

SUMMARY
Kinesin and dynein are opposite-polarity microtubule motors that drive the tightly regulated transport of a variety of cargoes. Both motors can bind to cargo but their overall composition on axonal vesicles and whether this composition directly modulates transport activity, is unknown. Here we characterize the intracellular transport and steady state motor subunit composition of mammalian prion protein (PrPC) vesicles. We identify Kinesin-1 and cytoplasmic dynein as major PrPC vesicle motor complexes, and show that their activities are tightly coupled. Regulation of normal retrograde transport by Kinesin-1 is independent of dynein-vesicle attachment, and requires the vesicle association of a complete Kinesin-1 heavy and light chain holoenzyme. Furthermore, motor subunits remain stably associated with stationary as well as with moving vesicles. Our data suggest a coordination model where PrPC vesicles maintain a stable population of associated motors whose activity is modulated by regulatory factors instead of by structural changes to motor-cargo associations.
INTRODUCTION
The viability and proper function of neurons depends on the active axonal transport of diverse cargoes (Goldstein et al., 2008; Hirokawa and Takemura, 2005; Verhey and Hammond, 2009). The microtubule (MT)-based motors driving these movements are kinesin and cytoplasmic dynein, which use the energy of ATP hydrolysis to translocate along MT tracks in plus- (anterograde), and minus-end (retrograde) directions. Cytoplasmic dynein consists of a core processive dynein heavy chain (DHC) motor that interacts with a large assembly of accessory subunits and with dynactin, to drive most retrograde transport (Kardon and Vale, 2009; Karki and Holzbaur, 1999). Kinesin-1 is a heterotetramer consisting of a homodimer of one of three kinesin heavy chains (KHC; Kinesin-1A, -1B, and -1C, formerly KIF5A, -B, and −C; Xia et al., 1998), that can interact in vitro with a homodimer of either of two accessory kinesin light chains (KLC1 and KLC2; Rahman et al., 1998). It is unknown what complexes of heavy and light chains form in vivo to drive the movement of any vesicular cargo studied to date (DeBoer et al., 2008; Rahman et al., 1998).
Intracellular transport is often bidirectional, as cargoes regularly reverse course en route to their final destinations. These dynamics have been observed for mitochondria, peroxisomes, melanosomes, endosomes, lipid droplets, synaptic vesicle precursors, and viral particles, where transport of opposite polarity motors is often coordinated (Gross et al., 2002; Kural et al., 2005; Lyman and Enquist, 2009; Plitz and Pfeffer, 2001; Sato-Yoshitake et al., 1992; Shubeita et al., 2008; Soppina et al., 2009; Welte, 2004). An important question in transport regulation is how motor activity is controlled in cells to achieve bidirectionality. Because Kinesin-1 and dynein are uni-directional motors, coordination could occur either by the alternating association/dissociation of motors of either polarity to/from cargo, which generates motor activation by cargo-binding; by the modulation of activity of both types of motors that simultaneously bind to cargo; or by generation of opposing forces of simultaneously cargo-bound motors in a tug-or-war (TOW) (Gross, 2004; Welte, 2004). It has been proposed that motor regulation by association/dissociation might be a generalized mechanism of transport regulation, as motors can exist in inactive, un-bound forms, and autoinhibition can be released by binding to cargo (Akhmanova and Hammer, 2010; Verhey and Hammond, 2009). Alternatively, there is evidence that certain neuronal cargoes in vitro or non-neuronal cargoes in vivo experience opposing TOW forces such that the total number of motors associated with cargo determines activity (Hendricks et al., 2010; Soppina et al., 2009). However, in coordination models of axonal transport, the extent of plus- and minus-end motor association with cargo, and whether cargo association relates to changes in motor activity, remains unclear. To test whether motor-cargo association modulates motor activity in axons and to build an in vivo model of bidirectional transport, it is imperative to characterize the steady-state composition of total motor assemblies on a single type of vesicular cargo, and relate this analysis to live movement data for the same cargo.
