

Abstract
Recent mutagenesis studies using the hydrophobic segment of Aβ suggest that aromatic π-stacking interactions may not be critical for fibril formation. We have tested this conjecture by probing the effect of Leu, Ile, and Ala mutation of the aromatic Phe residues at positions 19 and 20, on the double-layer hexametric chains of Aβ fragment Aβ16–22 using explicit solvent all-atom molecular dynamics. As these simulations rely on the accuracy of the utilized force fields, we first evaluated the dynamic and stability dependence on various force fields of small amyloid aggregates. These initial investigations led us to choose AMBER99SB-ILDN as force field in multiple long molecular dynamics simulations of 100 ns that probe the stability of the wild-type and mutants oligomers. Single-point and double-point mutants confirm that size and hydrophobicity are key for the aggregation and stability of the hydrophobic core region (Aβ16–22). This suggests as a venue for designing Aβ aggregation inhibitors the substitution of residues (especially, Phe 19 and 20) in the hydrophobic region (Aβ16–22) with natural and non-natural amino acids of similar size and hydrophobicity.
Keywords: amyloids, force fields, molecular dynamics, hydrophobicity
Introduction
Various degenerative diseases are associated with intercellular or extracellular presence of amyloid fibrils. For instance, Aβ peptide aggregates play a role in the pathogenesis of Alzheimer's disease (AD),1 with small soluble oligomers being the most likely cytotoxic entities.2 An understanding at atomic resolution of the formation of the amyloid oligomers, protofibrils and the factors that modulate aggregation, is crucial for the rational design of new therapeutic strategies to prevent Aβ aggregation into toxic structures.3 In order to design such targeted inhibitors, it is important to understand which features of the primary sequence lead peptides to aggregate in amyloid disorders.
A suitable test system to probe these questions is the segment Aβ16–22, which is among the shortest sequences that form amyloid fibrils in aqueous solutions at neutral pH.4 Proline scanning mutagenesis indicates that the aromatic residues at positions 19 and 20 in the central hydrophobic cluster (Leu17–Val18–Phe19–Phe20–Ala21) are particularly sensitive to replacement5 making this region is a prime target in the design of inhibitors.6, 7 A variety of studies indicate that aromatic residues are important in accelerating amyloidogenic process and for their stabilization. Although aromatic interactions are not crucial for the process of amyloid formation, they can substantially accelerate it, modulating the morphology of the assemblies and reducing the minimal association concentrations.8 It has been previously shown that very short aromatic peptide fragments, even penta- and tetrapeptides, can form amyloid fibrils sharing the same biophysical and structural properties of the assemblies formed by much larger polypeptides. Furthermore, diphenylalanine peptides are known to form well-ordered nanotubular assemblies with amyloid-like structural signatures.8 Adler-Abramovich et al.9 have also demonstrated that oligomers of single amino-acid phenylalanine can form well-ordered fibrillar assemblies at the nano scale. Although the prominent role of the two Phe of Aβ seems to indicate that aromatic π-stacking interactions are critical for fibril formation, this has been put recently to doubt by mutagenesis experiments. Armstrong et al.10 probed the relative contributions of π-stacking interactions versus hydrophobic packing in the aggregation of Aβ1-42 by examining variants in which the Phe at positions 19 and 20 are replaced by Ile and Leu residues, which are similar in sizes and hydrophobicities to Phe but are not capable of π-stacking. Interestingly, both the F19L/F20L and F19I/F20I mutants form amyloid fibrils and do so at higher levels than the wild-type peptide. Hence, not aromatic interactions but rather the hydrophobicity and β-sheet propensity of these residues at position 19 and 20 might be the requirement for the aggregation of Aβ. A similar result was also obtained by Senguen et al.11 on nine variant of Aβ16–22.
Increasingly, molecular dynamic simulations complement experiments as a tool to research the assembly of oligomer and protofibrils, effects of mutation, mechanism of toxicity and inhibition of amyloid aggregate.12–16 In principle, amyloid aggregates and fibril structure can be studied by atomistic molecular dynamics simulations of the aggregation and fibril formation process, including conformational changes, seed formation, and protofilament packing. However, such simulations are often not practical due to the long-time scales involved in seed formation and in the fibrillation process.17 Computationally more feasible are molecular dynamics simulations that start from fibril structural models and test their stability. This approach bypasses the slow fibril formation process and focuses on the chemical interactions that stabilize the fibril. The underlying assumption is that if the model aggregates do not disintegrate during a molecular dynamics run of sufficient length then the aggregates and protofibrils can be considered stable.18
Molecular dynamics simulations rely on empirically parameterized force fields that include explicit solvent.19, 20 Examples are AMBER,21 CHARMM,22 GROMOS,23 and OPLS.24 Discrepancies in simulation result, as the choice of force fields have been documented for a number of systems25–28, and Nguyen et al.29 recently compared the differences in structures and energetic of the monomer, dimer, and trimers of Aβ16–22 resulting from various all-atom force fields. However, although previous explicit solvent all-atom molecular dynamics studies of pre-formed aggregates used a large range of force fields, to our best knowledge, there are no numerical studies that have investigated the effect of the various force fields on the stability of amyloid aggregates. Hence, in order to put our investigations on a firm ground, we start with comparing between various force fields the stability and structural properties of the aggregates taken from fibril structures. We use fully atomic molecular dynamics simulations to investigate the effect of widely used force fields (AMBER 03, AMBER99SB, AMBER99SB-ILDN, CHARMM27, GROMOS96-53a6, and OPLS-AA/L) on structural properties of the Aβ16–22 aggregates from fibril model.
The distribution of Aβ monomers, the early stages of oligomerization, and their dependence on sequence (i.e., mutations) and environment,30–34 the mechanism of Aβ fibril disassembly,35–38 and the early steps of Aβ monomer deposition on fibril fragments39–42 have been studied extensively in silico. A wide range of models including protein coarse-grained lattice,43 off-lattice models,44 and all-atom force fields45 have been used to explore the different stages of oligomerization. It was found that the difference in the oligomerization propensity for different mutants can be related to the change in the stability of Aβ peptides upon mutation.46–48 A key factor is the exposure to solvent.49 Fernandez and Scheraga50 have noted that proteins that aggregate tend to have a significant number of backbone H-bond donors/acceptors exposed to the solvent, available for protein–protein interaction. Computational studies have been essential for understanding this complex interplay of molecular interactions. This is because early self-assembly stages involve transient prefibrillar species (e.g., oligomers) that are difficult to investigate in vivo or in vitro.45, 51 For instance, Rohrig et al.52 have simulated preformed KLVFFAE (Aβ16–22) oligomers, from the dimer to the 32-mer. They found that octamers and larger aggregates are stable on the simulation timescale (20–60 ns) which suggests a critical nucleus size.
Mutational studies of amyloidogenic peptides that utilize conservative aromatic-Leu/Ile mutations (relative to aromatic-Ala) indicate that Leu/Ile containing variants do self-assemble, although at attenuated rates relative to the parent aromatic sequences. The slower rate of aggregation in these types of variants is more consistent with differences in hydrophobicity and β-sheet propensity than with a loss of attractive π–π interactions.11
In this article, we examine the relative importance of attractive π–π interactions vs. hydrophobicity and β-sheet propensity through all-atom molecular dynamics simulations in explicit solvent. This is possible as classical force fields have been shown to be effective in modeling and exploring the dynamics of aromatic interactions in aggregation studies.53–55 Three mutations of Aβ16–22 implicated in the stability of the wild type are studied. Our results confirm that size and hydrophobicity are key for the aggregation and stability of the hydrophobic core region (Aβ16–22). This suggests the substitution of residues (especially, Phe 19 and 20) in the hydrophobic region (Aβ16–22) with such of similar size and hydrophobicity as a venue for designing Aβ aggregation inhibitors.
