

Abstract
Aim
Previous studies have shown transient decreases in heart rate (HR) following administration of sphingosine 1‐phosphate (S1P) receptor modulators including BAF312. This study was conducted to determine whether dose titration of BAF312 reduces or eliminates these effects.
Methods
Fifty‐six healthy subjects were randomized 1:1:1:1 to receive BAF312 in one of two dose titration (DT) regimens (DT1 and DT2: 0.25–10 mg over 9–10 days), no titration (10 mg starting dose) or placebo. Pharmacodynamic and pharmacokinetic parameters were assessed.
Results
Neither DT1 nor DT2 resulted in clinically significant bradycardia or atrioventricular conduction effects. Both titration regimens showed a favourable difference on each of days 1–12 vs. the non‐titration regimen on day 1 for HR effects (P < 0.0001). On day 1, the geometric mean ratio of the fraction from the previous day in minimum daily HR between DT1 and non‐titration was 1.18 (95% confidence interval [CI] 1.13, 1.23) and 1.14 (95% CI 1.09, 1.18) for DT2 (both P < 0.05) with significant differences noted through to day 12. Non‐titration HRs showed considerable separation from placebo throughout the study. There was no statistically significant reduction in HR vs. placebo on day 1 in either titration regimen. On days 3–7 subjects in DT1 and DT2 experienced minor reductions in HR vs. placebo (approximately 5 beats min−1; P ≤ 0.0001). From days 9–12, HRs in both titration regimens were comparable with placebo.
Conclusion
Both titration regimens effectively attenuated the initial bradyarrhythmia observed on day 1 of treatment with BAF312 10 mg.
Keywords: dose‐response relationship, heart rate, lysosphingolipid, phase I as topic, receptors, sphingosine/analogues and derivatives
What Is Already Known about This Subject
Sphingosine‐1‐phosphate (S1P) is a key regulator of numerous physiological functions, including the egress of lymphocytes from the lymph nodes into the circulation.
S1P receptors are the targets of a number of compounds currently in development for the treatment of a range of conditions, including multiple sclerosis.
A transient, dose‐dependent decrease in heart rate has been shown to occur following first dose administration of S1PR modulators.
What This Study Adds
Dose titration with BAF312 over 9 or 10 days attenuates the bradyarrhythmia typically seen at treatment onset with S1P receptor modulators.
Introduction
BAF312 is the next generation selective sphingosine 1‐phosphate (S1P) receptor modulator currently in clinical development for the treatment of multiple sclerosis (MS). BAF312 targets MS via effects on the immune system [1], and may also have direct effects on S1P receptor‐expressing cells in the central nervous system (CNS), including oligodendrocytes and astrocytes [2]. S1P receptors are expressed on lymphocytes and relevant cells in the CNS and S1P signalling plays a key role in neuroinflammatory processes [3]. BAF312 is a modulator of the S1P1 and S1P5 receptors, and therapeutic doses of BAF312 can significantly reduce neuroinflammation in preclinical models of MS [1,2]. In studies in healthy subjects, BAF312 reduces peripheral lymphocytes following once daily dosing and recovery lymphocyte count is rapid after cessation of treatment due to a short half‐life of approximately 30 h [1].
In addition to cells in the CNS, S1P1 receptors have been shown to be highly expressed in atrial, septal and ventricular cardiomyocytes, and in the endothelial cells of cardiac vessels in humans [4,5]. Transient, dose‐dependent decreases in heart rate have been observed at treatment initiation both in patients with MS and in healthy subjects receiving S1P receptor modulators [6–8]. Atrioventricular (AV) conduction effects have also been reported in patients with MS receiving S1P receptor modulators [6–8]. Preliminary results in a phase I multiple dose study with BAF312 have shown a rapid tachyphylaxis of the bradycardic effect, starting at low doses [7]. The maximum bradycardic effect occurred at 3 h post‐dose, with heart rate returning to placebo values by 10 h post‐dose. The magnitude of the effect was shown to decrease with each subsequent day of dosing.
The current hypothesis is that the heart rate effects previously observed with first dose (therapeutic dose) of BAF312 may be attenuated using a dose titration strategy. The concept of this dose titration, using the Fibonnaci ratio, is to induce the internalization (or down regulation) of S1P receptors at low doses that are known to have little or no cardiac effect. In addition, earlier data suggest that there may be a physiological compensatory mechanism that results in the correction of heart rate within 2 days of first dose administration of BAF312 at therapeutic dose [9]. It is considered that, if the dose titration regimen were to start at a pharmacologically, but not clinically active dose, this compensatory mechanism would also occur during the 10‐day titration period. Therefore, any heart rate effects would be minimal and occur at the lower doses. At the point patients receive therapeutic doses no cardiac effects would be seen. Finally, it was considered that dose titration over a period of 10 days would lead to a more gradual, and therefore less clinically significant, daily reduction in heart rate (for example, if, at therapeutic doses, the overall reduction in heart rate is 10 beats min–1 with the dose titration protocol, this reduction would occur over 10 days (i.e. 1 beat min–1 day−1), whereas with no dose titration, this reduction would occur on day 1 (i.e. 10 beats min–1 day−1).
