

Abstract
Host cell invasion by the obligate intracellular apicomplexan parasites, including Plasmodium (malaria) and Toxoplasma (toxoplasmosis), requires a step-wise mechanism unique among known host–pathogen interactions. A key step is the formation of the moving junction (MJ) complex, a circumferential constriction between the apical tip of the parasite and the host cell membrane that traverses in a posterior direction to enclose the parasite in a protective vacuole essential for intracellular survival. The leading model of MJ assembly proposes that Rhoptry Neck Protein 2 (RON2) is secreted into the host cell and integrated into the membrane where it serves as the receptor for apical membrane antigen 1 (AMA1) on the parasite surface. We have previously demonstrated that the AMA1-RON2 interaction is an effective target for inhibiting apicomplexan invasion. To better understand the AMA1-dependant molecular recognition events that promote invasion, including the significant AMA1-RON2 interaction, we present the structural characterization of AMA1 from the apicomplexan parasites Babesia divergens (BdAMA1) and Neospora caninum (NcAMA1) by X-ray crystallography. These studies offer intriguing structural insight into the RON2-binding surface groove in the AMA1 apical domain, which shows clear evidence for receptor–ligand co-evolution, and the hyper variability of the membrane proximal domain, which in Plasmodium is responsible for direct binding to erythrocytes. By incorporating the structural analysis of BdAMA1 and NcAMA1 with existing AMA1 structures and complexes we were able to define conserved pockets in the AMA1 apical groove that could be targeted for the design of broadly reactive therapeutics.
Keywords: parasite invasion, Apicomplexa, moving junction, structural plasticity, neosporosis, babesiosis
Introduction
Phylum Apicomplexa harbors more than five thousand parasitic protozoan species, many of which cause serious morbidity and mortality in humans and animals worldwide. Some of the most prevalent apicomplexans are Plasmodium, Toxoplasma, Babesia, and Neospora, the etiological agents of malaria, toxoplasmosis, babesiosis, and neosporosis, respectively. More than 250 million cases of malaria every year result in at least 1 million deaths,1, 2 and up to a third of the world's population is chronically infected with Toxoplasma.3, 4 Babesiosis is one of the most prevalent infections of free-living animals, particularly cattle,5, 6 while Neospora is an important veterinary pathogen causing premature abortion in cattle and fatal neurological defects in dogs.7, 8 Despite the diversity of hosts and disease outcomes, a unique feature of all apicomplexans is their obligate intracellular lifestyle. Consequently, these resourceful parasites have developed highly sophisticated mechanisms to invade a wide range of host cells.5, 9–11
Modes of host cell recognition and attachment vary across the phylum and likely play a key role in defining cellular tropism. However, the process of active invasion appears to be highly conserved and a major contributor to the virulence of the parasites.12 Importantly, invasion is largely independent of traditional host cell uptake processes and proceeds with the formation of the moving junction (MJ),13–15 a ring-like protein structure formed at the interface between parasite and host cell. During invasion, the parasite propels its way through the MJ resulting in its encapsulation in a protective vacuole within the host cell.16 Thus, assembly of a functional MJ is crucial to parasite survival. It has recently been postulated that formation of the MJ is initiated when Rhoptry Neck Protein 2 (RON2) is secreted from the parasite's rhoptry organelles in a preformed complex with RONs 4 and 5, and also RON8 in coccidia (Toxoplasma, Neospora, and Eimeria).15, 17–18 This complex is discharged into the host cell, where the leading MJ model suggests that RON2 integrates into the host cell membrane and serves as the receptor for apical membrane antigen 1 (AMA1) displayed on the parasite cell surface.19–23 Thus, it has been suggested that apicomplexan parasites are capable of providing both ligand and receptor to actively invade host cells. Although a recent study found that functional junctions could form in the absence of detectable AMA1,24 other studies, such as those showing that RON2-derived peptide inhibitors of the AMA1–RON2 interaction inhibit MJ assembly and parasite invasion,25–27 provide strong support for the leading MJ model with AMA1 as a structural component of the junction. Recent evidence suggests an altered composition of the Plasmodium sporozoite MJ, as well as the potential for yet unidentified proteins to participate in host cell invasion.24 Overall, the importance of the AMA1–RON2 interaction is clear, but the specific role in the invasion process remains ambiguous—whether this interaction provides the scaffold for the MJ or initiates a signal within the parasite for productive invasion will require further investigation.
Homologues of AMA1 exist in nearly every apicomplexan parasite,28 with the conserved role of RON2 binding as demonstrated in Toxoplasma and Plasmodium,20, 26 but also suspected roles in immune system evasion, early invasion events such as host cell adherence, engagement of the parasite motor complex, and signaling.24, 29–39 The importance of AMA1 in host cell invasion by the parasite is supported by the inability to generate ama1 knockouts in Plasmodium merozoites40 and Toxoplasma tachyzoites,41 as well as the severely attenuated invasion of conditional ama1 knock-downs in these parasite lifecycle stages.24, 42 Collectively, these studies support the classification of AMA1 as a leading malarial vaccine candidate, which initially prompted the structural characterization of the AMA1 ectodomain from Plasmodium falciparum (P. falciparum),43–44 Plasmodium vivax (P. vivax),45 and Toxoplasma gondii (T. gondii).46 Each structure revealed a stacked three-domain architecture (DI, DII, and DIII) with a hydrophobic groove framed by a network of divergent surface loops localized to the apical surface of DI. These structural features are targeted by invasion inhibitory antibodies and peptides,47–50 which presumably function through blocking AMA1–RON complex formation.51, 52 Subsequent studies localized the AMA1 binding region on RON2 to its C-terminal portion26, 53 and revealed the structural basis for complex formation.25, 27
From our recent structural characterization of the AMA1–RON2 complexes from T. gondii and P. falciparum,25, 27 we proposed a binding model where the N-terminal helix of the RON2 peptide is seated at one end of the AMA1 hydrophobic groove and extends through ordered coil to a disulfide-bound beta-hairpin structure (cystine loop), forming a U-shape in the apical groove. However, intriguing differences in anchor points of the AMA1-RON2 interaction were also observed and clearly represented a co-evolution between ligand and receptor.25, 27 These studies highlighted the complexity of defining the structural basis of cross species and strain selectively, which remains somewhat elusive. To more thoroughly define the structural features of AMA1 that promote host cell invasion, we present the crystal structures of AMA1 from B. divergens and N. caninum. In addition to addressing the contributions of structural plasticity and supporting the design of broadly reactive inhibitors to disrupt the AMA1–RON2 interaction, our study reveals intriguing insight into the highly variable DIII domain, which in the case of Plasmodium has been shown to directly coordinate a receptor on erythrocytes.33
Results
Sequence analysis reveals distinct features of Bd/NcAMA1
To establish domain boundaries and regions of divergence in B. divergens AMA1 (BdAMA1) and N. caninum AMA1 (NcAMA1) we generated sequence alignments comparing the fully processed ecto domains of BdAMA1 and NcAMA1 with the structurally characterized AMA1s from P. falciparum (PfAMA1), P. vivax (PvAMA1) and T. gondii (TgAMA1; Fig. 1). The alignment reveals that NcAMA1 is closely related to TgAMA1, with sequence identity of 75% over the entire ectodomain, while BdAMA1 appears intermediate between Tg/NcAMA1 (25% sequence identity) and Pf/PvAMA1 (30% sequence identity). Phylogenetic analysis supports both the close relationship between TgAMA1 and NcAMA1 and the distant clustering of BdAMA1 with Pf/PvAMA1 [Fig. 1 (bottom)]. For both BdAMA1 and NcAMA1, the DI and DII cystine network is conserved, suggesting a conserved structural core (Fig. 1). However, BdAMA1 displays unique features including large insertions and deletions corresponding to apical surface loops functionally important in related AMA1s and two fewer conserved cysteine residues in DIII, implying that it will not be able to form the ultra stable cystine knot observed in all AMA1 DIII structures to date43, 45, 46, 54, 55 (Fig. 1). It is noteworthy that, while the overall sequence identity between TgAMA1 and NcAMA1 is high, NcAMA1 displays divergence in residues shown to be critical in the interaction of Tg AMA1 with a TgRON2 synthetic peptide (TgRON2sp)25 (Fig. 1). To investigate the structural consequences of these unique sequence features, BdAMA1 and NcAMA1 were characterized using X-ray crystallography.
Figure 1.

