

Abstract
Histone deacetylases (HDACs) repress transcription by deacetylating acetyllysines on specific histone tails. HDAC3 is implicated in neurodegenerative diseases, certain leukemias, and even in disrupting HIV-1 latency. A recent crystal structure of HDAC3 in complex with the deacetylase-activating domain (DAD) of its corepressor complex revealed an inositol tetraphosphate (IP4) molecule at the protein–protein interface. IP4 was shown to play an important, yet mechanistically ambiguous, role in the activity of HDAC3. The goal of this article is to explore the conformational ensemble of HDAC3 in its inactive apo state and in the presence of each or both of DAD and IP4. Using triplicate, 100 ns molecular dynamic simulations, we study the apo, ternary, and intermediate DAD-bound or IP4-bound HDAC3 states. We find that a population-shift effect is induced by the presence of each corepressor, and is most notable in the presence of both. Our results offer new insights into the change in dynamics necessary for the activation of HDAC3 and highlight the roles of IP4 and DAD in this process.
Keywords: histone deacetylase 3, inositol tetraphosphate, molecular dynamics, population-shift
Introduction
Histone deacetylases (HDACs) play an important role in the regulation of gene expression by modifying local chromatin structure.1 HDACs directly oppose the activity of acetyl transferases by deacetylating acetyllysine residues on specific histone tails. Hypoacetylation of histone tails leads to compacted chromatin structure, limiting accessibility of DNA to necessary transcription factors, and thus repressing transcription of certain genes.2
Abnormal gene expression is often associated with cancer cells, and epigenetic routes to cancer therapy are of growing interest. Eleven of the known human HDACs (classes I, II, and IV) have known important roles in cancer cell biology3 and neurodegenerative diseases.4–6 Some HDAC inhibitors are already utilized in cancer therapies and targeted inhibition of HDACs may be key to less toxic treatments. In this study, we focus specifically on the activation mechanism of the class I deacetylase HDAC3. Inhibition of HDAC3 in particular has been shown to disrupt HIV-1 latency and reduce cancer cell proliferation.7–9
The catalytic activity of HDACs generally depends on their recruitment to large multiprotein complexes.10, 11 Structural data on active HDAC complexes are still sparse, and only recently Watson et al.12 reported the first crystal structure of HDAC3. Unlike the other class I deacetylases, HDAC3 is recruited to the NCoR/SMRT nuclear receptor complex. Both NCoR and SMRT have critical N-terminal deacetylase-activating domains (DAD), which is necessary for deacetylase activity.13–15 The SMRT DAD domain was also resolved in the crystal structure of HDAC3.12 In the crystal structure, an inositol (1,4,5,6) tetraphosphate (IP4) molecule was found at the interface of HDAC3 and DAD, adjacent to the mouth of a 12 Å tunnel that leads to the zinc center of the active site.12 Mutations that disrupt the interactions between HDAC3 and IP4 abolished both deacetylase activity and HDAC3:DAD complex formation, consistent with the necessity for DAD binding in the activation of HDAC3.12 Interestingly, a DAD mutation K449A which was inoffensive to HDAC3:DAD complex formation also abolished deacetylase activity.16 As K449 is close to the IP4 binding site, the K449A mutation may inactivate the deacetylase via mediation of IP4-HDAC3 interactions, which would suggest that IP4 plays an additional role in the activation of HDAC3, aside from stabilizing the HDAC3:DAD complex.
Here, we examine the structural ensemble of HDAC3, specifically: (i) how does this ensemble of HDAC3 structures change between the active HDAC3: DAD:IP4 complex and the inactive apo HDAC3 state? (ii) Does the presence of either IP4 or DAD alone have a significant impact on the dynamics of HDAC3? and (iii) What effect does IP4 have on the HDAC3:DAD complex?
Our approach is to use molecular dynamics simulations to explore the structural ensembles of apo HDAC3 as well as the HDAC3:DAD:IP4, HDAC3:DAD, and HDAC3:IP4 complexes. In the Results section, we will address each of the above questions. We will then discuss our findings about the HDAC3 conformational ensembles in terms of its HDAC activity.
Results
Dynamic regions in apo HDAC3 are stabilized by the presence of DAD and IP4
Without DAD and IP4, HDAC3 has intrinsic flexibility
There is currently only one crystal structure of HDAC3 available in the protein data bank (pdb 4A69), in which the deacetylase is in complex with DAD and IP4. We simulated the apo state of HDAC3 by removing both the DAD domain and the IP4 from the crystal structure. In these simulations, we observed a highly dynamic deacetylase backbone (Fig. 1(A) and Supporting Information Fig. 1(A)), particularly near the active site loops. In the ternary complex, on the other hand, we found the HDAC3 backbone maintained its crystal structure conformation (Fig. 1(D) and Supporting Information Fig. 1(D)). The only region with notable flexibility in the ternary complex was the random coil region (residues, 340–348).
Figure 1.