Results and Discussions
Comparison of the effect of various force fields on the stability of Aβ16–22 aggregates
We run two independent molecular dynamics simulations of the aggregate for each of the six different force fields in order to gain a better understanding of the influence of force fields on structural stability and dynamics of the aggregate models. The individual aggregates are examined using several structural metrics such as root mean square deviation, root mean square fluctuation, radius of gyration, and solvent accessible surface area (shown in Tables I and II and Fig. 1). The root mean square deviation is calculated for backbone heavy atoms against the initial energy-minimized coordinates, its value is listed in Table I for each force field and trajectory.
Table I.
Average Root-Mean-Square-Deviation (in Å) and Radius of Gyration (in Å) During 50 ns MD Simulations of Single Sheet and Double Sheet Models
| Force fields | Run | Cα RMSD (SH1-ST4) | Rg (Å) (SH1-ST4) | Cα RMSD (SH1-ST6) | Rg (Å) (SH1-ST6) | Cα RMSD (SH2-ST4) | Rg (Å) (SH2-ST4) | Cα RMSD (SH2-ST6) | Rg (Å) (SH2-ST6) |
|---|---|---|---|---|---|---|---|---|---|
| AMBER03 | 1 | 1.38(0.57) | 11.26(0.11) | 1.42(0.28) | 11.26(0.11) | 2.10(0.26) | 11.10(0.08) | 1.65(0.25) | 12.85(0.75) |
| 2 | 1.17(0.57) | 11.30(0.10) | 1.40(0.28) | 11.30(0.10) | 2.53(0.38) | 11.10(0.07) | 2.06(0.37) | 12.85(0.90) | |
| Average | 1.28 | 11.28 | 1.41 | 11.28 | 2.31 | 11.28 | 1.86 | 12.85 | |
| AMBER99SB | 1 | 1.00(0.20) | 11.30(0.09) | 1.18(0.27) | 11.30(0.09) | 2.36(0.59) | 11.04(0.10) | 1.54(0.26) | 12.76(0.46) |
| 2 | 1.00(0.25) | 11.34(0.09) | 1.15(0.26) | 11.34(0.09) | 1.52(0.18) | 11.10(0.59) | 1.65(0.19) | 12.68(0.69) | |
| Average | 1.00 | 11.32 | 1.17 | 11.32 | 1.94 | 11.32 | 1.60 | 12.72 | |
| AMBER99SB- ILDN | 1 | 3.23(1.94) | 11.34(0.10) | 1.17(0.28) | 11.34(0.10) | 3.40(0.93) | 11.22(0.10) | 1.66(0.23) | 12.76(0.05) |
| 2 | 1.05(0.28) | 11.29(0.09) | 1.16(0.28) | 11.29(0.09) | 1.60(0.28) | 11.10(0.06) | 1.59(0.22) | 12.73(0.06) | |
| Average | 2.14 | 11.32 | 1.17 | 11.31 | 2.5 | 11.31 | 1.62 | 12.74 | |
| CHARMM27 | 1 | 3.51(1.08) | 11.15(0.14) | 3.31(1.74) | 11.15(0.14) | 3.71(0.83) | 11.26(0.23) | 1.87(0.44) | 12.56(0.08) |
| 2 | 4.87(1.14) | 11.10(0.18) | 3.91(1.11) | 11.10(0.18) | 4.01(0.54) | 11.11(0.10) | 1.80(0.17) | 12.51(0.08) | |
| Average | 4.19 | 11.25 | 3.61 | 11.12 | 3.86 | 11.12 | 1.83 | 12.53 | |
| GROMOS96– 53a6 | 1 | 2.30(0.61) | 10.97(0.12) | 2.54(0.95) | 10.97(0.12) | 3.65(0.70) | 10.98(0.12) | 2.50(0.87) | 12.61(0.08) |
| 2 | 2.23(0.53) | 10.90(0.12) | 2.98(0.82) | 10.90(0.12) | 4.08(0.62) | 10.99(0.09) | 2.39(0.79) | 12.60(0.08) | |
| Average | 2.27 | 10.93 | 2.76 | 10.93 | 3.87 | 10.93 | 2.44 | 12.61 | |
| OPLS-AA/L | 1 | 3.36(0.62) | 10.81(0.23) | 5.09(1.59) | 10.81(0.23) | 3.25(0.48) | 10.93(0.07) | 2.09(0.28) | 12.67(0.06) |
| 2 | 3.39(0.68) | 10.74(0.20) | 5.30(1.43) | 10.74(0.20) | 3.29(0.32) | 10.90(0.06) | 2.24(0.22) | 12.60(0.08) | |
| Average | 3.38 | 10.77 | 5.19 | 10.77 | 3.27 | 10.77 | 2.17 | 12.63 | |
Mean values are calculated by using the two average values obtained for each model.
Table II.
Solvent Accessible Surface Area (nm2) of the Double Sheets and Six Strands Per Sheet of Aβ16–22 Model Under Various Force Fields
| Force fields | Run | SASA hydrophobic (g_sas calculation) | SASA hydrophilic (g_sas calculation) | SASA hydrophobic (sum over per residue) | SASA hydrophilic (sum over per residue) |
|---|---|---|---|---|---|
| AMBER03 | 1 | 43.77(1.24) | 20.71(0.78) | 27.49 | 47.27 |
| 2 | 44.02(1.56) | 20.97(0.99) | 27.93 | 48.77 | |
| Average | 48.89 | 20.84 | 27.71 | 48.02 | |
| AMBER99SB | 1 | 31.74(0.82) | 31.48(0.86) | 25.14 | 48.98 |
| 2 | 29.71(1.22) | 31.37(0.86) | 24.89 | 46.08 | |
| Average | 30.75 | 31.42 | 25.01 | 47.53 | |
| AMBER99SB-ILDN | 1 | 31.11(0.88) | 31.60(0.83) | 24.99 | 50.64 |
| 2 | 31.23(1.12) | 31.32(0.99) | 25.50 | 49.54 | |
| Average | 31.17 | 31.46 | 25.24 | 50.09 | |
| CHARMM27 | 1 | 19.45(0.96) | 40.41(1.17) | 29.28 | 47.99 |
| 2 | 18.93(1.15) | 40.18(0.95) | 27.36 | 48.14 | |
| Average | 19.19 | 40.30 | 28.32 | 48.09 | |
| GROMOS96–53a6 | 1 | 39.30(1.27) | 20.34(0.78) | 24.69 | 50.51 |
| 2 | 39.12(1.44) | 20.41(0.94) | 26.49 | 51.28 | |
| Average | 39.21 | 30.37 | 25.59 | 50.89 | |
| OPLS-AA/L | 1 | 43.14(1.17) | 18.29(0.86) | 24.40 | 49.30 |
| 2 | 43.56(1.09) | 17.78(1.06) | 24.20 | 44.25 | |
| Average | 43.35 | 18.04 | 24.30 | 46.78 |
Figure 1.