Analyzing motor composition of cargo in vivo has been experimentally challenging because of the difficulty in isolating populations of a single type of cargo, and the absence of quantitative methods to characterize motor composition on them. Biochemical purifications of heterogeneous membrane populations or of melanosomes have yielded estimates of co-fractionating plus- and minus-end motors (Gross et al., 2002; Hendricks et al., 2010). However, these represent indirect estimates of average levels of bound motors, as motor-cargo associations could vary from cargo to cargo and over time. Likewise, stall-force measurements have provided estimates of numbers of active motors (Kural et al., 2005; Shubeita et al., 2008; Soppina et al., 2009), but it is unclear whether bidirectionality is dictated by the rapid association/dissociation of these active motors, or whether these engaged motors represent a subgroup of a regulated but stable assembly of cargo-bound motors. Thus, the steady-state composition of motor assemblies on any single type of cargo remains undefined.
To distinguish between regulatory versus association/dissociation models of bidirectional transport, we characterized the mechanism of axonal transport of vesicles containing the cellular mammalian prion protein (PrPC). PrPC is a neuronally enriched glycosyl-phosphatidylinositol (GPI) anchored protein that follows the secretory pathway inside the lumen of vesicles toward the cell surface (Caughey et al., 2009; Harris, 2003). PrPC can convert to a pathogenic form called PrP-scrapie (PrPSc), which has been implicated in neurological disorders including Creutzfeldt-Jakob disease in humans (Caughey et al., 2009). The function of PrPC is unclear, but evidence suggests that while at the cell surface it can interact with proteins involved in cell adhesion and signaling (Malaga-Trillo et al., 2009; Mouillet-Richard et al., 2000), as well as with PrPSc (Caughey et al., 2009). Thus, trafficking of PrPC to the plasma membrane via an intact transport system might be relevant to PrPC function and for the initiation of neurodegenerative pathologies. While PrPC is transported in nerves (Butowt et al., 2006; Moya et al., 2004; Rodolfo et al., 1999), the mechanism of intracellular PrPC vesicular transport is unknown. Here, we developed assays to characterize relative motor subunit composition on individual PrPC vesicles, and used live imaging to identify Kinesin-1C/KLC1 and DHC1 as the major axonal motor complexes driving PrPC vesicle transport. Live tracking and motor composition analyses demonstrate that opposing motors positively coordinate each other’s activities independently of cargo-association mechanisms. This coordination mechanism requires the formation and vesicle association of an intact Kinesin-1 complex composed of heavy and light chains.
RESULTS
Mammalian PrPC Vesicles Move Bidirectionally in Hippocampal Axons
Previous studies showed that mammalian and avian PrPC are transported along peripheral and central nervous system nerves in anterograde and retrograde directions (Borchelt et al., 1994; Butowt et al., 2007; Moya et al., 2004; Rodolfo et al., 1999). We confirmed these observations using a protein accumulation paradigm in mouse sciatic nerves (Figures S1A and S1B).
To characterize the intracellular transport of PrPC vesicles in live neurons, we tracked individual moving vesicles in 10-day old cultured mouse hippocampal axons from neurons transfected with a YFP-PrPC fusion construct (Borchelt et al., 1996; Figures S1C-E). We restricted analyses to axons at day 10-post plating, which have a largely uniform microtubule polarity with plus-ends directed toward axonal termini and minus-ends toward cell bodies (Baas et al., 1988). To quantify YFP-PrPC vesicular movement, we used a MATLAB based custom particle tracking software (LAPTrack; Reis et al., unpublished data), to generate a comprehensive data set of trajectories at a spatial and temporal resolution of 0.126 um, and 10Hz, respectively.
