

Abstract
This review provides an overview of the structure, function, and catalytic mechanism of lacZ β-galactosidase. The protein played a central role in Jacob and Monod's development of the operon model for the regulation of gene expression. Determination of the crystal structure made it possible to understand why deletion of certain residues toward the amino-terminus not only caused the full enzyme tetramer to dissociate into dimers but also abolished activity. It was also possible to rationalize α-complementation, in which addition to the inactive dimers of peptides containing the “missing” N-terminal residues restored catalytic activity. The enzyme is well known to signal its presence by hydrolyzing X-gal to produce a blue product. That this reaction takes place in crystals of the protein confirms that the X-ray structure represents an active conformation. Individual tetramers of β-galactosidase have been measured to catalyze 38,500 ± 900 reactions per minute. Extensive kinetic, biochemical, mutagenic, and crystallographic analyses have made it possible to develop a presumed mechanism of action. Substrate initially binds near the top of the active site but then moves deeper for reaction. The first catalytic step (called galactosylation) is a nucleophilic displacement by Glu537 to form a covalent bond with galactose. This is initiated by proton donation by Glu461. The second displacement (degalactosylation) by water or an acceptor is initiated by proton abstraction by Glu461. Both of these displacements occur via planar oxocarbenium ion-like transition states. The acceptor reaction with glucose is important for the formation of allolactose, the natural inducer of the lac operon.
Keywords: lactose, allolactose, glucose, X-gal, α-complementation, binding, mechanism
Introduction
β-Galactosidase [3.2.1.23] (Escherichia coli) has a special place in both the history and the practice of molecular biology. It played a central role in Jacob and Monod's1 development of the operon model for the regulation of gene expression. Also, its ability to signal its presence by producing an easily recognizable blue reaction product has made it a workhorse in cloning and other such molecular biology procedures.
The purpose of this review is to provide an overview of the biochemical and other properties of lacZ β-galactosidase in light of the three-dimensional structure.
β-Galactosidase has three enzymatic activities (Fig. 1).2 First, it can cleave the disaccharide lactose to form glucose and galactose, which can then enter glycolysis. Second, the enzyme can catalyze the transgalactosylation of lactose to allolactose, and, third, the allolactose can be cleaved to the monosaccharides. It is allolactose that binds to lacZ repressor and creates the positive feedback loop that regulates the amount of β-galactosidase in the cell.
Figure 1.
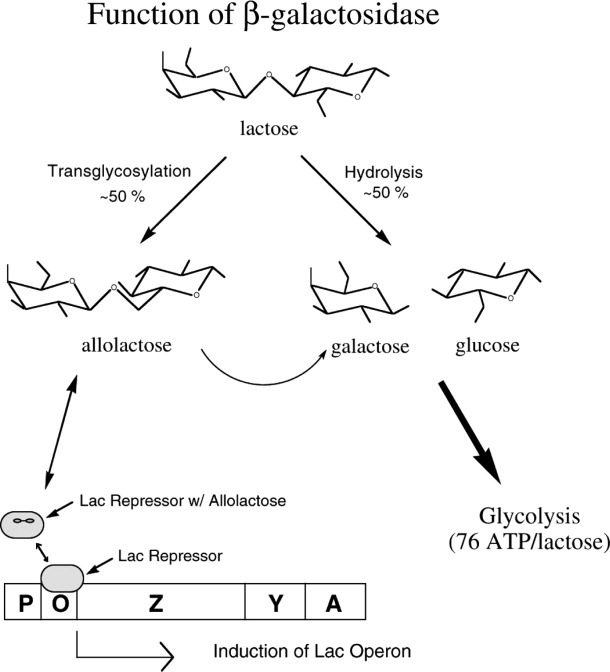
Schematic summarizing the roles of β-galactosidase in the cell. The enzyme can hydrolyze lactose to galactose plus glucose, it can transgalactosylate to form allolactose, and it can hydrolyze allolactose. The presence of lactose results in the synthesis of allolactose which binds to the lac repressor and reduces its affinity for the lac operon. This in turn allows the synthesis of β-galactosidase, the product of the lacZ gene.
In many respects, β-galactosidase is best recognized for its reaction with X-gal (5-bromo-4-chloro-3-indoyl-β-d-galactopyranoside), a soluble colorless compound consisting of galactose linked to a substituted indole. β-Galactose has high specificity for the galactose part of its substrates but low specificity for the remainder. Thus, it hydrolyzes X-gal, releasing the substituted indole that spontaneously dimerizes to give an insoluble, intensely blue product. On growth medium containing X-gal, colonies of E. coli that have an active β-galactosidase become blue because of this reaction.
As shown in Figure 2, the X-gal reaction can readily be performed in single crystals of the enzyme. β-Galactosidase, as proteins in general, forms crystals that include about 50% protein and 50% solvent by volume. The solvent-filled channels that extend throughout the crystal are much larger than the substrate and allow substrate to freely diffuse throughout the crystal. In early experiments on the nature of protein crystals, Wyckoff et al.3 used a flow cell to investigate the diffusion of ligands into a 0.4 mm crystal of ribonuclease S. When the concentration of ammonium sulfate surrounding the crystal was rapidly changed, the half-time for re-equilibration within the crystal was 90 s. Matthews4 used crystal density measurements to monitor the diffusion of ammonium sulfate solutions into crystals of γ-chymotrypsin and obtained very similar results. Based on these experiments, it can be estimated that a molecule the size of X-gal will diffuse through a 0.4 mm × 0.4 mm × 0.4 mm crystal of β-galactosidase in several minutes.
Figure 2.
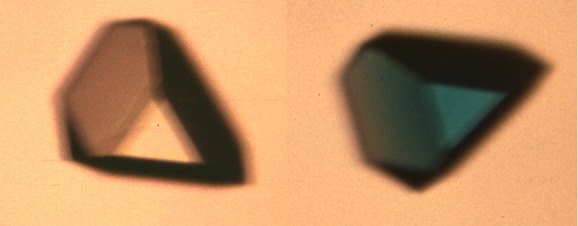
Demonstration that β-galactosidase in crystals is catalytically active. Crystal of β-galactosidase (orthorhombic; ca. 0.2 mm) in the absence (left) and in the presence, after about 2 h, of the substrate X-gal (right).
The blue color of a crystal of β-galactosidase exposed to X-gal (Fig. 2) confirms that the enzyme in the crystal is catalytically competent. It also tends to suggest, but does not prove, that catalysis proceeds via relatively modest changes in the conformation of the enzyme, that is, there is no suggestion of major structural changes which might destroy the crystals.
Structure of β-Galactosidase
β-Galactosidase is a tetramer of four identical polypeptide chains, each of 1023 amino acids.5, 6 The crystal structure was initially determined in a monoclinic crystal form with four tetramers in the asymmetric unit.7 Subsequently the structure was refined to 1.7 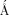 resolution in an orthorhombic crystal with a single tetramer in the asymmetric unit.8 The latter form is technically superior and has been used for subsequent structural and functional studies.
resolution in an orthorhombic crystal with a single tetramer in the asymmetric unit.8 The latter form is technically superior and has been used for subsequent structural and functional studies.
Within each monomer, the 1023 amino acids form five well-defined structural domains.7, 8 The third (central) domain (residues 334–627) is a so-called triose phosphate isomerase (TIM) or α8β8 barrel with the active site forming a deep pit at the C-terminal end of this barrel. As noted below, critical elements of the active site are also contributed by amino acids from elsewhere in the same polypeptide chain as well as from other chains within the tetramer.
The overall structure of the tetramer is illustrated in Figure 3(a) and in the associated interactive images.8 In Figure 3(a), there is one two-fold axis of symmetry that is horizontal, another that is vertical and a third that is perpendicular to the page. The horizontal two-fold axis forms the so-called “long” interface, while the vertical symmetry axis forms the “activating” interface.
Figure 3.
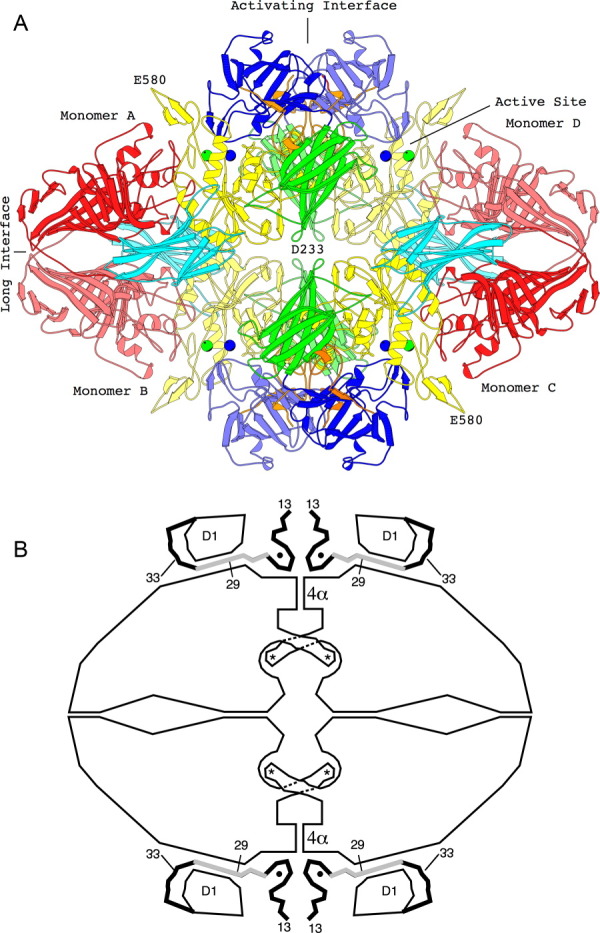
The tetrameric structure of β-galactosidase. (a) The backbone structure of the enzyme. Domain 1, blue; Domain 2, green; Domain 3, yellow; Domain 4, cyan; Domain 5, red. Lighter and darker shading is used to differentiate equivalent domains in different subunits. Metal ions are shown as spheres, Na+, green; Mg++, blue. Interactive views are available in the electronic version of the article (see below). (b) Sketch of the tetramer, aligned as in (a) showing features particularly relevant to α-complementation. The four active sites (each highlighted with an asterisk) are located toward the center of the figure. In each case a loop including residues 272–288 extends from one subunit to complete the active site of a neighboring subunit. The “activation interface” extends vertically through the center of the tetramer. Part of the interface comprises a bundle of four α-helices labeled 4α. Residues 13–50, shown as thick lines, pass through a tunnel between the first domain (labeled D1) and the rest of the protein. The region shaded gray (residues 23–31) is deleted in one of the α-donors. Magnesium ions (small solid circles) bridge between the complementation peptide and the rest of the protein (from Ref. 8). An interactive view is available in the electronic version of the article.
