
Results and Discussion
Identification of binding modes for palmitate in the barley LTP cavity
Initially a 20 ns simulation of barley LTP with palmitate was run starting from the NMR structure of this complex (1be2 model 121) with no NOE restraints included (WT_LA_310 simulation; Table I). Very quickly in this simulation the ligand moved and began to come out of the LTP internal cavity. This change is reflected in an increase, to ∼1 nm, of the atom positional root-mean-square deviation (RMSD) for the ligand atoms from the initial NMR structure (Fig. 1). Note that in calculating the RMSD values, the protein backbone atoms were superimposed and the ligand RMSD was then calculated. The RMSD values therefore reflect changes in both the ligand conformation and its position in the LTP cavity. There is also an increase in the solvent accessible surface area (SASA) of the palmitate ligand in the simulations from 0 nm2 in the initial NMR structure to an average of ∼0.65 nm2 (Fig. 1). This increase comes mostly from the charged carboxylate head group atoms, which become highly solvent exposed.
Table I.
Summary of the Barley LTP-Palmitate Simulations Reported in This Work
| Simulation name | Sequence | Starting structure | NOE restraints | Ligand temperature | Number water molecules | Simulation length (ns) |
|---|---|---|---|---|---|---|
| WT_LA_310 | WT | 1be2 | No | 310 K | 8428 | 20 |
| WT_LA_NOE_310 | WT | 1be2 | Yes | 310 K | 8428 | 20 |
| WT_LAH_310 | WT | LAH | No | 310 K | 8254 | 20 |
| WT_LB_NOE_400 | WT | LB | Yes | 400 K | 8428 | 10 |
| M12_LAH_310 | P12V | LAH | No | 310 K | 8635 | 10 |
| M12,70_LAH_310 | P12V, P70V | LAH | No | 310 K | 8330 | 10 |
The simulation name used, the protein sequence (either wild type (WT) or the mutations made), the starting structure for the simulation, whether NOE restraints (protein-ligand and intraligand) were applied, the ligand temperature in the simulations, the number of SPC water molecules in the rectangular periodic simulation box and the length of simulation used in the analysis are indicated. 1be2 refers to simulations that were started from the NMR structure of the LTP-palmitate complex. LB refers to simulations started from the final structure of a 5 ns simulation where the 54B7 (vacuum) force-field parameters were used for the ligand and the 54A7 parameters were used for the protein and solvent. LAH refers to simulations started from final structures taken from 20 ns simulations with protonated palmitate as a ligand. Full details are given in the Methods section.
Figure 1.
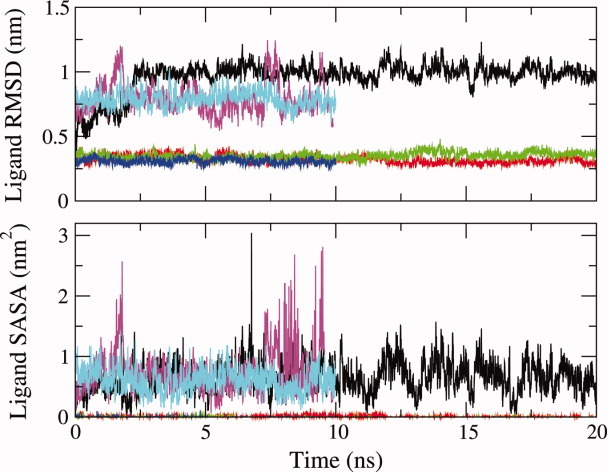
Time series showing variations in the atom-positional RMSD of the palmitate ligand atoms from the 1be2 NMR structure (upper panel) and the palmitate solvent accessible surface area (lower panel) through the simulations. The data for the different simulations are shown in the following colours: black, WT_LA_310; red, WT_LA_NOE_310; green, WT_LAH_310; blue, WT_LB_NOE_400; magenta, M12_LAH_310; cyan, M12,70_LAH_310. To calculate the RMSD values the protein backbone (N, CA, C) atoms were superimposed and the ligand RMSD was then calculated. These values therefore reflect changes both in the ligand conformation and its position in the LTP cavity.
There have been suggestions that the palmitate head group may make a hydrogen bond or salt bridge with the side chain of Lys 9 in the internal cavity of barley LTP.21 However, hydrogen bonds between the palmitate head group and Lys 9 have a population of less than 3% in the 20 ns WT_LA_310 simulation. More persistent hydrogen bonds (27–36% populations) are seen between the palmitate head group and the side chains of Gln 5 and Gln 37 (Table II). These two side chains are on the surface of the protein rather than in the internal cavity. In the experimental NMR structure determination 25 protein–ligand atom-atom NOE distance bounds defined the position of the palmitate ligand within the LTP cavity.21 All of these NOE bounds are satisfied in the NMR structure. However, comparisons of these NOE bounds with the simulation trajectory show a very poor agreement. Only six of the NOE bounds are satisfied and there are eight very large NOE bound violations greater than 0.5 nm (Table III).
Table II.