In wild-type neurons, YFP-PrPC vesicles moved in anterograde and retrograde directions, and a large percentage were stationary (Figures 1A and 1B; Movie S1). The remaining vesicles reversed directions at a frequency of 0.027 switches/sec (± 0.004 switches/sec SEM). Vesicle trajectories were broken into segments, defined as uninterrupted periods of movement framed by pauses (Supplemental Experimental Procedures). Mean anterograde and retrograde segmental velocities were 0.85 μm/sec (± 0.036 μm/sec SEM), and 0.86 μm/sec (± 0.06 μm/sec SEM; Figure 1C), respectively, similar to those reported for Kinesin-1 and cytoplasmic dynein in vitro (Howard, 2001; Mazumdar et al., 1996). Analysis of segmental velocity distributions showed a wide range of anterograde and retrograde velocities that included maximal velocities of 2.8 μm/sec and 2.6 μm/sec, respectively (Figure 1D). Retrograde particles had shorter mean run lengths (4.8 μm ± 0.4 μm SEM) than anterograde ones (6.2 μm ± 0.5 μm SEM), but paused as long and as frequently (Figures 1E-G). These run lengths were longer than the 1-2 μm reported for Kinesin-1 and dynein in vitro (King et al., 2003; Thorn et al., 2000). Thus, vesicles containing YFP-PrPC move bidirectionally en route to the synapse in primary hippocampal neurons with dynamics that are consistent with MT-dependent fast axonal transport mediated by kinesin and dynein motor proteins.
Figure 1. PrPC Vesicles are Transported Bidirectionally in Wild-type Hippocampal Axons.
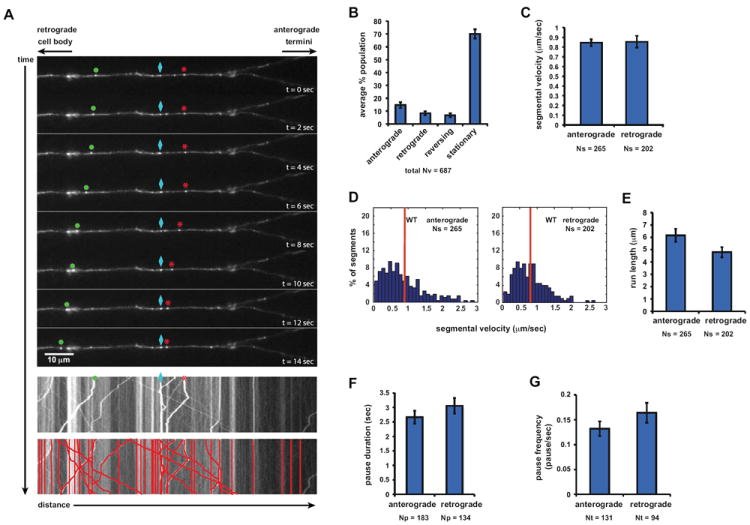
(A) Top panels: sequential images of YFP-PrPC vesicle movement in a hippocampal axon. Vesicles moving bidirectionally (*), in a retrograde direction (•), and a stationary one (◊) are followed for a period of 14 seconds. Middle panel: kymograph generated from movie in (A). Bottom panel: same kymograph depicting individual particle traces generated by particle tracking software.
(B) Population breakdown of YFP-PrPC vesicles.
(C) Mean segmental velocity; (D) Segmental velocity histograms. Red lines show mean.
(E) run length, (F) pause duration, and (G) pause frequency of YFP-PrPC vesicles.
Nv = # vesicles; Np = # pauses; Nt = # tracks; Ns = # segments. All values are shown as mean ± SEM. See also Figure S1.
Kinesin-1 and Cytoplasmic Dynein Associate with PrPC Vesicles in vivo
To understand the mechanism of axonal transport of PrPC vesicles, we sought to identify the motor proteins moving these vesicles in axons. Because PrPC and Kinesin-1 are predominantly expressed in brain, we tested the hypothesis that Kinesin-1 is a PrPC vesicle motor protein. We first tested whether the KLC1 cargo-binding subunit associated biochemically with PrPC vesicles in floated membrane fractions (Figure 2A). Using an antibody against KLC1 to pull-down associated membrane components, we found that PrPC and KHC (as detected by an antibody that recognizes primarily Kinesin-1C) immunoprecipitated with KLC1, as did the amyloid precursor protein (APP), previously identified in a complex with KLC1 (Kamal et al., 2000; Figure 2B). Because PrPC is in the vesicular lumen, the reverse immunoprecipitation experiment using PrPC antibodies to pull-down KLC1 was not possible without breaking vesicular membranes.
