

Abstract
Statistical potentials that embody torsion angle probability densities in databases of high-quality X-ray protein structures supplement the incomplete structural information of experimental nuclear magnetic resonance (NMR) datasets. By biasing the conformational search during the course of structure calculation toward highly populated regions in the database, the resulting protein structures display better validation criteria and accuracy. Here, a new statistical torsion angle potential is developed using adaptive kernel density estimation to extract probability densities from a large database of more than 106 quality-filtered amino acid residues. Incorporated into the Xplor-NIH software package, the new implementation clearly outperforms an older potential, widely used in NMR structure elucidation, in that it exhibits simultaneously smoother and sharper energy surfaces, and results in protein structures with improved conformation, nonbonded atomic interactions, and accuracy.
Keywords: knowledge-based torsion angle potential, adaptive kernel density estimation, NMR protein structure calculation, protein structure validation
Introduction
Over the years, the accumulation of high-resolution X-ray structures in the Protein Data Bank (PDB)1 has refined our knowledge of protein conformational preferences. Boundaries for the most favorable regions of the Ramachandran (ϕ, ψ) plot have shrunk,2 and side chain rotamer distributions have become sharper and narrower than ever before.3 Not only is this wealth of structural information exploited as validation criteria for newly generated models4 but also as a search bias during structure calculation. The latter approach aims at reproducing physically realistic conformational features of the structure database to alleviate the uncertainty associated with incomplete experimental information, such as that in low-resolution X-ray datasets. For instance, rotamer libraries can be used to fit side chain conformations to electron density,5, 6 and statistical potentials derived from database torsion angle distributions can supplement experimental restraints derived from NMR data. Driven by the relative sparseness of NMR data, the latter application was introduced more than 15 years ago7 and is at the center of this study.
Statistical torsion angle potentials can be succinctly described as follows.8 The probability density of torsion angles of interest is estimated from a database, and subsequently converted into potential energy by inversion of the Boltzmann equation. Thus (assuming a unit partition function as it cannot be directly obtained from the database),
| (1) |
where β is a constant, x, one or more torsion angles, and p(x|a), the probability density of x given another variable a. Ea(x) is a statistical potential that acts on x (given a). (It is also known as potential of mean force, and sometimes associated with other adjectives, such as empirical, database, and knowledge based.) For example, if x consists of ϕ and ψ, and a is the amino acid residue type “alanine,” then, during the calculation of a novel protein structure, EAla(ϕ, ψ) biases the backbone torsion angles of alanines toward the densest regions of the Ramachandran distribution of alanine in the database. A collection of such potentials (e.g., one per residue type) is needed to handle every possible protein sequence.
Although statistical torsion angle potentials may be implemented under different conformational sampling techniques, NMR structure elucidation is typically achieved by molecular dynamics-based simulated annealing and gradient minimization, both of which call for smoothness and differentiability of the potentials. In this regard, the latest and most advanced implementation9 relies on kernel density estimation (KDE) to obtain smooth, continuous probability densities involving all torsion angles within each residue type, the corresponding energy terms efficiently represented during structure calculations by cubic spline interpolation. However, since the main focus of that study was in structure prediction, NMR-relevant tests were reported as superficial, limited to the experimentally unrestrained minimization of previously solved NMR structures, omitting analysis of the accuracy of the resulting models, and their compatibility with the NMR data.
Possibly, the most thoroughly tested and most widely used statistical torsion angle potential in NMR is the so-called DELPHIC potential, developed by Kuszewski et al.,7 which has evolved over time.10–12 Included in the Xplor-NIH software package,13, 14 the latest version of this potential relies on a histogram-based approach for probability density estimation, the resulting energy (hyper)surfaces fit via an iterative protocol11 whereby a quartic function is fit to the global minimum and subtracted from the surface. This procedure is repeated until a desired tolerance is met, and the energy surface is then represented by the sum of the quartic functions. Under comprehensive analysis,15 the DELPHIC potential has been shown to significantly improve both structural validation criteria and accuracy, the former indicated by software tools such as WHAT IF,16 the latter, by better agreement of the models with residual dipolar couplings (RDCs) purposely excluded from structure calculations (i.e., RDC cross-validation).
Despite the above-described encouraging results, visual inspection of the DELPHIC potential energy surfaces reveals roughness and other features (or lack thereof) that seem unsupported by torsion angle populations in modern databases (see below). Here, such deficiencies are addressed by a completely reformulated statistical torsion angle potential. A database of more than 106 quality-filtered residues is used to generate probability densities via adaptive KDE. This results in density estimates that are not only continuous and smooth overall but also free of defects in regions of low density, where the noisy contribution of isolated points is automatically smoothed out. Finally, energy terms are efficiently represented during the course of structure calculation by cubic interpolation, from which forces are readily obtained. This new potential is incorporated into Xplor-NIH,13, 14 and tested on the structure calculation of 10 proteins of various folds and sizes, using publicly available NMR restraints. The latter include RDCs, omitted from the calculations for cross-validation. The quality of backbone and side chain conformations, as well as that of nonbonded atomic interactions, was assessed using MolProbity2, 17, 18 and WHAT IF.16
Results
The torsion angle database
The database on which this study relies consists of 1,005,827 residues, extracted from protein crystal structures solved at a resolution of 1.8 Å or better, all atoms with B factors < 35 Å2 and no serious atomic clashes reported by MolProbity. This database is a subset of the Top8000 database of almost 8000 nonhomologous protein chains (see Methods for details), kindly provided by Jane S. Richardson as a successor of the popular Top500 database.2
Residue type definitions and statistical approximations
The initial goal was to estimate the probability density function of all torsion angles within each residue type, starting from torsion angle instances in the database. Density estimates within a predefined grid were subsequently needed to obtain the corresponding energy values [via Eq. (1)], used in a cubic interpolation routine during structure computation (see Methods for details), where the statistical potential term (or terms; see below) was applied to all torsion angle degrees of freedom of residues with the corresponding type. Each residue was assigned only one type, following the order of precedence
| (2) |
where cis/trans refers to the peptide bond conformation, prePro denotes a residue immediately preceding a proline in the primary structure, and the lowest-precedence types consist of all amino acid names minus Gly and Pro. For example, to estimate the (ϕ, ψ) probability density of Gly, p(ϕ, ψ|Gly), all glycines in the database were used, regardless of whether they preceded a proline, whereas estimation of p(ϕ, ψ|Ala) omitted pre-proline alanines. This residue classification is based on well-known distinctive torsion angle distributions (e.g., the relatively large, steric clash-free areas accessible to glycine, afforded by the lack of Cβ), as well as the large size of the database. For example, whereas chemical similarity between tyrosine and phenylalanine has previously prompted their joint treatment to alleviate database scarcity (e.g., Ref. 10), here they yielded separate residue types, regardless of possible common features. Henceforth, Eq. (2) will be implied whenever a name in it is used (e.g., “Ala” stands for “non-pre-proline alanine”).
Use of Eq. (1) yields an energy term of the same dimensionality as the probability density function. Because all torsion angles within a residue type are involved, the highest possible dimensionality is six (e.g., Arg's ϕ, ψ, χ1, …, χ4). However, the number of coefficients needed to represent the interpolated energy term during structure calculations becomes excessively computationally expensive beyond three dimensions.9 Therefore, the problem is one of breaking probability densities of dimensionality >3 into components of dimensionality ≤ 3, a statistical task that can be achieved by assuming conditional independence.19 For example, in the case of Leu, χ2 was assumed conditionally independent of ϕ and ψ given χ1, which yields
| (3) |
It should be noted that Eq. (3) does not imply that χ2 is independent of the backbone torsion angles, but that its dependence is indirect, via χ1. Table I provides the probability density expressions for all residue types, including the statistical assumptions used to derive them. Such approximations rely on the number of atoms shared by torsion angles along the covalent framework of the residue: adjacent torsion angles (e.g., ϕ and χ1) share more atoms than nonadjacent ones (e.g., ϕ and χ2), and are, consequently, more likely to directly influence one another. Approximations similar to those in Table I have been previously used in another statistical torsion angle potential,9 although the latter made more assumptions in that no three-dimensional probability densities were used for side chain torsion angles.