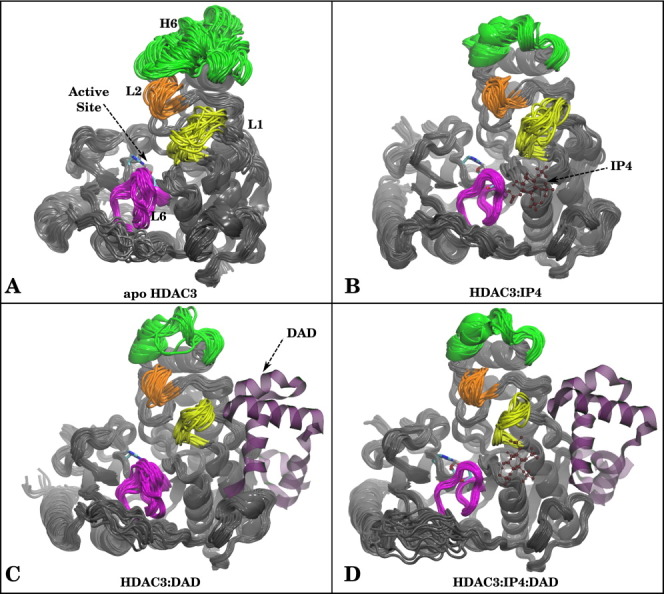
Superimposed snapshots from MD simulations for (A) apo HDAC3, (B) HDAC3:IP4, (C) HDAC3:DAD, and (D) HDAC3:IP4:DAD. Helix 6 (green), Loop1 (yellow), Loop2 (orange), and Loop 6 (magenta) are highlighted to show differences in dynamics. If present in the simulation, DAD and IP4 are shown as purple ribbons or red ball-and-stick representations, for reference. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
The increased fluctuations in the apo structure were localized to the active site loops and a nearby helix (Helix 6). Figure 2(A) shows the change in root mean square fluctuations (RMSF) per residue for the ternary and apo states. Negative ΔRMSF values indicate flexible regions of apo HDAC3 that are stabilized in the ternary complex. The ΔRMSF values are also projected onto the protein structure, using a Blue (negative ΔRMSF)-Gray-Red (positive ΔRMSF) color scale. The regions stabilized in the presence of both corepressors not surprisingly include Loops 1 and 6, which form part of the IP4-binding site at the protein–protein interface. Perhaps, less expected was the flexibility of Helix 6 (residues, 78–88) near Loop 2, which was most flexible and even unfolds in the apo HDAC3 simulation, but does not directly interact with either DAD or IP4. These results suggest an overall decreased flexibility of specific regions of the deacetylase backbone upon corepressor binding.
Figure 2.
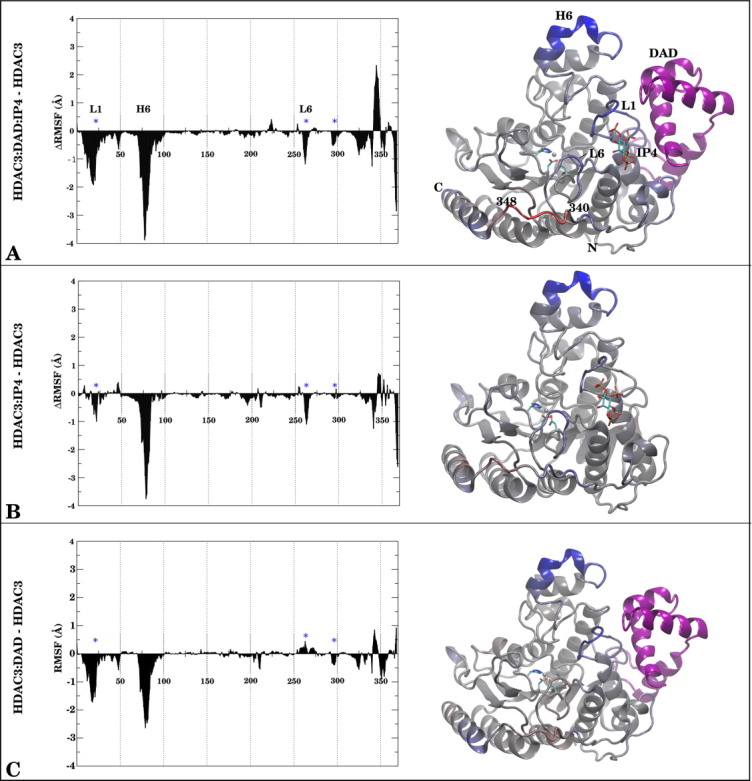
The ΔRMSF (Å) per residue in HDAC3 between (A) HDAC3:DAD:IP4, (B) HDAC3:IP4, (C) HDAC3:DAD and the apo state is shown on the left side of each panel. The same values are projected onto a structure of the protein for reference, using a Blue (Negative)→Gray (Zero)→Red (Positive) scale. A range from −3.00 to +3.00 Å was used to make the figure. DAD and IP4, when present in the simulations are shown for reference in purple ribbons and stick representations, respectively.