Protein–Ligand Hydrogen Bonds With Populations of Greater Than 10% in the Simulations of the Barley LTP-Palmitate Complex
| Hydrogen bond | Simulation | ||||||
|---|---|---|---|---|---|---|---|
| Protein donor group | PLM group | WT_LA_310 | WT_LA_ NOE_310 | WT_ LAH_310 | WT_LB_ NOE_400 | M12_ LAH_310 | M12,70_ LAH_310 |
| 5 Gln HE2 | O1 | 32 | |||||
| 5 Gln HE2 | O2 | 30 | |||||
| 8 Ser OH | O1 | 12 | |||||
| 8 Ser OH | O2 | 16 | |||||
| 11 Lys HN | O1 | 50 | |||||
| 11 Lys HN | O2 | 50 | |||||
| 13 Cys HN | O1 | 57 | |||||
| 13 Cys HN | O2 | 55 | |||||
| 14 Leu HN | O1 | 28 | |||||
| 14 Leu HN | O2 | 52 | |||||
| 15 Thr HN | O1 | 50 | |||||
| 15 Thr HN | O2 | 45 | 93 | ||||
| 15 Thr OH | O2 | 54 | 96 | ||||
| 16 Tyr NH | O1 | 36 | 90 | ||||
| 16 Tyr HN | O2 | 66 | |||||
| 17 Val HN | O1 | 80 | 96 | ||||
| 24 Ser HN | O2 | 93 | |||||
| 37 Gln HE2 | O1 | 27 | |||||
| 37 Gln HE2 | O2 | 36 | |||||
| 65 Asn ND2 | O1 | 18 | |||||
| 65 Asn ND1 | O2 | 18 | |||||
| 68 Ser OH | O1 | 24 | |||||
| 68 Ser OH | O2 | 26 | |||||
Table III.
Results of the Comparison of Average Distances Calculated From the Simulation Trajectories for the 25 Inter-Proton Distances Between Palmitate and Barley LTP Derived From the Experimental NOE Cross-Peak Intensities.
| Number of NOE bound violations | |||||||
|---|---|---|---|---|---|---|---|
| Structure or simulation | No violation | 0-0.05 | 0.05-0.1 | 0.1-0.3 | 0.3-0.5 | 0.5-0.7 | > 0.7 |
| 1be2 NMR | 25 | ||||||
| WT_LA_310 | 6 | 2 | 2 | 4 | 3 | 6 | 2 |
| WT_LA_NOE_310 | 23 | 1 | 1 | ||||
| WT_LAH_310 | 20 | 1 | 1 | 1 | 2 | ||
| WT_LB_NOE_400 | 23 | 2 | |||||
| M12_LAH_310 | 7 | 2 | 2 | 5 | 2 | 7 | |
| M12,70_LAH_310 | 7 | 1 | 2 | 4 | 6 | 4 | 1 |
The number of distance bound violations within given ranges (in nm) are listed for each simulation. These were calculated from the difference between the r−3 averaged distances in the simulation and the corresponding NMR-derived upper distance bounds. The data for the 1be2 NMR structure are given for comparison. A full list of the protein-ligand NOEs and the individual violations for each simulation are given in the Supporting Information.
From the initial simulation results it is clear that the original NMR structure of the barley LTP-palmitate complex does not exhibit a palmitate binding mode that is stable in the GROMOS 54A7 force field. To determine whether there is a ligand binding mode for palmitate in the LTP cavity that satisfies the experimental NOE data and can persist in an MD simulation, a series of MD simulations were run. In the first of these, 25 protein–ligand and 9 intraligand experimental NOEs were applied as NOE restraints throughout a 20 ns simulation (WT_LA_NOE_310 simulation). In this simulation the ligand only moves slightly from its original position in the NMR structure (ligand atom positional RMSD about 0.3 nm) and stays buried in the LTP internal cavity (mean ligand SASA 0.008 nm2) (Fig. 1). However, unlike in the NMR structure, the ligand carboxylate head group makes persistent hydrogen bonds to the main chain NH groups of Lys 11, Cys 13, and Leu 14 (population 28–57%; Table II). There is also a good agreement with the experimental data (no violations of the protein–ligand NOE bounds>0.1 nm; Table III). When the NOE restraints are removed and the simulation continued for another 10 ns this binding mode persisted. The hydrogen bonds to Lys 11, Cys 13, and Leu 14 retained populations of 88–98% and the mean ligand atom positional RMSD from the NMR structure was about 0.22 nm.
To counteract the affinity of the carboxylate head group of the palmitate ligand for the solvent (SPC water), which appears to drive the ligand out of the hydrophobic cavity, a further 20 ns simulation was run with the palmitate ligand protonated. This modification prevented the head group from being immediately expelled from the hydrophobic cavity, allowing the ligand time to search for and establish favourable hydrogen-bonding interactions. In this simulation hydrogen bonds formed between the carboxylate head group and the main chain NH groups of Tyr 16, Val 17, and Ser 24 with populations of 19, 45, and 25% respectively through the simulation trajectory. After 20 ns the proton was removed from the head group and a further 20 ns simulation was run with the normal palmitate ligand (simulation WT_LAH_310). The hydrogen bonds from the main chain NH groups of Tyr 16, Val 17 and Ser 24 to palmitate persist and there are also hydrogen bonds from the Thr 15 main chain NH and side chain OH groups to palmitate (populations of 36–93%; Table II). In this simulation the ligand stays buried in the cavity (mean ligand SASA 0.001 nm2) with a ligand atom positional RMSD to the starting NMR structure of about 0.35 nm (Fig. 1). No experimental NOE restraints were applied during the WT_LAH_310 simulation. Comparison of the 20 ns WT_LAH_310 trajectory with the 25 experimental protein–ligand NOE distance bounds shows that there are two violations greater than 0.3 nm (Table III). Both of these NOEs are from protons within the palmitate carbon chain to the side chain of Lys 9. To assess these violations the simulation was continued, applying the 25 protein–ligand and 9 intraligand experimental NOEs as distance restraints. The continuation simulation showed that rotation of the side chain of Lys 9 results in a good agreement with the NOE data (only one violation>0.05 nm and none>0.1 nm), whilst the ligand binding mode and protein–ligand hydrogen bonds are unchanged.