Sequence analysis of BdAMA1 and NcAMA1 compared with AMA1 proteins for which structures have been determined. Sequences are numbered from the initiation methionine in the signal sequence. Top secondary structure elements refer to TgAMA1, bottom elements refer to PvAMA1. Cyan arrows indicate DI limits; blue—DII, purple—DIII. Orange numbers indicate disulfide connectivity, with top DIII numbers referring to Tg/NcAMA1 and bottom DIII numbers referring to Pf/PvAMA1. The highly variable DII loop is indicated by a blue box. Bottom—phylogenetic tree of the five structurally characterized AMA1 proteins generated in MEGA4 with 500 bootstrap replicates from a MUSCLE alignment of the fully processed ectodomains. Domain boundaries of the fully processed Bd/NcAMA1 ectodomains were defined based on the paradigm established for previously characterized AMA1s.37, 43, 45, 46, 56 BdAMA1 DI spans residues from Pro84 to Pro305 (numbering based on the initiation methionine in the signal sequence), DII from Met307 to Pro445 and DIII from Phe446 to Lys523, while NcAMA1 DI spans residues from Thr62 to Pro281, DII from Asn282 to Ile410 and DIII from Ile411 to Ala481.
Overall structure of Bd/NcAMA1 ectodomains
Soluble forms of the fully processed three domain (DI, DII, and DIII) ectoplasmic region of BdAMA1 and NcAMA1 were produced recombinantly in insect cells, purified to homogeneity using nickel affinity, size exclusion, and ion exchange chromatography, and set in crystallization screens. Refinement of preliminary crystals required recombinant protein produced in the presence of tunicamycin to reduce N-linked glycosylations. BdAMA1 crystallized as thin clusters of sheets in space group P21212 with one molecule in the asymmetric unit, while NcAMA1 crystallized as thin sheets in C2 with four molecules in the asymmetric unit. The final model of BdAMA1 incorporates residue 93 through 510, with two apical surface loops and a portion of the DII loop disordered. NcAMA1 is modeled between residues 62(chain A)/64(chains B, D, and E) and 473, with localized regions of disorder near the tip of the DII loop and in the DII-DIII linker. Four-fold non-crystallographic symmetry averaging with NcAMA1 was required to define the structure of several surface loops. The overall structure of each chain is largely conserved with root-mean-square deviations (r.m.s.d) relative to chain A of 0.50 Å over 390 Cα atoms (chain B), 0.47 Å over 394 Cα atoms (chain D) and 0.56 Å over 389 Cα atoms (chain E). Chain A was the most extensively modeled (398 of 431 total residues), and used for all subsequent structural analyses unless otherwise noted.
The similarity of the BdAMA1 and NcAMA1 structures to previously characterized AMA1 proteins was probed using a DALI search,57 which revealed that while BdAMA1 is clearly structurally homologous with Pf/PvAMA1 (Z-scores of 34–36), it is unexpectedly also closely related to TgAMA1, with a Z-score of 32. As expected, NcAMA1 shows a high degree of structural similarity with TgAMA1 (Z-score 60), and lower, but still significant, similarity to Pf/PvAMA1 (Z-scores of 32–34). These observations confirm that BdAMA1 is a “hybrid” between TgAMA1 and Pf/PvAMA1, consistent with the distant phylogenetic clustering with Plasmodium AMA1s (Fig. 1). Intriguingly, NcAMA1 DI and DII are not classified as PAN module containing domains, similar to TgAMA1,46 while DI and DII of BdAMA1 are recognized as PAN domains, as was identified for PfAMA1 and PvAMA143, 45 (highest Z-score with PAN-containing protein—Tg/Nc/Bd/Pf/PvAMA1: 3.9/3.9/8.0/7.1/4.1). However, both NcAMA1 and BdAMA1 contain similar arrangements of secondary structure in DI and DII with a central alpha helix in each surrounded by a curved beta sheet of four anti-parallel strands, with the majority of insertions, deletions, and structural variations localized to surface loops (Fig. 1).
Divergence in the apical groove reveals structural consequences for the DII loop
The BdAMA1 and NcAMA1 ecto domains are approximately equivalent in size and predicted to extend more than 80 Å from the parasite cell membrane [Fig. 2(A)]. Both AMA1 proteins are structured with DI assembled on top of DII, a loop of DII extending up the side of DI to form an integral part of the apical surface, and DIII forming the membrane proximal base [Fig. 2(A)], analogous to the other structurally characterized AMA1 proteins.43, 45, 46 Both BdAMA1 and NcAMA1 present a hydrophobic groove that extends roughly 40 Å across the length of the apical surface [Fig. 2(B)]. The tip of the BdAMA1 DII loop is disordered [Fig. 2(B top)], which is not surprising given that the corresponding region is completely disordered in PvAMA1,45 only partially modeled in PfAMA1,43 and displaced from the apical groove in Tg/PfAMA1 upon Tg/PfRON2 binding.25, 27 In contrast, the DII loop is relatively well modeled in NcAMA1, likely due to its truncated size (15 residues shorter than BdAMA1) [Figs. 1 and 2(B bottom)]. However, despite the truncated structure, the NcAMA1 DII loop is markedly more mobile than observed in TgAMA1 [Fig. 2(C)] indicating that loop length is only partially responsible for order; the interactions formed by the tip of the DII loop also appear to play an influential role in its organization. The tip of the NcAMA1 DII loop is anchored in the apical groove by a pair of tryptophan residues, Trp347 and Trp348, which bury into pockets on either side of a central serine, Ser224 [Fig. 2(C left, top middle)]. This serine does not appear to serve as an effective anchor for the DII loop, as the two pockets are not well separated, leading to two observed conformations of Trp348 [Fig. 2(C left, top middle)]. The observed organization is in contrast with apoTgAMA1, where the DII loop is well ordered and modeled in its entirety, with the two Trp residues (Trp353, Trp354) anchored in a hydrophobic pocket bifurcated by Tyr230 [Fig. 2(C bottom middle, right)].46 The Ser-Tyr substitution in NcAMA1 may be advantageous as it could reduce the energy barrier for DII loop rearrangement upon RON2 binding.
Figure 2.
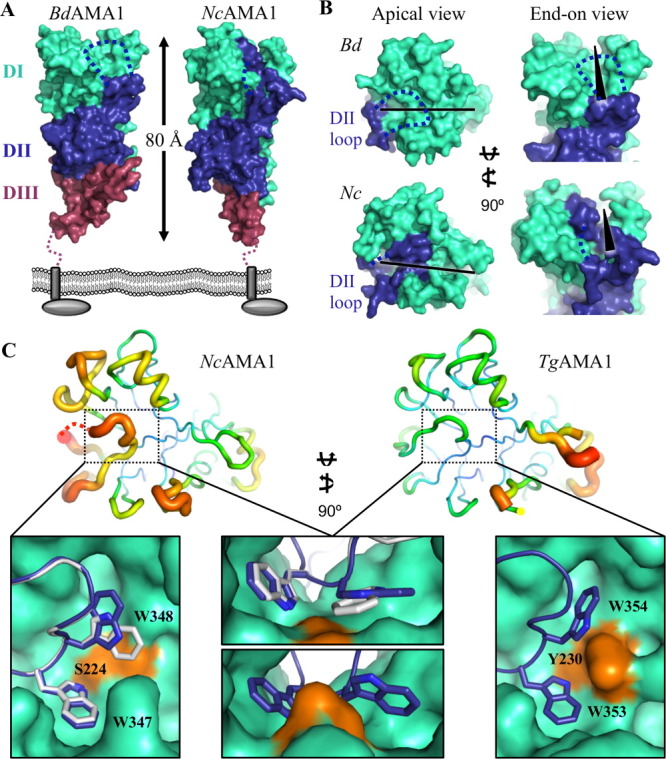
Initial structural characterization of BdAMA1 and NcAMA1.(A) Three-domain architecture of BdAMA1 and NcAMA1 shown in the predicted orientation to the parasite membrane, with DI cyan, DII blue, and DIII purple. Dotted blue line indicates the un-modeled portion of the DII loop. (B) Apical (left) and end-on (right) surface views of BdAMA1 (top) and NcAMA1 (bottom), colored as in (A). The DII loop extends up the side of DI to form part of the apical surface. Among the apical surface loops, a deep groove is present (black bar/wedge). (C) Top—B-factor analysis of the NcAMA1 and TgAMA1 apical surfaces, showing a relatively higher mobility in the DII loop (black box) of NcAMA1 (left) compared with TgAMA1 (right). Tubes colored blue to red and with thin to thick lines, indicating the scale of order to disorder within each structure. Bottom—the DII loop of NcAMA1 is held into the apical groove (cyan) by a pair of tryptophans on either side of a central serine (orange); overlay of NcAMA1 chains A (blue) and E (white) illustrates the two orientations of Trp348 (left, top middle). The DII loop of TgAMA1 (blue) is anchored into the apical groove (cyan) by a pair of Trp residues burying into pockets on either side of a central tyrosine (orange) (bottom middle, right; PDB ID: 2X2Z). All molecular figures generated in PyMol. An interactive view is available in the electronic version of the article.