Table I.
Probability Density Expressions Extracted from the Torsion Angle Database
| Residue type | Probability density functiona | Statistical approximationb |
|---|---|---|
| Gly, Ala | 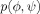 |
None |
| Thr, Val, Ser, Cys | 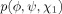 |
None |
| cis-Pro, trans-Pro | 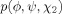 |
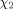 determines other ring torsion angles determines other ring torsion angles |
| Asp, Asn, Ile, Leu, His, Trp, Tyr, Phe | 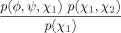 |
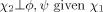 |
| Met, Glu, Gln | 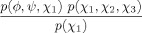 |
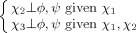 |
| Lys, Arg | 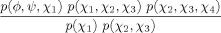 |
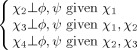 |
| prePro | 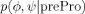 |
None |
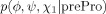 |
None | |
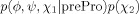 |
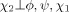 |
|
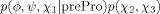 |
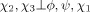 |
|
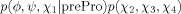 |
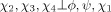 |
For simplicity, probability density functions omit conditionals whenever possible. For example, for residue types Gly and Ala, p(ϕ, ψ) implies p(ϕ, ψ|Gly) and p(ϕ, ψ|Ala), respectively. In the case of prePro, the explicit use of a conditional is informative. For example, the probability density function of pre-proline arginines is represented by p(ϕ, ψ, χ1|prePro) p(χ2, χ3, χ4), where p(ϕ, ψ, χ1|prePro) corresponds to all non-glycine, non-proline pre-proline residues (with at least one side chain torsion angle), and p(χ2, χ3, χ4) to all arginines, regardless whether they precede a proline (see text for another example).
The statistical approximations used to arrive at the different probability density expressions are indicated, where the orthogonality sign (⊥) means “independent of”.
Two residue types in Table I deserve particular attention. First, the side chain torsion angles of Pro are highly correlated to one another due to covalent restrictions imposed by the ring. As a result, a single torsion angle, χ2, can be used to determine the side chain conformation,20 and the entire conformational space captured by p(ϕ, ψ, χ2|Pro). Second, prePro is special in that it represents a diverse group [all non-glycine, non-proline residues immediately preceding a proline; Eq. (2)], with several residue subtypes. Because of insufficient pre-proline alanines (preProAla subtype) in the database, its (ϕ, ψ) density is represented by that of all prePro residues, p(ϕ, ψ|prePro). The (ϕ, ψ, χ1) distribution of the remaining prePro subtypes is captured by that of all prePro residues with at least χ1, p(ϕ, ψ, χ1|prePro). The distribution of torsion angles beyond χ1 is assumed to be that of the corresponding residue, regardless of whether it precedes a proline. For example, for pre-proline leucines
| (4) |
where p(χ2) is obtained from all leucines in the database. Although, Eq. (4) implies the not-necessarily-true assertion that χ2 is uncorrelated with the remaining torsion angles, such a correlation cannot be accurately obtained from the database due to the scarcity of pre-proline leucines, hence the current approximation. Similar considerations apply to the other prePro residue subtypes with torsion angles beyond χ1 (see Table I). A more detailed version of Table I is provided as Supporting Information (Table SI). It is noteworthy that while certain long-range correlations might be neglected by the above approximations, those based on atomic clashes are accounted for during structure calculations by repulsive interactions (see Methods for more details).
Adaptive KDE produces smooth, yet sharp potential surfaces
The general methodology chosen to extract the probability densities listed in Table I from the torsion angle database is KDE.21 It consists of centering “bumps” or kernel functions on top of each database point; the density at any arbitrary position in torsion angle space is then estimated by summing the contribution of all kernels. In this case, the kernels take the form of d-dimensional (d = 1, 2, or 3), symmetrical Gaussians, so that their overall smoothness is inherited by the density estimates. In particular, the adaptive version of KDE was used,21 where the width of each Gaussian adapts to the local density in that narrow ones are placed in regions of high density, and wide ones in regions of low density. This has the effect of reproducing features at high local density, while smearing sparsely populated areas of torsion angle space (i.e., the “tails” of the distribution). The latter would appear bumpy if fixed-width kernels were used, and yield false high-energy local minima [via Eq. (1)] that could hamper structure calculations. This shortcoming of nonadaptive KDE may be mitigated by removal of isolated database points, followed by padding low-density regions with artificial points.9 Here, the use of adaptive KDE rendered such modifications of the database unnecessary. Other examples of adaptive KDE in torsion angle space are provided elsewhere.2, 22
The energy terms that comprise the new statistical torsion angle potential, henceforth referred to as “torsionDBPot,” follow directly from Table I and Eq. (1). For example, Boltzmann inversion of Eq. (3) yields
| (5) |
The subtraction of ELeu(χ1) intuitively accounts for the overweighting of χ1 in the remainder of Eq. (5) (note χ1 appears in both ELeu(ϕ, ψ, χ1) and ELeu(χ1, χ2)). This is not an ad-hoc property of the potential, but one that arises naturally from the statistical treatment described in the previous section.
Figure 1 shows typical energy surfaces obtained from both the current version of the DELPHIC potential12 in Xplor-NIH [Fig. 1(A,C)] and torsionDBPot [Fig. 1(B, D)], introduced in Xplor-NIH as part of this work. Comparison of contour plots of the His (χ1, χ2) energy term reveals the absence of features in the DELPHIC potential [Fig. 1(A)], notably, a shallow minimum at (62°, 83°) (corresponding to the sparsely populated p80° rotamer3), which is apparent in torsionDBPot [Fig. 1(B)]. Moreover, the DELPHIC potential suffers from noise and unrealistic shapes of energy surfaces, a problem exacerbated at high dimensions, as exemplified by the (ϕ, ψ, χ1) energy term of Val [Fig. 1(C)], which contrasts with the both smoother and sharper surfaces of torsionDBPot [Fig 1(B,D)]. Visual inspection of several other surfaces indicates a prevalence of noise and multiple instances of missing features for the DELPHIC potential.
Figure 1.
Representative energy surfaces of the DELPHIC and torsionDBPot statistical torsion angle potentials in Xplor-NIH. (A and B) Contour plots of the His (χ1, χ2) energy term in the DELPHIC potential and torsionDBPot, respectively. (C and D) Single isoenergetic surfaces of the Val (ϕ, ψ, χ1) energy term in the DELPHIC potential and torsionDBPot, respectively. Panels A and B were generated with Matplotlib,23 C and D with Mayavi.24 All units are in degrees.