It has been suggested that β-galactosidase arose from a much simpler, single-domain TIM barrel enzyme that had an extended active-site cleft and could have cleaved extended oligosaccharides.9 The subsequent incorporation of additional domains could have reduced the size of the active-site cleft to a pocket commensurate with binding disaccharide substrates. Furthermore, some of these additional elements might promote the production of the inducer, allolactose.
α-Complementation
In early studies of β-galactosidase, it was found that deletion of certain residues near the amino-terminus such as 23–31 or 11–4110, 11 caused the tetrameric enzyme to dissociate into inactive dimers. Furthermore, by using peptides that included some or all the “missing” residues (e.g., 3–41 or 3–92), it was possible to reconstitute the active tetrameric form of the enzyme.12 This phenomenon of “α-complementation” is the basis for the common blue/white screening (with X-gal) used in cloning. It can now be rationalized in terms of the three-dimensional structure.
As can be seen in Figure 3(a) (see also the interactive image), residues from about 13 to 20 in adjacent subunits contact each other (at the bottom of the figure). A study13 of α-donors with substitutions showed that Glu17 is important for α-complementation. An equivalent interaction between the other two subunits occurs at the top of the figure. Removal of these residues weakens the vertical activating interface and the tetramer dissociates into dimers (or “α-acceptors”). At the same time, the residues that form the horizontal long interface are unchanged and allow the protein to remain as dimers. The dimers are somewhat unstable and tend to dissociate to monomers unless thiols and sufficient Na+ and Mg2+ are present. These additives stabilize the dimeric structure.14
When the complementation peptide (“α-donor”) is supplied to the α-acceptor, it binds at the site vacated by the removed N-terminal residues. Furthermore, α-donors that include residues 29–33 [Fig. 3(b)] can occupy a tunnel between Domain 1 and the rest of the protein. This will further stabilize the binding of the α-donor and help restore the tetrameric structure. As long as the N-terminal ∼41 amino acid residues are present, the length of the α-complementing peptide is not important. Even denatured whole wild type enzyme brings about complementation.15
The above transition between the tetrameric and dimeric states has major consequences for the activity of the enzyme. Although the active site is formed primarily by the TIM barrel of Domain 3, it includes critical catalytic residues of other domains. In particular, a loop from Domain 2 of Monomer A [Fig. 3(a)] extends across the activating interface to contribute to the active site of Monomer D. In total, there are four such interactions across the activating interface (A to D, D to A, B to C, and C to B) that form the four equivalent active sites per tetramer. Conversely, dissociation of the tetramer into the dimer disrupts all four active sites and completely abolishes the activity of the enzyme.
Activity of Individual β-Galactosidase Molecules
Craig and coworkers have developed a novel procedure to measure the activity of single β-galactosidase molecules.16, 17 It depends on the conversion of the weakly fluorescent substrate resorufin β-d-galactopyranoside to the highly fluorescent product resorufin. Individual β-galactosidase molecules produce several thousand product molecules per minute, and using a typical incubation of 15 min, the amount of product can be measured with an estimated error of about  15%.
15%.
The single-molecule activity measurements are performed using a purpose-designed capillary electrophoresis instrument that has a number of advantages. For example, a single protein molecule can be allowed to react with substrate for a desired period, and then moved away from the accumulated product into a new location, permitting a repeated measurement with the same protein molecule. Also, activity measurements for several protein molecules can be performed in a single experiment.
Molecules of β-galactosidase both before crystallization, and from dissolved crystals, displayed a range of activity of 20-fold or greater. The precrystallized protein had an overall activity distribution of 38,500  900 reactions per minute while that for molecules from a crystal was 31,600
900 reactions per minute while that for molecules from a crystal was 31,600  1100 reactions per minute.17
1100 reactions per minute.17
On one hand, it could be argued that the range of catalytic activities reflects oxidation or other such chemical modification of individual β-galactosidase molecules. Additional chemical modification during crystal growth could also explain why the crystallized proteins have slightly lower activity than those measured before crystallization.
On the other hand, the above scenario does not easily rationalize the observed distribution of activities.17 One might anticipate that there would be a relatively large population of “undamaged” β-galactosidase molecules with identical activities. The “damaged” molecules would then have a range of lower activities. This is not at all what is observed. As can be seen in Figure 3 of Shoemaker et al.,17 the bulk of the molecules (ca. 80%) have activities between 11,000 reactions/min and 50,000 reactions/min. A smaller number (ca. 20%) have activities that extend up to at least 100,000 reactions/min. The origin of these “superactive” molecules is not obvious. One possibility, discussed by Shoemaker et al.,17 is the presence of higher oligomeric forms of the enzyme. In this assay, a β-galactosidase octamer, for example, with eight active sites, would still be counted as a single molecule. On the other hand, if some molecules were tetramers and others octamers, these would be expected to give two distinct activity values.
Specificity
β-Galactosidase catalyzes reactions with β-d-galactopyranosides with an oxygen glycosidic bond.18, 19 The enzyme also reacts with substrates having other glycosidic linkages, including nitrogen,20 sulfur and fluorine, but with much reduced catalytic efficiency.21
The enzyme is very specific for d-galactose22–24 and the 2, 3, and 4 positions are especially important. The hydroxyls at those positions must each be present and in the correct orientation for the enzyme to catalyze the reaction. Studies25 with fluorinated and deoxy glycosides demonstrated that the fraction of binding energy released as a result of interactions at the 2 position was much larger than the fraction contributed from interactions with the other galactosyl hydroxyls. Reversion (reverse) β-galactosidase reactions26 done at very high concentrations of sugars having orientation changes and/or the absence of individual hydroxyls at different positions showed that only d-galactopyranose, l-arabinopyranose, d-fucopyranose, and d-galactal reacted in the reverse direction when d-glucose was the other reactant. The enzyme also hydrolyzes p-nitrophenyl-α-l-arabinopyranoside and p-nitrophenyl-β-d-fucopyranoside (which do not have O6 hydroxyls), but these substrates bind poorly and react slowly.27 Thus, sugars with modifications at the C6 hydroxyl position of d-galactose are still substrates, albeit poor ones—but sugars with changes elsewhere (except d-galactal) are unreactive. d-Galactal reacts because it forms a relatively stable covalent intermediate when Glu537 reacts with the C1 with a simultaneous proton addition at the C2 position.28, 29 This results in the formation of covalently attached 2-deoxy-galactose that is released on hydrolysis. In the reversion reaction26 at high concentrations of d-galactal and glucose, the covalent entity reacts with glucose.
Lactose is probably the natural substrate of β-galactosidase,30 but the enzyme is promiscuous for the nongalactose part of the substrate. Many different aglycones are tolerated. One example already mentioned is X-gal (Fig. 2). One of two substrates—o-nitrophenyl-β-d-galactopyranoside (oNPG) or p-nitrophenyl-β-d-galactopyranoside (pNPG)—is almost always used for routine assay of the enzyme. The nitrophenol products released on hydrolysis of these substrates are usually measured at 420 nm, and assays are rapid and straightforward at pH 7 (the optimal pH for the assay). As these two substrates have different rate-determining consequences, it is useful to study the kinetics of both in mechanistic studies. Assays of β-galactosidase reactions with lactose, best done with gas–liquid chromatography,2 are not routinely performed because the assay is technically cumbersome, requires scrutiny of the production of galactose, glucose, and allolactose, and must also account for the effects of these products as transgalactosidic acceptors and for allolactose being a substrate. Even lactose itself is a transgalactosidic acceptor. As the assays are much simpler and more accurate (because they are more sensitive) with pNPG than lactose, since the rate-determining step with pNPG as the substrate is the same as that with lactose and as the kinetic effects of varying assay conditions (pH, salt, metal, etc.) with lactose and pNPG are also very similar,2 it is much simpler to determine the effects of changes in the enzyme or assay mixtures using pNPG than lactose unless specific information about the reaction with lactose is needed.
Overview of the Enzymatic Reaction
Figure 4 shows the general overall reaction scheme. This scheme includes the movement of the substrate from a binding position (Es•Gal-OR) called the “shallow” mode (subscript s) to another position (Ed•Gal-OR) called the “deep” mode (subscript d). Binding in the “shallow” and “deep” modes are illustrated, respectively, in Figure 5(a,b) [see also Fig. 5(c) and the associated interactive image].
Figure 4.
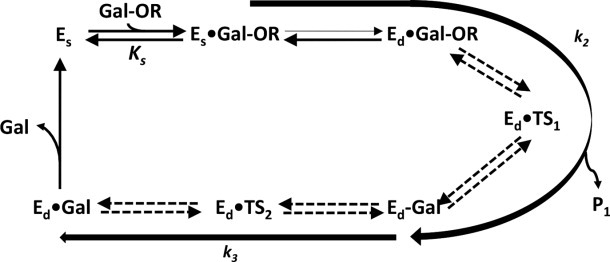
Overall reaction scheme for β-galactosidase. A galactoside (Gal-OR) binds to free enzyme (Es) initially in a shallow mode (Es•Gal-OR) which progresses to a deep mode (Ed•Gal-OR). The first chemical step (galactosylation, k2) is shown here to begin with Es•Gal-OR and follows through the transition state Ed•TS1 to a covalent intermediate, Ed-Gal, normally releasing the first product HOR (not shown). The intermediate is released to water (degalactosylation, k3) through the second transition state Ed•TS2 to a product complex Ed•Gal, finally releasing the second product, galactose. In the case of allolactose production, the first product (glucose) is not released and acts as the acceptor for the degalactosylation reaction, releasing the disaccharide, allolactose.
Figure 5.