DIII displays the most structural divergence of the three ectoplasmic AMA1 domains
Although the apical surface of AMA1 has a defined role in RON2 coordination,25, 27 the membrane proximal DIII has multiple potential roles, including functioning as an adhesin in Plasmodium attachment to erythrocytes33 and as a conduit for any signal passed from the ectoplasmic region through to the intracellular domain.35 The divergence of DIII indicated by sequence alignments (Fig. 1) is clearly retained at the structural level. NcAMA1 DIII consists of two beta-strands layering across the bottom of DI/DII and a short C-terminal helix, which is stabilized by a cystine knot [Fig. 3(A left)]. BdAMA1 DIII is comprised entirely of beta strands and extended connecting loops that form a single sheet beneath DI/DII [Fig. 3(A middle)]. The NcAMA1 and TgAMA1 minimalist DIII [Fig. 3(A left)] are highly conserved, overlaying with an r.m.s.d. of 0.69 Å over 51 Cα atoms (>80% sequence identity). Importantly, both the small Tg/NcAMA1 DIII and the large PvAMA1 DIII [Fig. 3(A right)] adopt the structurally ultra-stable cystine knot motif, with a single disulfide bond threading through two others within DIII. In contrast, BdAMA1 DIII contains only two disulfide bonds and is noticeably less compact, forming a more extended layer across the base of DI and DII [Fig. 3(A middle)]. This is evident from a comparison of the DIII—DI/DII interfaces. BdAMA1 DIII buries 1958 Å2 of surface area with DI/DII, with a complexation significance score (CSS)58 of 0.44, while Nc/TgAMA1 DIII, which is roughly the same size, buries 2236 Å2 with DI/DII, with the highest CSS of 1.0 indicating a more intimate association between the domains. By comparison, PvAMA1 DIII, which is 1.5 times the size, buries 3325 Å2 with DI/DII with a CSS of 1.0 and appears to have an additional role in stabilizing the DI N-terminal extension. Construction of a phylogenetic tree of the DIII domains for the five structurally characterized AMA1s supports the observation that BdAMA1 DIII is more closely related to Nc/TgAMA1 DIII [Fig. 3(B)].
Figure 3.
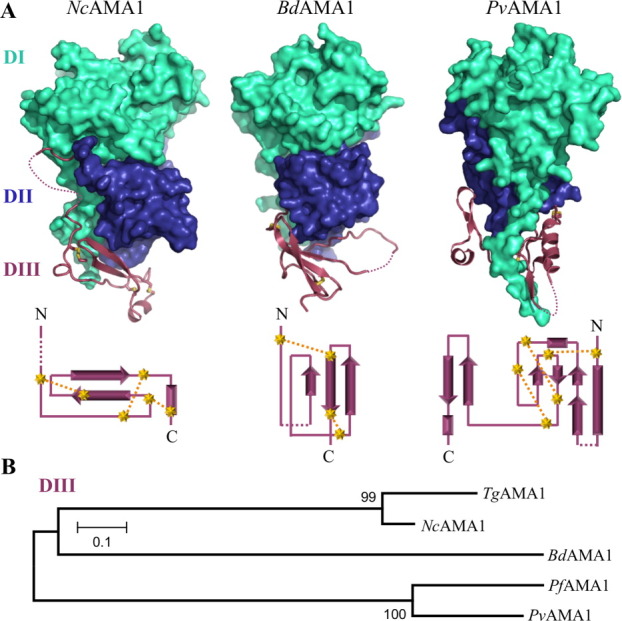
Extreme divergence in DIII of AMA1.(A) Top—solid surfaces of NcAMA1, BdAMA1, and PvAMA1 (PDB ID: 1W8K) DI (cyan) and DII (blue), with secondary structure of DIII (purple). Disulphide bonds in DIII shown as yellow sticks. Bottom—corresponding DIII topology diagrams with cysteines depicted as yellow stars, disulfide bonds as yellow dotted lines, and disordered regions of DIII as purple dotted lines. (B) Phylogenetic analysis in MEGA4 with 500 bootstrap replicates based off of MUSCLE alignments of DIII domains (as indicated by purple arrows in Fig. 1) supports divergence of the DIII domains and distant clustering of BdAMA1 DIII with Nc/TgAMA1 DIII.
NcAMA1 displays cross reactivity with a C-terminal region ofTgRON2
With the flexible DII loop removed, the apical surfaces of BdAMA1 and NcAMA1 maintain a deep groove, similar to the mature receptor binding grooves of TgAMA125 and PfAMA1,27 that is likely capable of accommodating RON2 [Fig. 4(A, black bar)]. The similarity of NcAMA1 and TgAMA1 coupled with the divergence between BdAMA1 and TgAMA1 provides an excellent opportunity to investigate the roles of divergent substructures in the RON2 binding region. Consequently, we tested the capacity of the TgRON2 peptide to bind BdAMA1 and NcAMA1 in a native gel electrophoresis assay [Fig. 4(B)—multiple AMA1 bands are due glycosylation heterogeneity]. Although no interaction between BdAMA1 and the TgRON2 peptide was observed [Fig. 4(B left)], NcAMA1 and TgRON2sp formed a stable complex [Fig. 4(B middle)] at the same molar ratio observed for TgAMA1 [Fig. 4(B right)].
Figure 4.

Cross-genera reactivity in the AMA1–RON2 interaction.(A) Apical surface views of BdAMA1 (left) and NcAMA1 (right) with the DII loop removed to the point of corresponding disorder in the TgAMA1-TgRON2sp co-structure. DI/DII are shown as semi-transparent cyan/blue surfaces, with secondary structure of loops visible, and un-modeled loops indicated by dotted lines. Black bars indicate the apical groove and black numbers represent the apical surface loops as defined by previous AMA1 structures. (B) Native gel electrophoresis assays reveal a level of structural plasticity in the AMA1–RON2 interaction. BdAMA1 does not interact with TgRON2sp (left), but NcAMA1 is able to bind TgRON2sp (middle) at the same molar ratio as TgAMA1 (right). (C) Fluorescence polarization assay with recombinant TgAMA1, NcAMA1, and BdAMA1, and 5FAM-TgRON2sp. Error bars indicate SEM for triplicate samples. EC50 values calculated from a sigmoidal nonlinear regression model in GraphPad Prism 5.0. mP—millipolarization units; ND—not determined.
To probe cross-reactivity at a more detailed level, a fluorescence polarization assay was developed to assess binding between a fluorescently labeled TgRON2 peptide [5-carboxyfluorescein (5FAM)–TgRON2sp] and TgAMA1, NcAMA1, and BdAMA1. Similar to previous studies, the EC50 value of the TgAMA1—5FAM-TgRON2sp interaction was determined to be in the low nanomolar range (51.7 ± 5.9 nM; [Fig. 4(C)]). As expected from our structural predictions, the NcAMA1—5FAM-TgRON2sp binding event occurs with an EC50 value similar to TgAMA1 (57.4 ± 3.8 nM; [Fig. 4(C)]). In contrast, no binding could be detected between BdAMA1 and 5FAM-TgRON2sp (ND; [Fig. 4(C)]) consistent with the native gel shift assay.
Discussion
Hyper-variability of DIII implies divergent functions
Based on sequence alignments (Fig. 1) and structural analyses (Fig. 3), DIII displays the most extensive divergence of the three AMA1 ectoplasmic domains. NcAMA1 and TgAMA1 DIII are notably compact and stabilized by the cystine knot, and may serve a simple function such as orienting the apical surface toward the host cell, or a more complex function in signal transduction. However, DIII of P. falciparum AMA1 has been implicated in attachment to the erythrocyte surface through the membrane protein Kx.33 In contrast, the attachment of BdAMA1 to erythrocyte surfaces is Kx-independent and has a similar adhesion profile to Plasmodium yoelii AMA1 DI/II: neuraminidase resistant, but trypsin and chymotrypsin sensitive.37, 59 The divergence between DIII of BdAMA1 and PvAMA1 (which likely has a similar structure to PfAMA1 DIII) is therefore not surprising as Plasmodium and Babesia have similar cellular tropisms, but AMA1-mediated attachment appears to occur through variable AMA1 domains and distinct erythrocyte receptors.33, 37, 60, 61 The loss of the cystine knot in BdAMA1 DIII leading to a more extended structure [Fig. 3(A middle)] suggests it does not have a major role in orienting DI/DII. Ultimately, additional studies are required to define the role of the DIII domains, though the structural variability suggests the potential for a broad range of functions.
Structural plasticity in the AMA1-RON2 interaction
Prior to this study cross reactivity between AMA1 from one species and RON2 from a different species had not been identified, which led us to perform a detailed comparative structural analysis to understand the basis for both plasticity and selectivity in this important interaction.