Effect of torsionDBPot on NMR structure calculations
The new statistical torsion angle potential, torsionDBPot, was tested on 10 protein structures of diverse sizes (54 to 259 residues in length) and topologies (α, β, and αβ folds), listed in Table II. Structure calculations were performed with Xplor-NIH, enforcing publicly available NMR distance and torsion angle restraint sets. These sets reflect heterogeneity in their derivation from the experimental data. For example, whereas the backbone torsion angle restraints for the protein KH325 were obtained from scalar couplings, those for DinI26 were derived from chemical shifts. The interpretation of nuclear Overhauser effects (NOEs) in terms of interproton distances also varies from one research group to another (six of which are represented here), a fact notably exemplified by SrtA,27 which relied heavily on automation for NOE analysis, as opposed to, for example, IIBMtl,28 which followed a more conventional manual iterative approach. Differences in NOE data are also quantitative as the average number of long-range NOEs (i.e., between residues separated by more than five in the amino acid sequence) ranges from 1.8 to 10.6 per residue in the 10-protein set. Thus, the systems tested aim at representing a range of situations that may be encountered during the determination of a novel protein structure by NMR.
Table II.
Proteins Used in Test Structure Calculations
| Protein (Name and Abbreviation) | Residues | Fold | NMR Data (PDB ID) | References |
|---|---|---|---|---|
| B1 domain of protein G (GB1) | 54 | αβ | 3GB1 | 29 |
| Ubiquitin (Ubi) | 76 | αβ | 1D3Z | 30 |
| DNA damage inducible protein 1 (DinI) | 81 | αβ | 1GHH | 26 |
| LM5-1 FYVE domain (LM5-1) | 84 | αβ | 1Z2Q | 31 |
| C-terminal KH domain of heterogeneous nuclear ribonucleoprotein K (KH3) | 89 | αβ | 1KHM | 25 |
| Barrier-to-autointegration factor (BAF, chain A only) | 89 | α | 2EZX | 32 |
| Cytoplasmic B domain of the mannitol transporter IImannitol (IIBMtl) | 97 | αβ | 1VKR | 28 |
| Sortease A in covalent complex with an LPXTG analog (SrtA) | 148 | β | 2KID | 27 |
| Apo dihydrofolate reductase (DHFR) | 162 | αβ | 2L28 | 33 |
| N-terminal domain of enzyme I (EIN) | 259 | αβ | 1EZA (distance, torsion angle) 3EZA (RDC) | 34, 35 |
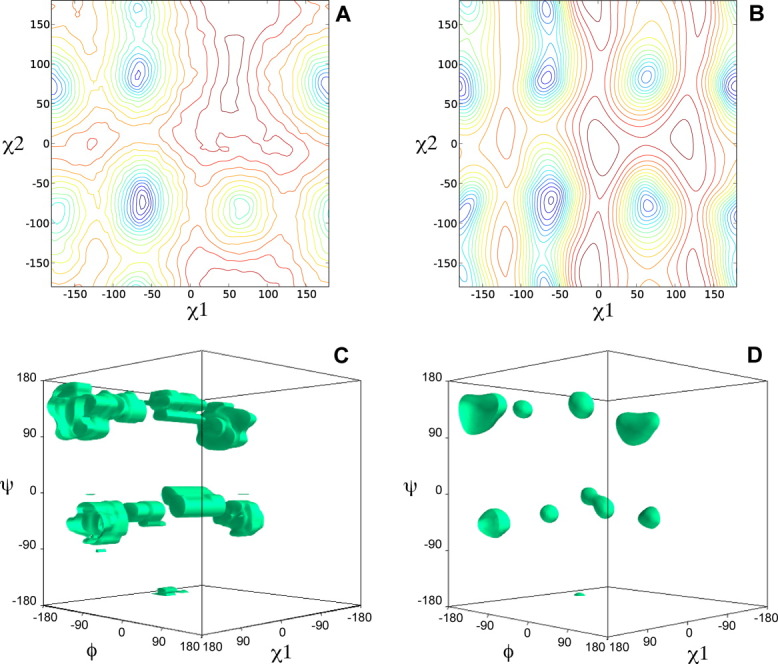
For each protein, three types of structure calculations were carried out, differing in the statistical torsion angle potential used: (i) none, (ii) the DELPHIC potential, or (iii) torsionDBPot. The generated structures were validated with MolProbity (Fig. 2) and WHAT IF (Fig. 3), which report that calculations using either the DELPHIC or the torsionDBPot potential outperform in every criterion those that exclude such potentials. Furthermore, torsionDBPot improves the quality of the backbone conformation relative to the DELPHIC potential. Indeed, MolProbity indicates that, with the sole exception of EIN, the percent of Ramachandran outliers drops to its lowest values with torsionDBPot [Fig. 2(A)], and the favored regions of the Ramachandran plot become more populated in every case, except for GB1, where a similar outcome is achieved with the DELPHIC potential [Fig. 2(B)]. These results agree with WHAT IF's Ramachandran plot appearance score, which improves throughout [Fig. 3(A)] upon use of torsionDBPot. With regards to side chain conformation, both potentials perform similarly according to MolProbity: the percent of poor rotamers is slightly smaller with torsionDBPot for seven proteins, the remaining three yielding slightly better statistics with the DELPHIC potential [Fig. 2(C)]. On the other hand, WHAT IF favors torsionDBPot, which results in better χ1/χ2 rotamer normality scores for all proteins [Fig. 3(B)].
Figure 2.
MolProbity validation. Each barplot displays a Molprobity validation statistic for structure ensembles of different proteins, with bars representing the mean ± standard deviation computed from 20 structures. Structure calculations without any statistical torsion angle potential (black), with the DELPHIC potential12 (gray), and with the new torsionDBPot potential (white) are included. Abbreviated protein names are used; for full names see Table II. The clashscore36 (panel D) and the MolProbity score18 (panel E) are costs: the lower the better. Barplots in this and all other figures were generated with Matplotlib.23
Figure 3.
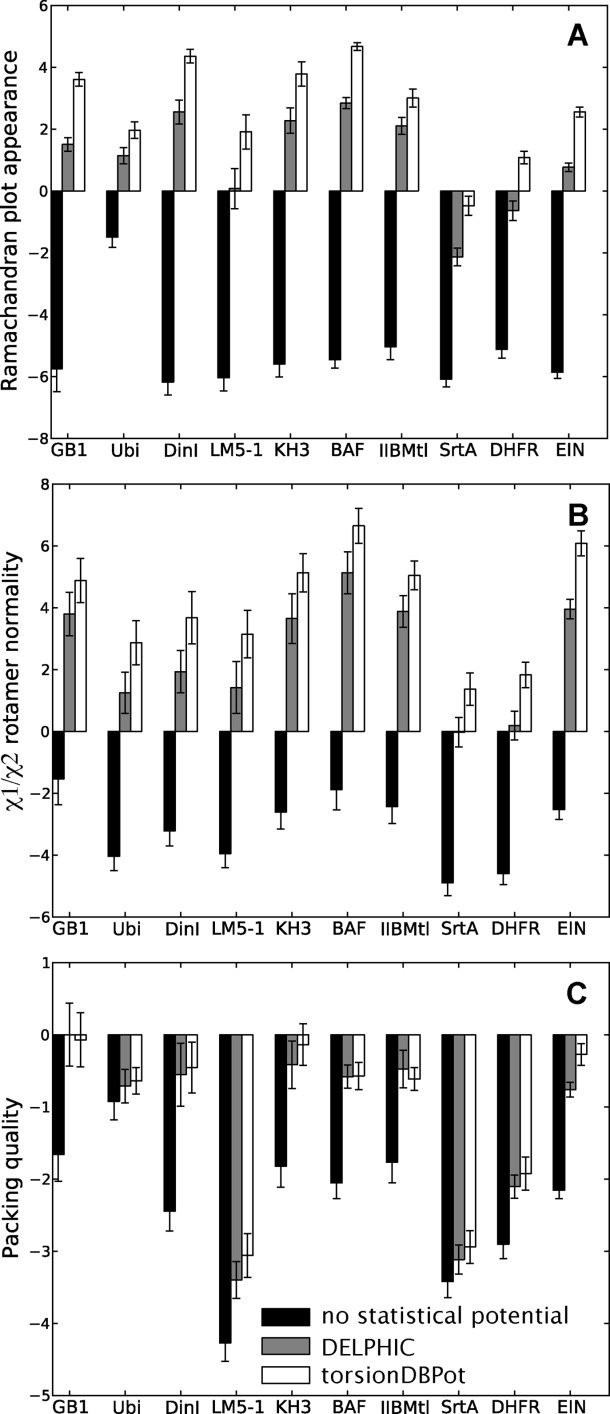
WHAT IF validation. Each barplot displays a WHAT IF validation statistic for structure ensembles of different proteins, with bars representing the mean ± standard deviation computed from 20 structures. Structure calculations without any statistical torsion angle potential (black), with the DELPHIC potential12 (gray), and with the new torsionDBPot potential (white) are included. Abbreviated protein names are used; for full names see Table II. Each statistic is a score: the larger the better. Packing quality (panel C) refers to the 2nd generation packing quality.