Figure 2. PrPC Vesicles Associate with Kinesin-1 and Dynein.
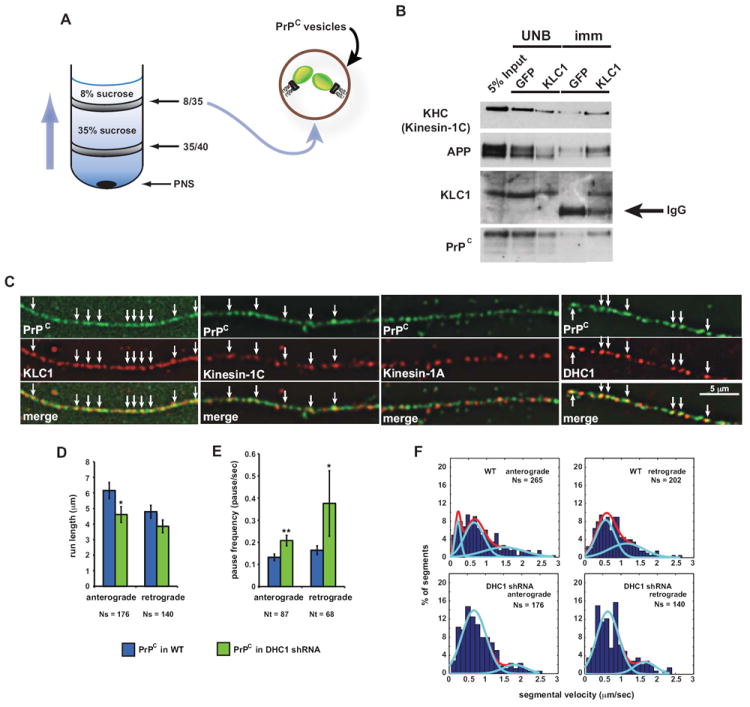
(A) Schematic diagram of a membrane flotation experiment showing the 8/35 fraction used as starting material for the vesicle immunoisolation in (B). Wild-type post-nuclear supernatant (PNS) obtained from wild-type mouse brain homogenate was bottom loaded. Buffers used did not contain detergent to prevent breaking of membranes.
(B) An antibody against KLC1 was used to pull down associated membrane components from 8/35 fractions, including PrPC-containing vesicles. KHC antibody recognizes mostly Kinesin-1C. UNB = unbound fraction; imm = immunoisolation. Anti-GFP was used as a control.
(C) Deconvolved images of vesicles stained with antibodies against PrPC and KLC1, Kinesin-1C, Kinesin-1A, or DHC1. Arrows point to some colocalization events.
(D) Run length and (E) pause frequency in DHC1 shRNA axons. All values are shown as mean ± SEM. **p<0.01, *p<0.05, permutation t-test.
(F) Segmental velocity histograms (shown as percent of segments) of YFP-PrPC transport in wild-type and DHC1 shRNA axons. Red and light blue curves represent the overall and predicted Gaussian modes, respectively.
Ns = # segments; Nt = # tracks. See also Figure S2.
To further test whether Kinesin-1 subunits interact with PrPC vesicles, we imaged fixed hippocampal cell axons stained with antibodies against PrPC, and KLC1, Kinesin-1C or Kinesin-1A. The fluorescent signal observed was punctate, suggesting that these proteins were localized to vesicular structures (Figure 2C). We observed significant colocalization between PrPC and KLC1 (58% PrPC vesicles colocalized with KLC1 ± 1.5% SEM; Nvesicles=510), and PrPC and Kinesin-1C (35% PrPC vesicles colocalized with Kinesin-1C ± 1.5% SEM; Nvesicles =80), but not between PrPC and Kinesin-1A. Complete colocalization was not expected as Kinesin-1 also mediates transport of other cargoes, and because other kinesin motors might also transport PrPC vesicles in addition to Kinesin-1.