In the T. gondii and P. falciparum AMA1–RON2 interactions, when the AMA1 DII loop is displaced a clear pocket is revealed formed by a trio of tyrosine residues (Tyr110/142, Tyr213/234, and Tyr215/236) into which a conserved RON2 proline (Pro1309/2033) docks [Fig. 5(A,B)]. Although BdAMA1 has a histidine (His131) substituted for Tyr110/142, the remaining two tyrosine residues (Tyr233 and Tyr235) are conserved, and together these three residues retain the generally conserved architecture of the surface pocket [Fig. 5(B 1 top)]. The aromatic imidazole ring of the histidine residue aligns with the corresponding benzene ring of Tyr110/142 and likely maintains a stacking interaction with the analogous BdRON2 proline (Pro953). In NcAMA1, as for TgAMA1, this pocket was previously occupied by the DII loop Trp353 [Fig. 2(C)]. The TgAMA1 tyrosine trio is conserved in both sequence and structure with NcAMA1 (Tyr104, Tyr207, and Tyr209) and the TgRON2 proline is conserved in sequence with NcRON2 [Fig. 5(A,B 1 bottom)], suggesting a similar anchor point is exploited during complex formation. Notably, in the co-structure of PfAMA1 with the invasion inhibitory phage display peptide R1, a Leu residue docks into this same pocket,27 suggesting that occupation of the Tyr trio defined pocket is an important component of complex formation, perhaps in maintaining the DII loop in its displaced conformation.
Figure 5.
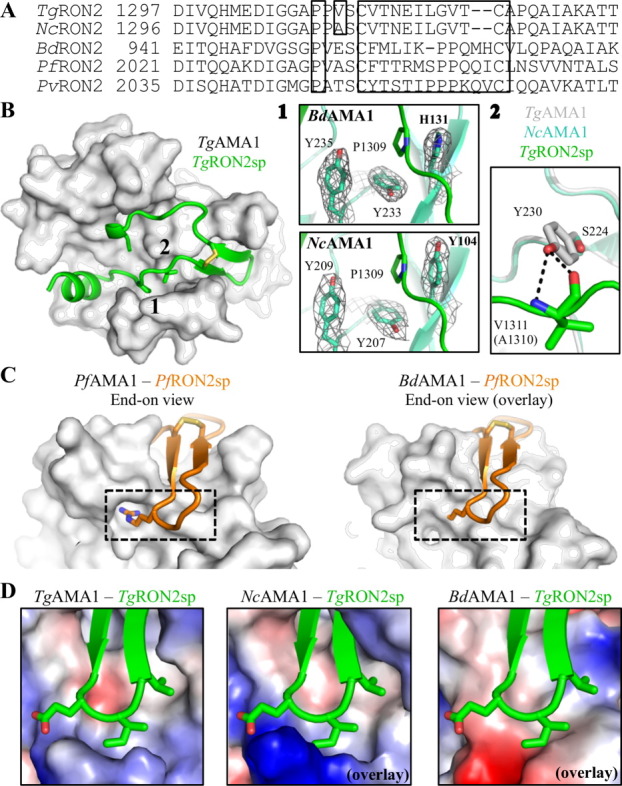
Structural plasticity and specificity of the AMA1–RON2 interaction. (A) Sequence alignment of RON2 sequences for the regions that align with TgRON2sp, with boxes around the conserved RON2 Pro, the single difference between TgRON2 and NcRON2 in this region, and the cystine loop. (B) TgAMA1 apical surface (gray) with TgRON2sp secondary structure (green) (PDB ID: 2Y8T) and residues of interest shown as sticks; black numbers indicate regions of conservation and divergence with Bd/NcAMA1 under investigation. 1—Top: overlay of TgRON2sp onto BdAMA1 shows that the AMA1 Tyr trio that accommodates a key RON2 Pro (green sticks) is substituted in BdAMA1 with a Tyr/Tyr/His (cyan sticks) that conserves the architecture of the pocket. Bottom: overlay of TgRON2sp (green) onto NcAMA1 shows that the complete Tyr trio is conserved in NcAMA1 (cyan sticks). Gray mesh: 2Fo-Fc electron density maps around the pocket defining residues contoured at 1.0 σ. 2—The central TgAMA1 Tyr230 (gray) forms a functionally important bifurcated hydrogen bond with the backbone of TgRON2 Val1311 (green). Plasticity may allow similar binding between NcAMA1 Ser224 (cyan) and the corresponding NcRON2 Ala1310. (C) Left—End-on view of PfRON2sp1 (orange secondary structure) bound to PfAMA1 (gray surface) (PDB ID: 3ZWZ) shows a deep pocket on the surface of AMA1 into which a critical RON2 arginine residue anchors (orange sticks, dotted box). Right—An overlay of PfRON2sp1 on BdAMA1 (gray surface) shows that a similar pocket is not present (dotted box). (D) Electrostatic surface, end-on views of TgAMA1 bound to TgRON2sp (green secondary structure with cystine loop tip side chains shown as sticks) (left; PDB ID: 2Y8T), and NcAMA1 (middle) and BdAMA1 (right) overlayed on the TgAMA1-TgRON2sp co-structure, reveal key differences in the chemical nature of the cystine loop binding region. An interactive view is available in the electronic version of the article.
Intriguingly, only one residue differs between the AMA1-binding regions of TgRON2 and NcRON2 [Fig. 5(A)]. In the T. gondii interaction, RON2 Val1311 forms two backbone hydrogen bonds with the AMA1 apical groove central tyrosine (Tyr230). The structurally equivalent residue to TgAMA1 Tyr230 in NcAMA1 is Ser224, and the sequence-aligned residue of Val1311 is NcRON2 Ala1310 [Fig. 5(A,B 2)]. It is possible that the NcAMA1 serine aids in destabilizing the DII loop to facilitate displacement [Fig. 2(C left)], while the NcRON2 alanine provides flexibility to maintain hydrogen bonding interactions important in complex formation. The ability of NcAMA1 to bind TgRON2sp with similar binding affinity as that observed for TgAMA1 reflects an intriguing level of structural plasticity [Fig. 4(C)]. The similar binding affinity fits well with the observations of NcAMA1 DII loop instability [Fig. 2(C right)] combined with the predicted absence of backbone hydrogen bonds between NcAMA1 Ser224 and TgRON2sp Val1311 [Fig. 5(B 2)], and is also consistent with the order of magnitude lower binding affinity of the TgAMA1 Y230A mutant for TgRON2sp.25 Collectively, these data indicate that interaction with the central tyrosine, and by extension the central serine, is important but not essential.25 Aside from the Tyr–Val interaction, every residue on TgAMA1 shown to play a critical role in binding TgRON2sp is conserved in NcAMA1. Spatially, Phe157 (TgAMA1 Phe163) likely needs to rotate in order to accommodate RON2, while loops 1 and 2 guarding the cystine loop binding surface need to be displaced by at least 3.0 Å to enable access to this region. A similar loop reorganization is observed between the apo and receptor bound forms of TgAMA1.
No binding could be detected between BdAMA1 and 5FAM-TgRON2sp [Fig. 4(C)]. This is not surprising based on the observation that neither PfAMA1 nor PvAMA1 are capable of binding TgRON2 in SPR assays,25 and is likely due to a different anchoring mechanism in the cystine loop binding region.
Selectivity in the AMA1-RON2 interaction
Based on a comparison of the TgAMA1-TgRON2sp and PfAMA1-PfRON2sp coordination events, it is clear that selectivity is governed by the cystine loop region.25, 27 Analysis of the TgAMA1-TgRON2sp co-structure did not reveal any particular hot spots of interaction, aside from the disulfide bond structure.25 However, a similar analysis of the co-structure from P. falciparum showed that a single arginine residue in the cystine loop is absolutely critical for complex formation27; very little of the PfRON2 cystine loop contacts the PfAMA1 surface, but Arg2041 buries into a deep pocket [Fig. 5(C left)], resulting in numerous polar contacts.27 The PfAMA1 surface pocket that accommodates this Arg residue is also exploited by the invasion inhibitory, species-specific phage display peptide, R1.27 From the alignment of RON2 sequences [Fig. 5(A)], and given that there are no well-defined pockets on the BdAMA1 surface within the cystine loop binding region [Fig. 5(C right)], BdRON2 is unlikely to have a singular anchor as was observed for the PfAMA1–PfRON2sp interaction. Therefore, although BdAMA1 is overall more similar to PfAMA1, the BdAMA1-BdRON2 interaction is more likely to resemble TgAMA1–TgRON2sp, with numerous contacts anchoring the cystine loop.