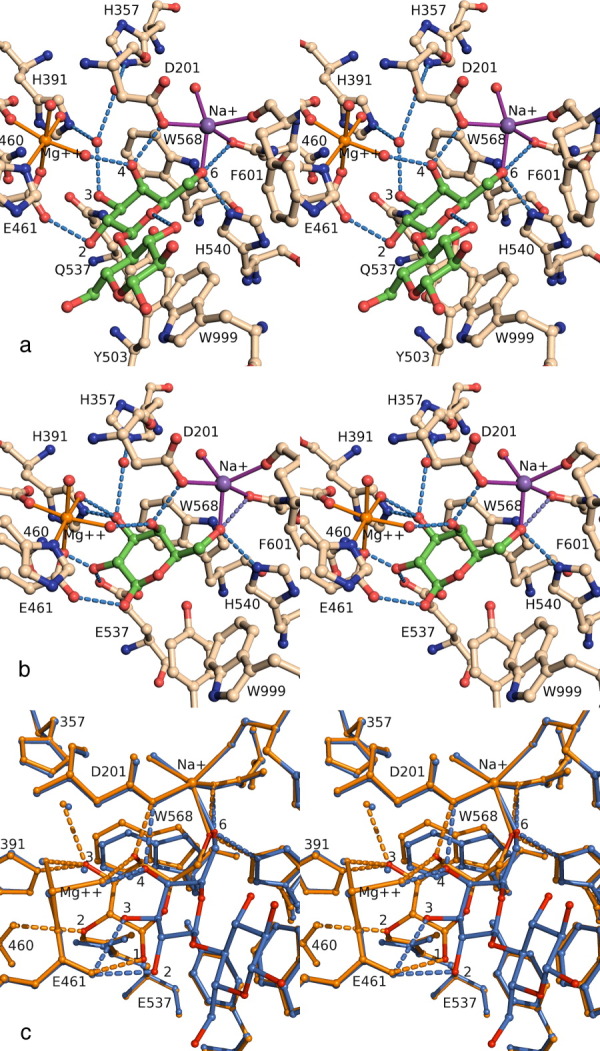
(a) Shallow mode binding illustrated by a stereoview of active site complex between E537Q β-galactosidase and lactose (PDB code 1jyn). H-bonds less than 3.0 Å involving the lactose molecule or key water molecules are shown as dashed lines. Protein residues are shown with wheat colored carbons and the ligand with green colored carbons. Figures 5 and 6 were prepared with PYMOL.31 (b) Deep mode binding of substrate illustrated by a stereoview of the active site complex between native enzyme and galactose (1jz7). (c) Comparison of shallow mode and deep mode binding. The complexes shown are E537Q/lactose (1jyn, shallow, in blue) and native/galactose (1jz7 deep, in orange). Progression from shallow to deep involves rotation of the galactosyl moiety. The 6- and 4-hydroxyls maintain their interactions, but the other hydroxyls shift. The 3-hydroxyl moves to occupy the position formerly occupied by a water molecule. An interactive view is available in the electronic version of the article.
Ed•TS1 and Ed•TS2 (Fig. 4) are the two transition states. They exist only in the deep mode. The first transition state forms after Glu461 donates a proton to the galactosidic oxygen and probably includes a partial bond to Glu537 (the nucleophile) and one to Glu461 (whilst it donates a proton). The second transition state also has the galactosyl moiety partially linked to Glu537 and water (or an acceptor) is involved. The Glu461 carboxyl acts as a base catalyst.
The dashed arrows in Figure 4 from Ed•Gal-OR to Ed•Gal represent the catalytic interactions. These reactions are shown as being reversible because the overall reaction can take place in the reverse direction,26 if large amounts of product are present. The rate constant k2 represents reactions encompassing the enzyme forms Es•Gal-OR to Ed-Gal. Because galactose becomes covalently linked to the enzyme (via Glu537), this catalytic step is called “galactosylation.” The rate constant k3 represents the catalytic step comprising the enzyme forms from Ed-Gal to Ed•Gal and is called “degalactosylation” because the covalent bond between galactose and Glu537 is broken. If a water is involved in the degalactosylation step, galactose is formed. Acceptors other than water can react to form galactosyl adducts. Such reactions are called “transgalactosylation.” When d-glucose is the acceptor, allolactose, the natural inducer of the lacZ operon is formed. When lactose is the substrate, allolactose forms intramolecularly with the glucose bond involving the O6 rather than the O4.2
Metal Requirements
β-Galactosidase requires Na+ or K+18, 19, 32–35 and Mg2+19, 27, 36–43 to be fully active [Fig. 5(a)]. The monovalent and divalent cations are important for both binding and reactivity, but the dependence on these cations is not absolute as there is some residual activity in their absence.
The ligands [Fig. 5(a)] coordinating with Na+ or K+ are the carboxyl of Asp201, the peptide oxygen of Phe601 and the side-chain oxygen of Asn604, and between 1 and 3 waters. In addition, Tyr100 interacts with Asp201 and may also be important. It is of interest that the π electron cloud of the benzyl group of Phe601 also becomes a “ligand” of the monovalent cation during the reaction. In addition, the O6 hydroxyl8 of galactose replaces one of the waters whenever substrate, transition states, or the covalent intermediate are at the active site. Interactions such as this, between the monovalent ion and a hydroxyl, seem to be unique to β-galactosidases. The functions of monovalent cations at the active sites of all other enzymes that are known to have monovalent metal requirements44–49 either help neutralize the negative charge of substrate phosphate groups, often in conjunction with a divalent cation, or they have structural importance, but in this case, Na+ interacts with a hydroxyl group.
The selectivity of β-galactosidase for Na+ or K+ and the roles of these ions derive mainly from the size dissimilarity between the two ions that manifests itself in a coordination number (CN) of either 5 or 6. Na+ is more likely to have a CN of 5 while K+ is more likely to have a CN of 6 because the ionic radius of Na+ is smaller than that of K+. The monovalent cation affects the affinity of substrates, the stability of the transition states, and the stability of the covalent intermediate and is part of a “pivot point” for the movement of substrate at the active site during the reaction.
Although several Mg2+ bind to each subunit,8, 29 only two directly affect activity. Some of the others may have structural importance. The Mg2+ that binds nearest to the substrate binding site (active site Mg2+) has the largest effect on the activity and will be the only one discussed in detail. The Kd for this Mg2+ is small (ca. 10−7 M). The second Mg2+, which interacts with Glu797, stabilizes a mobile loop at the active site.50 It does not bind as well (Kd ca. 10−4 M), and its effect on activity is smaller. The active site Mg2+ can be replaced by Mn2+ with only small differences in activity. The ligands for the active site Mg2+ are Glu416, His418, Glu461, and three waters [Fig. 5(a)]. One of these waters interacts with the O4 hydroxyl and is important for binding substrate in both the shallow and deep modes and for stabilizing the transition states and covalent intermediate. Another of the waters interacts with the O3 hydroxyl of the substrate when it is in position to react (deep mode), as well as with the transition states and the covalent intermediate.
Another role of the active site Mg2+ is to modulate the chemistry of active site components. Of these components, its effect on Glu461 is probably most important because this residue, a ligand to the Mg2+, plays several roles in the enzyme reaction. The active site Mg2+ does not seem to be very important for structure.51 The role of the second Mg2+ that modulates activity seems to be related to transition state stabilization.50
The Na+ site and active site Mg2+ site are in relative close proximity, separated by 8 Å [Fig. 5(a)] and linked via His418, Val103, Asn102, and Asp201.The affinity of the protein for one ion seems to be affected by the other.52
The Active-Site Loop
Langridge et al.53–55 created mutants of E. coli having β-galactosidases with high activity with some substrates. Later, a mutant having a β-galactosidase with similar properties was created56 that had an Asp replacing Gly794. The activity was not increased with all substrates—only those for which the first catalytic step (galactosylation) was rate determining. Further studies57 showed that β-galactosidases with several different substitutions for Gly794 had similar properties. The structure of the enzyme8 revealed that Gly794 is a “hinge” at one end of a loop near to the active site [Fig. 6(a)]. The loop (residues Gly794 to Pro803) assumes one of two positions (“open” and “closed” based on whether or not it closes onto the active site). It is open when the enzyme has no ligands bound and when substrate is bound. It is closed when some (not all) transition state analogs are bound. It is partly closed when the covalent intermediate is present.29
Figure 6.
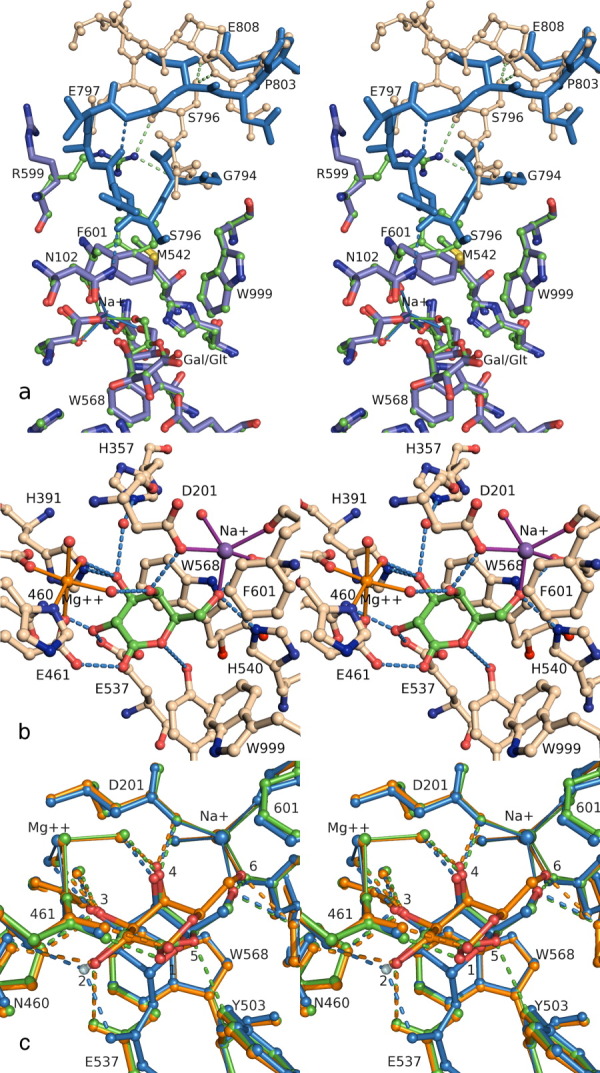
(a) Loop switching. Stereoview comparing the galactose complex (ball-and-stick) to the lactone complex (stick only), emphasizing the conformation changes in Arg599, Phe601, and the Gly794-Pro803 loop. In the galactose complex (1jz7), the loop is in the open conformation (wheat ball-and-stick), with H-bonds to Arg599 and intraloop H-bonds to Ser796. In the lactone complex (1jz5), the ligand is deeper in the active site, Phe601 closes toward the Na+, and the loop follows (blue sticks), which breaks the Arg599 and Ser796 H-bonds and creates a new H-bond to Asn102, and a new intraloop H-bond. The side chain for Arg800 has been omitted for clarity. (b) Transition state analog. Stereoview of active site complex between native enzyme and galactonolactone (1jz5). (c) Comparison of the binding modes for galactose (orange), galactonolactone (green) and a 2-F-galactosyl-enzyme intermediate (blue). All complexes are with native enzyme (1jz7, 1jz5, and 1jz4). Each hydroxyl makes similar interactions in the three different complexes, with the greatest differences at the 6-OH, 2-OH, ring oxygen, and C5 positions. Progressing through the three complexes, the galactosyl ring is deeper in the active site, and Phe601 is in the closed conformation with the lactone and the intermediate. An interactive view is available in the electronic version of the article.