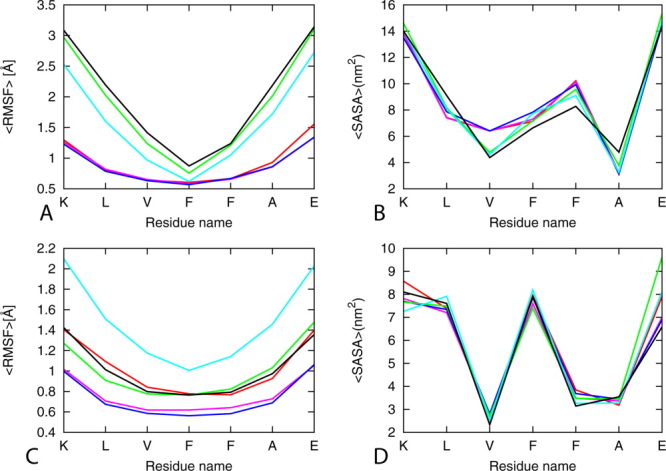
Average RMSF as a function of residue (left panels) and SASA per residue (right panels) of single layer and double Aβ16-22 models (6 stranded single and double layers). The results are calculated by using the two trajectories for each model using the force fields. Red, A03; pink, A99SB; blue, A99SB-ILDN; green, Charmm27; cyano, G95-53a6, black; OPLS-AA/L. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
We find that trajectories of simulations that rely on a version of AMBER force fields have smaller root mean square deviation from the initial configuration than such relying on one of the other force fields. Hence, in the simulations that rely on AMBER force fields, the peptide stays close to the initial conformation indicating that the Aβ16–22 aggregates are more stable. The largest root mean square deviation is found in the simulation performed using GROMOS96-53a6; the large instability of the aggregate is probably due to the united-atom approximation. The difference among the force fields becomes smaller as the size of the double-sheet aggregates increases suggesting that differences in stability decrease with increasing size of the system (Table I).
The radius of gyration of a system measures its compactness. Increasing values indicate an expanding conformation and decreasing values indicate shrinking conformation. We find that the average radius of gyration in simulations with AMBER force fields is larger than in such that rely on the GROMOS96-53a6 and OPLS-AA/L (Table I). As the root mean square deviation differences between the force fields decrease as the size of the aggregate model (i.e. SH2-ST6) grows (Table I).
We use the root mean square fluctuation to describe movement of individual atomic positions relative to the first frame, which is a measure for the flexibility of structures. The root mean square fluctuations for Cα atoms of the Aβ16–22 aggregates of our SH2-ST6 model in the six different force fields are shown in Figure 1, where higher value of RMSF indicates higher flexibility. Values for most of the residues, ignoring the terminal residues, are around 1 Å for the simulation with AMBER03, AMBER99SB, and AMBER99SB-ILDN, but the CHARMM27 force field, and even more pronounced the OPLS-AA/L GROMOS96 force fields, have larger fluctuations.
The solvent accessible surface area is defined by the surface area of the atoms in the peptide that is accessible by a water probe with a radius of 1.4 Å.56 Its value depends on the interactions of hydrophobic and hydrophilic amino acids with water, and the surface tension near the protein–solvent interface. Measured solvent accessible surface area values for each force field are listed in Table II, separated into contribution from polar and non-polar residues. The only noticeable difference is that in CHARMM simulations, the value for hydrophobic residues is lower than the ones observed in simulation relying on the other force fields, while the exposed surface area of the hydrophilic residues is higher in CHARMM simulations.
In the analysis tools in GROMACS packages for calculating SASA, the g_sas module, the polar and non-polar surface areas are often defined through atomic partial charges taken from the utilized molecular potential. These atomic partial charges differ significantly between force fields. In order to avoid this force field dependence, we have recalculated the mean polar and non-polar solvent accessible surface areas, summing up contributions not according to partial charges of atoms but according to whether these atoms belong to hydrophobic or hydrophilic residues. This can be done by using the per residues option in the g_sas tool. The recalculated values are reported again in Table II. Now, note that the solvent accessible surface values differ little between the various force fields. Hence, calculation of mean polar and non-polar solvent accessible surface area according to atomic partial charges is misleading and could lead to wrong conclusions. For instance, Todorova et al.25 reported a strong deviation in the hydrophobic/hydrophilic solvent accessible surface areas of insulin, measured with g_sas tool of GROMACS package, between various force fields. The solvent accessible surface area of hydrophobic residues was smaller in CHARMM simulations when compared with corresponding structures produced by the other force fields, while the exposed area of the hydrophilic residues was higher. The deviation can be explained again by differences in point charges between various force fields, that is, an artifact of incorrect use of the g_sas tool of GROMACS.
The solvent accessible surfaces of the terminal amino acids are much larger and do not depend on the force fields, indicating that they are largely exposed to the solvent (Fig. 1). The central residues that have the lowest root mean square fluctuation are also the residues that have the lowest solvent exposed area (Fig. 1) as they are buried in and thus protected from solvent. Comparing the average per residue solvent accessible surface area of the single sheet with six strands model with the double sheet with six strands model indicates that the double layer is favored by the partial desolvation of the peptides. This is predicted by all the tested force fields (Fig. 1).
In summary, a comparison of the various force fields indicates that one should test systematically force fields for their suitability for the system under investigation before using them for modeling amyloid aggregates derived from experimental structure. In our case, all monitored quantities indicate that the latest AMBER force field, AMBER99SB-ILDN, preserves best the integrity of the aggregate; and therefore, it is chosen by us in the following investigations.
Stability and aggregation tendency of wild type and mutants
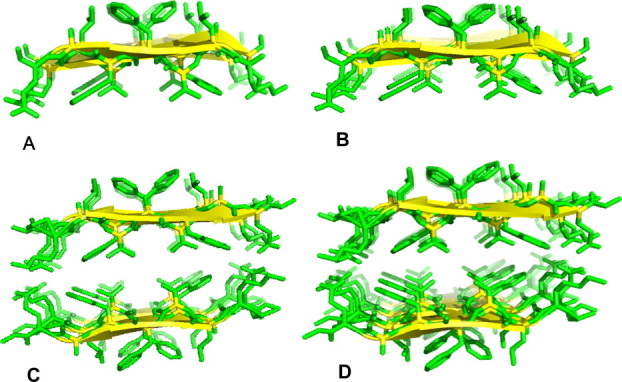
The stability and dynamics of the wild type and mutants systems are assessed by changes in root mean square deviation, the radius of gyration, root mean square fluctuation, solvent accessible surface area, end-to-end distance, inter-sheet distances, and number of native contacts. Our analysis relies on 100 ns of the molecular dynamics trajectory. The final configurations from the four independent simulation for the wild type and mutants (Fig. 2) differ from each other by either slight changes (for the double mutants 19I20I and 19L20L) or broad reductions in the degree of order in the aggregates (in the case of the mutant 19A) relative to the wild-type model.
Figure 2.