The surface of TgAMA1 engaged by the TgRON2sp cystine loop is largely hydrophobic, with a partially hydrophilic border [Fig. 5(D left)]. This surface is compatible with the Glu-Ile-Leu tip of the cystine loop, in which the Ile and Leu brace the interaction.25 The NcRON2 cystine loop region is conserved with TgRON2sp, yet the corresponding AMA1 region is notably more basic [Fig. 5(D middle)]. The increased alkalinity is due to an Asn-Lys substitution (TgAMA1 Asn184–NcAMA1 Lys178), which may result in a stabilizing salt bridge with NcRON2 Glu1316. Not surprisingly, the BdAMA1 surface likely capable of accommodating the BdRON2 cystine loop has a hydrophobic core, but with an acidic border that would coordinate the Ile-Lys-Pro-Pro loop tip [Fig. 5(A, D right)]. It is noteworthy that the electrostatic charges across the remainder of the groove are well conserved despite low sequence identity, with a central hydrophobic region, and a basic patch revealed by displacement of the DII loop. Together, these observations support the previously proposed model of specificity, wherein the RON2 cystine loop governs cross-species selectivity within an otherwise highly conserved biding paradigm.25, 27
Implications for generating cross genera inhibitors of AMA1-RON2 binding
In addition to better defining the basis for both plasticity and selectivity in the AMA1–RON2 interaction, the five AMA1 structures, two AMA1-RON2 co-structures, and AMA1-R1 co-structure serve as a robust model for the design of broadly reactive inhibitors. The first step in this process is the identification of well-conserved pockets that can be exploited by small molecules. Overlaying all three structurally characterized ligands—TgRON2sp, PfRON2sp, and R1—on the five AMA1 structures—TgAMA1, NcAMA1, BdAMA1, PfAMA1, and PvAMA1—[Fig. 6(A left), TgAMA1 alone shown for clarity] reveals three defined pockets on the surface of AMA1 [Fig. 6(A inset)]. Pocket 1 is very well conserved (Table I), and has been shown to be occupied by the tip of the NcAMA1 and TgAMA1 DII loops (Trp347 and Trp353), a conserved RON2 proline (TgRON2 Pro1309; PfRON2 Pro2033), and R1 Leu6.25, 27, 46 Clearly, occupation of Pocket 1 is a strategic target, but to be accessed by a small molecule the DII loop would need to be displaced. To overcome the energy barrier associated with DII loop rearrangement, additional anchor points within the AMA1 groove would be essential. TgRON2 Ile1328, PfRON2 Val2054, and R1 Leu8 all brace against the base of loop 4 in a shallow hydrophobic pocket, which constitutes Pocket 2 (backstop; Fig. 6, Table I). Binding to this pocket alone would not provide enough traction to displace the DII loop, but extending too far toward the cystine loop binding region would severely limit cross reactivity. However, the disulfide bond plays a critical and highly conserved role in complex formation,25, 27 and thus the pocket that binds this structural feature would be an excellent anchor point, and is presented here as Pocket 3 (Fig. 6 and Table I). These three pockets have a highly conserved chemical and structural architecture (Fig. 6 and Table I), are in close proximity, and maintain a triangular geometry within the apical groove of AMA1 [Fig. 6(A right)] that could be efficiently occupied by a broadly reactive small molecule inhibitor.
Figure 6.
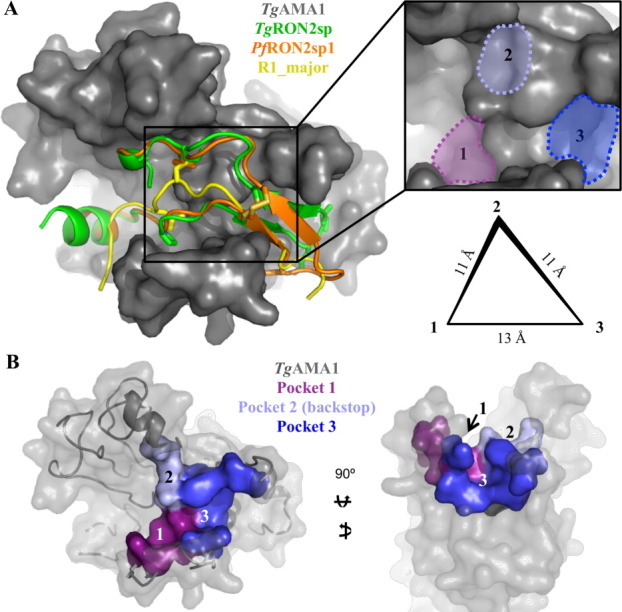
Identification of surface pockets ideal for targeting with therapeutics. (A) Left—apical view of an overlay of three structurally characterized ligands (shown as secondary structure, with residues occupying pockets of interest shown as sticks)—TgRON2sp (green; PDB ID: 2Y8T), PfRON2sp1 (orange; PDB ID: 3ZWZ), R1_major (yellow; PDB ID: 3SRJ)—on the surface of TgAMA1 (gray). Inset—enlarged apical view for the identification of three surface pockets with structural and chemical features conserved across all the structurally characterized AMA1 proteins. Right—schematic representation showing the relative positions of the three pockets in a triangular organization, with Pocket 2 in a higher plane than Pockets 1 and 3. Pocket 2 is equidistant from 1 and 3 (11 Å), while Pockets 1 and 3 are separated by roughly 13 Å. (B) Left—apical surface of TgAMA1 (gray) in the same orientation as (A), with the residues defining Pocket 1 shown purple; 2, light blue; 3, bright blue. Right—end-on view highlighting the depth of Pocket 3 and the backstop nature of Pocket 2.
Table I.
Residues Lining the Conserved Apical Surface Pockets of AMA1
| TgAMA1 | NcAMA1 | BdAMA1 | PfAMA1 | PvAMA1 |
|---|---|---|---|---|
| Pocket 1 | ||||
| Val105 | Val99 | Val126 | Val137 | Val82 |
| Tyr110 | Tyr104 | His131 | Tyr142 | Tyr87 |
| Ala203 | Ala197 | Pro225 | Pro226 | NDa |
| Tyr213 | Tyr207 | Tyr233 | Tyr234 | Tyr179 |
| Tyr215 | Tyr209 | Tyr235 | Tyr236 | His181 |
| Pocket 2 (backstop) | ||||
| Val142 | Ile136 | Ile157 | Val169 | Val114 |
| Pro143 | Pro137 | Ser158 | Ala170 | Ala115 |
| Tyr230 | Ser224 | Phe250 | Tyr251 | Tyr196 |
| Pocket 3 | ||||
| Leu161 | Met155 | Phe169 | Phe183 | Phe128 |
| Phe163 | Phe157 | Pro170 | Thr186 | Ala131 |
| Thr165 | Thr159 | NDa | Phe188 | His134 |
| Ile171 | Ile165 | Leu195 | Met190 | Ile135 |
| Thr201 | Thr195 | Ile223 | Met224 | Phe169 |
| Met204 | Met198 | Ala226 | Asp227 | NDa |
| Tyr215 | Tyr209 | Tyr235 | Tyr236 | His181 |
| Ile228 | Ile222 | Ile248 | Ile249 | Met194 |
Structurally equivalent residues are horizontally aligned.
Not determined (ND) due to flexible, unmodeled loop.
Materials and Methods
Bioinformatics
Boundaries for DI, DII, and DIII were defined based on the paradigm established for previously characterized AMA1s.43, 45, 46, 56 Phylogenetic analysis was performed using MEGA 462, 63 and multiple sequences were aligned using MUSCLE64 and illustrated in ESPript.65 Accession numbers for aligned AMA1 sequences are as follows: T. gondii (UniProt ID: B6KAM0), N. canium (UniProt ID: A2A114), B. divergens (UniProt ID: C0IR59), P. falciparum (UniProt ID: Q7KQK5), and P. vivax (UniProt ID: A5K4Z2; sequence modified to reflect crystallized sequence,45 with 3 N-linked glycosylation sites mutated—PDB ID: 1W8K).
RON2 sequences in the region of Tg/PfRON2sp were aligned using ClustalW,66 and manually edited based on structural evidence. Accession numbers and sequence limits for aligned RON2 sequences are as follows: T. gondii (UniProt ID: F6KDI4, residues 1297-1333), N. canium (UniProt ID: F0VQN9, residues 1296-1332), B. divergens (UniProt ID: G8Z7K1, residues 941-978), P. falciparum (UniProt ID: Q8IKV6, residues 2021-2059), and P. vivax (UniProt ID: A5K3N8, residues 2035-2073).
Protein expression and purification
Sequences encoding the fully processed ectoplasmic domains of BdAMA1 and NcAMA1 were synthesized by GenScript, codon optimized for insect cells, and subcloned into a modified pAcGP67b vector (Pharmingen) incorporating a C-terminal hexahistidine tag separated from ama1 by a thrombin cleavage site. Bd/NcAMA1 encoding viruses for insect cell protein production were generated and amplified using established protocols.25, 46 Selected protein preparations had a final concentration of 0.3 μg/mL tunicamycin. Following a 65 h infection the supernatant was harvested, concentrated, buffer exchanged into Buffer A1 (20 mM HEPES pH 8.0, 30 mM imidazole, 1 M NaCl) and allowed to batch bind with Ni-agarose beads at 4°C for 1 h. AMA1 proteins were eluted with Buffer B1 (20 mM HEPES pH 8.0, 250 mM imidazole, 1 M NaCl), and fractions were analyzed by SDS-PAGE and pooled based on purity. The hexahistidine tag was removed by thrombin cleavage and AMA1 proteins were further purified by size exclusion chromatography (Superdex 16/60 200) in HEPES buffered saline (HBS), followed by anion exchange chromatography (Source 30Q). The final yield of both BdAMA1 and NcAMA1 was approximately 2 mg of purified protein per liter of insect cell culture.