The WT_LA_NOE_310 and WT_LAH_310 simulations identified two different binding modes for palmitate in the LTP cavity (Fig. 2). In the WT_LA_NOE_310 simulation the palmitate head group hydrogen bonds with the main chain NH groups of Lys 11, Cys 13, and Leu 14 in helix A (mode 1) while in the WT_LAH_310 simulation the hydrogen bonds are with the main chain NH groups of Thr 15, Tyr 16, and Val 17 towards the C-terminus of helix A and with the Ser 24 NH and Thr 15 OH groups (mode 2).
Figure 2.
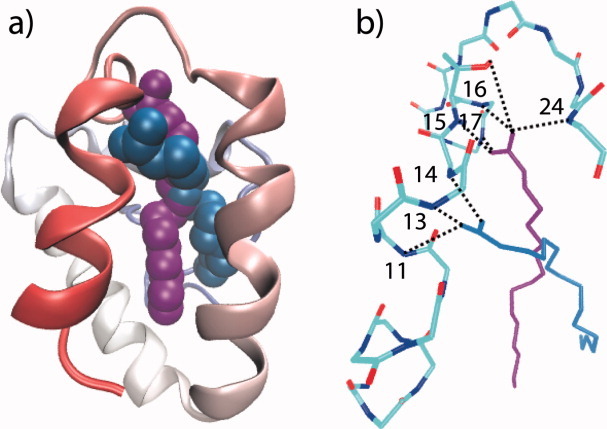
The two binding modes for palmitate in the internal cavity of barley LTP. Palmitate binding in Mode 1, where the head group hydrogen bonds with the main chain NH groups of Lys 11, Cys 13, and Leu 14, is shown in blue (structure taken from the WT_LA_NOE_310 trajectory after 20 ns of simulation). Palmitate binding in Mode 2, where the head group hydrogen bonds with the main chain NH groups of Thr 15, Tyr 16, Val 17, and Ser 24 and the side chain OH group of Thr 15, is shown in purple (structure taken from the WT_LAH_310 trajectory after 20 ns of simulation). In panel a) the protein backbone is shown schematically, coloured from red at the N-terminus (and helix A) through to blue at the C-terminus, and the palmitate ligands are shown in space filling representation. In panel b) the main chain of residues 4–24 and the side chain of Thr 15 in barley LTP are shown coloured according to atom type (carbon, cyan; nitrogen, blue; oxygen, red). Hydrogen bonds between the protein and palmitate that have a population of 50% or greater through the simulations are indicated by dotted lines. Note that in most cases hydrogen bonds involving both the palmitate O1 and O2 atoms are seen (Table II) but for clarity only one hydrogen bond is shown in each case. The figure was generated using the program VMD.41
To determine the best way of exploring these two alternative ligand binding modes, as well as the possibility of additional binding modes, a number of further simulation techniques were tested. These included applying synthetic distance restraints to keep the charged ligand in the LTP cavity and simulations in which only two of the experimental protein–ligand NOEs were applied as restraints, rather than the full set of 25, to facilitate more extensive conformational searching by the ligand. One more complex approach involved using the GROMOS 54A7 force-field parameters for protein and solvent but the 54B7 (vacuum) parameters for the ligand,27 with the aim of promoting its retention in the binding pocket during the initial stages of the search for a binding mode. After an initial 5 ns simulation, the ligand parameters were changed back to those of the 54A7 force field and the simulation was run for a further 10 ns. The 25 protein–ligand NOE restraints were applied throughout to ensure that the calculated structures would be in agreement with the experimental data. Additionally, the ligand was at an elevated temperature (400 K) to promote conformational sampling, while the rest of the system was at 310 K. Only the final 10 ns of this simulation, during which the 54A7 force-field parameters were used for the whole system, were used for analysis (WT_LB_NOE_400). The advantage of this approach is that it can be used for systems where the ligand cannot be protonated.
The ligand binding mode adopted in the WT_LB_NOE_400 simulation is closely similar to that seen in the WT_LAH_310 simulation (i.e., mode 2). The ligand atom positional RMSD to the starting NMR structure is about 0.3 nm and there are hydrogen bonds from Thr 15 NH, Thr 15 OH, Tyr 16 NH, and Val 17 NH to the head group of palmitate. In addition, there is good agreement with the experimental protein–ligand NOEs (no violations>0.05 nm).
To assess energetically the ligand binding modes for palmitate in the LTP cavity the protein and ligand interaction energies in the simulations were compared (Table IV). The original WT_LA_310 simulation, where the ligand head group starts to come out of the LTP cavity, is characterized by a very favorable ligand-water total noncovalent interaction energy (−394 kJ mol−1). The WT_LA_NOE_310 simulation, where Lys 11, Cys 13, and Leu 14 hydrogen bond to the palmitate head group (mode 1) and the WT_LAH_310 and WT_LB_NOE_400 simulations where Thr 15 NH, Thr 15 OH, Tyr 16 NH, and Val 17 NH (and Ser 24 NH) hydrogen bond to the palmitate head group (mode 2) have very similar protein–ligand van der Waals energies (−162, −164, and −158 kJ mol−1, respectively). The WT_LAH_310 and WT_LB_NOE_400 simulations (mode 2) have slightly more favorable protein–ligand electrostatic energies (−390 and −402 kJ mol−1, respectively) than the WT_LA_NOE_310 simulation (mode 1; −325 kJ mol−1). The ligand–water electrostatic energy is slightly more favorable for the WT_LA_NOE_310 simulation (−152 kJ mol−1). The overall similarities of the protein–ligand interaction energies for palmitate binding modes 1 and 2 in the simulations suggest that both binding modes may be observed with comparable populations.