The purpose of the current study was to investigate whether dose titration of BAF312 (0.25–10 mg) over a 9 or 10‐day period can attenuate the initial bradycardia typically seen with S1P receptor modulators.
Methods
Subject selection
Healthy male and female volunteers aged 18–55 years were eligible for this study. All volunteers were required to have a body mass index of 18–30 kg m−2 and a body weight of ≥50 kg. Female subjects were required to be post‐menopausal or surgically sterilized. Subjects with heart rate <55 beats min–1 or with clinically significant symptoms or pathological laboratory findings, vital signs or 12‐lead electrocardiogram (ECG) at screening were excluded from the study. Additional exclusion criteria included use of tobacco in the 3 months prior to the study, a positive test for human immunodeficiency virus or hepatitis, a history of cardiovascular disease, recent (within the previous 3 years) or recurrent history of autonomic dysfunction or acute or chronic bronchospastic disease as reported by the subject, a history or evidence of drug or alcohol abuse, donation or loss of blood ≥400 ml in the previous 8 weeks, any surgical or medical condition that might significantly affect pharmacokinetic (PK) parameters and pregnancy or lactation. Concurrent use of prescription or over‐the‐counter medicines (excluding hormonal replacement therapy or contraceptives and occasional use of paracetamol) was not permitted. Xanthine‐ (e.g. caffeine) and alcohol‐containing food or beverages were not allowed during the study or for 48 h prior to the first dosing.
Study design
This was a randomized, double‐blind, placebo‐controlled, parallel group study conducted in healthy adult subjects to assess the effect of two BAF312 titration schemes (0.25–10 mg) on heart rate vs. 12 days of BAF312 10 mg not titrated, or placebo. The primary pharmacodynamic (PD) variable was the fraction from previous day (FFPD) in minimum daily heart rate (HRmin). Secondary PD metrics included HRmin, mean hourly heart rate and daily maximum effect on hourly average heart rate (Emax). PK and safety assessments, which included evaluation of atrioventricular conduction effects and sinus pause events, were also done.
The study was carried out in accordance with the Declaration of Helsinki and International Conference on Harmonisation/Good Clinical Practice and applicable regulatory requirements. The protocol was approved by the Copernicus Group Institutional Review Board at Parkway Research Center, Miami, Florida, USA and all subjects gave written, informed consent prior to study initiation.
Study period and drug administration
The study period was a maximum of 44 days for each subject, comprising a 21‐day screening period, a 12‐day treatment period (days 1–12) and an end of study assessment 10 days after the last treatment day (day 22). Subjects remained in the study centre from day −2 until discharge on day 13 and returned for the end of study visit on day 22.
Prior to the treatment period (day −2 and day −1), subjects underwent baseline assessments, including a 24 h Holter monitoring recording (day −1). All subjects were required to have a resting heart rate of ≥50 beats min–1 at screening. This was an arbitrary safety precaution to avoid enrolment of subjects with pre‐existing bradycardic heart rates potentially associated with an increased risk of symptomatic bradycardia under BAF312 treatment. On day 1, subjects were randomized 1:1:1:1 to one of four treatment arms: two BAF312 dose‐titrated (0.25–10 mg) groups (DT1 and DT2), a BAF312 non‐dose titration group (10 mg starting dose: positive control) and a placebo group (negative control).
Study medication was administered with water following a 10 h fast and administration was followed by a further 2 h fast (including fluids). Dosing schedules for each of the treatment groups are described in Figure 1. In the DT1 and DT2 groups, doses were increased from 0.25–10 mg over 9 or 10 days in increment ratios of 2 and 1.68 (Fibonacci ratio), respectively. Subjects in both DT1 and DT2 were subsequently followed for a 2–3 day confirmation period at the 10 mg dose. BAF312 was supplied by Novartis Pharma AG, Basel, Switzerland.
Figure 1.

Study design and dosing schedule.  , 10 mg non‐titration;
, 10 mg non‐titration;  , DT1;
, DT1;  , DT2;
, DT2;  , placebo
, placebo
Study assessments
Pharmacodynamic assessments
Heart rate and cardiac rhythm were assessed at baseline and from days 1–12 using continuous 12‐lead digital Holter recordings (Mortara H12+, Mortara Instrument, Inc., 7865 North 86th Street, Milwaukee, Wisconsin, USA) taken over 24 h periods. Recordings were started between 15 and 5 min before dosing on days 1–12. Hourly average heart rates were derived from 24 h Holter recordings; the lowest value (i.e. the most clinically significant value in terms of monitoring the heart rate reducing effects of different BAF312 dosing regimens) was taken as the daily value for the analysis. Calculation methods for FFPD and Emax are described in the Statistical analyses section.
Pharmacokinetic assessments
Plasma concentrations of BAF312 were assessed pre‐dose and at 2, 3, 4, 6, 8 and 12 h post‐dose throughout the study and at 24 h post‐dose on day 12. Blood samples for drug quantification were taken from an indwelling venous cannula or by direct venipuncture. On each occasion, 2 ml of blood was collected and centrifuged within 30 min at 2500 g for 5 min at 3–5°C. The plasma was kept in polypropylene screw capped tubes at ≤−60°C until analyzed.