Crystallization and data collection
Using the sitting drop method, crystals of BdAMA1 were grown in 16% PEG8000, 300 mM calcium acetate, 100 mM sodium cacodylate pH 5.5 and 3% 2-methyl-2,4-pentanediol (MPD) and crystals of NcAMA1 were grown in 20% PEG3350, 300 mM sodium citrate pH 4.2 and 10 mM zinc chloride. The final drops consisted of 1.5 μL protein (BdAMA1 12 mg/mL; NcAMA1 15 mg/mL) with 1.5 μL reservoir solution and were equilibrated against 100 μL of reservoir solution. Cryo protection of the Bd/NcAMA1 crystals was performed in mother liquor supplemented with 12.5% glycerol for 20 s and the crystals were flash cooled at 100 K directly in the cryo stream. Diffraction data were collected on beamline 9-2 at Stanford Synchrotron Radiation Lightsource for BdAMA1 and on beamline CMCF-ID at Canadian Light Source for NcAMA1.
Data processing, structure solution and refinement
Diffraction data were processed using Imosflm67 and Scala68 in the CCP4 suite of programs.69 Initial phases for BdAMA1 were obtained by molecular replacement (MR) using PHASER70 with the DI and DII domains of PfAMA1 (PDB ID: 2Q8A) pruned with CHAINSAW.71 Despite multiple attempts, no MR solution was obtained for DIII, which was ultimately traced de novo into the electron density maps using COOT72 and shown to be divergent from other characterized DIIIs. Initial phases for NcAMA1 were obtained by MR using PHASER70 with chain B of TgAMA1 (PDB ID: 2X2Z) pruned with CHAINSAW,71 and improved with four-fold non-crystallographic symmetry averaging. Solvent molecules were selected using COOT72 and refinement performed using Refmac5.73 Stereochemical analysis performed with PROCHECK and SFCHECK in CCP469 showed good stereochemistry with more than 92% of the residues in the favored conformations and no residues modeled in disallowed orientations of the Ramachandran plot. Overall 5% of the reflections were set aside for calculation of Rfree. Data collection and refinement statistics are presented in Table II.
Table II.
Data Collection and Refinement Statistics
| BdAMA1 | NcAMA1 | |
|---|---|---|
| A. Data collection | ||
| Spacegroup | P21212 | C2 |
| a, b, c (Å) | 67.82, 139.18, 45.01 | 251.56, 51.00, 145.34 |
| α, β, γ (°) | 90, 90, 90 | 90, 90.93, 90 |
| Wavelength (Å) | 0.9791 | 0.9795 |
| Resolution range (Å) | 42.83–2.30 (2.42–2.30) | 50.00–2.90 (3.06–2.90) |
| Measured reflections | 90,064 | 161,770 |
| Unique reflections | 19,533 | 41,269 |
| Redundancy | 4.6 (4.5) | 3.9 (3.8) |
| Completeness (%) | 99.2 (96.4) | 99.1 (96.6) |
| I/σ(I) | 12.5 (3.7) | 9.8 (4.4) |
| Rmergea | 0.070 (0.389) | 0.112 (0.385) |
| B. Refinement | ||
| Resolution (Å) | 42.83–2.30 (2.36–2.30) | 49.98–2.90 (2.98–2.90) |
| Rcrystb/Rfreec | 0.190/0.251 (0.228/0.284) | 0.193/0.271 (0.273/0.396) |
| No. of atoms | ||
| Protein (A/B/D/E) | 2624 | 3155/3123/3132/3094 |
| Solvent | 166 | 142 |
| Calcium | 1 | N/A |
| Acetate/Glycerol/MPD | 12/36/8 | N/A |
| B-values (Å2) | ||
| Protein (A/B/D/E) | 38.9 | 36.3/39.0/34.7/45.1 |
| Solvent | 45.7 | 31.0 |
| Calcium | 30.2 | N/A |
| Acetate/Glycerol/MPD | 62.3/65.4/66.7 | N/A |
| r.m.s. deviation from ideality | ||
| Bond lengths (Å) | 0.01 | 0.01 |
| Bond angles (°) | 1.33 | 1.05 |
| Ramachandran statistics | ||
| Most favoured | 97.0% | 92.8% |
| Allowed | 3.0% | 7.2% |
| Disallowed | 0.0% | 0.0% |
Values in parentheses are for the highest resolution shell.
Rmerge = ∑hkl ∑i |Ihkl,i − [Ihkl]|/∑hkl ∑i Ihkl,i, where [Ihkl] is the is the average of symmetry related observations of a unique reflection.
Rcryst = ∑|Fobs − Fcalc|/∑Ffobs, where Fobs and Fcalc are the observed and the calculated structure factors, respectively.
Rfree is R using 5% of reflections randomly chosen and omitted from refinement.
Native gel electrophoresis assay
Purified AMA1 protein in HBS buffer pH 8.0 was incubated for 30 min at room temperature with or without TgRON2sp25 in a 1:2 protein to peptide molar ratio. Reactions were run on 8–25% gradient gels using native buffer strips and the PhastGel system (GE Healthcare).
Fluorescence polarization assay
In black 96-well assay plates (Nunc), AMA1 proteins in HBS were serially diluted from 1 μM to 0.042 nM and incubated with > 90% pure 10 nM TgRON2sp labeled at the N-terminus with 5FAM (Kinexus; Vancouver, Canada). The plate was mixed on a shaker for 15 min and incubated at room temperature for 30 min to reach equilibrium. Fluorescence polarization in millipolarization units (mP) was read using a SpectraMax 5M plate reader (Molecular Devices) with an excitation wavelength of 485 nm and emission wavelength of 538 nm. For determination of EC50 values, data were fit with a sigmoidal dose dependent nonlinear regression model (variable slope) in GraphPad Prism 5.0. Errors were calculated as the standard error of the mean (SEM) from triplicate samples.
Accession numbers
The atomic coordinates and structure factors have been deposited in the Protein Data Bank under the following codes: BdAMA1—PDB ID: 4APM, r4APMsf; NcAMA1—PDB ID: 4APL, r4APLsf.
Acknowledgments
The authors gratefully acknowledge the staff at the Stanford Synchrotron Radiation Lightsource (SSRL) and the Canadian Light Source (CLS).