Presence of IP4 alone reduces overall HDAC3 backbone flexibility
In an attempt to separate the contributions of DAD and IP4 to the observed stability of the deacetylase backbone in the ternary complex, we analyzed the dynamics of the intermediate HDAC3:IP4 and HDAC3:DAD complexes. Figure 1(B,C) shows superimposed snapshots from these simulations. Although the presence of the DAD domain resulted in less fluctuations in the deacetylase backbone than in the apo state [Fig. 2(C)], we were surprised to find that the presence of IP4 alone greatly reduced the flexibility of the deacetylase backbone [Fig. 2(B)]. The regions stabilized by either DAD or IP4 were the same regions stabilized in the ternary complex although their respective contributions seem to be complementary. The ΔRMSF data show that IP4 has a slightly greater stabilizing effect on Helix 6, Loop 6, and the C-terminal helix [Fig. 2(B)] than does the DAD domain [Fig. 2(C)]. The DAD domain, on the other hand, has a more notable stabilizing effect on Loop 1 (residues 19–25). Neither the binding of DAD nor IP4 appears to affect the random coil at residues 340–348 substantially, but the binding of both corepressors does seem to cause an increase in flexibility in this region. We should highlight the fact that the stabilized regions in the presence of IP4 were not necessarily near the IP4 binding site, suggesting the importance of allosteric regulation in HDAC3 activity.
Clustering of the apo HDAC3 structural ensemble suggests population-shift upon DAD and IP4-binding
Given the observed variations in the flexibility of the deacetylase backbone in the different corepressor-bound and apo states, we sought to answer the question: are the same HDAC3 conformations being stabilized in the different corepressor-bound HDAC3 ensembles? We first clustered the structural ensemble of the apo state, using an RMSD metric and the hierarchical clustering algorithm (for details, see Supporting Information and Shao et al.17 ). The main characteristics distinguishing each of the four clusters were the orientation and helicity of Helix 6, as well as the conformations of Loops 1 and 6. To compare the corepressor-bound states to these reference clusters, we then measured the RMSD of the HDAC3 backbone from the ternary, IP4-bound, and DAD-bound HDAC3 ensembles against each of the four apo cluster representatives. Nearly, all of the HDAC3 structures from the ternary simulations (∼90%) are within 1.4 Å of the Cluster2 representative structure (Table I), which comprises only ∼15% of the apo structural ensemble. In fact, none of the structures from the ternary simulation resembles (using a cutoff value of 1.4 Å) any of the other apo cluster representatives aside from Cluster2. The presence of DAD alone resulted in ∼70% of the structures, resembling the Cluster2 representative, and ∼10% resembling the Cluster0 representative. The presence of IP4 alone surprisingly resulted in ∼90% of the structural ensemble to lie within 1.4 Å of the Cluster2 representative as well. It appears, then, that the limited flexibility in each of the corepressor-bound deacetylase states actually shifts the population toward a single conformation, which was only minimally populated in the apo HDAC3 ensemble.
Table 1.
Summary of Clustering Analysis
| Cluster2 (%) | Cluster0 (%) | Cluster3 (%) | Cluster1 (%) | |
|---|---|---|---|---|
| HDAC3:DAD:IP4 | 90 | — | — | — |
| HDAC3:IP4 | 90 | — | — | — |
| HDAC3:DAD | 70 | 10 | — | — |
| HDAC3 | 14 | 20 | 27 | 39 |
Percentage of structures within 1.42 Å (see Supporting Information for rationale) of each of the four apo HDAC3 clusters is given.
Exact cluster occupancies are given for apo HDAC3 simulation.
DAD and IP4 shift HDAC3 population by restraining motions along the first two principal components in apo HDAC3 dynamics
To further characterize the dynamics of apo HDAC3 and the impact of DAD and IP4 on these dynamics, we used principal component analysis (PCA) to identify the most dominant modes in the dynamics. This analysis provides information about the low-frequency motions of the dynamics, which may complement the conformational analysis above. We computed the first two principal components (PC1 and PC2), which comprised just more than 50% of the total variance, from the apo HDAC3 simulations (using only the Cα atoms). Movies that show PC1 and PC2 motions are shown in Supporting Information. We then projected the HDAC3 dynamics from the apo, HDAC3:IP4, HDAC3:DAD, and HDAC3:DAD:IP4 simulations onto the PC space, which describes motions of the loops contracting around the active site. Figure 3 shows the projections of the HDAC3 dynamics from all four simulations, along with the projection of the crystal structure conformation for reference. The PCA analysis shows that the ternary complex is notably restrained along PC1 and PC2 [Fig. 3(D)], which was not surprising, given the low root mean square deviation from the crystal structure conformation (Supporting Information Fig. 1(D)). With the sole addition of IP4 to HDAC3, the motions characterized by both PC1 and PC2 were already restrained considerably [Fig. 3(B)]. The addition of the DAD domain to HDAC3 also notably reduces the motions along both principal components. The PCA results identify two dominant modes in the apo HDAC3 dynamics that are each limited in the presence of DAD and IP4, which we suspect results in the observed population-shift effect upon the binding of both IP4 and DAD to HDAC3.
Figure 3.