Safety assessments
Safety assessments included physical examinations, vital signs and body measurements, standard 12‐lead ECG evaluations, standard clinical laboratory evaluations, haematology, blood chemistry and urinalysis, and adverse event (AE) monitoring. Frequency and duration of sinus pauses (>2 s), AV blocks, atrial fibrillation and ventricular ectopy were also assessed.
Statistical analyses
The PD, PK and safety population consisted of all volunteers who received the study drug. All subjects were analyzed according to treatment received.
In the PD assessment, the primary study endpoint was calculated as the FFPD in HRmin, i.e. the ratio in HRmin between two consecutive days:
HRmin was calculated as the minimum average HR recorded over 24 1 h periods between two consecutive doses and summarized using descriptive statistics. This methodology allows for any large fluctuations or artefacts in heart rate measurements to be smoothed out at each hourly interval and potentially adds to the robustness of the study findings [b10]. The FFPD values were log‐transformed and presented as geometric means. The estimated and 95% confidence intervals (CIs) for the geometric mean ratios (dose titration regimen/non‐titration regimen) between the FFPD on each day in each of the two dose titration regimens and the FFPD on day 1 in the non‐titration regimen were calculated using a first order autoregression model (12 primary treatment comparisons). The study sample size of 56 was calculated on the basis of requiring 12 completers in each treatment group to provide ≥89% power for the hypothesis that the mean FFPD in the dose titration regimens during the 10 days of the treatment period was larger than the mean FFPD on day 1 in the non‐titration regimen. This assumed an observed coefficient of variation equal to 9% for the FFPD (comparable with that previously observed for a single dose of BAF312). The hypothesis was rejected if the 95% CI for each of the 12 primary treatment comparisons were entirely above 1.
Other PD variables were summarized using descriptive statistics and included mean hourly HR, HRmin and Emax. Emax was determined as the maximum of the time‐matched decreases in hourly average HR from baseline (day −1) for each of the 24‐hourly average HRs on each of days 1 to 12.
Holter endpoints were described according to numbers (%) of subjects with 0–10, 11–20, 21–50 and >50 supraventricular ectopies occurring during the treatment period, numbers (%) of subjects with 0–5, 6–10, 11–30 and >30 ventricular ectopies occurring during the treatment period and numbers (%) of subjects with 0, 1 and >1 pauses of >2 s occurring during the treatment period. Any atrioventricular conduction effects and sinus pause events were also described.
PK evaluations comprised AUC(0,tlast) (mean, SD, CV, min and max), and were summarized descriptively only.
Molecular target nomenclature
The nomenclature for molecular targets has been based on the British Journal of Pharmacology Guide to Receptors and Channels, 5th edition, throughout this report [b11].
Results
Fifty‐six healthy subjects were enrolled beginning 23 March 2009 at Parkway Research Center, Miami, Florida, USA and randomized (n = 14 for each treatment group; Figure 2). The last subject completed the study on 10 June 2009. All treatment groups were comparable at baseline for all analysis populations (Table 1).
Figure 2.

Subject flow diagram. *Two subjects withdrew consent
Table 1.
Demographic summary
| DT1 (n = 14) | DT2 (n = 14) | 10 mg (n = 14) | Placebo (n = 14) | Total (n = 56) | ||
|---|---|---|---|---|---|---|
| Age (years) | Mean (SD) | 35.6 (6.0) | 38.6 (8.8) | 34.4 (8.1) | 36.3 (9.7) | 36.2 (8.2) |
| Gender | Male (%) | 13 (92.9) | 13 (92.9) | 13 (92.9) | 12 (85.7) | 51 (91.1) |
| Race | Caucasian (%) | 13 (92.9) | 13 (92.9) | 14 (100.0) | 12 (85.7) | 52 (92.9) |
| Black (%) | 1 (7.1) | 1 (7.1) | 0 | 2 (14.3) | 4 (7.1) | |
| Height (cm) | Mean (SD) | 172.0 (8.8) | 173.3 (6.8) | 173.2 (5.1) | 170.6 (8.0) | 172.3 (7.2) |
| Weight (kg) | Mean (SD) | 80.0 (11.2) | 80.3 (8.7) | 81.1 (9.0) | 79.2 (10.2) | 80.1 (9.6) |
DT, dose titration; SD, standard deviation.
PD assessments
Mean hourly heart rate
On day 1, mean hourly HR of the BAF312 10 mg non‐titration group dropped sharply from 69.3 beats min–1 at the time of dosing to 52.9 beats min–1 at 2 h post‐dose. In comparison, mean hourly HR of placebo, DT1 and DT2 groups at 2 h post‐dose (day 1) remained similar to mean hourly HR at time of dosing (74.6 from 69.6 beats min–1, 70.2 from 70.4 beats min–1 and 69.3 from 69.5 beats min–1, respectively) (Figure 3).
Figure 3.