To test whether cytoplasmic dynein transports PrPC vesicles, we quantified YFP-PrPC vesicle movement in hippocampal cells co-transfected with YFP-PrPC and with a short hairpin RNA (shRNA)-mCherry construct targeted to reduce the amount of dynein heavy chain 1 (DHC1; referred to as DHC1 shRNA). Imaging was restricted to axons co-expressing YFP and mCherry markers. Using the live imaging assay, we found that 2 days after co-transfection, reduction of DHC1 (mRNA reduced by 80-90%, protein reduced by 66%; Figures S2A, S2B, S5D, and S5E), disrupted bidirectional YFP-PrPC vesicle transport, decreasing run lengths and increasing the frequency of pauses (Figures 2D and 2E). Mean segmental velocities remained unchanged (Figure S2C). To confirm DHC1 association with PrPC vesicles, we stained hippocampal axons and found that DHC1 partially colocalized with PrPC vesicles (43% PrPC vesicles colocalized with DHC1 ± 2.9% SEM; Nvesicles= 388; Figure 2C). Thus, KLC1, Kinesin-1C, and DHC1, but not Kinesin-1A, associate with PrPC vesicles in vivo. The interaction between Kinesin-1 and PrPC is not a direct one, as immunoisolations from vesicular fractions using a KLC1 antibody in the presence of detergent did not pull down PrPC (data not shown). Furthermore, disruptions of dynein inhibited bidirectional transport, indicating that dynein is required for normal retrograde movement, and is involved in the activation of the plus-end anterograde transport of these vesicles.
Kinesin-1 Light Chains Mediate Anterograde Transport and Activate Retrograde Movement of PrPC Vesicles
Previous work showed that either of two KLCs can form complexes with any of the three KHCs (Rahman et al., 1998). However, it is unknown what combinations of KLC and KHC subunits interact in vivo to transport any cargo. Having shown that KLC1 and Kinesin-1C interact with PrPC vesicles, we next tested whether these physical interactions translated into functional transport requirements. Thus, we systematically reduced the function of each Kinesin-1 subunit and assayed for defects in PrPC vesicle transport.
We tested KLC1 by analyzing YFP-PrPC vesicle transport in hippocampal cells from mice homozygous for a gene-targeted KLC1 deletion (referred to as KLC1 -/- neurons; Rahman et al., 1999). We tested KLC2 in wild-type hippocampal neurons co-transfected with YFP-PrPC and a KLC2 shRNA-mCherry construct, which reduced KLC2 by ~ 83% (referred to as KLC2 shRNA neurons; Supplemental Experimental Procedures). Reducing the function of each KLC subunit caused a significantly decreased percentage of anterograde-moving vesicles, and a higher frequency of stalled particles (Figures 3A and 3B). Noticeably, the percentage of retrograde moving PrPC particles was also reduced in the absence of KLC1, suggesting that this subunit might be involved in promoting dynein-based movement. Observed and estimated run lengths were reduced in KLC1-/- and shRNA KLC2 axons, respectively (Figure 3C and 3D; Supplemental Experimental Procedures), and vesicles paused more often (Figures 3E). Given the pronounced reductions of bidirectional movement, mean segmental velocities were surprisingly largely unaffected in KLC mutants, with the exception of slight increases in KLC1 -/- neurons contributed solely by the small number of reversing vesicles (Figures S3A-C). Thus, KLC1 and KLC2 mediate anterograde transport of YFP-PrPC vesicles and also activate retrograde motility.
Figure 3. PrPC Vesicular Transport is Inhibited in Kinesin Light Chain Mutant Axons.