Table IV.
Contributions Made by the Covalent and Noncovalent Terms to the Potential Energy (kJ mol−1) of the Barley LTP-Palmitate Systems During the MD Simulations.
| WT_LA_310 | WT_LA_NOE_310 | WT_LAH_310 | WT_LB_NOE_400 | M12_LAH_310 | M12,70_LAH_310 | |
|---|---|---|---|---|---|---|
| Covalent | ||||||
| Protein | 3016 (81) | 3015 (82) | 3025 (84) | 3019 (77) | 3026 (84) | 2939 (80) |
| Ligand | 40.7 (10) | 42.7 (10) | 40.6 (10) | 41.0 (10) | 41.2 (10) | 41.6 (10) |
| van der Waals | ||||||
| Protein–protein | −2168 (64) | −2114 (59) | −2140 (58) | −2105 (55) | −2104 (56) | −2150 (55) |
| Protein–ligand | −147 (11) | −162 (12) | −164 (14) | −158 (13) | −160 (13) | −160 (11) |
| Protein–water | −552 (78) | −580 (76) | −555 (76) | −577 (74) | −600 (79) | −591 (75) |
| Ligand–ligand | −17.4 (4) | −16.0 (4) | −17.9 (3) | −18.9 (3) | −19.0 (3) | −18.3 (3) |
| Ligand–water | 7.6 (12) | 1.4 (7) | −3.3 (4) | −4.5 (3) | 9.9 (12) | 13.5 (12) |
| Electrostatic | ||||||
| Protein–protein | −5412 (175) | −5323 (223) | −5233 (205) | −5472 (162) | −5502 (154) | −5404 (173) |
| Protein–ligand | −177 (74) | −325 (42) | −390 (57) | −402 (49) | −144 (71) | −168 (73) |
| Protein–water | −8128 (364) | −8231 (438) | −8322 (373) | −7653 (332) | −8053 (329) | −8183 (353) |
| Ligand–ligand | 0 | 0 | 0 | 0 | 0 | 0 |
| Ligand–water | −402 (82) | −152 (44) | −116 (43) | −77 (37) | −423 (77) | −378 (81) |
| Total noncovalent | ||||||
| Protein–protein | −7581 (172) | −7438 (215) | −7373 (191) | −7577 (156) | −7607 (153) | −7554 (165) |
| Protein–ligand | −324 (72) | −488 (40) | −553 (55) | −560 (45) | −303 (68) | −326 (70) |
| Protein–water | −8680 (345) | −8810 (413) | −8877 (352) | −8230 (303) | −8653 (301) | −8774 (325) |
| Ligand–ligand | −17.4 (4) | −16.0 (4) | −17.9 (3) | −18.9 (3) | −19.0 (3) | −18.3 (3.2) |
| Ligand–water | −394 (78) | −151 (41) | −118 (41) | −81 (35) | −413 (73) | −364 (77) |
In each case the mean value is given, averaged over the trajectory, with the root-mean-square fluctuation in brackets.
Proline 12 Is Required for Favorable Palmitate Binding
An interesting feature of both binding modes identified for palmitate in the LTP cavity is the involvement of hydrogen bonds to the ligand from the main chain NH groups of residues in helix A of the protein. Normally the main chain NH groups would be involved in the hydrogen bonds that stabilize the α-helical structure, but helix A (residues 4–19) has an irregular hydrogen-bonding pattern due to the presence of a proline at residue 12 (Table V). NMR hydrogen-exchange studies28 identified slow amide proton exchange rates for residues 6, 9, 10, 11, 13, 14, 17, and 18 in helix A. In the NMR structure determination21 NH(i)-CO(i-4) hydrogen bond restraints were included for the NH groups of residues 9, 10, 11, 17, 18, and 19 and an NH(i)-CO(i-3) hydrogen bond restraint for the NH group of residue 13. The NH groups of residues 14, 15, and 16 are not involved in helical hydrogen bonds in the NMR structure.
Table V.
Populations of Helical NH(i)-CO(i-4) and NH(i)-CO(i-3) Hydrogen Bonds in the Region of helix A (Residues 4-19) That Makes Contacts to the Ligand in the Simulations of the Barley LTP-Palmitate Complex.