Examination of Ramachandran plots showed that when the loop is in the open position, Gly794 is in a conformation that would be unfavorable for any non-Gly residue but becomes favorable for non-Gly residues when the loop closes. When Gly794 is replaced by Ala, the loop prefers the closed conformation, as anticipated.58 Kinetic studies indicated that this mutant enzyme binds substrates and substrate analogs poorly but transition state analogs well. The rate of the first catalytic step (galactosylation (k2)) increased while the rate of the second catalytic step (degalactosylation (k3)) decreased. As a result, the overall rates of reaction for substrates with k2 values much smaller than k3 increase whereas the rates of reactions decrease for substrates with small k3 values compared with k2.
Phe601, Met542, Arg599, and Glu808 play specific roles in the opening and closing of the loop59–62 [Fig. 6(a)]. One face of the benzyl side-chain of Phe601 forms a hydrophobic interaction with Met542 when the loop is open,59 while the edge of the Phe601 benzyl ring60 interacts with the positively charged guanidinium group of Arg599 via a cation-π interaction.50–52 In addition, the Arg599 side-chain forms an electrostatic interaction with the Glu797 side-chain of the loop and also has H-bonds with the peptide carbonyls of the loop residues Gly794 and Ser796. Also, there are H-bond interactions between the hydroxyl of Ser796 and both the carboxyl of Glu808 and the peptide nitrogen of Asp802. These interactions stabilize the open loop form.
When the loop closes, the Phe601 side-chain moves across the thiomethyl group of Met542 and the edge rather than the face of the benzyl group then interacts with the thiomethyl group.59 The face of Phe601 that was interacting with Met542 then interacts with the monovalent cation via a cation-π interaction. The Arg599 side-chain moves out of the way and its guanidinium group becomes mainly disordered, no longer interacting with Phe601, Glu797, or the carbonyls of Gly794 and Ser796. Because Gly794, Ser796, and Glu797 are no longer anchored by the Arg599 side-chain, the loop closes. Furthermore, in the closed state, the side-chain of Ser796 loses its interactions with Glu808 and Asp802. The Ser796 side chain forms hydrophobic interactions and becomes positioned on the side of Phe601 benzyl ring opposite to that interacting with the monovalent cation. The benzyl ring is then essentially sandwiched between the monovalent cation and the Ser796 side-chain.
Any substitutions for residues of the loop or the important residues in the vicinity of the loop61–63 that cause the closed conformation to be preferred, result in enzymes that bind substrates and substrate analogs less well and transition state analogs better. Substitutions that favor the open-state conformation cause substrates and substrate analogs to bind better and transition state analogs worse.
It is not yet clear what triggers cause the opening/closing of the loop. Phe601 closure is necessary but does not guarantee loop closure, while Phe601 closure seems to be simultaneous with movement of the substrate deeper into the active site. Present studies indicate that loop function seems to be related to the formation of allolactose but further analysis is needed.
Initial Substrate Binding
Substrate initially binds in the active site in the shallow mode, not totally buried29 [Fig. 5(a)]. The dissociation constant, Ks, is associated with this interaction (Fig. 4). The active site loop and Phe601 are open both before and after substrate binding but the open form of the loop and of Phe601 are defined a little better than the closed form by electron density when substrate is bound.63 This indicates that the loop is not fully open until substrate is bound.
The substrate in the shallow mode takes up a position roughly parallel to Trp999 with the galactosidic oxygen more or less centered over the indole8, 29, 64 [Fig. 5(a)]. The distances between Trp999 and most of the galactosyl and glucosyl carbons are in the van der Waals range.65 In these cases, the sides of the sugars having hydrogens rather than hydroxyls face towards the indole. These hydrogens are thought to have partial positive charges induced by electron pull by the hydroxyl groups on the opposite side. These partially charged hydrogens interact with the π electron cloud of the Trp999 indole. Hydrophobic aglycones of synthetic substrates were shown some time ago to bind strongly,66 and it is now seen that they interact with Trp999.29 In addition to the interaction with Trp999, there are specific bonds to each of the hydroxyls of the galactosyl component of the substrate in the shallow site. Glu461 forms an H-bond (∼2.6Å) with the C2 hydroxyl while Glu537 is close enough (∼3.1 Å) to form an H-bond with the C3 hydroxyl. Asn460 forms an indirect interaction with the C3 hydroxyl (via a water).67 The C4 hydroxyl of the galactosyl portion of the substrate interacts with Asp201 and with a water molecule ligated by the active site Mg2+. Asn604 interacts with the C6 hydroxyl. The C6 hydroxyl also seems to have strong interactions with both Na+29, 33 and His54068 and there is a hydrophobic interaction between the C6 and the benzyl group of Phe601. Except for contacts with Trp999, there are no specific bonds with the glucose portion of lactose, although there is an intralactose 2.9Å hydrogen bond between the C3 glucosyl hydroxyl and the galactosyl ring oxygen. The o-nitro group of oNPG interacts with Trp999 but also forms an interaction with His418.29 There is, however, no bond between the p-nitro group of pNPG and His418. Despite the extra bond, oNPG binds less well than pNPG because some of the intrinsic binding energy cannot be accessed.69 This interaction has significant effects on the galactosylation rate and the oNPG reaction rate is considerably different from the pNPG rate.
Movement of Substrate from the Shallow to the Deep Site
When the substrate is in the shallow site [Fig. 5(a)], it is bound preproductively and is not in the catalytically correct juxtaposition to Glu461 and Glu537. The substrate has to move about 3 Å deeper into the active site (Ed•Gal-OR) for catalysis to begin. The roles of Glu461 and Glu537 as the acid catalyst and the nucleophile, respectively, have been well established by chemical modification,70, 71 site-directed mutagenesis,72–74 and structural studies.29 Deep-site binding is visualized by the binding of galactose to the free enzyme,29 in which the galactose stacks with Trp568 [Fig. 5(b)]. Galactose is a product of the reaction but is also a substrate in the reverse direction.26 The evidence that it binds like a substrate is that the loop, Arg599 and Phe601 are in the same position as when substrate is present. Specifically, when galactose is in the deep site, Phe601 does not rotate, Arg599 still interacts with Glu797, Ser796, and Gly794, and the loop does not close [Fig. 6(a)]. Galactose binds in the deep mode because it does not have a glucose or a hydrophobic aglycone to restrict it from readily leaving the shallow mode. Normally lactose or the synthetic substrates are constrained (presumably by Trp999) and would require energy to shift to the deep site. The Ki for galactose binding is high showing that binding to this site is not good (albeit galactose binds better to the deep site than it does to the shallow site as noted by the lack of any shallow mode electron density for a galactose).
Progression to the deep mode [Fig. 5(c)] involves a rotation about an axis connecting the galactosyl C4 and C6 hydroxyls, which maintain similar interactions in the two modes—the C4 hydroxyl interacts with Asp201 and an Mg2+ ligated water molecule while the C6 hydroxyl interacts with Na+, His540, and Asn604. The energies of these interactions in the two modes are, however, probably different. The other three hydroxyls undergo substantial changes in environment. The C3 hydroxyl displaces a water molecule to interact with His391 and two waters—one ligated to the active site Mg2+ and another ligated to His357 [Fig. 5(a–c)]. The C2 hydroxyl becomes close enough (<3.2 Å) to Asn460, Glu461, and Glu537 to form interactions. Of these three, the geometry is the most ideal for an H-bond to Asn460,67 although the geometry is also good for an H-bond to Glu537. The C1 hydroxyl now occupies the position of the shallow mode C2 hydroxyl, H-bonding with Glu461. As Glu461 is the acid catalyst and the C1 hydroxyl is in the β conformation and therefore is equivalent to the galactosidic oxygen of the substrate, this is expected.
It is of interest that early kinetic studies40, 75, 76 led to the conclusion that a protein conformation change occurs after substrate binds. It was postulated that the acid catalytic group must move into position before it can interact with the galactosidic oxygen. Such a conformation change does not occur. However, the step with substrate moving from the shallow nonproductive site (Es•Gal-OR) to the deep productive site (Ed•Gal-OR) so that the general acid (Glu461) contacts the galactosidic bond would be kinetically identical to a conformation change in which the acid group moves towards the substrate. Thus, the concept that there is a conformation change that moves the acid group into place was not too far off the mark.
There are other enzymes besides β-galactosidase that have two-stage binding mechanisms.77–79 This may be a reasonably common phenomenon.
Galactosylation (first transition state)
The activation energy needed for galactosylation (k2) may include the energy needed for the movement of the substrate from the shallow to the deep mode and that movement is included in the galactosylation step (k2) that is depicted in Figure 4. This movement has an unfavorable equilibrium as indicated by the lack of any electron density in the deep mode when E537Q β-galactosidase is incubated with substrates such as lactose, allolactose, or oNPG, or when native enzyme is incubated with substrate analogs such as IPTG. However, despite the unfavorable equilibrium, the shifting of substrate from the shallow to the deep position could be fast. The equilibrium may be unfavorable because movement back to the shallow mode is even faster. If the shifting is indeed rapid, the movement of the substrate from the shallow to the deep state would be kinetically irrelevant, and the rate would mainly depend on the activation energy needed to form the activated complex as the energy needed to transfer substrate from the shallow to the deep mode would be negligible in comparison. On the other hand, the shifting of substrate to the deep mode could be slow enough so that it does affect the reaction rate. The formation of the highly activated transition state would certainly still be a much more difficult process but the shifting of the substrate could be of significance. There is some experimental evidence consistent with movement to the deep mode being at least partially rate limiting. Isotopic substitution of the galactosidic oxygen of pNPG with O-18 affects catalysis, reducing kcat more than kcat/Km. One possible explanation for the smaller effect on kcat/Km is that the approach of the substrate to the Michaelis complex (the deep mode) is partially rate limiting.80 Similarly, solvent isotope effects on catalysis of pNPG suggest that proton transfer is part of the rate-limiting step for kcat but not for kcat/Km.81 Proton transfer from Glu461 is expected to be involved in the chemical step for galactosylation but not for movement to the deep mode. These isotope effects can be at least partially explained by a rate-determining bond cleavage for kcat, and rate-determining progression to the deep mode for kcat/Km.