Measures of quality of nonbonded atomic interactions, or atomic packing, are more independent validation criteria than those discussed above, in that they usually do not rely on the variables directly affected by the statistical torsion angle potentials, i.e., the torsion angles. Further, packing encompasses long-range features outside the scope of both the torsionDBPot and DELPHIC potentials, which only act at the local residue level. MolProbity's “clashscore,”36 the number of serious atomic overlaps per thousand atoms (see Methods for details), and WHAT IF's packing quality score,37 which considers atomic distributions around different molecular fragments, are two such measures of packing. Relative to the DELPHIC potential, all structures generated with torsionDBPot display systematic improvements in the MolProbity clashscore, except for the slightly worse clashscore of IIBMtl, well within error bars [Fig. 2(D)]. This trend is also reflected by WHAT IF's packing quality score [Fig. 3(C)].
The introduction of an additional energy term usually causes the agreement between calculated structures and terms in the original target function to deteriorate. It is, therefore, important to confirm that the improvements in conformation and atomic interactions afforded by torsionDBPot do not come at significant cost to the remaining terms, particularly those associated with the NMR data. Indeed, torsionDBPot is more compatible than the DELPHIC potential with the experimentally determined distances, as suggested by slightly lower root mean square (RMS) deviations from the upper and lower bounds of the distance restraints [Fig. 4(A)]. With regards to torsion angle restraints, RMS statistics suggest that some proteins exhibit better agreement when generated with the DELPHIC potential, others with torsionDBPot, but in every case the agreement is comparable to that of structures generated without either potential (except for ubiquitin whose publicly released restraints have unrealistically narrow bounds, hence the large RMS deviations) [Fig. 4(B)].
Figure 4.
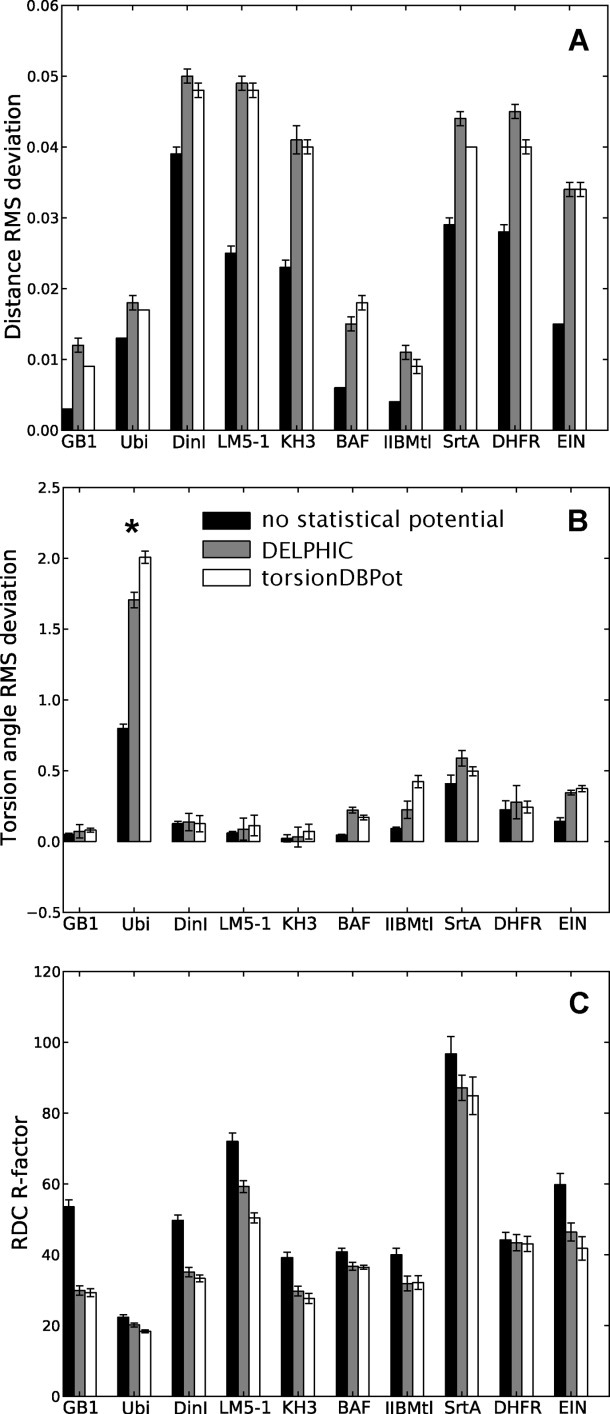
Fit to experimental data. Each barplot displays a figure of merit for the fit to a given experimental NMR observable of structure ensembles of different proteins, with bars representing the mean ± standard deviation computed from 20 structures (note that error bars associated with very small standard deviations may seem missing). Structure calculations without any statistical torsion angle potential (black), with the DELPHIC potential12 (gray), and with the new torsionDBPot potential (white) are included. Abbreviated protein names are used; for full names see Table II. Large torsion angle RMS deviations for ubiquitin (panel B, asterisk) stem from unrealistically narrow bounds in the publicly released restraints (PDB ID: 1D3Z). Each RDC R-factor (panel C) is an unweighted average over different alignment media and nuclei pairs (when applicable).
The compatibility with experimental data excluded from structure calculations represents an independent test of structural accuracy. RDCs depend on the orientation of interatomic vectors relative to an external alignment tensor, and are commonly used for cross-validation.38 Experimentally observed RDCs and those computed from the protein models were compared via an R-factor,39 which ranges from 0% (perfect correlation) to 100% (no correlation). Figure 4(C) shows that inclusion of the DELPHIC potential in structure calculations significantly improves the fit to RDCs, as previously reported elsewhere.15 torsionDBPot improves the fit even further in all cases, with the exception of IIBMtl, where a slightly worse fit is observed.
Discussion
The previously developed statistical torsion angle potential7, 10–12 in Xplor-NIH13, 14 (or its precursors, X-PLOR40 and CNS41) has become an important tool in NMR protein structure determination, inspiring similar implementations in other software packages.9,42, 43 Although originally applied to solution NMR, the statistical torsion angle potential in Xplor-NIH (named DELPHIC) has additionally made significant contributions with other types of experimental data, such as combination of solution and solid state NMR,44–46 combination of solution NMR and small- and wide-angle X-ray scattering,47, 48 combination of solid state NMR and X-ray diffraction,49 and purely solid state NMR data (e.g., Refs. 50–52). With this in mind, a “First, do no harm” approach was followed in the development of torsionDBPot, a new statistical torsion angle potential in Xplor-NIH. (Note that the DELPHIC potential remains available for backwards compatibility.) Indeed, while accomplishing similar or better fit to experimental restraints relative to the DELPHIC potential, torsionDBPot improves the quality of protein conformation and nonbonded atomic interactions. This is summarized by the overall MolProbity score18 (the lower the better), which improves in every case tested [Fig. 2(E)]. Moreover, such benefits are concomitant with enhanced structural accuracy, as suggested by better agreement with cross-validated RDCs. Albeit relatively moderate, the above-reported improvements are consistent across the entire protein test set.