Individual hourly mean heart rate (HR), and group hourly mean HR (bold line) for (A) non‐titration, (B) DT2 and (C) placebo groups on days −1, 1 and 2.  , 10 mg non‐titration mean HR;
, 10 mg non‐titration mean HR;  , DT2 mean HR;
, DT2 mean HR;  , placebo mean HR
, placebo mean HR
Minimum daily heart rate
On day 1, clinically significant bradycardia was observed in the BAF312 10 mg non‐titration group, indicated by a reduction in HRmin from 59.9 beats min–1 (95% CI 55.6, 64.1) to 50.6 beats min–1 (95% CI 45.8, 55.5) vs. 59.5 beats min–1 (95% CI 55.6, 63.4) to 60.4 beats min–1 (95% CI 56.1, 64.6) for the placebo group (P < 0.0001). On day 2, HRmin in the non‐titration group recovered to 56.4 beats min–1 (95% CI 52.7, 60.2) and on day 3 to end of study, HRmin ranged from 57.3 to 59.5 beats min–1. On day 12, HRmin for the BAF312 10 mg non‐titration group remained lower than placebo (59.3 beats min−1, 95% CI 56.4, 62.1 vs. 64.0 beats min–1, 95% CI 60.1, 67.9; P > 0.05) (Figure 4).
Figure 4.
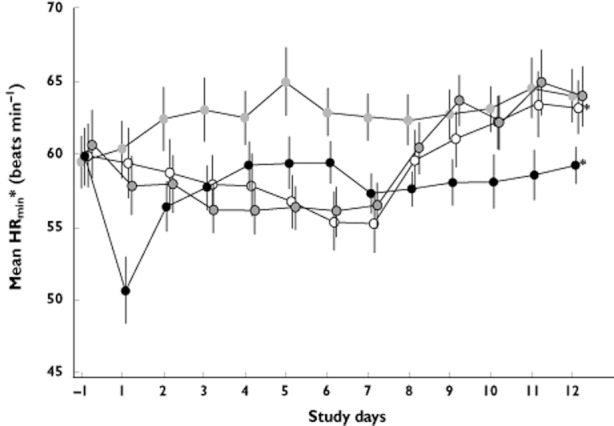
Summary of mean (± SEM) daily minimum heart rate (HRmin) data. * Individual heart rate profiles displayed in Figure 2.  , 10 mg non‐titration;
, 10 mg non‐titration;  , DT1;
, DT1;  , DT2;
, DT2;  , placebo
, placebo
Clinically significant bradycardia was not observed at any point in the study with either dose titration regimen. On day 1, HRmin was reduced from 59.9 beats min–1 to 59.4 beats min–1 in DT1, and from 60.7 beats min–1 to 57.9 beats min–1 in DT2. On days 2–7, the trend of gradual reductions in HRmin for both dose titration regimens continued until day 7 (DT1 range 55.3–58.7 beats min–1; DT2 range 56.1–58.0 beats min–1). On days 8–10, HRmin for both dose titration regimens gradually increased, reaching HRmin similar to placebo by day 10 (DT1 62.2 beats min–1; DT2 62.2 beats min–1; placebo 63.1 beats min–1). A HRmin similar to placebo was then maintained on days 11–12 in both groups.
Fraction from previous day in minimum daily heart rate
FFPD data (untransformed) for all four treatment groups throughout the study are shown in Figure 5. Log‐transformed geometric mean ratios between the FFPD on each day in each of the two dose titration regimens and the FFPD on day 1 in the non‐titration regimen were calculated. Both titration regimens showed a significant treatment difference (P < 0.0001) on each of days 1–12 in favour of the titration regimens vs. the non‐titration regimen on day 1 for FFPD in HRmin ratios.
Figure 5.
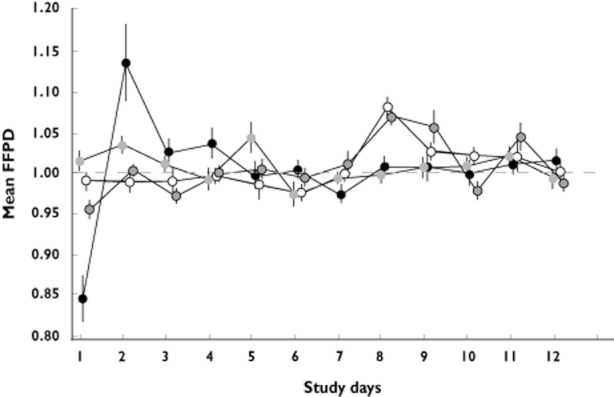
Summary (mean ± SEM) of fraction from previous day (FFPD) in heart rate minimum by day.  , 10 mg non‐titration;
, 10 mg non‐titration;  , DT1;
, DT1;  , DT2;
, DT2;  , placebo
, placebo
Daily maximum effect
Subjects in the non‐titration treatment group showed a statistically and clinically significant reduction in mean Emax compared with placebo (difference from placebo 15.64 beats min–1; 95% CI 11.36, 19.92; P < 0.0001) on day 1. From day 2 onwards this reduction in mean Emax recovered and remained in the range 6.15–10.03 beats min–1 (with statistically significant separation from placebo) on all days throughout the study.