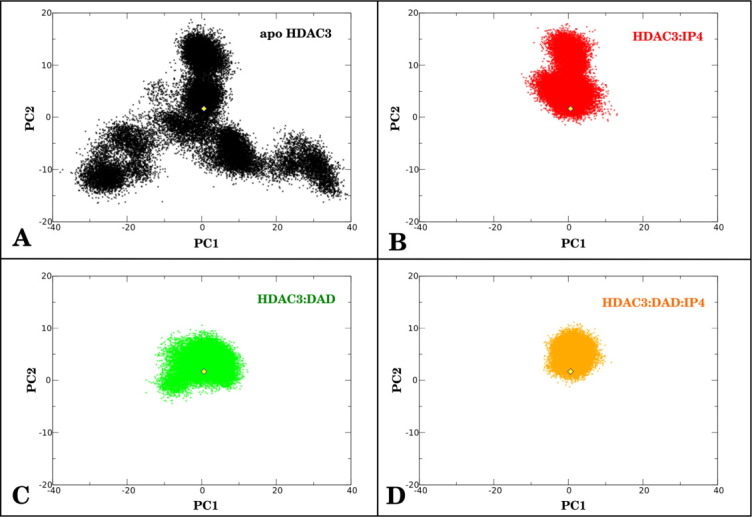
PCA of HDAC3. First two principal components were calculated from apo HDAC3 Cα dynamics. Structural ensembles of HDAC3 Cα atoms from (A) apo HDAC3, (B) HDAC3:IP4, (C), HDAC3:DAD, and (D) HDAC3:DAD:IP4 simulations were projected onto these two principal components. A yellow diamond in each graph marks the crystal structure conformation of HDAC3 for reference. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
I4 stabilizes the protein–protein interface as well as the deacetylase backbone
Given the presence of a number of basic residues at the protein–protein interface where IP4 binds, we expected a major contribution of IP4 to stabilize the HDAC3:DAD complex in this region, as was also suggested previously by Watson et al.12 Upon closer comparison of the HDAC3:DAD complexes with and without the small molecule, we found that IP4 does more than simply stabilize the protein–protein interface near the binding site. The entire DAD backbone was quite flexible when bound to HDAC3 without IP4 (Fig. 4(A) and Supporting Information Fig. 3(A)). Surprisingly, the stabilizing effect of IP4-binding affected the DAD backbone nearly uniformly, regardless of proximity to the IP4-binding site (Fig. 5).
Figure 4.
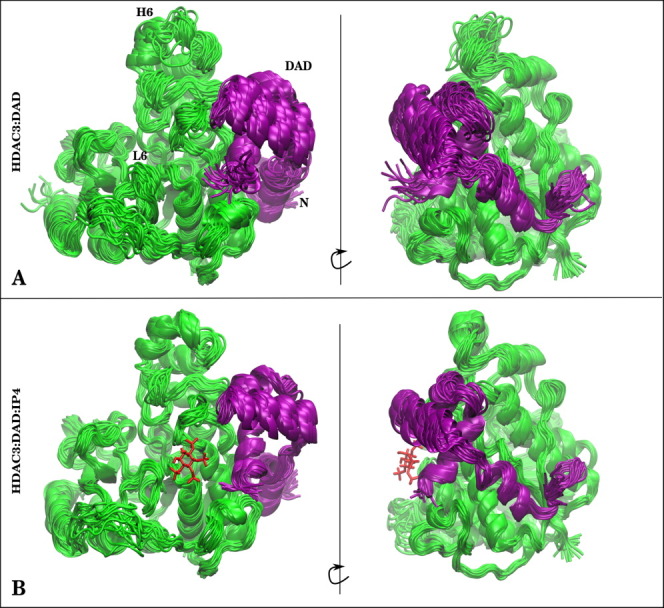
Structures from (A) HDAC3:DAD and (B) HDAC3:DAD:IP4 simulations are presented to show difference in stabilities of the protein–protein complex owing to the presence of IP4. HDAC3 (green) is shown with the DAD domain (purple). If IP4 was present in the simulation, a single snapshot of IP4 is shown as red sticks for reference. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 5.
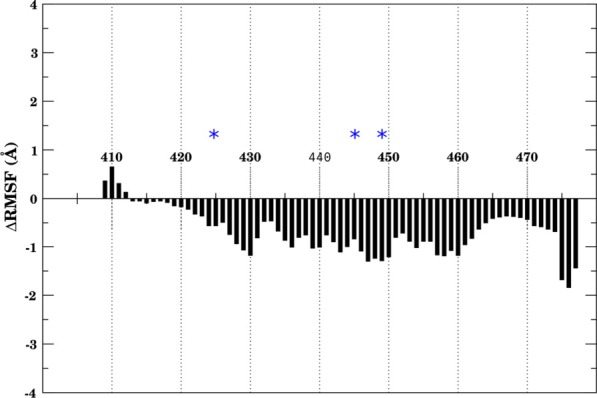
ΔRMSF of DAD backbone (HDAC3:DAD:IP4 minus HDAC3:DAD). Negative values indicate stabilization of backbone owing to the presence of IP4. Blue asterisks indicate residues near IP4-binding site. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Changes in active site loop dynamics are expected to affect substrate recognition
Accessibility to the active site is determined by loop conformations
The remarkable stability of the active site loops in the ternary structure helps to maintain a very clear tunnel to the zinc metal center (Fig. 6). In its stable conformations, the tunnel is lined with hydrophobic residues, with the exception of Tyr298. In the alternative loop conformations, this access to the active site is distorted into a wider opened, yet unblocked, conformation. The tunnel to the active site is likely to play an important role in substrate recognition. In the crystal structure of the ternary complex, an acetate ion was resolved in the active site along with a methionine side chain from an adjacent DAD domain in the crystal lattice. The authors suggested that the resolved structure mimicked the presence of the substrate in the active site.12 It is important to note that we observed the same tunnel without any ligands in the active site, given the presence of both DAD and IP4 bound to HDAC3.