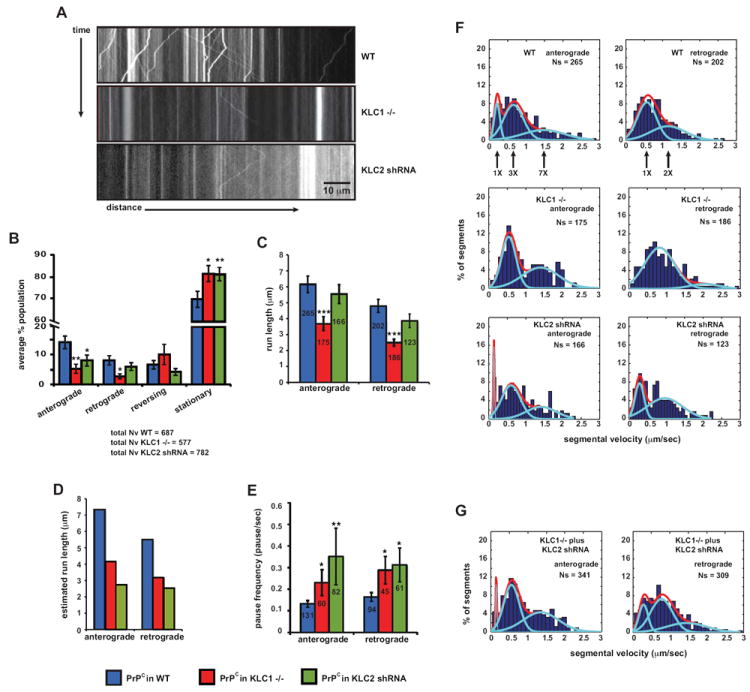
(A) Representative kymograph of YFP-PrPC vesicle movement in wild-type (top panel), KLC1 -/- (middle panel), and KLC2 shRNA (bottom panel) hippocampal axons.
(B-E) Transport parameters in KLC1 -/- and KLC2 shRNA axons. (B) Population breakdown of YFP-PrPC vesicles (Nv = # vesicles), (C) run length, (D) estimated run length, and (E) pause frequency. Numbers inside bars are segments (run length in C), and tracks (pause frequency in E). All values are shown as mean ± SEM. ***p<0.001, **p<0.01, *p<0.05, permutation t-test.
(F) Segmental velocity histograms (shown in percent of segments) in wild-type, KLC1 -/-, and KLC2 shRNA axons. Red and light blue curves represent the overall and predicted Gaussian modes, respectively.
(G) Anterograde and retrograde wild-type segmental velocity histograms (shown as percent of segments) were reconstituted from adding together histograms of KLC1 -/- and KLC2 shRNA axons (in F).
See also Figure S3.
Activation of Bidirectional Transport by a Neuronal Kinesin-1 Heavy Chain
To test whether KHC is required for the transport of PrPC vesicles, we analyzed YFP-PrPC vesicle movement in hippocampal neurons from Kinesin-1A -/-, and Kinesin-1C -/- mice (Figures S4A and S4B), and from conditional Kinesin-1B mice (Cui et al., 2010). Conditional Kinesin-1B neurons were treated post-plating with a cre-recombinase adenovirus at a multiplicity of infection (MOI) of 100 or 400, to remove Kinesin-1B genomic DNA flanked by two loxP sites and to create functional null cells (Figures S4C and S4D; Supplemental Experimental Procedures).
In Kinesin-1C -/- axons, the proportion of anterograde moving PrPC vesicles declined, run lengths in both directions were significantly decreased, and these vesicles paused more frequently when moving in both directions (Figures 4A-C). As was the case for DHC1 and KLC reduction, mean segmental velocities were unchanged (Figure 4D). In Kinesin-1B-cre axons, increasing adenoviral-cre MOI resulted in an increase in stationary vesicles (Figure 4E). However, anterograde movement was either unchanged (pause frequencies; Figure 4G), or activated as demonstrated by longer runs, and strikingly faster mean segmental velocities (Figures 4F and 4H). While the basis for the enhanced mean velocities is uncertain, perhaps a faster Kinesin-1C motor, which we showed above is required for normal YFP-PrPC anterograde motion, could be responsible for these increases. We did not observe major changes in YFP-PrPC transport in Kinesin-1A -/- axons, as only a minor decrease in retrograde run lengths and slight changes in segmental velocity distributions were observed (Figures 4A-D, and 4I; see next section). These results suggest that Kinesin-1A, and -1B are not major components of the PrPC vesicular transport machinery, and are consistent with our immunofluorescence data that showed no significant colocalization between PrPC and Kinesin-1A. We conclude that Kinesin-1C is required for normal anterograde YFP-PrPC transport, and can act as an activator of retrograde movement. The requirements of both the neuronal-specific Kinesin-1C and of DHC1 to activate each other’s transport suggest that the activities of these motors are tightly coupled.