Subjects in the two dose titration treatment regimens showed no difference from placebo in mean Emax on day 1. On days 3–7 subjects in both titration regimens showed statistically significant differences in mean Emax vs. placebo of approximately 5 beats min–1 (P ≤ 0.0001). By day 8, mean Emax in both dose titration groups was similar to placebo, and this was maintained until the end of study.
Pharmacokinetic assessments
Daily geometric mean plasma concentrations of BAF312 are shown in Figure 6. Steady‐state was reached in the non‐titration group after approximately 6 days for most subjects. In the two dose groups, steady‐state concentrations were reached at day 10. PK data for each group at steady‐state (day 12) are summarized in Table 2.
Figure 6.
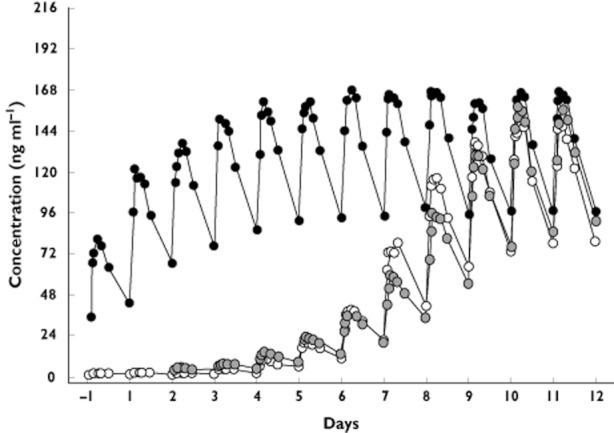
Daily geometric mean plasma concentrations of BAF312 after administrations once a day over 12 days to healthy subjects under fasted conditions.  , 10 mg non‐titration;
, 10 mg non‐titration;  , DT1;
, DT1;  , DT2
, DT2
Table 2.
Summary of pharmacokinetic data for each treatment group at steady state (day 12)
| DT1 (n = 14) | DT2 (n = 14) | 10 mg (n = 13)* | |
|---|---|---|---|
| tmax (h)† | 1.29 (2.00–6.00) | 1.68 (2.00–8.00) | 2.05 (2.00–8.00) |
| Cmax (ng ml−1)‡ | 155 [23] | 161 [33] | 175 [27] |
| AUC(0,24 h) (ng ml−1 h)‡ | 2814 [29] | 3032 [36] | 3255 [32] |
| Racc | ND | ND | 2.33 [26] |
AUC(0,24 h), area under the plasma concentration–time curve 0–24 h; Cmax, peak plasma concentration; DT, dose titration; ND, not determined; Racc mean accumulation ratio, ratio of AUC(0,24 h) on day 12 over AUC(0,24 h) on day 1); tmax, time to maximum plasma concentration.
One outlier excluded. †Median (min−max);
Geometric mean [%CV geometric mean].
Safety assessments
Atrioventricular conduction effects and sinus pause events
There was no apparent difference between treatment groups for ventricular ectopy and supraventricular ectopy. Events were not equally distributed among the subjects and some were cumulating episodes that did not allow statistical comparison. Episodes of type 1 (Mobitz I/Wenckebach) and/or type 2 (Mobitz II/Hay) second‐degree AV block were reported in all treatment groups except DT2 (Table 3). No subjects in the placebo group reported episodes of sinus pause >2 s. A total of 638 episodes were experienced by five subjects in DT1 and DT2 compared with 1522 episodes experienced by four subjects in the 10 mg non‐titration group (see Table 4 for details).
Table 3.
Incidence of type 1 (Mobitz I/Wenckebach) and type 2 (Mobitz II/Hay) second‐degree AV block episodes: number of episodes/number of subjects
| Mobitz I/Wenckebach | Mobitz II/Hay | |||
|---|---|---|---|---|
| Day −1 | Days 1–12 | Day −1 | Days 1–12 | |
| Placebo | 0/0 | 1/1 | 0/0 | 0/0 |
| DT1 | 2/1 | 36/2* | 0/0 | 0/0 |
| DT2 | 0/0 | 0/0 | 0/0 | 1/1‡ |
| 10 mg | 0/0 | 3/1† | 0/0 | 5/1† |
AV, atrioventricular; DT, dose titration.
One subject accumulated 36 episodes over 8 days.
All episodes on day 1.
One episode on day 2.
Table 4.
Overall incidence of sinus pause events
| Day | DT1 (n = 14) | DT2 (n = 14) | 10 mg (n = 14) | Placebo (n = 14) | ||||
|---|---|---|---|---|---|---|---|---|
| Number of episodes | Number of subjects | Number of episodes | Number of subjects | Number of episodes | Number of subjects | Number of episodes | Number of subjects | |
| Day −1 | 1 | 1 | 632* | 4 | 0 | 0 | 0 | 0 |
| Days 1–12 | 0 | 0 | 6† | 1 | 1522‡ | 4 | 0 | 0 |
DT, dose titration.
One subject accumulated 622 episodes.
All episodes on day 6.
One subject cumulated 834 episodes; one subject cumulated 685 episodes.