| HN-CO | NMR structure | WT_LA_310 | WT_LA_NOE_310 | WT_LAH_310 | WT_LB_NOE_400 | M12_LAH_310 | M12,70_LAH_310 |
|---|---|---|---|---|---|---|---|
| 10-6 | 100 | 91 | 78 | 96 | 99 | 94 | |
| 10-7 | 5 | 15 | 2 | 1 | 13 | ||
| 11-7 | 100 | 30 | 93 | 91 | 96 | 87 | |
| 11-8 | 30 | 1 | 1 | 1 | 3 | ||
| 12-8 | Pro | Pro | Pro | Pro | Pro | 61 | 85 |
| 12-9 | Pro | Pro | Pro | Pro | Pro | 9 | 2 |
| 13-9 | 5 | 6 | 64 | 88 | |||
| 13-10 | 100 | 32 | 42 | 11 | 3 | ||
| 14-10 | 54 | 1 | 34 | 16 | 80 | ||
| 14-11 | 11 | 6 | 2 | 4 | |||
| 15-11 | 1 | 31 | 15 | ||||
| 15-12 | 29 | 17 | |||||
| 16-12 | 15 | 45 | |||||
| 16-13 | 15 | 30 | 18 | 6 | |||
| 17-13 | 100 | 46 | 57 | 57 | 27 | ||
| 17-14 | 5 | 5 | 1 | 14 | 4 | ||
| 18-14 | 100 | 88 | 63 | 24 | 54 | 51 | 91 |
| 18-15 | 5 | 14 | 28 | 1 | 15 | 3 | |
| 19-15 | 100 | 58 | 71 | 58 | 2 | 79 | 54 |
| 19-16 | 27 | 17 | 25 | 30 | 9 | 29 |
The hydrogen bonds in the 1be2 NMR structure (model 1) are included for comparison and the presence and position of the proline residue is shown (Pro).
In the simulations, the hydrogen-bonding pattern in the centre of helix A is quite dynamic. In the WT_LA_310 simulation all the amide protons in helix A are involved in NH(i)-CO(i-4) and/or NH(i)-CO(i-3) hydrogen bonds. However, the populations of some of the hydrogen bonds in the centre of the helix are quite low (Table V). In the WT_LA_NOE_310 simulation (binding mode 1) the NH groups of residues 11, 13, and 14 have very low helical hydrogen-bonding populations (≤6%) while the populations of helical hydrogen bonds for residues 15, 16, and 17 is higher (30–57%). In contrast in the WT_LAH_310 and WT_LB_NOE_400 simulations (binding mode 2) the NH groups of residues 11, 13, and 14 are involved in more persistent helical hydrogen bonds (populations 16–93%) while helical hydrogen bonds for residues 15, 16, and 17 are virtually absent (≤1%) (Table V). Thus fluctuations in the intrahelical hydrogen bond populations along the helix enable the NH groups of residues 11, 13, and 14 in binding mode 1 and residues 15, 16, and 17 in binding mode 2 to be available for hydrogen-bonding to palmitate. The inclusion of the helical hydrogen-bond restraints in the NMR structure determination21 may have been one of the reasons why these palmitate binding modes were not initially identified.
Overall, it appears that palmitate binding to barley LTP exploits the irregular intrahelical hydrogen-bonding in helix A around Pro 12. To test whether Pro 12 is required in the helix A sequence to stabilize these ligand binding modes we ran two further simulations. In the first Pro 12 was mutated to valine (M12_LAH_310) and in the second both Pro 12 and Pro 70 (in helix D) were changed to valines (M12,70_LAH_310). The same simulation protocol was followed as for the WT_LAH_310 simulation, with the palmitate ligand protonated for the first 20 ns, followed by a 10 ns continuation simulation (used for analysis) with the ligand deprotonated.
In the simulation of the single P12V mutant with protonated palmitate, the ligand stays in the LTP cavity hydrogen-bonding with the main chain NH of Ile 69 in helix D (76% population). The intrahelical hydrogen-bonding pattern in helix D is irregular due to the presence of Pro 70, which allows the persistent hydrogen bond from the Ile 69 NH to the ligand to form. Upon deprotonation the hydrogen bond to the Ile 69 NH group is quickly lost. The palmitate ligand starts to leave the LTP cavity with a ligand atom positional RMSD from the starting structure of about 0.80 nm and a ligand solvent accessible surface area of about 0.72 nm2 (Fig. 1). In the double mutant P12V P70V simulation the protonated ligand forms no persistent hydrogen bonds with the protein and directly begins to exit the LTP cavity. It remains solvent exposed on deprotonation with the carboxylate head group forming fluctuating hydrogen bonds to the side chain of Ser 8 on the protein surface (16% hydrogen-bond population). Thus for both variant proteins no persistent binding mode was found for palmitate in the LTP cavity with the simulation protocol used. These results therefore support the proposal that the presence of Pro 12 in the sequence is essential for allowing the favorable binding of palmitate to barley LTP.
Conclusions
Although the NMR structure determination for the barley LTP-palmitate complex used 25 protein–ligand NOE restraints,21 these were not sufficient to identify the details of the ligand binding site with a standard calculation protocol. Indeed, in an MD simulation starting from the NMR structure the ligand binding was not stable. The palmitate ligand moved out of the internal cavity and became significantly solvent exposed. To be able to search for and find the stable ligand binding mode(s) for palmitate in an MD simulation, the ligand must be kept in the largely hydrophobic internal cavity. This work has identified three approaches that can be used to achieve this. These are applying the experimental protein–ligand NOEs as restraints during the simulation; protonating the ligand head group in the initial stages of the simulation; and using the 54B7 vacuum force-field parameters for the ligand. The advantage of the third strategy is that it can be used in situations where the ligand's actual charge cannot be changed by protonation. With respect to the use of NMR distance restraints, we note that the use of hydrogen-bond restraints, which do not represent observable quantities, may inhibit proper sampling of ligand binding configurations.