Despite their usefulness in protein structure prediction and experiment-based determination, a general concern with statistical potentials of any kind is that their inherent average nature may bias structures away from features that are real although poorly represented in the database. Use of experimental data, however, as in this case, ameliorates this bias because the data are allowed to trump the statistical potential whenever possible. Thus, a rarely observed conformation firmly supported by NMR restraints should prevail over torsionDBPot in structure calculations (otherwise, restraint violations would arise highlighting the unusual region). In this light, flexibly disordered regions appear problematic as they are both poorly represented in the structured, low-B factor database, and usually sparsely restrained by NMR data. Future work may address the need for alternate descriptions of nonregular regions. For the 10 proteins studied here, the current protocol is clearly a step forward in generating high-quality NMR structures.
Methods
The torsion angle database
The starting point for the compilation of the torsion angle database used in this study is the Top8000 database (kindly provided by Jane S. Richardson, Duke University), of almost 8000 chains with X-ray structure resolution better than 2.0 Å, less than 70% sequence identity, and other satisfied filters, notably: chain MolProbity score <2.0, ≤5% of residues with bond lengths and angles outside four standard deviations from standard geometry, ≤5% of residues with Cβ deviations >0.25 Å, and best average of resolution and MolProbity score among the 70% homology cluster the chain represents. In addition, similar to previous versions of this database,2, 3 the Top8000 contains flipped planar side chain terminal groups of asparagines, glutamines, and histidines, when justified by analysis of atomic clashes and H-bonding.3 Further details are provided at the Richardson Lab's website (http://kinemage.biochem. duke.edu/databases/top8000.php). The Top8000 database was obtained as a table, each row containing information on a single residue, such as its torsion angles, resolution, atomic clashes (if any), etc.
As discussed in the Results section, the Top8000 database was subjected to more stringent filters to generate the custom database used by our new statistical torsion angle potential, torsionDBPot. Specifically, only chains with X-ray resolution of 1.8 Å or better were considered, residues from which were included in the custom database only if all their atoms had B factors < 35 Å2 and no serious clashes reported by MolProbity.2, 17, 18 Moreover, leucines with (χ1, χ2) pairs within regions that represent misfit rotamers3, 10 were avoided; a total of 553 such misfits were encountered and removed after resolution, B factor, and clash filtering. The resulting torsion angle database contains 1,005,827 residues.
A new Python module, torsionDBTools, has been added to Xplor-NIH to facilitate the extraction of torsion angles from Cartesian coordinates (i.e., PDB files). Although thoroughly tested, this module was not used here, as the Top8000 database already provided torsion angles, along with other useful information (see above). However, the module should prove useful in the derivation of new statistical torsion angle potentials from arbitrary subsets of the PDB (e.g., coil databases).
Torsion angle probability densities via adaptive KDE
The goal is to accurately estimate the probability density function of torsion angles of interest from the database, with the additional requirement that the estimate be smooth. The torsion angles under consideration can be represented by a column vector x, which defines a d-dimensional space where n database points X1, …, Xn are found (e.g., if xT = (ϕ, ψ), d = 2, where T denotes vector transpose). A first approximation toward extracting the probability density from the database is to perform KDE, by summing over “bumps” or kernels centered at the observed database points.21 KDE with a symmetrical Gaussian kernel function and window width h is defined by
| (6) |
where the N-notation is used for the d-dimensional (or d-variate) Gaussian, with mean vector μ and covariance matrix Σ (I is the identity matrix). Explicitly,
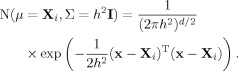 |
(7) |
In one dimension, for example, the left-hand side of Eq. (7) simply becomes 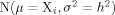 , where the boldface vector/matrix notation is no longer necessary, and the variance, σ2, replaces the covariance matrix. It is noteworthy that, for the sake of simplicity, all probability density functions in this section [including Eqs. (6) and (7) above] tacitly imply the residue type conditional, explicit elsewhere in the text [cf. Eq. (1)].
, where the boldface vector/matrix notation is no longer necessary, and the variance, σ2, replaces the covariance matrix. It is noteworthy that, for the sake of simplicity, all probability density functions in this section [including Eqs. (6) and (7) above] tacitly imply the residue type conditional, explicit elsewhere in the text [cf. Eq. (1)].
Gaussians are nonnegative and integrate to one, leading 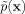 to also be nonnegative and integrate to one, as any probability density function must. Also, since Gaussians are continuous and smooth, so is
to also be nonnegative and integrate to one, as any probability density function must. Also, since Gaussians are continuous and smooth, so is 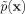 . The degree of smoothing is additionally controlled by the choice of window width h (i.e., the standard deviation in the one-dimensional case). Throughout our work, the periodicity of angular values is dealt with by augmenting the database with shifted copies of the original.21 Following the one-dimensional example, if the torsion angle under study is defined in the interval [-180°, 180°), adding copies of the database at intervals [-540°, -180°) and [180°, 540°) results in a new database {X1 - 360°, …, Xn - 360°, X1, …, Xn, X1 + 360°, …, Xn + 360°}. Performing KDE on this augmented database with Eq. (6) (where n is still the original number of points) accounts for the boundary condition.
. The degree of smoothing is additionally controlled by the choice of window width h (i.e., the standard deviation in the one-dimensional case). Throughout our work, the periodicity of angular values is dealt with by augmenting the database with shifted copies of the original.21 Following the one-dimensional example, if the torsion angle under study is defined in the interval [-180°, 180°), adding copies of the database at intervals [-540°, -180°) and [180°, 540°) results in a new database {X1 - 360°, …, Xn - 360°, X1, …, Xn, X1 + 360°, …, Xn + 360°}. Performing KDE on this augmented database with Eq. (6) (where n is still the original number of points) accounts for the boundary condition.
Despite its obvious advantages over simpler density estimation methods such as the histogram, KDE has the tendency to produce noise in regions of low density, arising from individual, isolated bumps, a problem exacerbated in high dimensions. Here, the solution chosen was the use of kernels with variable window width—as opposed to the fixed-width kernels of Eq. (6)—so that narrow kernels are placed in regions of high density, and wide ones in regions of low density. This method is called adaptive KDE,21 as the window width adapts to the local density, which is preliminary estimated via standard KDE [(Eq. (6), in this context usually referred to as the pilot estimate]. Using again symmetrical Gaussians, adaptive KDE takes the mathematical form
| (8) |
where local bandwidth factors 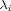 are given by
are given by
| (9) |
In Eq. (9), 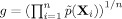 , the geometric mean of the
, the geometric mean of the 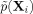 (a constant), and α is set to 0.5 as recommended elsewhere.21 The formulas for the variable-width, symmetrical Gaussians in Eq. (8) can be readily obtained from Eq. (7) by replacing h by
(a constant), and α is set to 0.5 as recommended elsewhere.21 The formulas for the variable-width, symmetrical Gaussians in Eq. (8) can be readily obtained from Eq. (7) by replacing h by 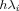 . Once the
. Once the 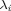 s are determined for a joint probability density estimate [e.g., p(ϕ, ψ\χ1)], the marginal probability density estimate of one (or more) torsion angles [e.g., p(χ1)] can be computed in a straightforward manner. When more than one joint probability density estimate is available, the marginal probability density of a common torsion angle is extracted from each joint distribution, and an average density computed.
s are determined for a joint probability density estimate [e.g., p(ϕ, ψ\χ1)], the marginal probability density estimate of one (or more) torsion angles [e.g., p(χ1)] can be computed in a straightforward manner. When more than one joint probability density estimate is available, the marginal probability density of a common torsion angle is extracted from each joint distribution, and an average density computed.