Figure 4. PrPC Vesicular Transport is Inhibited in Kinesin-1C Mutant Axons.
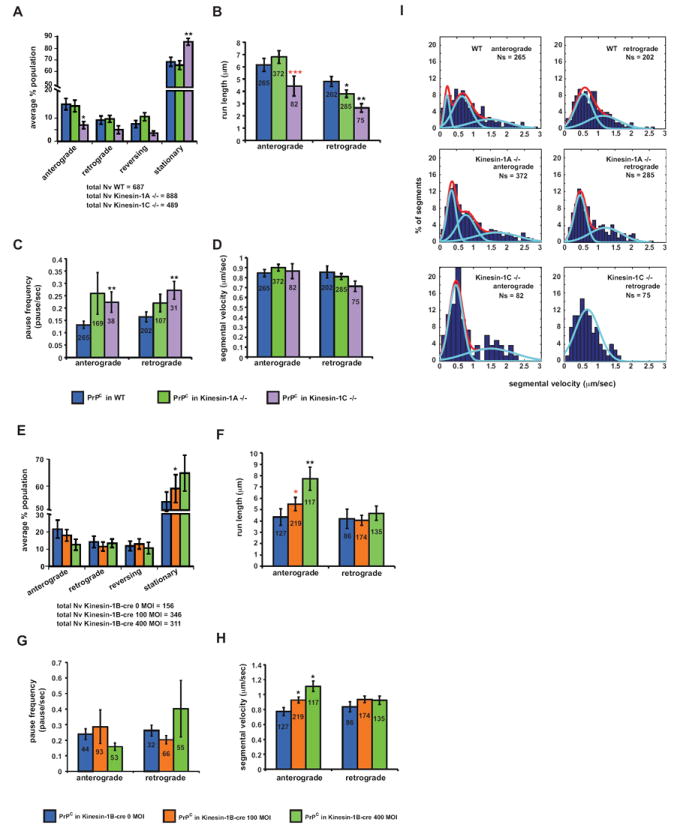
Transport parameters in Kinesin-1A -/-, Kinesin-1C -/-, and Kinesin-1B-cre axons. (A,E) Population breakdown of YFP-PrPC vesicles (Nv = # vesicles), (B, F) run length, (C, G) pause frequency, (D, H) segmental velocity. ***p<0.001, **p<0.01, *p<0.05, permutation t-test (black asterisks), Wilcoxon-Mann-Whitney test (red asterisks). Numbers inside bars are segments (run length in B and F, segmental velocity in D and H), and tracks (pause frequency in C and G). All values are shown as mean ± SEM. (I) Segmental velocity histograms (shown as percent of segments) in wild-type, Kinesin-1A -/-, and Kinesin-1C -/- axons. Red and light blue curves represent the overall and predicted Gaussian modes, respectively. See also Figure S4.
Reduction of Kinesin-1 Does Not Affect Global Transport in Axons
To test whether transport defects caused by reducing Kinesin-1 were specific to YFP-PrPC vesicles and not due to global disruptions of axonal transport, we characterized the movement of synaptophysin, a synaptic vesicle protein previously identified as a Kinesin-3 cargo (Okada et al., 1995). We tracked the movement of synaptophysin-mCherry vesicles in hippocampal cultured cells following identical conditions as described above for YFF-PrPC vesicles. Reducing the function of any of the Kinesin-1 subunits either did not change, or modestly stimulated bidirectional synaptophysin-mCherry transport, as observed by increased mean segmental velocities and reduced percentage of stationary vesicles (Figures S3D-I). Thus, while Kinesin-1 reduction alters transport dynamics of synaptophysin-mCherry vesicles, it is clearly not required for synaptophysin-mCherry transport.