Glossary
Abbreviations
- AMA1
apical membrane antigen 1
- B.divergens
Babesiadivergens
- CSS
complexation significance score
- DI
Domain I
- DII
Domain II, DIII, Domain III
- 5FAM
5-carboxyfluorescein
- HBS
HEPES buffered saline
- MJ
moving junction
- MR
molecular replacement
- P. falciparum
Plasmodium falciparum
- P. vivax
Plasmodium vivax
- RON
rhoptry neck protein
- RON2sp
RON2 synthetic peptide
- T. gondii
Toxoplasma gondii
- N. caninum
Neospora caninum
References
- 1.World Health Organization. World malaria report. Geneva, Switzerland: World Health Organization; 2010. [Google Scholar]
- 2.Snow RW, Guerra CA, Noor AM, Myint HY, Hay SI. The global distribution of clinical episodes of Plasmodium falciparum malaria. Nature. 2005;434:214–217. doi: 10.1038/nature03342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jackson MH, Hutchison WM. The prevalence and source of Toxoplasma infection in the environment. Adv Parasitol. 1989;28:55–105. doi: 10.1016/s0065-308x(08)60331-0. [DOI] [PubMed] [Google Scholar]
- 4.Tenter AM, Heckeroth AR, Weiss LM. Toxoplasma gondii: from animals to humans. Int J Parasitol. 2000;30:1217–1258. doi: 10.1016/s0020-7519(00)00124-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zintl A, Mulcahy G, Skerrett HE, Taylor SM, Gray JS. Babesia divergens, a bovine blood parasite of veterinary and zoonotic importance. Clin Microbiol Rev. 2003;16:622–636. doi: 10.1128/CMR.16.4.622-636.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Homer MJ, Aguilar-Delfin I, Telford SR, 3rd, Krause PJ, Persing DH. Babesiosis. Clin Microbiol Rev. 2000;13:451–469. doi: 10.1128/cmr.13.3.451-469.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dubey JP, Schares G, Ortega-Mora LM. Epidemiology and control of neosporosis and Neospora caninum. Clin Microbiol Rev. 2007;20:323–367. doi: 10.1128/CMR.00031-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garosi L, Dawson A, Couturier J, Matiasek L, de Stefani A, Davies E, Jeffery N, Smith P. Necrotizing cerebellitis and cerebellar atrophy caused by Neospora caninum infection: magnetic resonance imaging and clinicopathologic findings in seven dogs. J Vet Intern Med. 2010;24:571–578. doi: 10.1111/j.1939-1676.2010.0485.x. [DOI] [PubMed] [Google Scholar]
- 9.Kim K, Weiss LM. Toxoplasma gondii: the model apicomplexan. Int J Parasitol. 2004;34:423–432. doi: 10.1016/j.ijpara.2003.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sibley LD. Intracellular parasite invasion strategies. Science. 2004;304:248–253. doi: 10.1126/science.1094717. [DOI] [PubMed] [Google Scholar]
- 11.Desimone TM, Jennings CV, Bei AK, Comeaux C, Coleman BI, Refour P, Triglia T, Stubbs J, Cowman AF, Duraisingh MT. Cooperativity between Plasmodium falciparum adhesive proteins for invasion into erythrocytes. Mol Microbiol. 2009;72:578–589. doi: 10.1111/j.1365-2958.2009.06667.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morisaki JH, Heuser JE, Sibley LD. Invasion of Toxoplasma gondii occurs by active penetration of the host cell. J Cell Sci. 1995;108:2457–2464. doi: 10.1242/jcs.108.6.2457. [DOI] [PubMed] [Google Scholar]
- 13.Aikawa M, Miller LH, Johnson J, Rabbege J. Erythrocyte entry by malarial parasites. A moving junction between erythrocyte and parasite. J Cell Biol. 1978;77:72–82. doi: 10.1083/jcb.77.1.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Michel R, Schupp K, Raether W, Bierther FW. Formation of a close junction during invasion of erythrocytes by Toxoplasma gondii in vitro. Int J Parasitol. 1980;10:309–313. doi: 10.1016/0020-7519(80)90012-0. [DOI] [PubMed] [Google Scholar]
- 15.Besteiro S, Dubremetz JF, Lebrun M. The moving junction of apicomplexan parasites: a key structure for invasion. Cell Microbiol. 2011;13:797–805. doi: 10.1111/j.1462-5822.2011.01597.x. [DOI] [PubMed] [Google Scholar]
- 16.Suss-Toby E, Zimmerberg J, Ward GE. Toxoplasma invasion: the parasitophorous vacuole is formed from host cell plasma membrane and pinches off via a fission pore. Proc Natl Acad Sci USA. 1996;93:8413–8418. doi: 10.1073/pnas.93.16.8413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alexander DL, Mital J, Ward GE, Bradley P, Boothroyd JC. Identification of the moving junction complex of Toxoplasma gondii: a collaboration between distinct secretory organelles. PLoS Pathog. 2005;1:e17. doi: 10.1371/journal.ppat.0010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Straub KW, Cheng SJ, Sohn CS, Bradley PJ. Novel components of the Apicomplexan moving junction reveal conserved and coccidia-restricted elements. Cell Microbiol. 2009;11:590–603. doi: 10.1111/j.1462-5822.2008.01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Silvie O, Franetich JF, Charrin S, Mueller MS, Siau A, Bodescot M, Rubinstein E, Hannoun L, Charoenvit Y, Kocken CH, Thomas AW, Van Gemert GJ, Sauerwein RW, Blackman MJ, Anders RF, Pluschke G, Mazier D. A role for apical membrane antigen 1 during invasion of hepatocytes by Plasmodium falciparum sporozoites. J Biol Chem. 2004;279:9490–9496. doi: 10.1074/jbc.M311331200. [DOI] [PubMed] [Google Scholar]
- 20.Besteiro S, Michelin A, Poncet J, Dubremetz JF, Lebrun M. Export of a Toxoplasma gondii rhoptry neck protein complex at the host cell membrane to form the moving junction during invasion. PLoS Pathog. 2009;5:e1000309. doi: 10.1371/journal.ppat.1000309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cao J, Kaneko O, Thongkukiatkul A, Tachibana M, Otsuki H, Gao Q, Tsuboi T, Torii M. Rhoptry neck protein RON2 forms a complex with microneme protein AMA1 in Plasmodium falciparum merozoites. Parasitol Int. 2009;58:29–35. doi: 10.1016/j.parint.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 22.Riglar DT, Richard D, Wilson DW, Boyle MJ, Dekiwadia C, Turnbull L, Angrisano F, Marapana DS, Rogers KL, Whitchurch CB, Beeson JG, Cowman AF, Ralph SA, Baum J. Super-resolution dissection of coordinated events during malaria parasite invasion of the human erythrocyte. Cell Host Microbe. 2011;9:9–20. doi: 10.1016/j.chom.2010.12.003. [DOI] [PubMed] [Google Scholar]
- 23.Shen B, Sibley LD. The moving junction, a key portal to host cell invasion by apicomplexan parasites. Curr Opin Microbiol. 2012;15:449–455. doi: 10.1016/j.mib.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giovannini D, Spath S, Lacroix C, Perazzi A, Bargieri D, Lagal V, Lebugle C, Combe A, Thiberge S, Baldacci P, Tardieux I, Menard R. Independent roles of apical membrane antigen 1 and rhoptry neck proteins during host cell invasion by apicomplexa. Cell Host Microbe. 2011;10:591–602. doi: 10.1016/j.chom.2011.10.012. [DOI] [PubMed] [Google Scholar]
- 25.Tonkin ML, Roques M, Lamarque MH, Pugniere M, Douguet D, Crawford J, Lebrun M, Boulanger MJ. Host cell invasion by apicomplexan parasites: insights from the co-structure of AMA1 with a RON2 peptide. Science. 2011;333:463–467. doi: 10.1126/science.1204988. [DOI] [PubMed] [Google Scholar]
- 26.Lamarque M, Besteiro S, Papoin J, Boulanger MJ, Tomavo S, Lebrun M. The RON2–AMA1 interaction is a critical step in the moving-junction-dependent invasion by Apicomplexa parasites. PLoS Pathog. 2011;7:e1001276. doi: 10.1371/journal.ppat.1001276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vulliez-Le Normand B, Tonkin ML, Lamarque MH, Langer S, Hoos S, Roques M, Saul FA, Faber BW, Bentley GA, Boulanger MJ, Lebrun M. Structural and functional insights into the malaria parasite moving junction complex. PLoS Pathog. 2012;8:e1002755. doi: 10.1371/journal.ppat.1002755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chesne-Seck ML, Pizarro JC, Vulliez-Le Normand B, Collins CR, Blackman MJ, Faber BW, Remarque EJ, Kocken CH, Thomas AW, Bentley GA. Structural comparison of apical membrane antigen 1 orthologues and paralogues in apicomplexan parasites. Mol Biochem Parasitol. 2005;144:55–67. doi: 10.1016/j.molbiopara.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 29.Srinivasan P, Beatty WL, Diouf A, Herrera R, Ambroggio X, Moch JK, Tyler JS, Narum DL, Pierce SK, Boothroyd JC, Haynes JD, Miller LH. Binding of Plasmodium merozoite proteins RON2 and AMA1 triggers commitment to invasion. Proc Natl Acad Sci USA. 2011;108:13275–13280. doi: 10.1073/pnas.1110303108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sheiner L, Santos JM, Klages N, Parussini F, Jemmely N, Friedrich N, Ward GE, Soldati-Favre D. Toxoplasma gondii transmembrane microneme proteins and their modular design. Mol Microbiol. 2010;77:912–929. doi: 10.1111/j.1365-2958.2010.07255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Polley SD, Conway DJ. Strong diversifying selection on domains of the Plasmodium falciparum apical membrane antigen 1 gene. Genetics. 2001;158:1505–1512. doi: 10.1093/genetics/158.4.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Remarque EJ, Faber BW, Kocken CH, Thomas AW. Apical membrane antigen 1: a malaria vaccine candidate in review. Trends Parasitol. 2008;24:74–84. doi: 10.1016/j.pt.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 33.Kato K, Mayer DC, Singh S, Reid M, Miller LH. Domain III of Plasmodium falciparum apical membrane antigen 1 binds to the erythrocyte membrane protein Kx. Proc Natl Acad Sci USA. 2005;102:5552–5557. doi: 10.1073/pnas.0501594102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Santos JM, Ferguson DJ, Blackman MJ, Soldati-Favre D. Intramembrane cleavage of AMA1 triggers Toxoplasma to switch from an invasive to a replicative mode. Science. 2011;331:473–477. doi: 10.1126/science.1199284. [DOI] [PubMed] [Google Scholar]