Adverse events
All three BAF312 dosing regimens were well tolerated. Subjects in the BAF312 treatment groups experienced more AEs than the placebo group. All AEs were generally of mild‐to‐moderate severity. A total of 16 subjects experienced AEs. The most commonly reported AEs were headache (n = 8, 14.3%) and increased alanine aminotransferase (SGPT) (n = 6, 10.7%) (Table 5). There were no deaths or serious AEs, and no subject required medical treatment for AEs. One subject experienced clinically significant elevations in SGPT, lactate dehydrogenase (LDH), aspartate aminotransferase (SGOT) and creatine kinase (CK) at the end of study visit, which were considered to be related to a skeletal muscular injury and not to study medication. Three subjects in the non‐titration group experienced mild bradycardia which started approximately 2 h after dosing and resolved without intervention. No clinically significant changes in blood pressure occurred for any subject in any treatment group.
Table 5.
Summary of all adverse events reported by at least 10% of subjects in any of the treatment regimens
| DT1 (n = 14) | DT2 (n = 14) | 10 mg (n = 14) | Placebo (n = 14) | |
|---|---|---|---|---|
| n (%) | n (%) | n (%) | ||
| Headache, n (%) | 0 | 1 (7.1) | 7 (50.0) | 0 |
| Pallor, n (%) | 0 | 0 | 2 (14.3) | 0 |
| Increased SGPT, n (%) | 3 (21.4) | 2 (14.3) | 1 (7.1) | 0 |
| Increased SGOT, n (%) | 2 (14.3) | 0 | 0 | 0 |
DT, dose titration; SGOT, aspartate aminotransferase; SGPT, alanine aminotransferase.
Discussion
All of the S1P receptor modulators currently in development for the treatment of MS are either selective or non‐selective agonists of S1P1 [b12,b13]. Studies have shown that S1P1 is highly expressed in atrial, septal and ventricular cardiomyocytes, and in the endothelial cells of cardiac vessels in humans [4,5]. Furthermore, S1P1 is considered to play a dominant role in the regulation of atrial myocyte function and heart rate [b14]. As a result, administration of S1P receptor modulators such as fingolimod (FTY720) has been shown to cause transient, dose‐related effects on heart rate in patients with MS and in healthy subjects [6,7,8,15]. However, differences in specificity and pharmacokinetics, along with a paucity of available data, make it difficult to generalize cardiac effects across compounds in this class. Data on BAF312 preclinical models and healthy subjects have shown species‐specific differences in S1P receptor specificity for first dose cardiac effects [9].
A previous study has demonstrated that initiation of BAF312 at a therapeutic dose of 10 mg resulted in clinically relevant bradycardia (as determined by measurement of mean HRmin) [7]. The purpose of the current study was to investigate whether dose titration of BAF312 (0.25–10 mg) over a 9 or 10‐day period can attenuate the initial bradycardia typically seen with S1P receptor modulators.
No instances of clinically relevant bradycardia were observed in any individual at any point during the study in either of the dose titration regimens studied. On day 1 (when cardiac effects had been observed previously), both HR and HRmin of the subjects in the two dose titration regimens remained comparable with those in the placebo group. In comparison, all subjects in the 10 mg non‐titration group experienced a marked decrease in both HR and HRmin within 2 h of dosing on day 1. Three subjects in the non‐titration group experienced bradycardia on day 1 ranging from 34 to 54 beats min–1. However, none of these instances required treatment. These results confirmed the findings of the previous study [9]. Over the first 7 days, both dose titration regimens resulted in a decrease of 4 beats min–1, with limited or no clinical impact. On day 8, mean HRmin began to recover and by day 10 was comparable with observed placebo rates. In contrast, mean HRmin in the BAF312 10 mg non‐titration group remained low compared with placebo until the end of study (day 10). BAF312 was well tolerated throughout the study in all regimens, and no subject required medical treatment for any AEs.
The inclusion of FFPD was based on the hypothesis that a limited daily decrease in HRmin spread out over a 10 day period would be preferable to a significant decrease on day 1. Assessment of FFPD allowed the comparison of the difference of the log‐transformed HRmin between two consecutive days. It should be noted that in addition to attenuating the HR reducing effect (HRmin and FFPD) observed on day 1 in the BAF312 10 mg group, both titration regimens demonstrated a limited decrease in HRmin (Emax) every day compared with the non‐titration regimen. The favourable Emax profiles obtained for each of the dose titration regimens (compared with the Emax profile for the non‐titration regimen), are of greater clinical relevance than the HRmin profiles obtained for the dose titration regimens.
No sinus pauses or AV conduction with clinically symptomatic effects were triggered at any point during the study in subjects in the two dose titration regimens.
Subjects in DT1 reached the 10 mg dose in 9 days, and those in DT2 in 10 days. The comparability of mean Emax in both titration regimens was maintained until end of treatment (day 12), demonstrating that a PD effect plateau was reached and was stable. DT2 appeared to have a more favourable profile than the other active treatment groups in number of subjects for total number of sinus pauses of >2 s. However, the sample size was too low to show a conclusive difference between the two titration schemes. In addition, the absence of sinus pauses in the placebo group does not reflect the normal physiological presentation of these pauses in the general population [b16–b18].