With these approaches two different binding modes, characterized by different hydrogen-bonding patterns, have been identified for palmitate in the barley LTP cavity. The two modes have similar protein–ligand interaction energies, suggesting that both are significantly populated. In these binding modes the palmitate head group is not involved in a salt bridge or hydrogen bond to the side chain of Lys 9 as was originally suggested.21 Instead the ligand forms hydrogen bonds with main-chain amide groups of residues in helix A. The ligand binding exploits irregularities in the helical hydrogen-bonding pattern in this helix due to the presence of Pro 12. Simulations of single mutant P12V and double mutant P12V P70V variants of barley LTP suggest that nature may have included the proline in the helix A sequence to stabilize this ligand binding mode in barley LTP.
Computational Methods
The MD simulations were carried out using the GROMOS biomolecular simulation software29–32 and, unless otherwise stated, the GROMOS 54A7 force-field parameter set.27 The coordinates for the barley LTP-palmitate complex were taken from model 1 in the NMR structure21 deposited in the PDB with entry code 1be2. The barley LTP-palmitate simulations reported in this work are summarized in Table I. For the WT_LA_310 and WT_LA_NOE_310 simulations the starting structure was the 1be2 NMR structure. The WT_LB_NOE_400 simulation started from the final structure of a 5 ns simulation where the 54B7 (vacuum) force-field parameters29 were used for the ligand and the 54A7 parameters were used for the protein and solvent (LB). The WT_LAH_310, M12_LAH_310, and M12,70_LAH_310 simulation were started from final structures taken from 20 ns simulations with the palmitate ligand protonated (LAH). For the M12_LAH_310 and M12,70_LAH_310 simulations Pro 12, and both Pro 12 and Pro 70, respectively in the LTP sequence were changed to valines.
In all the simulations the Asp and Glu side chains in the protein were unprotonated and the two histidine side chains were singly protonated at Nδ1 to correspond to the pH 7.2 conditions used in the experimental NMR study.21 In all the simulations used for analysis the palmitate ligand was unprotonated and one chloride ion was added to achieve overall neutrality of the system. In each case the protein was solvated in a rectangular box and periodic boundary conditions were applied. The minimum solute-box wall distance was set to between 1.2 and 1.4 nm to give similar numbers of simple point charge (SPC) water molecules33 in each simulation (Table I). All simulations were performed at a temperature of 310 K for the protein and solvent, and the temperatures used for the ligand are given in Table I. For each simulation an initial equilibration scheme comprising six 20 ps simulations at temperatures of 60, 120, 180, 240, 280, and 310 K was used. During the first 80 ps of this equilibration the solute atoms were restrained to their positions in the starting structure. Following equilibration, in the WT_LA_NOE_310 and WT_LB_NOE_400 simulations, 25 protein–ligand and 9 intraligand NOE distance restraints were imposed31 as instantaneous restraints using a force constant of 2000 kJ mol−1 nm−2.
All simulations were performed at constant pressure (1 atm), the temperature and pressure being maintained using the weak coupling algorithm,34 with relaxation times of τT = 0.1 ps and τp = 0.5 ps and an isothermal compressibility of 4.575 × 10−4 (kJ mol−1 nm−3)−1. Protein and solvent were separately coupled to the heat bath. The SHAKE algorithm35 was used with a geometric tolerance of 10−4 nm to constrain bond lengths and the geometry of the water molecules, allowing for an integration time step of 2 fs. The centre of mass motion was removed every 1000 time steps. Nonbonded interactions were calculated using a triple-range cutoff scheme with cutoff radii of 0.8 and 1.4 nm. Interactions within 0.8 nm were evaluated every time step and intermediate range interactions were updated every fifth time step. To account for the influence of the dielectric medium outside the cutoff sphere, a reaction-field force36 based on a relative dielectric permittivity ε of 61 was used.37
Analysis was performed with the GROMOS++ suite of analysis programs,29 using coordinates and energies trajectories written to disk every 5 ps. The atom-positional root-mean-square deviation (RMSD) between indicated atoms of two structures was calculated after the superposition of the heavy atoms of the protein backbone. The RMSD values for the ligand therefore reflect both changes in the ligand position within the LTP cavity and changes in its conformation. 25 inter-proton distances between the ligand and protein, derived from the NOE cross-peak intensities,21 were compared with the average inter-proton distances <r−3>−1/3 calculated from the simulated trajectories using r−3 averaging. Because the GROMOS force fields make use of united atoms, positions of aliphatic hydrogen atoms were constructed based on standard geometries.32, 38 If an NOE upper bound involved nonstereospecifically assigned protons, a pseudo atom was constructed.32, 38 Regions of secondary structure were identified using the rules defined by Kabsch and Sander in the program DSSP.39 Hydrogen bonds were identified according to a geometric criterion: a hydrogen bond was assumed to exist if the hydrogen-acceptor distance is smaller than 0.25 nm and the donor-hydrogen acceptor angle is larger than 135°. The solvent accessible surface areas were calculated using the algorithm of Lee and Richards.40
Acknowledgments
The authors thank Flemming Poulsen for providing the list of experimental NOE restraints used to determine the NMR structure of the barley LTP-palmitate complex.
Glossary
Abbreviations
- LTP
nonspecific lipid transfer protein 1
- MD
molecular dynamics
- NMR
nuclear magnetic resonance
- NOE
nuclear Overhauser effect
- PDB
protein data bank
- RMSD
root-mean-square deviation
- SASA
solvent accessible surface area
- SPC
simple point charge
Supplementary material
Additional supporting Information may be found in the online version of this article.