In this study, adaptive KDE was performed as described above, for one, two, and three dimensions, where h was given the values of 4°, 5°, and 6°, respectively—as prescribed elsewhere,21h takes the same value in Eqs. (6) and (8). All calculations were performed using the Python module densityEstimation implemented within Xplor-NIH13, 14 for the present purposes.
Cubic interpolation of energy terms
The different energy terms that stem from the application of Eq. (1) to the adaptive KDE-based probability density functions in Table I (see Supporting Information Table SI for a more detailed version of Table I) were evaluated on a grid used for cubic interpolation with periodic boundary conditions. In one and two dimensions, a uniform grid with 10°-spacing was used. Extending this strategy to the construction of a three-dimensional grid results in an unacceptable increase of computer memory requirements. Consequently, a nonuniform grid was devised with 10°-spacing around each energy minimum, and wider spacing elsewhere. Specifically, the axis along one of the three dimensions is uniformly marked every 10°, and a tick mark retained only if within a distance r of the coordinate of a minimum along that dimension. Subsequently, to avoid under-sampling, if two adjacent tick marks are farther apart than 10°, a new one is added equidistant from the two. The same procedure is performed with the axes along the remaining two dimensions, and the grid constructed with the three sparsely sampled axes. r = 23° for all residue types [Eq. (2)], with the exception of cis/trans-Pro, where r = 30°, the denser sampling afforded by the fact that the minima are confined to a small region of torsion angle space.
Within Xplor-NIH, cubic interpolation routines in one and two dimensions53 were already present and were previously exploited by another potential term.50 Three-dimensional cubic interpolation capabilities, as described elsewhere,54 were added to Xplor-NIH (as the spline3D Python module) for the present purposes and have already been successfully applied to a recent unrelated problem.55 The interpolated energy terms make up the new statistical torsion angle potential, torsionDBPot, which is set up with the newly added module torsionDBPotTools. In addition, torsionDBPotTools contains the auxiliary function find_minima, which characterizes a queried torsionDBPot surface by listing the number, location, and depth of all its minima, and is useful for the comparison of different surfaces.
Structure calculations
Structures were calculated with Xplor-NIH,13, 14 using two conventional simulated annealing protocols: a first one for folding an initially extended conformation and a second one for the subsequent refinement of a selected folded model. Both protocols, based on the internal variable module,56 share the same basic scheme, comprising the following stages (respecting their order during calculations): (i) high-temperature torsion angle dynamics (3500 K for folding, 3000 K for refinement), the smallest of 15 ps or 15,000 timesteps in length, subject to torsion angle restraints (kta = 10 kcal mol–1 rad–2), distance restraints (kdist = 2 kcal mol–1 Å–2), and van der Waals-like repulsions57 (kvdw = 0.004 kcal mol–1 Å–4; only Cα–Cα interactions active, with a van der Waals radius scale factor svdw = 1.2), where kη represents the force constant of energy term η; (ii) torsion angle dynamics with simulated annealing, where the temperature is reduced from the initial value (see above) to 25 K in steps of 12.5 K (the smallest of 0.2 ps/step or 200 timesteps/step for folding, the smallest of 0.63 ps/step or 630 timesteps/step for refinement), kta = 200 kcal mol–1 rad–2, and kdist, kvdw, and svdw are geometrically increased from 2 to 30 kcal mol–1 Å–2, 0.004 to 4 kcal mol–1 Å–4, and 0.9 to 0.8, respectively (all van der Waals interactions active (see exceptions below) in this stage, a feature maintained in subsequent stages, as well as the final values of force constants and svdw); (iii) 500 steps of Powell torsion angle minimization; (iv) 500 steps of Powell Cartesian minimization. When including either the DELPHIC12 or the torsionDBPot statistical torsion angle potential term, its force constant is set to 0.002 kcal mol–1 rad–2 in stage (i), from which it geometrically increases to 1 (DELPHIC) or 2 kcal mol–1 rad–2 (torsionDBPot) in stage (ii), values maintained until the end of the protocol. Although many steric interactions are already accounted for by both the DELPHIC and torsionDBPot potentials (e.g., those in eclipsed conformations that result in staggered side chain rotamer distributions), the prevention of any atomic overlap is an essential part of any force field. Here, a compromise is achieved by allowing repulsions only between atoms separated by more than three covalent bonds whenever the DELPHIC or torsionDBPot potentials are used; when they are not used, repulsions are allowed only between atoms separated by more than two covalent bonds.
The folding protocol generated 100 structures, from which the one with the lowest experimental energy (i.e., energy from distance and torsion angle restraints) was selected for refinement. The refinement protocol generated 100 structures, and the top 20 ranked by the experimental energy were selected for further analysis. Computer memory requirements for torsionDBPot were similar to those of the DELPHIC potential.
Experimentally determined distance, torsion angle, and RDC restraints (the latter excluded from the structure calculations, and used only for purposes of cross-validation; see below for details) were obtained from the PDB for the 10 proteins listed in Table II.
Structure validation
The quality of backbone and side chain conformations, as well as that of nonbonded interatomic interactions in the calculated protein structures were assessed with MolProbity2, 17, 18 and WHAT IF.16 The increase of a WHAT IF score was considered an improvement of the associated quality criterion (a proper “score” behavior). On the other hand, MolProbity's overall score18 and clashscore36 are actually costs whose decrease reflect improvement. It is noteworthy that the clashscore (number of serious atomic overlaps per thousand atoms) ignores clashes between pairs of heavy atoms within three or fewer covalent bonds, and between pairs of atoms where one or both are hydrogens within four or fewer bonds. In other words, the MolProbity clashscore ignores clashes between atoms whose relative positions are directly affected by the statistical torsion angle potentials during structure calculations, thus making it a more independent measure of structure quality as opposed to, for example, the percentage of poor side chain rotamers.
Agreement between structures and RDCs
RDCs were fit to calculated structures by singular value decomposition58 with Xplor-NIH, which additionally reports the R-factor measure of fit,39
| (10) |
where 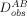 is the experimentally observed and
is the experimentally observed and 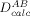 the structure-calculated RDC for nuclei pair type A–B (e.g., 1HN–15N) in a given molecular alignment medium,
the structure-calculated RDC for nuclei pair type A–B (e.g., 1HN–15N) in a given molecular alignment medium, 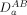 and Rh are the axial component and the rhombicity of the alignment tensor, respectively, and angular brackets denote averaging over the entire A–B RDC dataset. A single unweighted R-factor average over all nuclei pair types and media was used to assess the overall fit.
and Rh are the axial component and the rhombicity of the alignment tensor, respectively, and angular brackets denote averaging over the entire A–B RDC dataset. A single unweighted R-factor average over all nuclei pair types and media was used to assess the overall fit.
Availability
The new statistical torsion angle potential, torsionDBPot, is part of the Xplor-NIH software suite (as of version 2.31), downloadable from the web (http://nmr.cit.nih.gov/xplor-nih/).