The BAF312 10 mg non‐titration and placebo regimens used in this study provide positive and negative controls, respectively, to validate the results from the two dose titration regimens. The dose titration regimens for this study were designed so that half of the study duration was below 1.25 mg, with both regimens rising to 2 mg at day 6 and then rising rapidly over the next 3 or 4 days. The starting dose of 0.3 mg was based on a phase I study which showed BAF312 0.3 mg dose to have a minimal cardiac effect [9]. As there were no animal models available to help calculate the appropriate ratio for dose increases, a factor of 2 was selected, based on the pharmacological consideration that doubling the exposure remains safe. The Fibonacci ratio (1.68) was also selected as it is often used in first in man studies, as it allows a safe incrementation of dose. One possible limitation of the study is that the two titration schemes used were similar (this was because greater differences were expected). However, the similarities in the data obtained for the two titration schemes vs. the 10 mg non‐titration scheme in fact confirm the consistency of the observed results.
The PK profiles for BAF312 were consistent across the dose regimens. Steady‐state was reached in all three regimens after 10 days of treatment. The increasing PK profile in conjunction with a decreasing HR profile in the titration groups clearly indicates development of tolerance. At low doses during days 1–6, this tolerance may have been due to internalization of the S1P receptors, which has been previously demonstrated in in vivo studies [1,7]. Compensation via other mediators could explain the longer term effects seen from day 8. However, further work is needed to understand the underlying mechanism for tolerance.
All BAF312 dosing regimens investigated were well tolerated. Transient and asymptomatic elevations in liver enzymes were observed for BAF312, in line with previous reports for BAF312 and other S1P receptor modulators [b19–b21].
In conclusion, titration over 9 or 10 days to a BAF312 dose of 10 mg successfully attenuates the initial bradycardia observed on day 1 of treatment with BAF312 10 mg. These results support implementation of an initial dose titration regimen in future clinical studies of BAF312. However, it should be noted that these data are applicable to BAF312 because of its short elimination half‐life and unique pharmacological profile and this titration regimen cannot be extrapolated to other S1P receptor modulators with a different profile or a longer half‐life.
Acknowledgments
The authors would like to thank Sasha Mitchell and Kerrie O'Rourke of iMed Comms for their assistance in the manuscript preparation. Funding for this study and medical writing services from iMed Comms was provided by Novartis Pharma AG, Basel, Switzerland.
Competing Interests
EL, AG and DJ are all employees of Novartis and hold stock or shares in Novartis.
References
- 1.Nuesslein‐Hildesheim B, Gergely P, Wallström E, Luttringer A, Groenewegen A, Howard L, Pan S, Gray N, Chen YA, Bruns C, Zécri F, Cooke N. The S1P1/S1P5 receptor modulator BAF312 reverses neurological deficits in ongoing EAE, reduces specific lymphocyte subsets in healthy volunteers and is a potential new multiple sclerosis treatment. Mult Scler. 2009;15:S126. [Google Scholar]
- 2.Seabrook T, Smith P, Schweitzer A, Wallström E, Nuesslein‐Hildesheim B. 2010. Efficacy of the selective S1P1/5 modulator, BAF312 in established EAE and redistribution of S1P1 and S1P5 in the inflamed human CNS tissue. ECTRIMS.
- 3.Brinkmann V, Davis MD, Heise CE, Albert R, Cottens S, Hof R, Bruns C, Prieschl E, Baumruker T, Hiestand P, Foster CA, Zollinger M, Lynch KR. The immune modulator FTY720 targets sphingosine 1‐phosphate receptors. J Biol Chem. 2002;277:21453–21457. doi: 10.1074/jbc.C200176200. [DOI] [PubMed] [Google Scholar]
- 4.Mazurais D, Robert P, Gout B, Berrebi‐Bertrand I, Laville MP, Calmels T. Cell type‐specific localization of human cardiac S1P receptors. J Histochem Cytochem. 2002;50:661–670. doi: 10.1177/002215540205000507. [DOI] [PubMed] [Google Scholar]
- 5.Means CK, Brown JH. Sphingosine‐1‐phosphate receptor signalling in the heart. Cardiovasc Res. 2009;82:193–200. doi: 10.1093/cvr/cvp086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cohen JA, Barkhof F, Comi G, Hartung HP, Khatri BO, Montalban X, Pelletier J, Capra R, Gallo P, Izquierdo G, de Tiel‐Wilck K, Vera A, Jin J, Stites T, Wu S, Aradhye S, Kappos L. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362:402–415. doi: 10.1056/NEJMoa0907839. [DOI] [PubMed] [Google Scholar]