- 35.Treeck M, Zacherl S, Herrmann S, Cabrera A, Kono M, Struck NS, Engelberg K, Haase S, Frischknecht F, Miura K, Spielmann T, Gilberger TW. Functional analysis of the leading malaria vaccine candidate AMA-1 reveals an essential role for the cytoplasmic domain in the invasion process. PLoS Pathog. 2009;5:e1000322. doi: 10.1371/journal.ppat.1000322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leykauf K, Treeck M, Gilson PR, Nebl T, Braulke T, Cowman AF, Gilberger TW, Crabb BS. Protein kinase a dependent phosphorylation of apical membrane antigen 1 plays an important role in erythrocyte invasion by the malaria parasite. PLoS Pathog. 2010;6:e1000941. doi: 10.1371/journal.ppat.1000941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Montero E, Rodriguez M, Oksov Y, Lobo CA. Babesia divergens apical membrane antigen 1 and its interaction with the human red blood cell. Infect Immun. 2009;77:4783–4793. doi: 10.1128/IAI.00969-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tyler JS, Treeck M, Boothroyd JC. Focus on the ringleader: the role of AMA1 in apicomplexan invasion and replication. Trends Parasitol. 2011;27:410–420. doi: 10.1016/j.pt.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parussini F, Tang Q, Moin SM, Mital J, Urban S, Ward GE. Intramembrane proteolysis of Toxoplasma apical membrane antigen 1 facilitates host-cell invasion but is dispensable for replication. Proc Natl Acad Sci USA. 2012;109:7463–7468. doi: 10.1073/pnas.1114661109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Triglia T, Healer J, Caruana SR, Hodder AN, Anders RF, Crabb BS, Cowman AF. Apical membrane antigen 1 plays a central role in erythrocyte invasion by Plasmodium species. Mol Microbiol. 2000;38:706–718. doi: 10.1046/j.1365-2958.2000.02175.x. [DOI] [PubMed] [Google Scholar]
- 41.Hehl AB, Lekutis C, Grigg ME, Bradley PJ, Dubremetz JF, Ortega-Barria E, Boothroyd JC. Toxoplasma gondii homologue of plasmodium apical membrane antigen 1 is involved in invasion of host cells. Infect Immun. 2000;68:7078–7086. doi: 10.1128/iai.68.12.7078-7086.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mital J, Meissner M, Soldati D, Ward GE. Conditional expression of Toxoplasma gondii apical membrane antigen-1 (TgAMA1) demonstrates that TgAMA1 plays a critical role in host cell invasion. Mol Biol Cell. 2005;16:4341–4349. doi: 10.1091/mbc.E05-04-0281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bai T, Becker M, Gupta A, Strike P, Murphy VJ, Anders RF, Batchelor AH. Structure of AMA1 from Plasmodium falciparum reveals a clustering of polymorphisms that surround a conserved hydrophobic pocket. Proc Natl Acad USA. 2005;102:12736–12741. doi: 10.1073/pnas.0501808102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gupta A, Bai T, Murphy V, Strike P, Anders RF, Batchelor AH. Refolding, purification, and crystallization of apical membrane antigen 1 from Plasmodium falciparum. Protein Expr Purif. 2005;41:186–198. doi: 10.1016/j.pep.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 45.Pizarro JC, Vulliez-Le Normand B, Chesne-Seck ML, Collins CR, Withers-Martinez C, Hackett F, Blackman MJ, Faber BW, Remarque EJ, Kocken CH, Thomas AW, Bentley GA. Crystal structure of the malaria vaccine candidate apical membrane antigen 1. Science. 2005;308:408–411. doi: 10.1126/science.1107449. [DOI] [PubMed] [Google Scholar]
- 46.Crawford J, Tonkin ML, Grujic O, Boulanger MJ. Structural characterization of apical membrane antigen 1 (AMA1) from Toxoplasma gondii. J Biol Chem. 2010;285:15644–15652. doi: 10.1074/jbc.M109.092619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Coley AM, Parisi K, Masciantonio R, Hoeck J, Casey JL, Murphy VJ, Harris KS, Batchelor AH, Anders RF, Foley M. The most polymorphic residue on Plasmodium falciparum apical membrane antigen 1 determines binding of an invasion-inhibitory antibody. Infect Immun. 2006;74:2628–2636. doi: 10.1128/IAI.74.5.2628-2636.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harris KS, Casey JL, Coley AM, Masciantonio R, Sabo JK, Keizer DW, Lee EF, McMahon A, Norton RS, Anders RF, Foley M. Binding hot spot for invasion inhibitory molecules on Plasmodium falciparum apical membrane antigen 1. Infect Immun. 2005;73:6981–6989. doi: 10.1128/IAI.73.10.6981-6989.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Coley AM, Gupta A, Murphy VJ, Bai T, Kim H, Foley M, Anders RF, Batchelor AH. Structure of the malaria antigen AMA1 in complex with a growth-inhibitory antibody. PLoS Pathog. 2007;3:1308–1319. doi: 10.1371/journal.ppat.0030138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Henderson KA, Streltsov VA, Coley AM, Dolezal O, Hudson PJ, Batchelor AH, Gupta A, Bai T, Murphy VJ, Anders RF, Foley M, Nuttall SD. Structure of an IgNAR-AMA1 complex: targeting a conserved hydrophobic cleft broadens malarial strain recognition. Structure. 2007;15:1452–1466. doi: 10.1016/j.str.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 51.Richard D, MacRaild CA, Riglar DT, Chan JA, Foley M, Baum J, Ralph SA, Norton RS, Cowman AF. Interaction between Plasmodium falciparum apical membrane antigen 1 and the rhoptry neck protein complex defines a key step in the erythrocyte invasion process of malaria parasites. J Biol Chem. 2010;285:14815–14822. doi: 10.1074/jbc.M109.080770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Collins CR, Withers-Martinez C, Hackett F, Blackman MJ. An inhibitory antibody blocks interactions between components of the malarial invasion machinery. PLoS Pathog. 2009;5:e1000273. doi: 10.1371/journal.ppat.1000273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tyler JS, Boothroyd JC. The C-terminus of Toxoplasma RON2 provides the crucial link between AMA1 and the host-associated invasion complex. PLoS Pathog. 2011;7:e1001282. doi: 10.1371/journal.ppat.1001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Igonet S, Vulliez-Le Normand B, Faure G, Riottot MM, Kocken CH, Thomas AW, Bentley GA. Cross-reactivity studies of an anti-Plasmodium vivax apical membrane antigen 1 monoclonal antibody: binding and structural characterisation. J Mol Biol. 2007;366:1523–1537. doi: 10.1016/j.jmb.2006.12.028. [DOI] [PubMed] [Google Scholar]
- 55.Nair M, Hinds MG, Coley AM, Hodder AN, Foley M, Anders RF, Norton RS. Structure of domain III of the blood-stage malaria vaccine candidate, Plasmodium falciparum apical membrane antigen 1 (AMA1) J Mol Biol. 2002;322:741–753. doi: 10.1016/s0022-2836(02)00806-9. [DOI] [PubMed] [Google Scholar]
- 56.Hodder AN, Crewther PE, Matthew ML, Reid GE, Moritz RL, Simpson RJ, Anders RF. The disulfide bond structure of Plasmodium apical membrane antigen-1. J Biol Chem. 1996;271:29446–29452. doi: 10.1074/jbc.271.46.29446. [DOI] [PubMed] [Google Scholar]
- 57.Holm L, Sander C. Mapping the protein universe. Science. 1996;273:595–603. doi: 10.1126/science.273.5275.595. [DOI] [PubMed] [Google Scholar]
- 58.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 59.Fraser TS, Kappe SH, Narum DL, VanBuskirk KM, Adams JH. Erythrocyte-binding activity of Plasmodium yoelii apical membrane antigen-1 expressed on the surface of transfected COS-7 cells. Mol Biochem Parasitol. 2001;117:49–59. doi: 10.1016/s0166-6851(01)00326-7. [DOI] [PubMed] [Google Scholar]
- 60.Cowman AF, Crabb BS. Invasion of red blood cells by malaria parasites. Cell. 2006;124:755–766. doi: 10.1016/j.cell.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 61.Sun Y, Moreau E, Chauvin A, Malandrin L. The invasion process of bovine erythrocyte by Babesia divergens: knowledge from an in vitro assay. Vet Res. 2011;42:62. doi: 10.1186/1297-9716-42-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kumar S, Nei M, Dudley J, Tamura K. MEGA: a biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief Bioinform. 2008;9:299–306. doi: 10.1093/bib/bbn017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 64.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gouet P, Courcelle E, Stuart DI, Metoz F. ESPript: multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- 66.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Leslie AGW. Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4 + ESF-EAMCB Newsletter on Protein Crystallography. 1992:26. [Google Scholar]
- 68.Evans PR. Scaling and assessment of data quality. Acta Crystallogr Sect D. 2005;62:72–78. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 69.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Overview of the CCP4 suite and current developments. Acta Crystallogr Sect D. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schwarzenbacher R, Godzik A, Grzechnik SK, Jaroszewski L. The importance of alignment accuracy for molecular replacement. Acta Crystallogr Sect D. 2004;60:1229–1236. doi: 10.1107/S0907444904010145. [DOI] [PubMed] [Google Scholar]
- 72.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr Sect D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 73.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr Sect D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]