Figure 6.
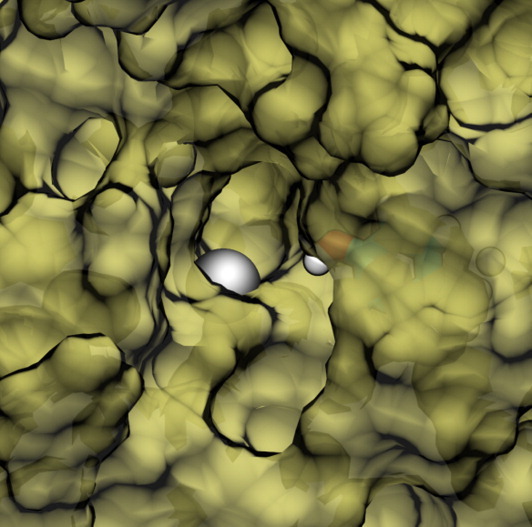
Surface representation of stable tunnel observed in ternary complex simulation. Zinc metal center is shown as a silver sphere. Tyr298 side-chain lining the tunnel is shown as sticks. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Binding of corepressors to HDAC3 shifts Tyr298 side chain from a solvent exposed to an inward conformation
The amino acid residues near the zinc center (D170, H172, D259, H134, H135, and Q255) maintained the same side-chain conformations, regardless of the observed loop motions (Fig. 7). One notable exception, however, was Tyr298, located at the rim of the active site. We found that Tyr298 sampled two rotameric states, either solvent exposed or poised inward toward the active site (as observed in the crystal structure).12 Figure 7(E) shows the populations of the two rotameric states for the ternary (HDAC3:DAD:IP4), DAD-bound, IP4-bound, and apo deacetylase states. Binding of each of the two corepressors to HDAC3 biases the inward conformation of Tyr298, and in the ternary complex the Tyr298 side chain is exclusively inward (throughout the triplicate 100-ns simulations). This population shift of the Tyr298 side chain may be necessary for substrate recognition, as it also forms part of the tunnel to the active site (Fig. 6).
Figure 7.
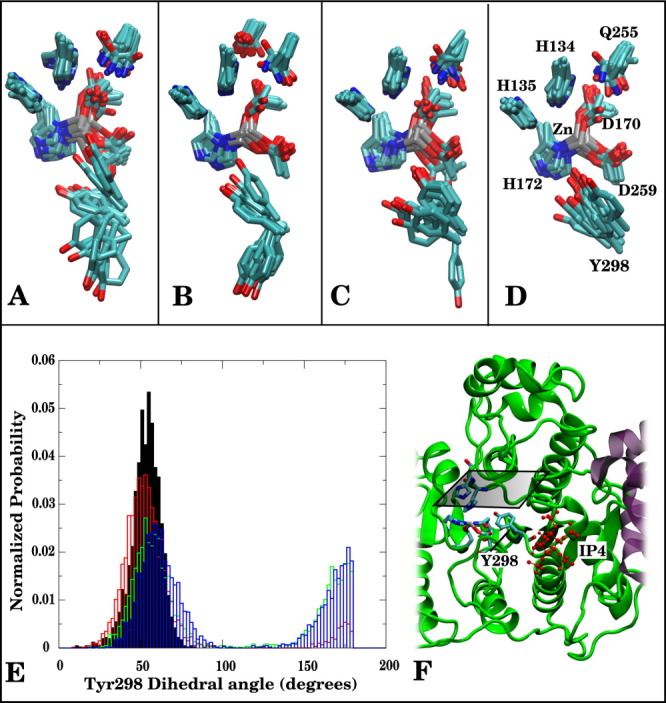
Superimposed snapshots from MD simulations show displacements of active site residues for (A) HDAC3, (B) HDAC3:IP4, (C) HDAC3:DAD, (D) HDAC3:DAD:IP4. (E) Tyr298 (N[sbond]Cα[sbond]Cβ[sbond]Cγ) dihedral angle probability distributions shown for HDAC3:DAD:IP4 (solid, black), HDAC3:DAD (red), HDAC3:IP4(green), and HDAC3 (blue). (F) Cartoon representation of HDAC3:IP4:DAD complex, showing active site resides as sticks; the horizontal plane above the active site marks the top-down perspective of (A–D).