- 7.Gergely P, Wallström E, Nuesslein‐Hildesheim B, Bruns C, Zécri F, Cooke N, Traebert M, Tuntland T, Rosenberg M, Saltzman M. Phase I study with the selective S1P1/S1P5 receptor modulator BAF312 indicates that S1P1 rather than S1P3 mediates transient heart rate reduction in humans. Mult Scler. 2009;15:S125–126. [Google Scholar]
- 8.Kappos L, Radue EW, O'Connor P, Polman C, Hohlfeld R, Calabresi P, Selmaj K, Agoropoulou C, Leyk M, Zhang‐Auberson L, Burtin P. A placebo‐controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362:387–401. doi: 10.1056/NEJMoa0909494. [DOI] [PubMed] [Google Scholar]
- 9.Gergely P, Nuesslein‐Hildesheim B, Guerini D, Brinkmann V, Traebert M, Bruns C, Pan S, Gray NS, Hinterding K, Cooke NG, Groenewegen A, Vitaliti A, Sing T, Luttringer O, Yang J, Gardin A, Wang N, Crumb WJ, Jr, Saltzman M, Rosenberg M, Wallström E. The selective S1P receptor modulator BAF312 redirects lymphocyte distribution and has species‐specific effects on heart rate: translation from preclinical to clinical studies. Br J Pharmacol. 2012 doi: 10.1111/j.1476-5381.2012.02061.x. . doi: 10.1111/j.1476‐5381.2012.02061.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Badilini F, Maison‐Blanche P. Holter monitoring for QT: types of analyses and endpoints the RR bin method in depth. In: Morganroth J, Gussak I, editors. Totowa, NJ: Humana Press; 2005. pp. 167–188. . In: Cardiac Safety of Noncardiac Drugs, eds. [Google Scholar]
- 11.Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Soliven B, Miron V, Chun J. The neurobiology of sphingosine 1‐phosphate signaling and sphingosine 1‐phosphate receptor modulators. Neurology. 2011;76:S9–14. doi: 10.1212/WNL.0b013e31820d9507. [DOI] [PubMed] [Google Scholar]
- 13.Bolli MH, Lescop C, Nayler O. Synthetic sphingosine 1‐phosphate receptor modulators – opportunities and potential pitfalls. Curr Top Med Chem. 2011;11:726–757. doi: 10.2174/1568026611109060726. [DOI] [PubMed] [Google Scholar]
- 14.Brinkmann V, Billich A, Baumruker T, Heining P, Schmouder R, Francis G, Aradhye S, Burtin P. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov. 2010;9:883–897. doi: 10.1038/nrd3248. [DOI] [PubMed] [Google Scholar]
- 15.Schmouder R, Serra D, Wang Y, Kovarik JM, DiMarco J, Hunt TL, Bastien MC. FTY720: placebo‐controlled study of the effect on cardiac rate and rhythm in healthy subjects. J Clin Pharmacol. 2006;46:895–904. doi: 10.1177/0091270006289853. [DOI] [PubMed] [Google Scholar]
- 16.Talan DA, Bauernfeind RA, Ashley WW, Kanakis C, Jr, Rosen KM. Twenty‐four hour continuous ECG recordings in long‐distance runners. Chest. 1982;82:19–24. doi: 10.1378/chest.82.1.19. [DOI] [PubMed] [Google Scholar]
- 17.Brodsky M, Wu D, Denes P, Kanakis C, Rosen KM. Arrhythmias documented by 24 hour continuous electrocardiographic monitoring in 50 male medical students without apparent heart disease. Am J Cardiol. 1977;39:390–395. doi: 10.1016/s0002-9149(77)80094-5. [DOI] [PubMed] [Google Scholar]
- 18.Adams SL, Roxe DM, Weiss J, Zhang F, Rosenthal JE. Ambulatory blood pressure and Holter monitoring of emergency physicians before, during, and after a night shift. Acad Emerg Med. 1998;5:871–877. doi: 10.1111/j.1553-2712.1998.tb02816.x. [DOI] [PubMed] [Google Scholar]
- 19.Cohen JA, Chun J. Mechanisms of fingolimod's efficacy and adverse effects in multiple sclerosis. Ann Neurol. 2011;69:759–777. doi: 10.1002/ana.22426. [DOI] [PubMed] [Google Scholar]
- 20.Selmaj K, Li D, Stüve O, Kappos L, Hartung HP, Hemmer B, Freedman MS, Rieckmann P, Montalban X, Zhang‐Auberson L, Pohlmann H, Mercier F, Dahlke F, Wallström E. 2011. BAF312, a selective sphingosine 1‐phosphate receptor modulator, effectively suppresses MRI lesion activity in relapsing‐remitting multiple sclerosis: findings of an adaptive dose‐ranging phase 2 study. 5th Joint triennial congress of the European and Americas Committees for Treatment and Research in Multiple Sclerosis. ; 17:(10) P1107. 19‐10‐2011. [DOI] [PubMed]
- 21.Moberly JB, Ford DM, Zahir H, Chen S, Mochizuki T, Truitt KE, Vollmer TL. Pharmacological effects of CS‐0777, a selective sphingosine 1‐phosphate receptor‐1 modulator: results from a 12‐week, open‐label pilot study in multiple sclerosis patients. J Neuroimmunol. 2012;246:100–107. doi: 10.1016/j.jneuroim.2012.03.007. [DOI] [PubMed] [Google Scholar]