References
- 1.Nelson D, Cox M. Lehninger Principles of Biochemistry. USA: W. H. Freeman; 2008. [Google Scholar]
- 2.Gohlke H, Klebe G. Approaches to the description and prediction of the binding affinity of small-molecule ligands to macromolecular receptors. Angew Chemie (Int Ed. 2002;41:2644–2676. doi: 10.1002/1521-3773(20020802)41:15<2644::AID-ANIE2644>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 3.Mobley DL, Dill KA. Binding of small-molecule ligands to proteins: “What you see” is not always “what you get”. Structure. 2009;17:489–498. doi: 10.1016/j.str.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Warren GL, Andrews CW, Capelli A-M, Clarke B, LaLonde J, Lambert MH, Lindvall M, Nevins N, Semus SF, Senger S, Tedesco G, Wall ID, Woolven JM, Peishoff CE, Head MS. A critical assessment of docking programs and scoring functions. J Med Chem. 2006;49:5912–5931. doi: 10.1021/jm050362n. [DOI] [PubMed] [Google Scholar]
- 5.Jorgensen WL. The many roles of computation in drug discovery. Science. 2004;303:1813–1818. doi: 10.1126/science.1096361. [DOI] [PubMed] [Google Scholar]
- 6.Pertinhez TA, Ferrari E, Casali E, Patel JA, Spisni A, Smith LJ. The binding cavity of mouse major urinary protein is optimised for a variety of ligand binding modes. Biochem Biophys Res Commun. 2009;390:1266–1271. doi: 10.1016/j.bbrc.2009.10.133. [DOI] [PubMed] [Google Scholar]
- 7.Myslinski JM, DeLorbe JE, Clements JH, Martin SF. Protein-ligand interactions: thermodynamic effects associated with increasing nonpolar surface area. J Am Chem Soc. 2011;133:18518–18521. doi: 10.1021/ja2068752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nobeli I, Favia AD, Thornton JM. Protein promiscuity and its implications for biotechnology. Nat Biotechnol. 2009;27:157–167. doi: 10.1038/nbt1519. [DOI] [PubMed] [Google Scholar]
- 9.Han GW, Lee JY, Song HK, Chang CS, Min K, Moon J, Shin DH, Kopka ML, Sawaya MR, Yuan HS, Kim TD, Choe J, Lim D, Moon HJ, Suh SW. Structural basis of non-specific lipid binding in maize lipid-transfer protein complexes revealed by high-resolution X-ray crystallography. J Mol Biol. 2001;308:263–278. doi: 10.1006/jmbi.2001.4559. [DOI] [PubMed] [Google Scholar]
- 10.Davis AM, St-Gallay SA, Kleywegt GJ. Limitations and lessons in the use of X-ray structural information in drug design. Drug Discov Today. 2008;13:831–841. doi: 10.1016/j.drudis.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pellecchia M, Bertini I, Cowburn D, Dalvit C, Giralt E, Jahnke W, James TL, Homans SW, Kessler H, Luchinat C, Meyer B, Oschkinat H, Peng J, Schwalbe H, Siegal G. Perspectives on NMR in drug discovery: a technique comes of age. Nature Rev Drug Dis. 2008;7:738–745. doi: 10.1038/nrd2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Dijk ADJ, Boelens R, Bonvin A. Data-driven docking for the study of biomolecular complexes. FEBS J. 2005;272:293–312. doi: 10.1111/j.1742-4658.2004.04473.x. [DOI] [PubMed] [Google Scholar]
- 13.Tuffery P, Derreumaux P. Flexibility and binding affinity in protein-ligand, protein-protein and multi-component protein interactions: limitations of current computational approaches. J Roy Soc Interface. 2012;9:20–33. doi: 10.1098/rsif.2011.0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kader JC. Lipid-transfer proteins in plants. Annu Rev Plant Physiol Plant Mol Biol. 1996;47:627–654. doi: 10.1146/annurev.arplant.47.1.627. [DOI] [PubMed] [Google Scholar]
- 15.Garcia-Olmedo F, Molina A, Segura A, Moreno M. The defensive role of nonspecific lipid-transfer proteins in plants. Trends Microbiol. 1995;3:72–74. doi: 10.1016/s0966-842x(00)88879-4. [DOI] [PubMed] [Google Scholar]
- 16.Kader JC. Lipid-transfer proteins: a puzzling family of plant proteins. Trends Plant Sci. 1997;2:66–70. [Google Scholar]
- 17.Yeats TH, Rose JKC. The biochemistry and biology of extracellular plant lipid-transfer proteins (LTPs. Protein Sci. 2008;17:191–198. doi: 10.1110/ps.073300108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salcedo G, Sanchez-Monge R, Barber D, Diaz-Perales A. Plant non-specific lipid transfer proteins: an interface between plant defence and human allergy. Biochim Biophys Acta. 2007;1771:781–791. doi: 10.1016/j.bbalip.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 19.Douliez JP, Michon T, Elmorjani K, Marion D. Structure, biological and technological functions of lipid transfer proteins and indolines, the major lipid binding proteins from cereal kernels. J Cereal Sci. 2000;32:1–20. [Google Scholar]
- 20.Lerche MH, Kragelund BB, Bech LM, Poulsen FM. Barley lipid-transfer protein complexed with palmitoyl CoA: The structure reveals a hydrophobic binding site that can expand to fit both large and small lipid-like ligands. Structure. 1997;5:291–306. doi: 10.1016/s0969-2126(97)00186-x. [DOI] [PubMed] [Google Scholar]