Discussion
Apo HDAC3 has flexible regions near the active site
HDAC3 has recently been shown to depend on IP4 in addition to the DAD domain for its HDAC activity.12 The goal of this article was to characterize any changes in the dynamics of HDAC3 owing to the presence of its corepressors. We observed from simulations of the apo state of HDAC3 that it samples a range of conformations, which we clustered into four groups. In the active ternary complex, we found that the structural ensemble of HDAC3 consisted predominantly of just one of these four conformations. Interestingly, the presence of IP4 or DAD alone could also effect a similar and substantial population shift toward the stable conformation found in the ternary complex. Differences between RMSFs by residue suggest that DAD and IP4 stabilize flexible regions of apo HDAC3 differentially (Fig. 2). Most of the stabilized regions not surprisingly are near the protein–protein interface where IP4 binds. Some significantly stabilized regions, however, for example Helix 6, or destabilized regions, such as the random coil (residues 340–348), in the ternary complex are not at all close to the DAD interface or the IP4-binding site, indicating allosteric effects of the corepressors on HDAC3. Given that HDAC3 has no HDAC activity in its flexible apo state, it would be tempting to suggest that the stabilized conformation is indeed the active conformation. This idea is reasonable as the observed conformational shifts, perhaps sufficiently described by the first two principal components, comprise motions that involve a contractile motion around the active site (see Supporting Information). The well-formed tunnel to the active site is distorted as a result of changes in the loop conformations. It is quite possible that this stable access route to the zinc center is important for the recognition of acetyllysines on histone tails. The observed shift in Tyr298 (which lines the tunnel) rotameric populations is also relevant to substrate recognition. In the crystal structure, Tyr298 was poised inward, hydrogen bonding with an acetate ion resolved in the crystal structure. The corresponding Tyr306 residue in HDAC8 has already been shown to be critical for both substrate recognition and in the reactivity of the deacetylase.18–20
Making the connection from structure to activity
If the stabilized conformation of HDAC3 can indeed be thought of as the active conformation, we must also acknowledge that both DAD and IP4 independently induce a population shift toward this very structure, albeit to a lesser extent than in the presence of both corepressors. It would seem not unreasonable than that HDAC3 have considerable deacetylase activity in the presence of just one or the other corepressor. Experimental mutagenesis experiments, however, seem to suggest otherwise.
Mutations disrupting the HDAC3:DAD complex also inactivate HDAC3,16 confirming that the binding to DAD is necessary for activity. Conversely, mutations of a DAD residue (K449A) near the IP4-binding site did not perturb the protein–protein association but still inactivated HDAC3,16 suggesting that IP4 is also necessary for HDAC activity. Recent inactivating mutations to HDAC3 Loop 1 and Loop 6 residues, that specifically interact with IP4 but not DAD, on the other hand, still disrupted complex formation with DAD, suggesting an interdependence between IP4 binding, DAD binding, and HDAC3 deacetylase activity.12 The observation that HDAC3 is active in the presence of both corepressors may suggest (i) that the subtle change in dynamics from one corepressor bound to both corepressors bound is actually critical to deacetylase activity and (ii) that the inability to bind one of the corepressors, for example IP4, reduces the affinity for the second corepressor, that is DAD, thereby increasing the equilibrium concentration of apo HDAC3. Future binding studies or free energy calculations would be needed to better evaluate the interdependence of DAD and IP4, but from our results it is clear that IP4 stabilizes the HDAC3:DAD complex as well as key regions (e.g., Loop 6) of the deacetylase backbone. It is also important to recall that DAD, in addition to stabilizing other regions (in particular Loop 1) of the deacetylase backbone, is critical in localizing HDAC3 near histones.
Conclusions
From a thorough analysis of multiple independent 100-ns MD simulations, we discussed the conformational ensemble of HDAC3, both in the apo state, and bound to its two corepressors necessary for activity: the DAD of the SMRT/NCoR nuclear complex and IP4. We showed that IP4 stabilizes the entire HDAC3:DAD protein–protein interface, well beyond its binding region. We further studied the independent effects of IP4 and DAD binding to HDAC3 and found that each is capable of stabilizing key regions of the deacetylase backbone. Together, IP4 and DAD restrain the conformation of HDAC3 to that found in the crystal structure, with a clear tunnel-like pathway to the zinc center. Based on a discussion of our results and available mutagenesis data, it is likely that activation of HDAC3 relies on a delicate interdependence between DAD and IP4, which only together can induce a sufficient population shift and activate HDAC3.
Methods
System preparation
Zinc metal center
Instead of assigning a formal charge of +2 to the zinc metal center, charges were assigned by performing quantum mechanical calculations on a simplified model of the active site. The active site model included the side chains of the zinc-coordinated ASP and HIS residues capped with methyl groups. To maintain the tetrahedral geometry and simulate the apo state of the protein, we added a water molecule where an acetate ion was found in the crystal structure. A geometry optimization was performed using Gaussian (g03),21 with B3LYP and the 6-31G(d) basis set. Electrostatic potential fits were also obtained from Gaussian, using the optimized geometry, and Hartree Fock level of theory with the 6-31G* basis set. The Restrained Electrostatic Potential module within the Antechamber program in AmberTools22 was used to assign charges to the atoms using the Gaussian output.