The initial and final configurations of the fibrils at the end of the (100 ns) MD simulations performed of Aβ16-22 double-sheet wild-type and mutant models. (A) Wild-type model with sequence KLVFFAE16-22, (B) variant with F19 substituted with alanine, (C) double substitution F19I/F20I, (D) double substitution F19L/F20L. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Root mean square deviation measures the fluctuations of protein atoms with respect to the reference structure, here, the first conformation from the production simulations obtained at 0 ns. It remains below 1.5 Å for the wild type. For the two mutants models (19I20I and 19L20L), the root mean square deviation to the reference structure stayed below 4.0 Å within the first 40 ns, indicating relative stability of the structures when compared with the 19A mutant that exhibited large fluctuations up to 7.0 Å [Fig. 3(A)]. Hence, replacement of Phe at position 19 by Ala destabilizes the aggregate while the double Leu and Ile mutants remain stable.
Figure 3.
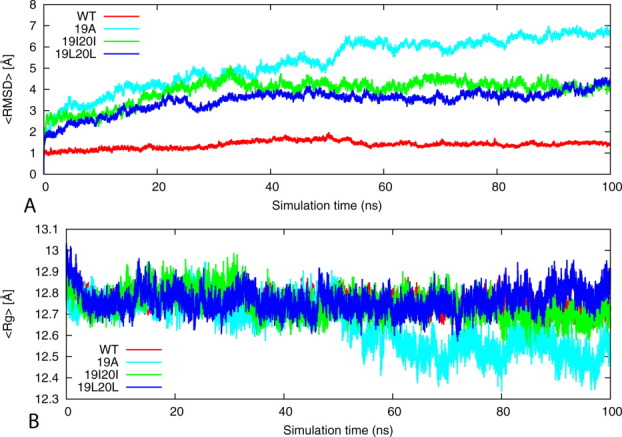
Root mean square deviation (RMSD) and Radius of gyration (Rg) of the wild type KLVFFAE16-22 and its mutants. (A) Time evolution of the backbone RMSD of the wild-type KLVFFAE16-22 segments of Aβ peptide and its phenylalanine to alanine, isoleucine, and leucine mutants of double-layer sheet six strands per sheet models during the 100 ns MD simulations. (B) Variation of the Rg with time in the wild-type and mutant models. The results are calculated by using the four trajectories for each model. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
The time evolution of the radius of gyration for the wild type and mutants is displayed in Figure 3(B). Values of the wild type and double mutants (19I20I and 19L20L) remain around 12.7 to 12.7 Å, indicating that these models are stable [Fig. 3(B)]. In the simulations of 19A mutant, we find that the aggregate radius of gyration is decreasing by more than 3 Å. The result is consistent with the measurements of root mean square deviation.
In order to investigate peptide fluctuations, we have measured the standard deviation of Cα carbons in a residue from averaged position during the 100 ns simulation time. These root mean square fluctuations are calculated as an average of four independent simulations [Fig. 4(A)]. Its value is largest for the single alanine mutant 19A and about twice as large as the wild type. In general, hydrophobic residues have smaller values as they are more buried in the core where their motions are more restricted. The edge N-terminal, C-terminal hydrophilic residues have much larger values than central residues because they tend to be more exposed on the surface where they can have greater mobility.
Figure 4.
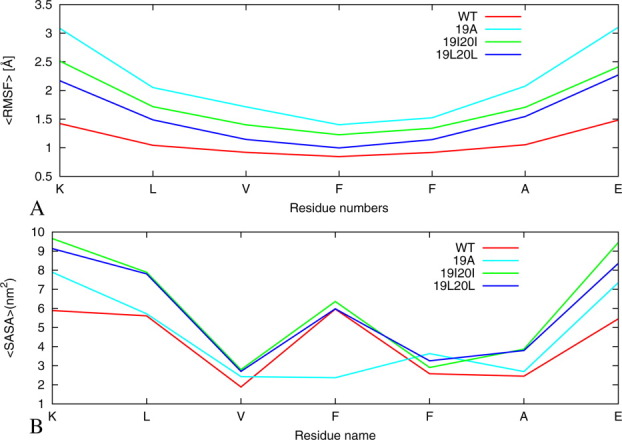
Comparison of all-atom root mean square deviation and solvent accessible surface areas of the wild-type KLVFFAE16-22 and its mutants. (A) The RMSF for the two-layer/six-strand models of the wild type and its mutants are average values for hexamers averaged for four trajectories. (B) The average SASA per amino acid of six β-strands double-sheet model of wild-type and its mutants computed on backbone atoms as an average of four trajectory 100 ns each. The results are calculated by using the four trajectories for each model. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
An exposed hydrophobic surface area is thought to be crucial to the peptide's ability to recognize and adhere to the fibril end.57, 58 Amino acids that are buried inside (shielded from the solvent) have lower solvent accessible surface area than amino acids exposed to the solvent. The exposure of polar residues is energetically favorable, whereas for hydrophobic residues, it typically increases the free energy of the system. Therefore, the solvent accessible surface area is a useful quantity to evaluate the stability of protein conformations. We plot the solvent accessible surface area per amino acid for the wild type and mutants in Figure 4(B). It is defined again as the surface area of the atoms in the peptide that is accessible by water probe with a radius of 1.4 Å. The solvation properties of the wild type and the two mutants 19I20I and 19L20L are similar. Residues V18, F20 (I20 and L20 in mutants 19I20I and 19L20L) have low solvent-accessible area, whereas K16, L17, and 19F (I20 and L20 in mutants 19I20I and 19L20L) are largely exposed. On the other hand, the hydrophobic region (Val18, Ala19, Phe20, and 21Ala) of the A19F mutant has little solvent exposure, especially when compared with the 19I20I and 19L20L mutated peptides, which in turn have similar values to the wild-type Aβ16–22. Hence, our simulations indicate that in the wild-type and both 19I20I and 19L20L mutants, the hydrophobic residues Val18–21Ala are more exposed to water than those of the 19A mutant [Fig. 4(B)]. We conjecture that the low solvent exposure of the hydrophobic core region (Val18–21Ala) in the 19A mutant reduces the interactions with “like” peptides and contributes to the slower fibril formation.10, 11 In contrast, in the wild-type and the other two mutant systems, the hydrophobic region is more exposed to solvent. This increases the probability of these residues to interact with other hydrophobic residues facilitating aggregation and possibly leading to faster formation of fibrils as observed in previous experiments.10, 11
Aβ16–22 has positively charged Lys at the N-terminus and negatively charged Glu at the C-terminus. The anti-parallel orientation is favored by electrostatic interactions between the C- and N-termini of neighboring molecules in β-sheet salt-bridges between positively charged Lys and negatively charged Glu side chains. These electrostatic interactions are most important for the stability of Aβ16–22 organization.59 The salt bridge distance is calculated as the averaged distance of N atom of the NH3+ in Lys 16 to the C=O bonds carboxyl group of Glu22. Direct salt bridges are assumed to be around 4.3 Å, whereas indirect or water-mediated salt bridges have a distance between 4.3 and 7.0 Å. In our case, the distance between Lys 16 and Glu 22 remained relatively stable and below the salt bridge distance threshold for the wild type (∼6 Å) 19I20I and 19L20L (7Å) mutants, leading to the association of the adjacent β-strands. On the other hand, no salt bridge between Lys 16 and Glu 22 is observed in the 19A mutant where the distance increased from about 6 to 10 Å during the course of simulation [Fig. 5(A)].
Figure 5.