- 21.Lerche MH, Poulsen FH. Solution structure of barley lipid transfer protein complexed with palmitate. Two different binding modes of palmitate in the homologous maize and barley nonspecific lipid transfer proteins. Protein Sci. 1998;7:2490–2498. doi: 10.1002/pro.5560071202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Charvolin D, Douliez JP, Marion D, Cohen-Addad C, Pebay-Peyroula E. The crystal structure of a wheat nonspecific lipid transfer protein (ns-LTP1) complexed with two molecules of phospholipid at 2.1 Ångstrom resolution. Eur J Biochem. 1999;264:562–568. doi: 10.1046/j.1432-1327.1999.00667.x. [DOI] [PubMed] [Google Scholar]
- 23.Tassin-Moindrot S, Caille A, Douliez JP, Marion D, Vovelle F. The wide binding properties of a wheat nonspecific lipid transfer protein—solution structure of a complex with prostaglandin B-2. Eur J Biochem. 2000;267:1117–1124. doi: 10.1046/j.1432-1327.2000.01109.x. [DOI] [PubMed] [Google Scholar]
- 24.Cheng HC, Cheng PT, Peng PY, Lyu PC, Sun YJ. Lipid binding in rice nonspecific lipid transfer protein-1 complexes from Oryza sativa. Protein Sci. 2004;13:2304–2315. doi: 10.1110/ps.04799704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shin DH, Lee JY, Hwang KY, Kim KK, Suh SW. High resolution crystal structure of the nonspecific lipid transfer protein from maize seedlings. Structure. 1995;3:189–199. doi: 10.1016/s0969-2126(01)00149-6. [DOI] [PubMed] [Google Scholar]
- 26.Bakan B, Hamberg M, Larue V, Prange T, Marion D, Lascombe MB. The crystal structure of oxylipin-conjugated barley LTP1 highlights the unique plasticity of the hydrophobic cavity of these plant lipid-binding proteins. Biochem Biophys Res Commun. 2009;390:780–785. doi: 10.1016/j.bbrc.2009.10.049. [DOI] [PubMed] [Google Scholar]
- 27.Schmid N, Eichenberger AP, Choutko A, Riniker S, Winger M, Mark AE, van Gunsteren WF. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur Biophys J Biophys Lett. 2011;40:843–856. doi: 10.1007/s00249-011-0700-9. [DOI] [PubMed] [Google Scholar]
- 28.Heinemann B, Andersen KV, Nielsen PR, Bech LM, Poulsen FM. Structure in solution of a four-helix lipid binding protein. Protein Sci. 1996;5:13–23. doi: 10.1002/pro.5560050103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eichenberger AP, Allison JR, Dolenc J, Geerke DP, Horta BAC, Meier K, Oostenbrink C, Schmid N, Steiner D, Wang D, van Gunsteren WF. GROMOS ++ Software for the analysis of biomolecular simulation trajectories. J Chem Theory Comput. 2011;7:3379–3390. doi: 10.1021/ct2003622. [DOI] [PubMed] [Google Scholar]
- 30.Schmid N, Christ CD, Christen M, Eichenberger AP, van Gunsteren WF. Architecture, implementation and parallelisation of the GROMOS software for biomolecular simulation. Comp Phys Comm. 2012;183:890–903. [Google Scholar]
- 31.Schmid N, Allison JR, Dolenc J, Eichenberger AP, Kunz A-P E, van Gunsteren WF. Biomolecular structure refinement using the GROMOS simulation software. J Biomol NMR. 2011;51:265–281. doi: 10.1007/s10858-011-9534-0. [DOI] [PubMed] [Google Scholar]
- 32. Available at: http://www.gromos.net.
- 33.Berendsen HJC, Postma JPM, Van Gunsteren WF, Hermans J. In: Interaction models for water in relation to protein hydration. Pullman B, editor. Reidel; Dordrecht, The Netherlands: Intermolecular forces; 1981. pp. 331–342. [Google Scholar]
- 34.Berendsen HJC, Postma JPM, van Gunsteren WF, Dinola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684–3690. [Google Scholar]
- 35.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical integration of cartesian equations of motion of a system with constraints—molecular dynamics of n-alkanes. J Comp Phys. 1977;23:327–341. [Google Scholar]
- 36.Tironi IG, Sperb R, Smith PE, van Gunsteren WF. A generalised reaction field method for molecular dynamics simulations. J Chem Phys. 1995;102:5451–5459. [Google Scholar]
- 37.Heinz TN, van Gunsteren WF, Hunenberger PH. Comparison of four methods to compute the dielectric permittivity of liquids from molecular dynamics simulations. J Chem Phys. 2001;115:1125–1136. [Google Scholar]
- 38.van Gunsteren WF, Billeter S, Eising A, Hünenberger P, Krüger P, Mark A, Scott W, Tironi I. Biomolecular simulation: the GROMOS96 manual and user guide. Switzerland: Vdf Hochschulverlag AG an der ETH Zürich. 1996 [Google Scholar]
- 39.Kabsch W, Sander C. Dictionary of protein secondary structure—pattern recognition of hydrogen bonded and geometical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 40.Lee B, Richards FM. Interpretation of protein structures—estimation of static accessibility. J Mol Biol. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- 41.Humphrey W, Dalke A, Schulten K. VMD—visual molecular dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.