The force constants for bond, angle, and torsions for the atoms bonded to zinc were taken from the literature or by analogy to similar active sites.23 Equilibrium angles and bond distances involving zinc were taken from the crystal structure.12
Inositol (1,4,5,6) tetraphosphate parameters
Charges for the small molecule InsP4 were obtained using the same procedure described for the zinc metal center. The Amber ff99SB force field was used (CT, OS, P, O2 atom types) for all parameters except for the C–O–P–O− dihedral which was treated like the C–O–P–OH dihedral by analogy. Equilibrium bonds and angles were adjusted slightly to agree with the crystal structure.
Side-chain protonation states
PROPKA 3.124 was run on the web-server (pdb 4A69). Only two histidine residues had particularly low pKas in HDAC3 and were modified to be charged HIP residues. Other histidines were designated as either HIE or HID (with epsilon or delta nitrogen protonated, respectively) by evaluating the local environments of each side chain in the crystal structure.
Molecular dynamics details
Solvation and equilibration
The systems were built using the AMBER ff99SB force field, with the ildn modification for ILE, LEU, ASP, and ASN residues.25 Each system was solvated in a 10.0 Å TIP3P octahedron, and potassium ions were added to neutralize the charge in each case: HDAC (9), HDAC:InsP4 (17), HDAC:InsP4:DAD (13). Additional potassium and chloride ions were added to simulate physiological salt concentration (0.15M). The updated Lennard Jones parameters for ions were used.26 Systems were heated to 300 K at constant volume (NVT) with restraints on the protein that were gradually reduced from 200 to 0 kcal/mol/Å2 over a period of 150 ps. The Langevin thermostat was used with a collision frequency of 1.0 ps−1. The SHAKE algorithm was used to constrain bonds to nonpolar hydrogens, and a 1.0 fs time step was used during dynamics.27 An 8.0 Å cutoff was used for nonbonded interactions. In brief, 100-ps equilibration runs were done in the NPT ensemble, using isotropic pressure scaling and a pressure relaxation time of 2.0 ps.
Production simulations
For each system, three independent simulations were started from different snapshots from the equilibration simulations (taken at arbitrary intervals after density of water box was equilibrated) with randomized velocities. All production runs were done in the same NVT conditions described in Solvation and equilibration section, with the exception of a 2.0 fs time step. All simulations were done using the PMEMD module within the Amber11 simulation package.22 Production runs were performed on GPUs using Amber11 pmemd. CUDA, each for 100 ns.
Acknowledgments
The authors thank Leonardo Boechi for thoughtful discussions and editing of the manuscript. RT was a summer scholar in the UCSD Pharmacology SURF program, sponsored by ASPET.
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Bhaskara S, Knutson SK, Jiang G, Chandrasekharan MB, Wilson AJ, Zheng S, Yenamandra A, Locke K, Yuan J, Bonine-Summers AR, Wells CE, Kaiser JF, Washington MK, Zhao Z, Wagner FF, Sun ZW, Xa F, Holson EB, Khabele D, Hiebert SW. Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer Cell. 2010;18:436–447. doi: 10.1016/j.ccr.2010.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pogo BG, Allfrey VG, Mirsky AE. RNA synthesis and histone acetylation during the course of gene activation in lymphocytes. Proc Natl Acad Sci USA. 1966;55:805. doi: 10.1073/pnas.55.4.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Witt O, Deubzer HE, Milde T, Oehme I. HDAC family. what are the cancer relevant targets? Cancer Lett. 2009;277:8–21. doi: 10.1016/j.canlet.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 4.Jia H, Pallos J, Jacques V, Lau A, Tang B, Cooper A, Syed A, Purcell J, Chen Y, Sharma S, Sangrey GR, Darnell SB, Plasterer H, Sadri-Vakili G, Gottesfeld JM, Thompson LM, Rusche JR, Marsh JL, Thomas EA. Histone deacetylase (HDAC) inhibitors targeting HDAC3 and HDAC1 ameliorate polyglutamine-elicited phenotypes in model systems of Huntington's disease. Neurobiol Dis. 2012;46:351–361. doi: 10.1016/j.nbd.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Debacker K, Frizzell A, Gleeson O, Kirkham-McCarthy L, Mertz T, Lahue RS. Histone deacetylase complexes promote trinucleotide repeat expansions. PLoS Biol. 2012;10:e1001257. doi: 10.1371/journal.pbio.1001257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Y, Fang H, Jiao J, Xu W. The structure and function of histone deacetylases: the target for anti-cancer therapy. Curr Med Chem. 2008;15:2840–2849. doi: 10.2174/092986708786242796. [DOI] [PubMed] [Google Scholar]