The results of a theoretical study of the mechanism of β-galactosidase82 suggest that the galactose part of the substrate in the deep site is found in a “pretransition state” form with a 4H3 conformation. The report also suggests that the transition state is in a 4E conformation. The projections of bond lengths within the transition state are also of interest. It is predicted that the length of the galactosidic bond (from the anomeric carbon to the galactosidic oxygen) increases from 1.47 Å in the reactant to 2.25 Å in the first transition state. At the latter distance, the bond is almost broken. This is despite the fact that the study predicts that the proton of Glu461 is not totally transferred. The nucleophile (Glu537) also moves closer (from 3.01 Å in the enzyme substrate complex to 2.45 Å in the first transition state). The length of this bond is 1.53 Å in the covalent form.
If galactose binding in the deep site is indeed representative of the manner in which substrate binds in the deep mode, the lack of distortion noted for the galactose29 suggests that the transition state probably really only begins to form when Glu461 donates a proton to the galactosidic bond of the substrate in the deep site and that significant amounts of “pretransition state form” do not exist. As a result of the protonation, the oxygen of the scissile bond has a positive charge and attracts electrons from C1. This carbon with a partial positive charge and with partial sp2 hybridization in turn attracts electrons from the ring oxygen giving the galactosyl moiety oxo-carbenium ion characteristics. A partial double bond would then be present between the O5 and C1 atoms. This would impose an element of planarity on the pyranose ring, a likely feature of the transition state. For SN1 cleavage, stereoelectronic theories require that a ring oxygen lone pair be antiperiplanar to the bond that breaks,83 which necessitates some distortion away from a chair configuration. For SN2 cleavage, planarity is an inherent structural aspect of the mechanism. In either case, both galactonolactone and GTZ (transition state analogs) have planar properties because of partial double bonds between the O5 and C1 and between the N5 and C1, respectively. Their good binding as well as other transition state analog properties that they possess (see below) is evidence that the transition state has planar tendencies.
It is useful to provide evidence that d-galactonolactone [Fig. 6(b)] and GTZ are indeed legitimate transition state analog inhibitors and thus that their binding indicates the manner in which this enzyme stabilizes the transition state. The Ki of d-galactonolactone is ∼0.6 mM, much higher than expected of a transition state analog. The binding is, however, very strong when it is noted that δ-1,5-galactonolactone, the only structural isomer of d-galactonolactone that binds at the active site,29 is present in solution in such very small amounts that it is not detectable by NMR even in very concentrated d-galactonolactonesolutions.84 The γ-1,4-galactonolactone form overwhelmingly predominates. Thus, δ-1,5-galactonolactone must bind with very high affinity. GTZ binds very strongly to the enzyme when presented as a competitive inhibitor. Other evidence that these are good transition state analogs is that when β-galactosidases with substitutions for His357, His391, and Asn460 (residues that stabilize the transition state) were studied, the binding of these two transition state analogs was decreased more or less in parallel with the kcat/Km values of these substituted enzymes.67, 85–87 In addition, the C1 of these two transition state analogs have trigonal character as is expected of the transition state. To help neutralize the positive charge of the trigonal carbons in these transition state analogs, electrons are thought to move somewhat from the ring oxygen or nitrogen towards the C1 to form a partial double bond. Thus, these compounds have a partial planar structure. Again this is an expected property of an analog mimicking an oxocarbenium ion. In addition, the partial positive charge of the C1 of these two compounds is a property expected of the transition state, mimicking an oxocarbenium ion. Finally, they both bind well in the deep mode29 as expected for transition state analogs of this enzyme.10
If galactose binding is indicative of substrate binding in the deep mode while d-galactonolactone or GTZ are representative of the transition states, it is of interest that most of the H-bond interactions that stabilize the transition state are similar to those that hold galactose in the deep site [see Fig. 6(c) and the associated interactive image]. However, the Ki of galactose is about 25 mM while the binding of δ-1,5-galactonolactone and GTZ are orders of magnitude better. It has been suggested that active sites can be thought of as “designer solvents”88 that drive the development of the cognate transition state. One can think of the residues of the active site pocket as the solvent to partially explain the effects. The main differences in the positioning of galactose compared with galactonolactone and GTZ in the pocket [Fig. 6(c)] are in respect to Trp568, Tyr503, Phe601, and Na+. The C5 atoms of d-galactonolactone and GTZ are much closer (∼0.8 Å) to Trp568 than is the C5 atom of galactose while the O5 of d-galactonolactone is ∼1 Å closer to Tyr503 while the N5 of GTZ is about 0.5 Å closer. The O6 maintains its ligation to the Na+, but also shifts deeper into the active site as Ph601 swings closed to interact with Na+ and pack against the galactosyl C6 [Figs. 5(b) and 6(a,b)]. The transition state may form29 partly because the C5 atom is hydrophobically attracted to Trp568, because the O5 atom is attracted to Tyr503, because the C6 is attracted to Phe601 and because the O6 is better attracted to the Na+. Trp568, Tyr503, Phe601, and Na+ may be the components of the designer solvent that ‘dissolve’ the C5, O5, C6, and O6. C3 and C4 also edge 0.1–0.2 Å closer to Trp568. This occurs even though His391, His357, Asn460, Asp201, and the two waters attached to Mg2+ hold O2, O3, and O4 in place by roughly the same distances for both galactose and the transition state analogs. Asn460 seems to be especially important67 for interaction with the O2, although Glu537 probably also plays an important role. All the bonds with the transition state are obviously of importance. Possibly the bonds to the hydroxyls of C2, C3, and C4 are important to hold the rest of galactose in place while C5, O5, and C6 move. In addition to the above, the positive charge on the trigonal C1 group is close enough to interact with and be stabilized by the negative charge of Glu537. This interaction is at the same distance in galactose as in galactonolactone but the C1 would have very little charge in galactose compared with that in galactonolactone. As shall be seen below, the positive charge of the C1 is stabilized to such an extent that a covalent bond forms.
The Tyr503 interactions noted with galactonolactone and GTZ may be significant in relation to the transition states. The electrons of Tyr503 probably interact with the ring oxygen of the transition state and help “push” the ring oxygen electrons towards C1 and aid in the formation of a partial double bond between O5 and C1. This also implies that planarity is important. The electrostatic interaction with Tyr503 could be especially strong as it takes place after the substrate is in position with no water interactions. Electrostatic interactions are stronger in a less-polar environment. Furthermore, pucker in the sugar ring in both the deep mode galactose complex and the covalent intermediate seems to create suboptimal geometry for bonding between Tyr503 and the galactosyl ring oxygen. Planarity of the galactosyl group in the transition state would improve this geometry.
l-Ribose, a pentose, also binds in the deep mode. Although the furanose form of l-ribose is structurally similar to other deep mode inhibitors (e.g., d-galactonolactone) with hydroxyls in the same orientations and planarity in the sugar ring, it is the pyranose form of l-ribose that binds to the active site, in the 1C4 rather than the 4C1 chair configuration. l-Ribose does not bind as well as d-galactonolactone or d-galactotetrazole but relative to the binding of other pentapyranoses it has a very low Ki. In addition, its Ki value increases in proportion to the changes to kcat/Km when residues that are important for transition state stabilization are substituted.85, 87l-Ribose binding puts hydroxyls within 0.4 Å of the positions of the lactone C6, C4, and C3 hydroxyls and within 1 Å of the lactone C2 hydroxyl, as well as stacking a hydrophobic surface of the sugar on Trp568. Thus, l-ribose is able to bind with hydroxyls in four of the five deep mode hexapyranose hydroxyl positions. Because l-ribose is a pentapyranose, the fifth deep mode hydroxyl position (the C1 hydroxyl, contacting Glu461) is unoccupied. Furthermore, l-ribose only uses a hydroxyl group to ligate the Na+ rather than a hydroxymethyl group. The combination of the 1C4 configuration with a rotation of the ring by ∼45° relative to the other deep mode inhibitors, puts the l-ribose ring oxygen in a perfect position to make 2.9 Å polar contacts to both Tyr503 and Glu537. Thus, of the inhibitor complexes whose structures have been determined, l-ribose is the only one whose ring oxygen makes two enzyme contacts, and it seems to take the greatest advantage of the H-bonding possibilities provided by Glu537.
Covalent intermediate
As already implied, Glu537 is close to the C1 of the transition state and reacts to form a quasi-stable covalent bond. The physical presence of a covalent bond to Glu537 was first shown71 by reacting β-galactosidase with 2,4-dintrophenyl-2-F-β-d-galactopyranoside. 2,4-Dinitrophenol is a very good leaving group while the fluorine at the O2 position slows the degalactosylation reaction. 2-Fluoro-d-galactose is covalently bound to Glu537 as a normal chair in an α configuration.29 The enzyme is totally inactive upon substituting for Glu537.61 In general, the same interactions29 needed to stabilize the transition state also stabilize the covalent intermediate. The positions of the galactosyl hydroxyls and enzyme groups are very similar between the two complexes, with the exception of Glu537, which rotates slightly in response to forming the covalent bond. The interaction between Tyr503 and the galactosyl ring oxygen is less optimal than in the lactone complex, due to the pucker in the sugar.
d-Galactal also interacts with Glu537. d-Galactal was at first thought to bind as a competitive inhibitor because of its planar shape.89 However, it was shown that 2-deoxy-d-galactose forms when β-galactosidase is incubated with d-galactal90 and kinetic experiments28 also suggested that d-galactal reacts with the enzyme.
Before the studies mentioned here, kinetic findings20, 76, 91, 92 had also suggested that a covalent intermediate exists.