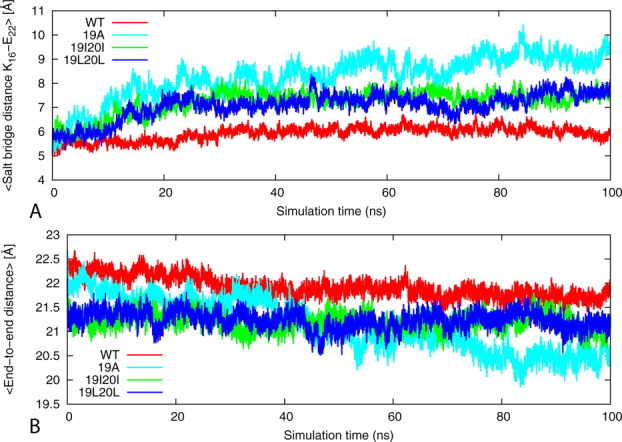
Comparison of the average intra-chain salt bridge distance and end-to-end distance. (A) The average intra-chain salt bridge distance (Lysn16/Glun-122) along the 100 ns simulation of the wild-type KLVFFAE16-22 and its mutants. The results are the average of four independent salutation of each system. (B) The average end-to-end distance along the 100 ns simulation of the wild-type KLVFFAE16-22 and its mutants. The results are the average of four independent simulations of each system. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
The end-to-end distance is calculated using g_dist tool in Gromacs.56 The average end-to-end distance of the peptides is taken as the distance between the centers of mass of the N-terminal amino acid Lys16 and the C-terminal residues of Glu22. The end-to-end distance varies considerably in the 19A from the initial 22 to 20 Å (with a reduction of about 2 Å) with shrinkage of the strand due to the folding of the strands upon itself [Figs. 2 and 5(B)]. The end-to-end distance of the strands in the wild type and 19I20I and 19L20L remain within the range of 22 to 21.5 Å, which can be compared with 22 Å for the initial energy minimized structure conforming that the three models are relatively stable than the 19A mutant [Fig. 5(B)].
In order to explore further the interaction responsible for the stability of the wild type and mutants, we have calculated also the percentage of native contacts defined as those present in the 0-ps time frame with the distance between the center mass of two side chains less than 6.0 Å. The reference structure for calculating the percentage of native contacts at 330 K is calculated based on the corresponding energy-minimized structures. The percentage of native contacts for wild type and mutants are shown in Figure 6(A). The result shows that the relatively stable systems (wild type, 19I20I, and 19L20L) maintain 80% to 60% of their native contacts, whereas the unstable system of mutant 19A loses 70% of the native contacts after 40 ns. Note that the native contacts for double mutants (19I20I and 19 L 20 L) are slightly lower than that of the wild type but larger than the single mutant 19A. This confirms that replacement of F19 and F20 with Ile and Leu (with similar size and hydrophobicity as Phe) keeps the aggregates stable.
Figure 6.
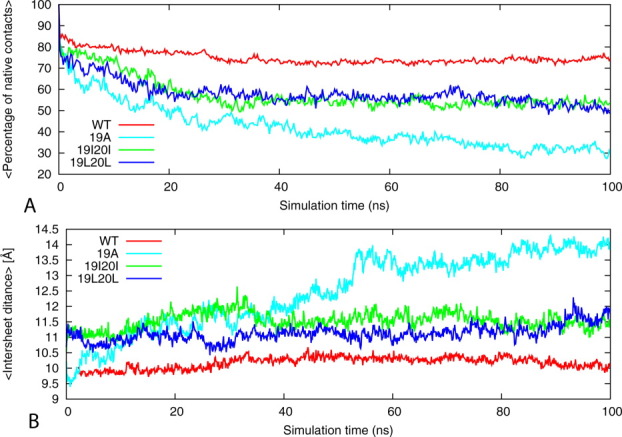
Variation of the percentage native contacts and distance between sheets. (A) The time evolution of sheet-to-sheet distances of wild-type KLVFFAE16-21 segments of Aβ peptide and its phenylalanine to alanine, isoleucine, and leucine mutants of double-layer sheet six strands per sheet models during the 100-ns MD simulations. (B) Percentage of native contacts, with respect to the energy minimized structure as a function of time. The results were calculated by using the four trajectories for each model. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
We assess the stability of the sheet-to-sheet associations' of the double-layered forms of the wild type and mutants by monitoring the inter-sheet distance across the interface. Figure 6(B) shows the averaged distances between the mass centers of two facing β-sheets. The wild type has an inter-sheet distance of about 10.5 Å. Its value for the two mutants 19I20I and 19L20L (between 10 and 11.5 Å) is smaller than one for the 19A mutant (ranging between 9.55 and 14 Å). This is due to the reduced hydrophobic interactions at interface. Hence, the stabilization of the sheet to sheet association is due to good geometrical fit between side chains at the interface leading to a favorable interaction that favors tight packing between β-sheets. The inter-sheet distance for the single residue mutation, 19A, grows in the first 50 ns from an initial of 9.5Å to 13.5 Å suggesting that the F19A mutation leads to disaggregation. The correlation between the stability of the aggregates, number of native contacts, and inter-sheet distance indicates that the inter-sheet interactions play an important role in stabilizing the aggregates.
Conclusion
We have tested for several molecular mechanical potentials their relative accuracy in simulations of a preformed β-sheet aggregate derived from an experimental model of Aβ16–22. The differences between the various force fields decrease with size of the systems; however, for small-sized aggregates, the structural differences are significant. Our results suggest that the AMBER99SB-ILDN is most suitable for our study of the effect of hydrophobicity of amino acid in the hydrophobic core. Using this force field in our simulations, we have researched a series of amyloid fibril models built from wild-type and three mutant sequences, F19A, F19L/F20L, and F19I/F20I. Although all three mutants disable attractive π–π interactions, they differ in hydrophobicity and β-sheet propensity. In the mutant F19A, these two quantities are reduced while enlarged in F19L/F20L and F19I/F20I. In our simulation, these differences lead to a reduced aggregation propensity for the mutant F19A. On the other hand, in the mutants F19L/F20L and F19I/F20I, the increase in peptide hydrophobicity [Phe (1.79), Ile (1,8), Leu(1.70)]60 and β-sheet propensity of [Phe (1.43), Ile (1,8), Leu (1.70)]61 enhances exposure of the hydrophobic region, and their probability to interact with other hydrophobic residues. The resulting larger number of nucleation events leads to a greater number of fibrils in solution. However, note that the relation between hydrophobicity and stability of aggregates is complex. Takeda and Klimov40 have shown that moderate reduction in hydrophobicity can in some circumstance lead to increased stability, while strong reduction destabilizes them. However, even with this caveat, the results from our molecular dynamics simulations explain the pervious experimental observation that size and hydrophobicity rather than aromatic π-stacking interactions are the key element in determining the aggregation tendency of mutants. Our results suggest considering size similarity and hydrophobicity as key factors in molecular modeling-guided design of Aβ aggregation inhibitors.
Methods and Models
The KLVFFAE16–22 aggregates are derived from the fibril model of Aβ reported by Eisenberg group62 and are based on microcrystal structures of KLVFFAE16–21 (pdb code, 3OW9) and the NMR structure of the fibril.63 For force field comparison, we use both single and double sheets aggregates of the wild type (the details are shown in Table III). Starting configurations of the mutants are built from the wild type62 by replacing the side chains of the targeted residues with alanine, isoleucine, and leucine.