- 7.Archin NM, Liberty AL, Kashuba AD, Choudhary SK, Kuruc JD, Crooks AM, Parker DC, Anderson EM, Kearney MF, Strain MC, Richman MD, Hudgens MG, Bosch RJ, Coffin JM, Eron JJ, Hazuda DJ, Margolis DM. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature. 2012;487:482–485. doi: 10.1038/nature11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huber K, Doyon G, Plaks J, Fyne E, Mellors JW, Sluis-Cremer N. Inhibitors of histone deacetylases: correlation between isoform specificity and reactivation of HIV type 1 (HIV-1) from latently infected cells. J Biol Chem. 2011;286:22211–22218. doi: 10.1074/jbc.M110.180224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spurling CC, Godman CA, Noonan EJ, Rasmussen TP, Rosenberg DW, Giardina C. HDAC3 overexpression and colon cancer cell proliferation and differentiation. Mol Carcinog. 2008;47:137–147. doi: 10.1002/mc.20373. [DOI] [PubMed] [Google Scholar]
- 10.Li J, Wang J, Wang J, Nawaz Z, Liu JM, Qin J, Wong J. Both corepressor proteins SMRT and N-CoR exist in large protein complexes containing HDAC3. EMBO J. 2000;19:4342–4350. doi: 10.1093/emboj/19.16.4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oberoi J, Fairall L, Watson PJ, Yang J-C, Czimmerer Z, Kampmann T, Goult BT, Greenwood JA, Gooch JT, Kallenberger BC, Nagy L, Neuhaus D, Schwabe JWR. Structural basis for the assembly of the SMRT/NCoR core transcriptional repression machinery. Nat Struct Mol Biol. 2011;18:177–184. doi: 10.1038/nsmb.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Watson PJ, Fairall L, Santos GM, Schwabe JWR. Structure of HDAC3 bound to corepressor and inositol tetraphosphate. Nature. 2012;481:335–340. doi: 10.1038/nature10728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wen Y-D, Perissi V, Staszewski LM, Yang W-M, Krones A, Glass CK, Rosenfeld MG, Seto E. The histone deacetylase-3 complex contains nuclear receptor corepressors. Proc Natl Acad Sci USA. 2000;97:7202–7207. doi: 10.1073/pnas.97.13.7202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang J, Kalkum M, Chait BT, Roeder RG. The N-CoR-HDAC3 nuclear receptor corepressor complex inhibits the JNK pathway through the integral subunit GPS2. Mol Cell. 2002;9:611–623. doi: 10.1016/s1097-2765(02)00468-9. [DOI] [PubMed] [Google Scholar]
- 15.Guenther MG, Barak O, Lazar MA. The SMRT and N-CoR corepressors are activating cofactors for histone deacetylase 3. Mol Cell Biol. 2001;21:6091–6101. doi: 10.1128/MCB.21.18.6091-6101.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Codina A, Love JD, Li Y, Lazar MA, Neuhaus D, Schwabe JWR. Structural insights into the interaction and activation of histone deacetylase 3 by nuclear receptor corepressors. Proc Natl Acad Sci USA. 2005;102:6009–6014. doi: 10.1073/pnas.0500299102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shao J, Tanner SW, Thompson N, Cheatham TE. Clustering molecular dynamics trajectories: 1. characterizing the performance of different clustering algorithms. J Chem Theory Comput. 2007;3:2312–2334. doi: 10.1021/ct700119m. [DOI] [PubMed] [Google Scholar]
- 18.Dowling DP, Gantt SL, Gattis SG, Fierke CA, Christianson DW. Structural studies of human histone deacetylase 8 and its site-specific variants complexed with substrate and inhibitors. Biochemistry. 2008;47:13554–13563. doi: 10.1021/bi801610c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu R, Wang S, Zhou N, Cao Z, Zhang Y. A proton-shuttle reaction mechanism for histone deacetylase 8 and the CATALYTIC ROLE OF METAL IONS. J Am Chem Soc. 2010;132:9471–9479. doi: 10.1021/ja103932d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vannini A, Volpari C, Gallinari P, Jones P, Mattu M, Carfí A, De Francesco R, Steinkühler C, Di Marco S. Substrate binding to histone deacetylases as shown by the crystal structure of the HDAC8–substrate complex. EMBO Rep. 2007;8:879–884. doi: 10.1038/sj.embor.7401047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03, Revision C.02.
- 22.Case DA, Darden TA, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM, Roberts BP, Wang B, Hayik S, Roitberg A, Seabra G, Kolossvai I, Wong KF, Paesani F, Vanicek J, Liu J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Cai Q, Ye X, Wang J, Hsieh MJ, Cui G, Roe DR, Mathews DH, Seetin MG, Sagui C, Babin V, Luchko T, Gusarov S, Kovalenko A, Kollman PA. Amber 11. San Francisco: University of California; 2010. [Google Scholar]
- 23.Peters MB, Yang Y, Wang B, Füsti-Molnár L, Weaver MN, Merz KM. Structural survey of zinc containing proteins and the development of the zinc AMBER force field (ZAFF. J Chem Theory Comput. 2010;6:2935–2947. doi: 10.1021/ct1002626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H, Robertson AD, Jensen JH. Very fast empirical prediction and rationalization of protein pKa values. Proteins. 2005;61:704–721. doi: 10.1002/prot.20660. [DOI] [PubMed] [Google Scholar]
- 25.Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO, Shaw DE. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins. 2010;78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Joung IS, Cheatham TE. Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J Phys Chem B. 2008;112:9020–9041. doi: 10.1021/jp8001614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comp Phys. 1977;23:327–341. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.