Degalactosylation (second transition state)
Water comes into position either when the galactose is in the covalent form or during the formation of the second oxocarbenium ion-like transition state (Ed•TS2). Glu-461 activates either the water or acceptor by general base catalysis. d-Galactose is formed when water reacts while galactosyl adducts form when acceptors are present. Secondary deuterium isotope effects for degalactosylation indicate greater sp2 hybridization in the transition state than the starting intermediate, while the dependence of the rate on the type of acceptor indicates that acceptors (including water) are an integral part of the second transition state. One would expect that greater precision in lining up the acceptor with the anomeric center is needed for the formation of the second transition state, which is supported experimentally by greater entropic requirements for the formation of the second transition state than the first.69 Flattening of the galactosyl ring from the chair conformation of the intermediate should improve the geometry of bonding to Tyr503. Theoretical studies of the second transition state82 indicate a similarity to the first. The distance between the anomeric carbon and the nucleophilic oxygen in the covalent intermediate is 1.53 Å, increasing to 2.25 Å in the second transition state. The attacking water is also 2.25 Å away—at the very edge of still being a bond. One proton of the water is 1.31 Å from the Glu461 carboxylic oxygen. In addition to its possible role in stabilizing the transition state, Tyr503 probably acts as an acid catalyst93, 94 to facilitate cleavage of the covalent bond.
Transgalactosidic reactions (allolactose formation)
Almost all alcohols and sugars can act as acceptors of galactose.2, 95–98 They react in place of water. Bis-tris, a component of the buffer, binds nonproductively in the putative “acceptor” site when the enzyme has 2-deoxy-d-galactose covalently bound. Although it was not well defined, a glucose was also visualized in this enzyme form29 when a high concentration of glucose was added. These findings suggest that the acceptor comes into position to react when the enzyme is in the covalent form.
The intramolecular galactosyl transfer reaction of β-galactosidase with lactose2 that produces allolactose is of physiological importance because allolactose is the natural lac operon inducer.99, 100 In that reaction, the β-1-4 linkage of lactose is broken and the C6 hydroxyl of the glucose acceptor reacts with the C1 of the galactose to form allolactose. After the 1–4 bond is broken, glucose takes up a position so the O6 hydroxyl interacts with the anomeric carbon of galactose with Glu461 being the base catalyst. About 50% of the lactose molecules that react with β-galactosidase are initially hydrolyzed while 50% form allolactose intramolecularly. A physiological experiment demonstrated the role of β-galactosidase in the production of the inducer inside E. coli cells. A null mutant of lacZ β-galactosidase can still produce lacY permease when grown with IPTG but not when grown with lactose.100 Thus, the processing of lactose to allolactose (catalyzed by β-galactosidase) is needed to produce the inducer inside the cell. Allolactose is itself a substrate of the enzyme97 and when reaction with lactose has run its course, essentially only galactose and glucose are present. Thus, allolactose is a transient intermediate of the overall reaction with lactose. The intramolecular reaction occurs with glucoses that do not leave the active site after the β-1-4 galactosidic bond is broken but intermolecular allolactose production can also take place with glucose that has been hydrolyzed and subsequently rebinds and reacts as a transgalactosidic acceptor.2
Glossary
Abbreviations
- Ed•Gal-OR
galactoside substrate (Gal-OR) bound to the free enzyme (E) in the “deep” mode (Ed)
- Es•Gal-OR
substrate bound in the “shallow” mode
- GTZ
galactotetrazole
- IPTG
isopropyl-β-d-1-thiogalactopyranoside
- lacZ
gene coding for β-galactosidase
- oNPG
o-nitrophenyl-β-d-galactopyranoside
- pNPG
p-nitrophenyl-β-d-galactopyranoside
- X-gal
5-bromo-4-chloro-indoyl-β-d-galactopyranoside
References
- 1.Jacob F, Monod J. Genetic regulatory mechanisms in the synthesis of proteins. J Mol Biol. 1961;3:318–356. doi: 10.1016/s0022-2836(61)80072-7. [DOI] [PubMed] [Google Scholar]
- 2.Huber RE, Kurz G, Wallenfels K. A quantitation of the factors which affect the hyrolase and trangalactosylase activities of β-galactosidase (E. coli) on lactose. Biochemistry. 1976;15:1994–2001. doi: 10.1021/bi00654a029. [DOI] [PubMed] [Google Scholar]
- 3.Wyckoff HW, Doscher M, Tsernoglou D, Inagami T, Johnson LN, Hardman KD, Allewell NM, Kelly DM, Richards FM. Design of a diffractometer and flow cell system for X-ray analysis of crystalline proteins with applications to the crystal chemistry of ribonuclease-S. J Mol Biol. 1967;27:563–578. doi: 10.1016/0022-2836(67)90059-9. [DOI] [PubMed] [Google Scholar]
- 4.Matthews BW. Determination of molecular weight from protein crystals. J Mol Biol. 1974;82:513–526. doi: 10.1016/0022-2836(74)90245-9. [DOI] [PubMed] [Google Scholar]
- 5.Fowler A, Zabin I. Amino acid sequence of β-galactosidase. XI. Peptide ordering procedures and the complete sequence. J Biol Chem. 1978;253:5521–5525. [PubMed] [Google Scholar]
- 6.Kalnins A, Otto K, Ruther U, Muller-Hill B. Sequence of the lacZ gene of Escherichia coli. EMBO J. 1983;2:593–597. doi: 10.1002/j.1460-2075.1983.tb01468.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jacobson RH, Zhang XJ, DuBose RF, Matthews BW. Three-dimensional structure of β-galactosidase from E. coli. Nature. 1994;369:761–766. doi: 10.1038/369761a0. [DOI] [PubMed] [Google Scholar]
- 8.Juers DH, Wigley RH, Zhang X, Huber RE, Tronrud DE, Matthews BW. High resolution refinement of β-galactosidase in a new crystal form reveals multiple metal binding sites and provides a structural basis for α-complementation. Protein Sci. 2000;9:1685–1699. doi: 10.1110/ps.9.9.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Juers DH, Huber RE, Matthews BW. Structural comparisons of TIM barrel proteins suggest functional and evolutionary relationships between β-galactosidase and other glycohydrolases. Protein Sci. 1999;8:122–136. doi: 10.1110/ps.8.1.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ullmann A. Complementation in β-galactosidase: from protein structure to genetic engineering. Bioessays. 1992;14:201–205. doi: 10.1002/bies.950140311. [DOI] [PubMed] [Google Scholar]
- 11.Ullmann A, Jacob F, Monod J. Characterization by in vitro complementation of a peptide corresponding to an operator-proximal segment of the β-galactosidase structural gene of Escherichia coli. J Mol Biol. 1967;24:339–343. doi: 10.1016/0022-2836(67)90341-5. [DOI] [PubMed] [Google Scholar]
- 12.Welply JK, Fowler AV, Zabin I. β-Galactosidase alpha-complementation; overlapping sequences. J Biol Chem. 1981;256:6804–6810. [PubMed] [Google Scholar]
- 13.Welply JK, Fowler A, Zabin I. β-Galactosidase α-complementation; effect of single amino acid substitutions. J Biol Chem. 1981;256:6811–6816. [PubMed] [Google Scholar]
- 14.Gallagher CN, Huber RE. Monomer-dimer equilibrium of uncomplemented M15 β-galactosidase from Escherichia coli. Biochemistry. 1997;36:1281–1286. doi: 10.1021/bi961347a. [DOI] [PubMed] [Google Scholar]
- 15.Gallagher CN, Huber RE. Studies of the M15 β-galactosidase complementation process. J Protein Chem. 1998;17:131–141. doi: 10.1023/a:1022579416300. [DOI] [PubMed] [Google Scholar]
- 16.Craig DB, Dovichi NJ. E. coli β-galactosidase is heterogeneous with respect to the activity of individual molecules. Can J Chem. 1998;76:623–626. [Google Scholar]
- 17.Shoemaker GK, Juers DH, Coombs JML, Matthews BW, Craig DB. Crystallization of β-galactosidase does not reduce the range of activity of individual molecules. Biochemistry. 2003;42:1707–1710. doi: 10.1021/bi0204138. [DOI] [PubMed] [Google Scholar]
- 18.Lederberg J. The beta-d-galactosidase of Escherichia coli, strain K-12. J Bacteriol. 1950;60:381–392. doi: 10.1128/jb.60.4.381-392.1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cohn M, Monod J. Purification et proprietes de la β-galactosidase (lactase) d'Echerichia coli. Biochim Biophys Acta. 1951;7:153–174. doi: 10.1016/0006-3002(51)90013-3. [DOI] [PubMed] [Google Scholar]
- 20.Sinnott ML, Withers SG. The β-galactosidase-catalysed hydrolyses of β-d-galactopyranosyl salts. Rate limiting generation of an enzyme-bound galactopyranosyl cation in a process dependent only on aglycone acidity. Biochem J. 1974;143:751–762. doi: 10.1042/bj1430751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wallenfels K, Weil R. Beta-galactosidase. In: Boyer P, editor. The enzymes VII. Academic Press, New York: 1972. [Google Scholar]
- 22.Huber RE, Gaunt MT. Importance of hydroxyls at positions 3, 4, and 6 for binding to the galactose site of β-galactosidase (Escherichia coli. Arch Biochem Biophys. 1983;220:263–271. doi: 10.1016/0003-9861(83)90409-5. [DOI] [PubMed] [Google Scholar]
- 23.Nam Shin JE, Maradufu A, Marion J, Perlin AS. Specificity of alpha- and β-d-galactosidase towards analogs of d-galactopyranosides modified at C-4 or C-5. Carbohydr Res. 1980;84:328–335. [Google Scholar]
- 24.Brockhaus M, Dettinger HM, Kurz G, Lehmann J, Wallenfels K. Participation of HO-2 in the cleavage of β-galactosides by the β-d-galactosidase from E. coli. Carbohydr Res. 1979;69:264–268. doi: 10.1016/s0008-6215(00)85773-0. [DOI] [PubMed] [Google Scholar]
- 25.McCarter JD, Adam MJ, Withers SG. Binding energy and catalysis. Fluorinated and deoxygenated glycosides as mechanistic probes of Escherichia coli (LacZ) beta-galactosidase. Biochem J. 1992;286:721–727. doi: 10.1042/bj2860721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huber RE, Hurlburt KL. Reversion reactions of β-galactosidase (Escherichia coli. Arch Biochem Biophys. 1986;246:411–418. doi: 10.1016/0003-9861(86)90487-x. [DOI] [PubMed] [Google Scholar]
- 27.Loeffler RS, Sinnott ML, Sykes BD, Withers SG. Interaction of the lac Z β-galactosidase of Escherichia coli with some β-d-galactopyranoside competitive inhibitors. Biochem J. 1979;177:145–152. doi: 10.1042/bj1770145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wentworth DF, Wolfenden R. Slow binding of d-galactal, a “reversible” inhibitor of bacterial β-galactosidase. Biochemistry. 1974;13:4715–4720. doi: 10.1021/bi00720a006. [DOI] [PubMed] [Google Scholar]
- 29.Juers DH, Heightman TD, Vasella A, McCarter JD, Mackenzie L, Withers SG, Matthews BW. A structural view of the action of Escherichia colilacZ) β-galactosidase. Biochemistry. 2001;40:14781–14794. doi: 10.1021/bi011727i. [DOI] [PubMed] [Google Scholar]
- 30.Egal R. The lac-operon for lactose degradation, or rather for the utilization of galactosylglycerols from galactolipids? J Theor Biol. 1979;79:117–119. doi: 10.1016/0022-5193(79)90260-1. [DOI] [PubMed] [Google Scholar]
- 31.DeLano WL. 2002. The PyMOL molecular graphics system on the World Wide Web. Available at: http://www.pymol.org, Jan. 4, 2011.