Table III.
Summary of Simulated System and Condition of the Molecular Dynamics Simulation of Aβ16–22 and Its Mutants
| System | No. of atoms | No. of water molecules | Simulation box size (A°) | Simulated time, ns | T (K) | Strand/Sheet organization |
|---|---|---|---|---|---|---|
| Wild type (one sheet and two sheet models) | ||||||
| 2 Sheet, 6 strands | 1608 | 8726 | 65.45 × 65.45 × 65.45 | 50 ns (2) | 310 | Anti-parallel/Anti-parallel |
| 2 Sheet, 4 strands | 1072 | 6738 | 59.87 × 59.87 × 59.87 | 50 ns (2) | 310 | Anti-parallel/Anti-parallel |
| 1 Sheet, 6 strands | 804 | 7111 | 60.47 × 60.47 × 60.47 | 50 ns (2) | 310 | Anti-parallel |
| 1 Sheet, 4 strands | 536 | 5104 | 54.38 × 54.38 × 54.38 | 50 ns (2) | 310 | Anti-parallel |
| Wild type and mutants (two sheet, six strands models) | ||||||
| WT | 1608 | 8726 | 65.45 × 65.45 × 65.45 | 100 ns (4) | 350 | Anti-parallel/Anti-parallel |
| 19A | 1488 | 8808 | 65.45 × 65.45 × 65.45 | 100 ns (4) | 350 | Anti-parallel/Anti-parallel |
| 19I20I | 1584 | 8793 | 65.45 × 65.45 × 65.45 | 100 ns (4) | 350 | Anti-parallel/Anti-parallel |
| 19L20L | 1584 | 8800 | 65.45 × 65.45 × 65.45 | 100 ns (4) | 350 | Anti-parallel/Anti-parallel |
In order to compare the AMBER 03/TIP3P, AMBER99SB/TIP3P, AMBER99SB-ILDN/TIP3P, CHARMM27/TIP3P, GROMOS96-53a6/SPC, and OPLS-AAL/TIP3P energy functions, we run two independent molecular dynamics simulations for each of the four models of the Aβ16–22 peptide (Fig. 7) using the GROMACS program version 4.5.323 and a time step of 2 fs. Note that the water models depend on the tested force field; we have chosen the recommended water models for each force field. Hydrogen atoms are added using the pdb2gmx module of the GROMACS suite. The starting configurations of all four proteins are set in the center of a cubical box where the distance between the solute and the edge of the box is at least 12 Å. Periodic boundary conditions are employed, and electrostatic interactions are calculated with the PME algorithm.64, 65 Hydrogen atoms are constrained using the LINCS66 algorithm, and for water, the Settle algorithm is used.67 The temperature of 310 K is kept constant by the V-rescale algorithm68 (τ = 0.1 fs) and pressure with the Parrinello–Rahman algorithm69 (τ = 1 fs) is 1 bar.
Figure 7.
The atomic representation of the initial energy minimized structure of the Aβ16-22 single-sheet and double-sheet models: (A) single sheet, 4 strand; (B) single sheet, 6 strand; (C) double sheet, 4 strand; and (D) double sheet, 6 strand. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
The solvated start configuration is first energy minimized using the steepest descent method, followed by conjugate gradient. Afterward, the system is equilibrated in two steps of 500 ps, the first step in an NVT ensemble and the second phase in an NPT ensemble at 1 bar. After equilibration, 50 ns of trajectories are analyzed for each system to examine the structural changes of the oligomers aggregates. Data are saved at 4.0 ps intervals for further analysis. Two independent simulations with different initial velocity distributions are performed for each system to test for thermalization and guarantee at least two independent sets of measurements.
For each mutant and the wild type (two sheets with six strands per sheets, see Table III), we run four independent molecular dynamics simulations of 100 ns using the molecular dynamics protocol described above. These simulations rely on the AMBER ff99SB-ILDN70 force field. The temperature of 330 K is chosen as a compromise between experimental stability of the amyloid fibrils71 and thermally enhanced sampling.12, 13
The molecular dynamics trajectories are analyzed using the tool set of the GROMACS package. Especially, we monitor conformational changes and stability of the aggregates through the time evolution of the root mean square deviations of the Cα atoms, radius of gyration, distance between the two sheets, persistence of salt bridge, end-to-end distance, and native contacts. Structural changes are visualized with the help of Visual Molecular Dynamic (VMD) software version 1.9.72
Acknowledgments
Graphs were made using gnuplot (http://www.gnuplot.org).
References
- 1.Glenner GG, Wong CW. Alzheimers disease—initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 2.Kirkitadze MD, Bitan G, Teplow DB. Paradigm shifts in Alzheimer's disease and other neuro degenerative disorders: the emerging role of oligomeric assemblies. J Neurosci Res. 2002;69:567–577. doi: 10.1002/jnr.10328. [DOI] [PubMed] [Google Scholar]
- 3.Eisenberg DJM. The amyloid state of proteins in human diseases. Cell. 2012;148:1188–1203. doi: 10.1016/j.cell.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balbach JJ, Ishii Y, Antzutkin ON, Leapman RD, Rizzo NW, Dyda F, Reed J, Tycko R. Amyloid fibril formation by A beta(16–22), a seven-residue fragment of the Alzheimer's beta-amyloid peptide, and structural characterization by solid state NMR. Biochemistry. 2000;39:13748–13759. doi: 10.1021/bi0011330. [DOI] [PubMed] [Google Scholar]
- 5.Williams AD, Portelius E, Kheterpal I, Guo JT, Cook KD, Xu Y, Wetzel R. Mapping A beta amyloid fibril secondary structure using scanning proline mutagenesis. J Mol Biol. 2004;335:833–842. doi: 10.1016/j.jmb.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 6.Findeis MA, Musso GM, Arico-Muendel CC, Benjamin HW, Hundal AM, Lee JJ, Chin J, Kelley M, Wakefield J, Hayward NJ, Molineaux SM. Modified-peptide inhibitors of amyloid beta-peptide polymerization. Biochemistry. 1999;38:6791–6800. doi: 10.1021/bi982824n. [DOI] [PubMed] [Google Scholar]
- 7.Soto C, Sigurdsson EM, Morelli L, Kumar RA, Castano EM, Frangione B. Beta-sheet breaker peptides inhibit fibrillogenesis in a rat brain model of amyloidosis: implications for Alzheimer's therapy. Nat Med. 1998;4:822–826. doi: 10.1038/nm0798-822. [DOI] [PubMed] [Google Scholar]
- 8.Gazit E. Self assembly of short aromatic peptides into amyloid fibrils and related nanostructures. Prion. 2007;1:32–35. doi: 10.4161/pri.1.1.4095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adler-Abramovich L, Vaks L, Carny O, Trudler D, Magno A, Caflisch A, Frenkel D, Gazit E. Phenylalanine assembly into toxic fibrils suggests amyloid etiology in phenylketonuria. Nat Chem Biol. 2012;8:701–706. doi: 10.1038/nchembio.1002. [DOI] [PubMed] [Google Scholar]
- 10.Armstrong AH, Chen J, McKoy AF, Hecht MH. Mutations that replace aromatic side chains promote aggregation of the Alzheimer's A beta peptide. Biochemistry. 2011;50:4058–4067. doi: 10.1021/bi200268w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Senguen FT, Doran TM, Anderson EA, Nilsson BL. Clarifying the influence of core amino acid hydrophobicity, secondary structure propensity, and molecular volume on amyloid-beta 16–22 self-assembly. Mol Biosyst. 2011;7:497–510. doi: 10.1039/c0mb00210k. [DOI] [PubMed] [Google Scholar]