Acknowledgments
Jane S. Richardson provided pre-release access to the Top8000 database, and, along with Daniel A. Keedy and Vincent B. Chen, participated in useful discussions. Robert T. Clubb provided Xplor-NIH topology and parameter files for Sortease A; Nuttee Suree and Albert Chan helped with their setup.
Glossary
Abbreviations
- BAF
barrier-to-autointegration factor
- CNS
crystallography and NMR system
- DHFR
apo dihydrofolate reductase
- DinI
DNA damage inducible protein
- EIN
N-terminal domain of enzyme I
- GB1
B1 domain of protein G
- IIBMtl
cytoplasmic B domain of the mannitol transporter IImannitol
- KDE
kernel density estimation
- KH3
C-terminal KH domain of heterogeneous nuclear ribonucleoprotein K
- LM5-1
LM5-1 FYVE domain
- NMR
nuclear magnetic resonance
- NOE
nuclear Overhauser effect
- PDB
protein data bank
- RDC
residual dipolar coupling
- RMS
root-mean-square
- SrtA
sortease A in covalent complex with an LPXTG analog
- Ubi
ubiquitin
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Berman H, Henrick K, Nakamura H. Announcing the worldwide Protein Data Bank. Nat Struct Biol. 2003;10:980–980. doi: 10.1038/nsb1203-980. [DOI] [PubMed] [Google Scholar]
- 2.Lovell SC, Davis IW, Adrendall WB, de Bakker PIW, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Cα geometry: φ, Ψ and Cβ deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 3.Lovell SC, Word JM, Richardson JS, Richardson DC. The penultimate rotamer library. Proteins. 2000;40:389–408. [PubMed] [Google Scholar]
- 4.Read RJ, Adams PD, Arendall WB, III, Brunger AT, Emsley P, Joosten RP, Kleywegt GJ, Krissinel EB, Lütteke T, Otwinowski Z, Perrakis A, Richardson JS, Sheffler WH, Smith JL, Tickle IJ, Vriend G, Zwart PH. A new generation of crystallographic validation tools for the protein data bank. Structure. 2011;19:1395–1412. doi: 10.1016/j.str.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 6.Headd JJ, Immormino RM, Keedy DA, Emsley P, Richardson DC, Richardson JS. Autofix for backward-fit sidechains: using MolProbity and real-space refinement to put misfits in their place. J Struct Funct Genomics. 2009;10:83–93. doi: 10.1007/s10969-008-9045-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuszewski J, Gronenborn AM, Clore GM. Improving the quality of NMR and crystallographic protein structures by means of a conformational database potential derived from structure databases. Protein Sci. 1996;5:1067–1080. doi: 10.1002/pro.5560050609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sippl MJ. Calculation of conformational ensembles from potentials of mean force—an approach to the knowledge-based prediction of local structures in globular proteins. J Mol Biol. 1990;213:859–883. doi: 10.1016/s0022-2836(05)80269-4. [DOI] [PubMed] [Google Scholar]
- 9.Amir ED, Kalisman N, Keasar C. Differentiable, multi-dimensional, knowledge-based energy terms for torsion angle probabilities and propensities. Proteins. 2008;72:62–73. doi: 10.1002/prot.21896. [DOI] [PubMed] [Google Scholar]
- 10.Kuszewski J, Gronenborn AM, Clore GM. Improvements and extensions in the conformational database potential for the refinement of NMR and X-ray structures of proteins and nucleic acids. J Magn Reson. 1997;125:171–177. doi: 10.1006/jmre.1997.1116. [DOI] [PubMed] [Google Scholar]
- 11.Kuszewski J, Clore GM. Sources of and solutions to problems in the refinement of protein NMR structures against torsion angle potentials of mean force. J Magn Reson. 2000;146:249–254. doi: 10.1006/jmre.2000.2142. [DOI] [PubMed] [Google Scholar]
- 12.Clore GM, Kuszewski J. χ1 Rotamer populations and angles of mobile surface side chains are accurately predicted by a torsion angle database potential of mean force. J Am Chem Soc. 2002;124:2866–2867. doi: 10.1021/ja017712p. [DOI] [PubMed] [Google Scholar]
- 13.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J Magn Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 14.Schwieters CD, Kuszewski JJ, Clore GM. Using Xplor-NIH for NMR molecular structure determination. Prog Nucl Mag Res Sp. 2006;48:47–62. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 15.Mertens HDT, Gooley PR. Validating the use of database potentials in protein structure determination by NMR. FEBS Lett. 2005;579:5542–5548. doi: 10.1016/j.febslet.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 16.Vriend G. What If—a molecular modeling and drug design program. J Mol Graph. 1990;8:52–56. doi: 10.1016/0263-7855(90)80070-v. [DOI] [PubMed] [Google Scholar]
- 17.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Cryst. 2010;D66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Russell SJ, Norvig P. Artificial intelligence: a modern approach. Upper Saddle River, N.J: Prentice Hall/Pearson Education; 2003. [Google Scholar]
- 20.DeTar DF, Luthra NP. Conformations of proline. J Am Chem Soc. 1977;99:1232–1244. doi: 10.1021/ja00446a040. [DOI] [PubMed] [Google Scholar]
- 21.Silverman BW. Density estimation for statistics and data analysis. London, New York: Chapman and Hall; 1986. [Google Scholar]
- 22.Shapovalov MV, Dunbrack RL. A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure. 2011;19:844–858. doi: 10.1016/j.str.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hunter JD. Matplotlib: a 2D graphics environment. Comput Sci Eng. 2007;9:90–95. [Google Scholar]
- 24.Ramachandran P, Varoquaux G. Mayavi: 3D visualization of scientific data. Comput Sci Eng. 2011;13:40–50. [Google Scholar]
- 25.Baber JL, Libutti D, Levens D, Tjandra N. High precision solution structure of the C-terminal KH domain of heterogeneous nuclear ribonucleoprotein K, a c-myc transcription factor. J Mol Biol. 1999;289:949–962. doi: 10.1006/jmbi.1999.2818. [DOI] [PubMed] [Google Scholar]
- 26.Ramirez BE, Voloshin ON, Camerini-Otero RD, Bax A. Solution structure of DinI provides insight into its mode of RecA inactivation. Protein Sci. 2000;9:2161–2169. doi: 10.1110/ps.9.11.2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suree N, Liew CK, Villareal VA, Thieu W, Fadeev EA, Clemens JJ, Jung ME, Clubb RT. The structure of the Staphylococcus aureus sortase-substrate complex reveals how the universally conserved LPXTG sorting signal is recognized. J Biol Chem. 2009;284:24465–24477. doi: 10.1074/jbc.M109.022624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Legler PM, Cai ML, Peterkofsky A, Clore GM. Three-dimensional solution structure of the cytoplasmic B domain of the mannitol transporter IIMannitol of the Escherichia coli phosphotransferase system. J Biol Chem. 2004;279:39115–39121. doi: 10.1074/jbc.M406764200. [DOI] [PubMed] [Google Scholar]
- 29.Kuszewski J, Gronenborn AM, Clore GM. Improving the packing and accuracy of NMR structures with a pseudopotential for the radius of gyration. J Am Chem Soc. 1999;121:2337–2338. [Google Scholar]
- 30.Cornilescu G, Marquardt JL, Ottiger M, Bax A. Validation of protein structure from anisotropic carbonyl chemical shifts in a dilute liquid crystalline phase. J Am Chem Soc. 1998;120:6836–6837. [Google Scholar]