- 32.Neville MC, Ling GN. Synergistic activation of β-galactosidase by Na+ and Cs+ Arch Biochem Biophys. 1967;118:596–610. doi: 10.1016/0003-9861(67)90395-5. [DOI] [PubMed] [Google Scholar]
- 33.Xu J, McRae MA, Harron S, Rob B, Huber RE. A study of the relationship of interactions between Asp-201, Na+ or K+, and galactosyl C6-hydroxyl and their effects on binding and reactivity of β-galactosidase. Biochem Cell Biol. 2004;82:275–284. doi: 10.1139/o04-004. [DOI] [PubMed] [Google Scholar]
- 34.Wallenfels K, Malhotra OP, Dabich D. Untersuchungen uber milchsuckerspaltende enzyme. VIII Der einfluss des kationen-milieus auf die aktivitat der β-galaktosidase von E. coli ML 308. Biochemi Zeit. 1960;333:377–394. [PubMed] [Google Scholar]
- 35.Becker VE, Evans HJ. The influence of monovalent cations and hydrostatic pressure on β-galactosidase activity. Biochim Biophys Acta. 1969;191:95–104. doi: 10.1016/0005-2744(69)90318-0. [DOI] [PubMed] [Google Scholar]
- 36.Case GS, Sinnott ML. The role of magnesium ions in β-galactosidase-catalysed hydrolyses. Biochem J. 1973;133:99–104. doi: 10.1042/bj1330099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hill JA, Huber RE. Effects of various concentrations of Na+ and Mg2+ on the activity of β-galactosidase. Biochim Biophys Acta. 1971;250:530–537. doi: 10.1016/0005-2744(71)90253-1. [DOI] [PubMed] [Google Scholar]
- 38.Huber RE, Parfett C, Woulfe-Flanagan H, Thompson DJ. Interaction of divalent cations with β-galactosidase (Escherichia coli. Biochemistry. 1979;18:4090–4095. doi: 10.1021/bi00586a005. [DOI] [PubMed] [Google Scholar]
- 39.Sinnott ML, Viratelle OM, Withers SG. pH and magnesium ion-dependence of the hydrolysis of β-d-galactopyranosyl pyridinium salts catalysed by Escherichia coli β-galactosidase. Biochem Soc Trans. 1975;3:1006–1009. [Google Scholar]
- 40.Sinnott ML, Withers SG, Viratelle OM. The necessity of magnesium cation for acid assistance of aglycone departure in catalysis by Escherichia coli (lacZ) β-galactosidase. Biochem J. 1978;175:539–536. doi: 10.1042/bj1750539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tenu JP, Viratelle OM, Yon J. Kinetic study of the activation process of β-galactosidase from Escherichia coli by Mg2+ Eur J Biochem. 1972;26:112–118. doi: 10.1111/j.1432-1033.1972.tb01746.x. [DOI] [PubMed] [Google Scholar]
- 42.Reithel FJ, Kim JC. Studies on the β-galactosidase isolated from Escherichia coli ML308. I. The effect of some ions on enzymatic activity. Arch Biochem Biophys. 1960;90:271–277. doi: 10.1016/0003-9861(60)90579-8. [DOI] [PubMed] [Google Scholar]
- 43.Strom R, Attardi DG, Forsen S, Turini P, Celada F, Antonini E. The activation of β-galactosidase by divalent and monovalent cations. Eur J Biochem. 1971;23:118–124. doi: 10.1111/j.1432-1033.1971.tb01598.x. [DOI] [PubMed] [Google Scholar]
- 44.Flaherty KM, DeLuca-Flaherty C, McKay DB. Three-dimensional structure of the ATPase fragment of a 70K heat-shock cognate protein. Nature. 1990;346:623–628. doi: 10.1038/346623a0. [DOI] [PubMed] [Google Scholar]
- 45.Kayne FJ. Thallium (I) activation of pyruvate kinase. Arch Biochem Biophys. 1971;143:232–239. doi: 10.1016/0003-9861(71)90204-9. [DOI] [PubMed] [Google Scholar]
- 46.Larsen TM, Laughlin LT, Holden HM, Rayment I, Reed GH. Structure of rabbit muscle pyruvate kinase complexed with Mn2+, K+, and pyruvate. Biochemistry. 1994;33:6301–6309. doi: 10.1021/bi00186a033. [DOI] [PubMed] [Google Scholar]
- 47.O'Brien MC, McKay DB. How potassium affects the activity of the molecular chaperone Hsc70. I. Potassium is required for optimal ATPase activity. J Biol Chem. 1995;270:2247–2250. doi: 10.1074/jbc.270.5.2247. [DOI] [PubMed] [Google Scholar]
- 48.Villeret V, Huang S, Fromm HJ, Lipscomb WN. Crystallographic evidence for the action of potassium, thallium, and lithium ions on fructose-1,6-bisphosphatase. Proc Natl Acad Sci USA. 1995;92:8916–8920. doi: 10.1073/pnas.92.19.8916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wilbanks SM, McKay DB. How potassium affects the activity of the molecular chaperone Hsc70. II. Potassium binds specifically in the ATPase active site. J Biol Chem. 1995;270:2251–2257. doi: 10.1074/jbc.270.5.2251. [DOI] [PubMed] [Google Scholar]
- 50.Sutendra G, Wong S, Fraser ME, Huber RE. β-Galactosidase (Escherichia coli) has a second catalytically important Mg2+ site. Biochem Biophys Res Commun. 2007;352:566–570. doi: 10.1016/j.bbrc.2006.11.061. [DOI] [PubMed] [Google Scholar]
- 51.Lo S, Dugdale ML, Jeerh N, Ku T, Roth NJ, Huber RE. Studies of Glu-416 variants of β-Galactosidase (E. coli) show that the active site Mg2+ is not important for structure and indicate that the main role of Mg2+ is to mediate optimization of active site chemistry. Protein J. 2010;29:26–31. doi: 10.1007/s10930-009-9216-x. [DOI] [PubMed] [Google Scholar]
- 52.Juers DH, Rob B, Dugdale ML, Rahimzadeh N, Giang C, Lee M, Matthews BW, Huber RE. Direct and indirect roles of His-418 in metal binding and in the activity of β-galactosidase (E. coli. Protein Sci. 2009;18:1281–1292. doi: 10.1002/pro.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Langridge J. Genetic evidence for the disposition of the substrate binding site of β-galactosidase. Genetics. 1968;60:1260–1267. doi: 10.1073/pnas.60.4.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Langridge J. Mutations conferring quantitative and qualitative increases in β-galactosidase activity in Escherichia coli. Mol Gen Genet. 1969;105:74–83. doi: 10.1007/BF00750315. [DOI] [PubMed] [Google Scholar]
- 55.Langridge J, Campbell JH. Classification and intragenic position of mutations in the β-galactosidase gene of Escherichia coli. Mol Gen Genet. 1968;103:339–347. doi: 10.1007/BF00383484. [DOI] [PubMed] [Google Scholar]
- 56.Martinez-Bilbao M, Holdsworth RE, Edwards LA, Huber RE. A highly reactive β-galactosidase (Escherichia coli) resulting from a substitution of an aspartic acid for Gly-794. J Biol Chem. 1991;266:4979–4986. [PubMed] [Google Scholar]
- 57.Martinez-Bilbao M, Huber RE. Substitutions for Gly-794 show that binding interactions are important determinants of the catalytic action of β-galactosidase (Escherichia coli. Biochem Cell Biol. 1994;72:313–319. doi: 10.1139/o94-044. [DOI] [PubMed] [Google Scholar]
- 58.Juers DH, Hakda S, Matthews BW, Huber RE. Structural basis for the altered activity of Gly794 variants of Escherichia coli β-galactosidase. Biochemistry. 2003;42:13505–13511. doi: 10.1021/bi035506j. [DOI] [PubMed] [Google Scholar]
- 59.Dugdale ML, Dymianiw DL, Minhas BK, D'Angelo I, Huber RE. Role of Met-542 as a guide for the conformational changes of Phe-601 that occur during the reaction of β-galactosidase (E. coli. Biochem Cell Biol. 2010;88:861–869. doi: 10.1139/O10-009. [DOI] [PubMed] [Google Scholar]
- 60.Dugdale ML, Vance ML, Wheatley RW, Driedger MR, Nibber A, Tran A, Huber RE. The importance of Arg-599 of β-galactosidase (E. coli) as an anchor for the open conformations of Phe-601 and the active site loop. Biochem Cell Biol. 2010;88:969–979. doi: 10.1139/O10-144. [DOI] [PubMed] [Google Scholar]
- 61.Jancewicz L. Biological sciences, Ph.D thesis. Calgary: University of Calgary; 2008. The role of Ser-796 in the loop (residues 794–803) of β-galactosidase of Escherichia coli; p. 142. [Google Scholar]
- 62.Sutendra G. Biological sciences. Calgary: University of Calgary; 2005. The role of residues 795–802 of the loop with emphasis on Glu-797 and a second important Mg2+ site in the catalytic mechanism of β-galactosdiase in Escherichia coli; p. 166. [Google Scholar]
- 63.Jancewicz LJ, Wheatley RW, Sutendra G, Lee M, Fraser ME, Huber RE. Ser-796 of β-galactosidase (Escherichia coli) plays a key role in maintaining a balance between the opened and closed conformations of the catalytically important active site loop. Arch Biochem Biophys. 2012;517:111–122. doi: 10.1016/j.abb.2011.11.017. [DOI] [PubMed] [Google Scholar]
- 64.Huber RE, Hakda S, Cheng C, Cupples CG, Edwards RA. Trp-999 of β-galactosidase (Escherichia coli) is a key residue for binding, catalysis, and Synthesis of allolactose, the natural Lac operon inducer. Biochemistry. 2003;42:1796–1803. doi: 10.1021/bi0270642. [DOI] [PubMed] [Google Scholar]