- 12.Berhanu WM, Masunov AM. Unique example of amyloid aggregates stabilized by main chain H-bond instead of the steric zipper: molecular dynamics study of the amyloidogenic segment of amylin wild-type and mutants. J Mol Model. 2012;18:891–903. doi: 10.1007/s00894-011-1030-7. [DOI] [PubMed] [Google Scholar]
- 13.Berhanu WM, Masunov AM. Controlling the aggregation and rate of release in order to improve insulin formulation: molecular dynamics study of full-length insulin amyloid oligomer models. J Mol Model. 2012;18:1129–1142. doi: 10.1007/s00894-011-1123-3. [DOI] [PubMed] [Google Scholar]
- 14.Workalemahu M, Berhanu AM, Masunov AE. Alternative packing modes leading to amyloid polymorphism in five fragments studied with molecular dynamics. Peptide Sci. 2012;98:131–144. doi: 10.1002/bip.21731. [DOI] [PubMed] [Google Scholar]
- 15.Berhanu WM, Masunov AE. Natural polyphenols as inhibitors of amyloid aggregation. Molecular dynamics study of GNNQQNY heptapeptide decamer. Biophys Chem. 2010;149:12–21. doi: 10.1016/j.bpc.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 16.Berhanu WM, Masunov AE. Can molecular dynamics simulations assist in design of specific inhibitors and imaging agents of amyloid aggregation? Structure, stability and free energy predictions for amyloid oligomers of VQIVYK, MVGGVV and LYQLEN. J Mol Model. 2011;1:2423–2442. doi: 10.1007/s00894-010-0912-4. [DOI] [PubMed] [Google Scholar]
- 17.Friedman R. Aggregation of amyloids in a cellular context: modelling and experiment. Biochem J. 2011;438:415–426. doi: 10.1042/BJ20110369. [DOI] [PubMed] [Google Scholar]
- 18.Jun-tao Guo CKH, Xu Y, Wetzel R. Modeling protein aggregate assembly and structure. In: Ying Xu, Dong Xu, Jie Liang., editors. Computational Methods for Protein Structure Prediction and Modeling. New York: Springer; 2007. pp. 279–318. [Google Scholar]
- 19.Adcock SA, McCammon JA. Molecular dynamics: Survey of methods for simulating the activity of proteins. Chem Rev. 2006;106:1589–1615. doi: 10.1021/cr040426m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Durrant JD, McCammon JA. Molecular dynamics simulations and drug discovery. BMC Biol. 2011;9:9. doi: 10.1186/1741-7007-9-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 23.Christen M, Hunenberger PH, Bakowies D, Baron R, Burgi R, Geerke DP, Heinz TN, Kastenholz MA, Krautler V, Oostenbrink C, Peter C, Trzesniak D, Van Gunsteren WF. The GROMOS software for biomolecular simulation: GROMOS05. J Comput Chem. 2005;26:1719–1751. doi: 10.1002/jcc.20303. [DOI] [PubMed] [Google Scholar]
- 24.Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J Phys Chem B. 2001;105:6474–6487. [Google Scholar]
- 25.Todorova N, Legge FS, Treutlein H, Yarovsky I. Systematic comparison of empirical forcefields for molecular dynamic simulation of insulin. J Phys Chem B. 2008;112:11137–11146. doi: 10.1021/jp076825d. [DOI] [PubMed] [Google Scholar]
- 26.Feig M, MacKerell AD, Brooks CL. Force field influence on the observation of pi-helical protein structures in molecular dynamics simulations. J Phys Chem B. 2003;107:2831–2836. [Google Scholar]
- 27.Mu YG, Kosov DS, Stock G. Conformational dynamics of trialanine in water. 2. Comparison of AMBER, CHARMM, GROMOS, and OPLS force fields to NMR and infrared experiments. J Phys Chem B. 2003;107:5064–5073. [Google Scholar]
- 28.Yoda T, Sugita Y, Okamoto Y. Comparisons of force fields for proteins by generalized-ensemble simulations. Chem Phys Lett. 2004;386:460–467. [Google Scholar]
- 29.Nguyen PH, Li MS, Derreumaux P. Effects of all-atom force fields on amyloid oligomerization: replica exchange molecular dynamics simulations of the A beta(16–22) dimer and trimer. Phys Chem Chem Phys. 2011;13:9778–9788. doi: 10.1039/c1cp20323a. [DOI] [PubMed] [Google Scholar]
- 30.Anand P, Hansmann UHE. Internal and environmental effects on folding and dimerisation of Alzheimer's beta-amyloid peptide. Mol Simulat. 2011;37:440–448. doi: 10.1080/08927022.2011.551879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huet A, Derreumaux P. Impact of the mutation A21G (Flemish variant) on Alzheimer's beta-amyloid dimers by molecular dynamics simulations. Biophys J. 2006;91:3829–3840. doi: 10.1529/biophysj.106.090993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baumketner A, Krone MG, Shea JE. Role of the familial Dutch mutation E22Q in the folding and aggregation of the 15–28 fragment of the Alzheimer amyloid-beta protein. Proc Natl Acad Sci USA. 2008;105:6027–6032. doi: 10.1073/pnas.0708193105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fawzi NL, Kohlstedt KL, Okabe Y, Head-Gordon T. Protofibril assemblies of the arctic, Dutch, and Flemish mutants of the Alzheimer's A beta(1–40) peptide. Biophys J. 2008;94:2007–2016. doi: 10.1529/biophysj.107.121467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berhanu WMHU. Structure and dynamics of amyloid-β segmental polymorphisms. PLoS One. 2012;7:e41479. doi: 10.1371/journal.pone.0041479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chebaro Y, Derreumaux P. Targeting the early steps of A beta 16–22 protofibril disassembly by N-methylated inhibitors: A numerical study. Proteins. 2009;75:442–452. doi: 10.1002/prot.22254. [DOI] [PubMed] [Google Scholar]
- 36.Takeda T, Klimov DK. Temperature-induced dissociation of A beta monomers from amyloid fibril. Biophys J. 2008;95:1758–1772. doi: 10.1529/biophysj.108.131698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takeda T, Klimov DK. Dissociation of A beta(16–22) amyloid fibrils probed by molecular dynamics. J Mol Biol. 2007;368:1202–1213. doi: 10.1016/j.jmb.2007.02.066. [DOI] [PubMed] [Google Scholar]
- 38.Chebaro YJP, Zang T, Mu Y, Nguyen PH, Mousseau N, Derreumaux P. Structures of Aβ17−42 trimers in isolation and with five small-molecule drugs using a hierarchical computational procedure. J Phys Chem B. 2012;116:8412–8422. doi: 10.1021/jp2118778. [DOI] [PubMed] [Google Scholar]