- 31.Mertens HDT, Callaghan JM, Swarbrick JD, Mcconville MJ, Gooley PR. A high-resolution solution structure of a trypanosomatid FYVE domain. Protein Sci. 2007;16:2552–2559. doi: 10.1110/ps.073009807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cai M, Huang Y, Zheng R, Wei SQ, Ghirlando R, Lee MS, Craigie R, Gronenborn AM, Clore GM. Solution structure of the cellular factor BAF responsible for protecting retroviral DNA from autointegration. Nat Struct Biol. 1998;5:903–909. doi: 10.1038/2345. [DOI] [PubMed] [Google Scholar]
- 33.Feeney J, Birdsall B, Kovalevskaya NV, Smurnyy YD, Peran EMN, Polshakov VI. NMR structures of Apo L. casei dihydrofolate reductase and its complexes with trimethoprim and NADPH: Contributions to positive cooperative binding from ligand-induced refolding, conformational changes, and interligand hydrophobic interactions. Biochemistry. 2011;50:3609–3620. doi: 10.1021/bi200067t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garrett DS, Seok YJ, Liao DI, Peterkofsky A, Gronenborn AM, Clore GM. Solution structure of the 30 kDa N-terminal domain of enzyme I of the Escherichia coli phosphoenolpyruvate: sugar phosphotransferase system by multidimensional NMR. Biochemistry. 1997;36:2517–2530. doi: 10.1021/bi962924y. [DOI] [PubMed] [Google Scholar]
- 35.Garrett DS, Seok YJ, Peterkofsky A, Gronenborn AM, Clore GM. Solution structure of the 40,000 M-r phosphoryl transfer complex between the N-terminal domain of enzyme I and HPr. Nat Struct Biol. 1999;6:166–173. doi: 10.1038/5854. [DOI] [PubMed] [Google Scholar]
- 36.Word JM, Lovell SC, LaBean TH, Taylor HC, Zalis ME, Presley BK, Richardson JS, Richardson DC. Visualizing and quantifying molecular goodness-of-fit: small-probe contact dots with explicit hydrogen atoms. J Mol Biol. 1999;285:1711–1733. doi: 10.1006/jmbi.1998.2400. [DOI] [PubMed] [Google Scholar]
- 37.Vriend G, Sander C. Quality control of protein models: directional atomic contact analysis. J Appl Cryst. 1993;26:47–60. [Google Scholar]
- 38.Bax A, Grishaev A. Weak alignment NMR: a hawk-eyed view of biomolecular structure. Curr Opin Struct Biol. 2005;15:563–570. doi: 10.1016/j.sbi.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 39.Clore GM, Garrett DS. R-factor, free R, and complete cross-validation for dipolar coupling refinement of NMR structures. J Am Chem Soc. 1999;121:9008–9012. [Google Scholar]
- 40.Brünger AT. X-PLOR, Version 3.1: a system for X-ray crystallography and NMR. New Haven: Yale University Press; 1992. [Google Scholar]
- 41.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. and others. [DOI] [PubMed] [Google Scholar]
- 42.Bertini I, Cavallaro G, Luchinat C, Poli I. A use of Ramachandran potentials in protein solution structure determinations. J Biomol NMR. 2003;26:355–66. doi: 10.1023/a:1024092421649. [DOI] [PubMed] [Google Scholar]
- 43.Yang JS, Kim JH, Oh S, Han G, Lee S, Lee J. STAP refinement of the NMR database: a database of 2405 refined solution NMR structures. Nucleic Acids Res. 2012;40:D525–D530. doi: 10.1093/nar/gkr1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shi L, Traaseth NJ, Verardi R, Cembran A, Gao JL, Veglia G. A refinement protocol to determine structure, topology, and depth of insertion of membrane proteins using hybrid solution and solid-state NMR restraints. J Biomol NMR. 2009;44:195–205. doi: 10.1007/s10858-009-9328-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Traaseth NJ, Shi L, Verardi R, Mullen DG, Barany G, Veglia G. Structure and topology of monomeric phospholamban in lipid membranes determined by a hybrid solution and solid-state NMR approach. Proc Natl Acad Sci USA. 2009;106:10165–10170. doi: 10.1073/pnas.0904290106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Verardi R, Shi L, Traaseth NJ, Walsh N, Veglia G. Structural topology of phospholamban pentamer in lipid bilayers by a hybrid solution and solid-state NMR method. Proc Natl Acad Sci USA. 2011;108:9101–9106. doi: 10.1073/pnas.1016535108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schwieters CD, Suh JY, Grishaev A, Ghirlando R, Takayama Y, Clore GM. Solution structure of the 128 kDa enzyme I dimer from Escherichia coli and its 146 kDa complex with HPr using residual dipolar couplings and small- and wide-angle X-ray scattering. J Am Chem Soc. 2010;132:13026–13045. doi: 10.1021/ja105485b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Takayama Y, Schwieters CD, Grishaev A, Ghirlando R, Clore GM. Combined use of residual dipolar couplings and solution X-ray scattering to rapidly probe rigid-body conformational transitions in a non-phosphorylatable active-site mutant of the 128 kDa enzyme I dimer. J Am Chem Soc. 2011;133:424–427. doi: 10.1021/ja109866w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tang M, Sperling LJ, Berthold DA, Schwieters CD, Nesbitt AE, Nieuwkoop AJ, Gennis RB, Rienstra CM. High-resolution membrane protein structure by joint calculations with solid-state NMR and X-ray experimental data. J Biomol NMR. 2011;51:227–233. doi: 10.1007/s10858-011-9565-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wylie BJ, Schwieters CD, Oldfield E, Rienstra CM. Protein structure refinement using 13Cα chemical shift tensors. J Am Chem Soc. 2009;131:985–992. doi: 10.1021/ja804041p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sengupta I, Nadaud PS, Helmus JJ, Schwieters CD, Jaroniec CP. Protein fold determined by paramagnetic magic-angle spinning solid-state NMR spectroscopy. Nat Chem. 2012;4:410–417. doi: 10.1038/nchem.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tian Y, Schwieters CD, Opella SJ, Marassi FM. AssignFit: a program for simultaneous assignment and structure refinement from solid-state NMR spectra. J Magn Reson. 2012;214:42–50. doi: 10.1016/j.jmr.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Press WH, Teukolsky SA, Vetterling WT, Flannery BP. Numerical recipes: the art of scientific computing. Cambridge, New York: Cambridge University Press; 2007. [Google Scholar]
- 54.Lekien F, Marsden J. Tricubic interpolation in three dimensions. Int J Numer Meth Eng. 2005;63:455–471. [Google Scholar]
- 55.Hu KN, Qiang W, Bermejo GA, Schwieters CD, Tycko R. Restraints on backbone conformations in solid state NMR studies of uniformly labeled proteins from quantitative amide 15N-15N and carbonyl 13C-13C dipolar recoupling data. J Magn Reson. 2012;218:115–127. doi: 10.1016/j.jmr.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schwieters CD, Clore GM. Internal coordinates for molecular dynamics and minimization in structure determination and refinement. J Magn Reson. 2001;152:288–302. doi: 10.1006/jmre.2001.2413. [DOI] [PubMed] [Google Scholar]
- 57.Nilges M, Clore GM, Gronenborn AM. Determination of 3-dimensional structures of proteins from interproton distance data by hybrid distance geometry-dynamical simulated annealing calculations. FEBS Lett. 1988;229:317–324. doi: 10.1016/0014-5793(88)81148-7. [DOI] [PubMed] [Google Scholar]
- 58.Losonczi JA, Andrec M, Fischer MW, Prestegard JH. Order matrix analysis of residual dipolar couplings using singular value decomposition. J Magn Reson. 1999;138:334–342. doi: 10.1006/jmre.1999.1754. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.