- 65.Drickamer K. Making a fitting choice: common aspects of sugar-binding sites in plant and animal lectins. Structure. 1997;5:465–468. doi: 10.1016/s0969-2126(97)00202-5. [DOI] [PubMed] [Google Scholar]
- 66.De Bruyn CK, Yde M. Binding of alkyl 1-thio-β-d-galactopyranosides to β-d-galactosidase from E. coli. Carbohydr Res. 1977;56:153–164. doi: 10.1016/s0008-6215(00)84247-0. [DOI] [PubMed] [Google Scholar]
- 67.Wheatley RW, Kappelhoff JC, Hahn JN, Dugdale ML, Dutkoski MJ, Tamman SD, Fraser ME, Huber RE. Substitution for Asn460 cripples β-galactosidase (E. coli) by increasing substrate affinity and decreasing transition state stability. Arch Biochem Biophys. 2012;521:51–61. doi: 10.1016/j.abb.2012.03.014. [DOI] [PubMed] [Google Scholar]
- 68.Roth NJ, Huber RE. The beta-galactosidase (Escherichia coli) reaction is partly facilitated by interactions of His-540 with the C6 hydroxyl of galactose. J Biol Chem. 1996;271:14296–14301. doi: 10.1074/jbc.271.24.14296. [DOI] [PubMed] [Google Scholar]
- 69.Kappelhoff JC, Liu SYJ, Dugdale ML, Dymianiw DL, Linton LR, Huber RE. Practical considerations when using temperature to obtain rate constants and activation thermodynamics of enzymes with two catalytic steps: native and N460T-β-galactosidase (E. coli) as examples. Protein J. 2009;28:96–103. doi: 10.1007/s10930-009-9168-1. [DOI] [PubMed] [Google Scholar]
- 70.Herrchen M, Legler G. Identification of an essential carboxylate group at the active site of lacZ β-galactosidase from Escherichia coli. Eur J Biochem. 1984;138:527–531. doi: 10.1111/j.1432-1033.1984.tb07947.x. [DOI] [PubMed] [Google Scholar]
- 71.Gebler JC, Aebersold R, Withers SG. Glu-537, not Glu-461, is the nucleophile in the active site of (lacZ) β-galactosidase from Escherichia coli. J Biol Chem. 1992;267:11126–11130. [PubMed] [Google Scholar]
- 72.Cupples CG, Miller JH, Huber RE. Determination of the roles of Glu-461 in β-galactosidase (E. coli) using site-specific mutagenesis. J Biol Chem. 1990;265:5512–5518. [PubMed] [Google Scholar]
- 73.Huber RE, Chivers PT. β-Galactosidase of Escherichia coli with substitutions for Glu-461 can be activated by nucleophiles and can form β-d-galactosyl adducts. Carbohydr Res. 1993;250:9–18. doi: 10.1016/0008-6215(93)84150-5. [DOI] [PubMed] [Google Scholar]
- 74.Yuan J, Martinez-Bilbao M, Huber RE. Substitutions for Glu-537 of β-galactosidase from Escherichia coli cause large decreases in catalytic activity. Biochem J. 1994;299:527–531. doi: 10.1042/bj2990527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fink AL. Cryoenzymology: The study of enzyme mechanisms at subzero temperatures. Acc Chem Res. 1977;10:233–239. [Google Scholar]
- 76.Sinnott ML, Souchard IJL. The mechanism of action of β-galactosidase: effect of aglycone nature and alpha deuterium substitution on the hydrolysis of some β-d-galactopyranosides: rate limiting degalactosylation; the pH-dependence of galactosylation and degalactosylation. Biochem J. 1973;133:89–98. doi: 10.1042/bj1330089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gruninger RJ, Dobing S, Smith AD, Bruder LM, Selinger LB, Wieden H-J, Mosimann SC. Substrate binding in protein tyrosine phosphatase-like inositol polyphosphatases. J Biol Chem. 2012;287:9722–9730. doi: 10.1074/jbc.M111.309872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Isin EM, Guengerich FP. Kinetics and thermodynamics of ligand binding by cytochrome P450 3A4. J Biol Chem. 2006;281:9127–9136. doi: 10.1074/jbc.M511375200. [DOI] [PubMed] [Google Scholar]
- 79.Wang L-H, Tsai A-L, Hsu P-Y. Substrate binding is the rate-limiting step in thromboxane synthase catalysis. J Biol Chem. 2001;276:14737–14743. doi: 10.1074/jbc.M009177200. [DOI] [PubMed] [Google Scholar]
- 80.Rosenberg S, Kirsch JF. Oxygen-18 leaving group isotope effects on the hydrolysis of nitrophenyl glycosides. 1. β-galactosidase-catalyzed hydrolysis. Biochemistry. 1981;20:3189–3196. doi: 10.1021/bi00514a031. [DOI] [PubMed] [Google Scholar]
- 81.Selwood T, Sinnott M. A solvent-isotope-effect study of protein transfer during catalysis by Escherichia coli (lacZ) β-galactosidase. Biochem J. 1990;268:317–323. doi: 10.1042/bj2680317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bras NF, Fernandes PA, Ramos MJ. QM/MM studies on the β-galactosidase catalytic mechanism: hydrolysis and transglycosylation reactions. J Chem Theory Comput. 2010;6:421–433. doi: 10.1021/ct900530f. [DOI] [PubMed] [Google Scholar]
- 83.Deslongchamps P. Intramolecular strategies and stereoelectric effects—glycosides hydrolysis revisited. Pure Appl Chem. 1993;65:1161–1178. [Google Scholar]
- 84.Huber RE, Brockbank RL. Strong inhibitory effect of furanoses and sugar lactones on β-galactosidase of Escherichia coli. Biochemistry. 1987;26:1526–1531. doi: 10.1021/bi00380a005. [DOI] [PubMed] [Google Scholar]
- 85.Huber RE, Hlede IY, Roth NJ, McKenzie KC, Ghuman KK. His-391 of beta-galactosidase (Escherichia coli) promotes catalyses by strong interactions with the transition state. Biochem Cell Biol. 2001;79:183–193. doi: 10.1139/o00-101. [DOI] [PubMed] [Google Scholar]
- 86.Kappelhoff JC. Biological sciences, Ph.D. thesis. Calgary, AB: University of Calgary; 2005. The roles of Asn-460 in the active site of β-galactosidase from Escherichia coli; p. 188. [Google Scholar]
- 87.Roth NJ, Rob B, Huber RE. His-357 of beta-galactosidase (Escherichia coli) interacts with the C3 hydroxyl in the transition state and helps to mediate catalysis. Biochemistry. 1998;37:10099–10107. doi: 10.1021/bi972796t. [DOI] [PubMed] [Google Scholar]
- 88.Schramm VL. Enzyme transition state poise and transition state analogues. Acc Chem Res. 2003;36:588–596. doi: 10.1021/ar0200495. [DOI] [PubMed] [Google Scholar]
- 89.Lee YM. Inhibition of β-galactosidases by d-galactal. Biochem Biophys Res Commun. 1969;35:161–167. doi: 10.1016/0006-291x(69)90499-9. [DOI] [PubMed] [Google Scholar]
- 90.Lehmann J, Schroter E. Reaktionen enolisher zuckerderivate. VIII. Die wirkung von β-d-glucosidase and β-d-galaktosidase auf d-glucal und d-galactal. Carbohydr Res. 1972;23:359–368. doi: 10.1016/s0008-6215(00)82706-8. [DOI] [PubMed] [Google Scholar]
- 91.Fink AL, Angelides KJ. The β-galactosidase-catalyzed hydrolysis of o-nitrophenol-β-d-galactoside at subzero temperatures: evidence for a galactosyl-enzyme intermediate. Biochem Biophys Res Commun. 1975;64:701–708. doi: 10.1016/0006-291x(75)90377-0. [DOI] [PubMed] [Google Scholar]
- 92.Sinnott ML. Ions, ion-pairs and catalysis by the lacZ β-galactosidase of Escherichia coli. FEBS Lett. 1978;94:1–9. doi: 10.1016/0014-5793(78)80894-1. [DOI] [PubMed] [Google Scholar]
- 93.Penner RM, Roth NJ, Rob B, Lay H, Huber RE. Tyr-503 of beta-galactosidase (Escherichia coli) plays an important role in degalactosylation. Biochem Cell Biol. 1999;77:229–236. doi: 10.1139/o99-042. [DOI] [PubMed] [Google Scholar]
- 94.Roth NJ, Penner RM, Huber RE. Beta-galactosidases (Escherichia coli) with double substitutions show that Tyr-503 acts independently of Glu-461 but cooperatively with Glu-537. J Protein Chem. 2003;22:663–668. doi: 10.1023/b:jopc.0000008731.16884.22. [DOI] [PubMed] [Google Scholar]
- 95.Deschavanne PJ, Viratelle OM, Yon JM. Conformational adaptability of the active site of β-galactosidase. J Biol Chem. 1978;253:833–837. [PubMed] [Google Scholar]
- 96.Huber RE, Gaunt MT, Hurlburt KL. Binding and reactivity at the “glucose” site of galactosyl-beta-galactosidase (Escherichia coli. Arch Biochem Biophys. 1985;234:151–160. doi: 10.1016/0003-9861(84)90336-9. [DOI] [PubMed] [Google Scholar]
- 97.Huber RE, Wallenfels K, Kurz G. The action of β-galactosidase (Escherichia coli) on allolactose. Can J Biochem. 1975;53:1035–1038. doi: 10.1139/o75-142. [DOI] [PubMed] [Google Scholar]
- 98.Viratelle OM, Yon JM. Nucleophilic competition in some β-galactosidase reactions. Eur J Biochem. 1973;33:110–116. doi: 10.1111/j.1432-1033.1973.tb02661.x. [DOI] [PubMed] [Google Scholar]
- 99.Burstein C, Cohn M, Kepes A, Monod J. Role of lactose and its metabolic products in the induction of the lactose operon in Escherichia coli. Biochim Biophys Acta. 1965;95:634–639. [PubMed] [Google Scholar]
- 100.Jobe A, Bourgeois S. Lac repressor-operator interaction. VI. The natural inducer of the lac operon. J Mol Biol. 1972;69:397–408. doi: 10.1016/0022-2836(72)90253-7. [DOI] [PubMed] [Google Scholar]