- 39.Takeda T, Klimov DK. Replica exchange simulations of the thermodynamics of a beta fibril growth. Biophys J. 2009;96:442–452. doi: 10.1016/j.bpj.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takeda T, Klimov DK. Probing energetics of a beta fibril elongation by molecular dynamics simulations. Biophys J. 2009;96:4428–4437. doi: 10.1016/j.bpj.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Han M, Hansmann UHE. Replica exchange molecular dynamics of the thermodynamics of fibril growth of Alzheimer's A beta(42) peptide. J Chem Phys. 2011;135:6. doi: 10.1063/1.3617250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rojas A, Liwo A, Browne D, Scheraga HA. Mechanism of fiber assembly: Treatment of a beta peptide aggregation with a coarse-grained united-residue force field. J Mol Biol. 2010;404:537–552. doi: 10.1016/j.jmb.2010.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li MS, Co NT, Reddy G, Hu CK, Straub JE, Thirumalai D. Factors governing fibrillogenesis of polypeptide chains revealed by lattice models. Phys Rev Lett. 2010;105:4. doi: 10.1103/PhysRevLett.105.218101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wei GH, Mousseau N, Derreumaux P. Computational simulations of the early steps of protein aggregation. Prion. 2007;1:3–8. doi: 10.4161/pri.1.1.3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ma BY, Nussinov Simulations as analytical tools to understand protein aggregation and predict amyloid conformation. Curr Opin Struct Biol. 2006;10:445–452. doi: 10.1016/j.cbpa.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 46.Blinov N, Dorosh L, Wishart D, Kovalenko A. Association thermodynamics and conformational stability of beta-sheet amyloid beta(17–42) oligomers: Effects of E22Q (Dutch) mutation and charge neutralization. Biophys J. 2010;98:282–296. doi: 10.1016/j.bpj.2009.09.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cruz L, Urbanc B, Borreguero JM, Lazo ND, Teplow DB, Stanley HE. Solvent and mutation effects on the nucleation of amyloid beta-protein folding. Proc Natl Acad Sci USA. 2005;102:18258–18263. doi: 10.1073/pnas.0509276102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Massi F, Klimov D, Thirumalai D, Straub JE. Charge states rather than propensity for beta-structure determine enhanced fibrillogenesis in wild-type Alzheimer's beta-amyloid peptide compared to E22Q Dutch mutant. Protein Sci. 2002;11:1639–1647. doi: 10.1110/ps.3150102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Berhanu WM, Mikhailov IA, Masunov AE. Are density functional theory predictions of the Raman spectra accurate enough to distinguish conformational transitions during amyloid formation? J Mol Model. 2010;16:1093–1101. doi: 10.1007/s00894-009-0610-2. [DOI] [PubMed] [Google Scholar]
- 50.Fernandez A, Scheraga HA. Insufficiently dehydrated hydrogen bonds as determinants of protein interactions. Proc Natl Acad Sci USA. 2003;100:113–118. doi: 10.1073/pnas.0136888100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Straub JE, Thirumalai D. Principles governing oligomer formation in amyloidogenic peptides. Curr Opin Struct Biol. 2010;20:187–195. doi: 10.1016/j.sbi.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rohrig UF, Laio A, Tantalo N, Parrinello M, Petronzio R. Stability and structure of oligomers of the Alzheimer peptide A beta(16–22): from the dimer to the 32-mer. Biophys J. 2006;91:3217–3229. doi: 10.1529/biophysj.106.088542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chelli R, Gervasio FL, Procacci P, Schettino V. Stacking and T-shape competition in aromatic–aromatic amino acid interactions. J Am Chem Soc. 2002;124:6133–6143. doi: 10.1021/ja0121639. [DOI] [PubMed] [Google Scholar]
- 54.Kuznicki T, Masliyah JH, Bhattacharjee S. Aggregation and partitioning of model asphaltenes at toluene-water interfaces: Molecular dynamics simulations. Energy Fuels. 2009;23:5027–5035. [Google Scholar]
- 55.Zgarbova M, Otyepka M, Sponer J, Hobza P, Jurecka P. Large-scale compensation of errors in pairwise-additive empirical force fields: comparison of AMBER intermolecular terms with rigorous DFT-SAPT calculations. Phys Chem Chem Phys. 2010;12:10476–10493. doi: 10.1039/c002656e. [DOI] [PubMed] [Google Scholar]
- 56.Van der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 57.Massi F, Peng JW, Lee JP, Straub JE. Simulation study of the structure and dynamics of the Alzheimer's amyloid peptide congener in solution. Biophys J. 2001;80:31–44. doi: 10.1016/S0006-3495(01)75993-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Berhanu WM, Masunov AE. Molecular dynamic simulation of wild type and mutants of the polymorphic amyloid NNQNTF segments of elk prion: Structural stability and thermodynamic of association. Biopolymers. 2011;95:573–590. doi: 10.1002/bip.21611. [DOI] [PubMed] [Google Scholar]
- 59.Ma BY, Nussinov R. Stabilities and conformations of Alzheimer's beta-amyloid peptide oligomers (A beta16–22, A beta16–35 and A beta10–35): Sequence effects. Proc Natl Acad Sci USA. 2002;99:14126–14131. doi: 10.1073/pnas.212206899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fauchere JL, Pliska V. hydrophobic parameters-pi of amino-acid side-chains from the partitioning of n-acetyl-amino-acid amides. Eur J Med Chem. 1983;18:369–375. [Google Scholar]
- 61.Costantini S, Colonna G, Facchiano AM. Amino acid propensities for secondary structures are influenced by the protein structural class. Biochem Biophys Res Commun. 2006;342:441–451. doi: 10.1016/j.bbrc.2006.01.159. [DOI] [PubMed] [Google Scholar]
- 62.Colletier JP, Laganowsky A, Landau M, Zhao ML, Soriaga AB, Goldschmidt L, Flot D, Cascio D, Sawaya MR, Eisenberg D. Molecular basis for amyloid-beta polymorphism. Proc Natl Acad Sci USA. 2011;108:16938–16943. doi: 10.1073/pnas.1112600108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tycko R, Sciarretta KL, Orgel J, Meredith SC. Evidence for novel beta-sheet structures in Iowa mutant beta-amyloid fibrils. Biochemistry. 2009;48:6072–6084. doi: 10.1021/bi9002666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Darden T, York D, Pedersen L. Particle Mesh Ewald—an N.log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 65.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 66.Hess B. P-LINCS: A parallel linear constraint solver for molecular simulation. J Chem Theory Comput. 2008;4:116–122. doi: 10.1021/ct700200b. [DOI] [PubMed] [Google Scholar]
- 67.Miyamoto S, Kollman PA. Settle—an analytical version of the shake and rattle algorithm for rigid water models. J Comput Chem. 1992;13:952–962. [Google Scholar]
- 68.Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys. 2007;126:7. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 69.Parrinello M, Rahman A. Polymorphic transitions in single-crystals—a new molecular-dynamics method. J Appl Phys. 1981;52:7182–7190. [Google Scholar]
- 70.Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO, Shaw DE. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins. 2010;78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Meersman F, Dobson CM. Probing the pressure-temperature stability of amyloid fibrils provides new insights into their molecular properties. Biochim Biophys Acta. 2006;1764:452–460. doi: 10.1016/j.bbapap.2005.10.021. [DOI] [PubMed] [Google Scholar]
- 72.Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]