Disrupting Kinesin-1 or Dynein Decreases Velocity Distributions Consistent with Downregulation of Motor Activity
Reducing Kinesin-1 or DHC1 results in bidirectional decreases in run lengths, and increases in pause frequencies, consistent with downregulation of opposing motor activity (Figures 3 and 4). Surprisingly however, mean segmental velocity, a parameter also influenced by motor activity, was largely unaffected (Figure 4D; Figures S2C and S3A). Because possible redundancy among Kinesin-1 subunits or incomplete removal of DHC1 might mask differences in average velocities, we analyzed segmental velocity distributions to test if these reflected reductions in opposing motor activity.
Wild-type anterograde and retrograde segmental velocity distributions were non-normal and showed a right-skewed bias (Figure 1D). To analyze these distributions further, we performed cluster mode analysis by fitting non-normal distributions observed in wildtype and in Kinesin-1 and DHC1 mutant axons, with predicted Gaussian modes using the MCLUST package in the R statistical computing environment (Fraley, 1999). Optimal mode fits were generated by the Bayesian Information Criterion (BIC; Supplemental Experimental Procedures). In wildtype, three and two modes best fit the anterograde and retrograde distributions, respectively (Figures 2F, 3F, and 4I; Table 1). Strikingly, reduction of KLC1 or Kinesin-1C reduced velocity distributions in both directions, suggesting that these two subunits pair to form the main holoenzyme that normally drives anterograde, and activates retrograde PrPC vesicle movement. Decreasing DHC1 also resulted in bidirectional shifts from higher to lower velocity modes and in a decreased number of modes (Figure 2F; Table S1). Notably, reducing Kinesin-1A and -1B also slightly reduced a mode or the proportion of vesicles within higher anterograde modes (Figure 4I; Table S1), suggesting that these motors might also play a role in the anterograde transport of PrPC vesicles, albeit a minor one since we did not observe major defects in other movement parameters. We conclude that segmental velocities represent a measure of the activity of motors, and that velocity distributions are reduced as a result of removal or decreases of opposite polarity motors, suggesting that regulation of motors can contribute to these activity changes.
Kinesin-1C is not Required for the Vesicle-Association of DHC1 and KLC1
Our tracking data suggest that Kinesin-1C and DHC1 activities are tightly coupled, since disruption of either inhibited opposite-polarity PrPC vesicle transport. Moreover, both KLC1 and Kinesin-1C are required for normal retrograde motion. To investigate the role of Kinesin-1 in bidirectional motion, we tested whether reduction of retrograde activity following removal of Kinesin-1C was the result of the dissociation of the primary retrograde motor DHC1, from PrPC vesicles. We also tested whether KLC1 association with vesicles is needed for normal retrograde motion. We developed a robust imaging method to quantify association of motor subunits on individual endogenous PrPC vesicles of hippocampal axons in the presence or absence of Kinesin-1C (Supplemental Experimental Procedures). Wild-type and Kinesin-1C -/- neurons were fixed and stained with antibodies against PrPC, DHC1, and KLC1 (Figure 5A), and immunofluorescence images of diffraction limited PrPC, DHC1, and KLC1 vesicle point sources were fitted with 2D Gaussians to estimate their point spread function, and to precisely map their coordinates and intensity amplitudes (Jaqaman et al., 2008). A custom-built ‘motor colocalization’ algorithm quantified presence or absence and intensity amplitudes of each detected DHC1 and/or KLC1 puncta within 300nm of each PrPC vesicle (L.S., S.E.E. et al., unpublished data). DHC1 and KLC1 antibody specificities were evaluated as described (Figures S2B, S5B-E; Supplemental Experimental Procedures).
Figure 5. Composition of Motor Subunits on PrPC Vesicles.
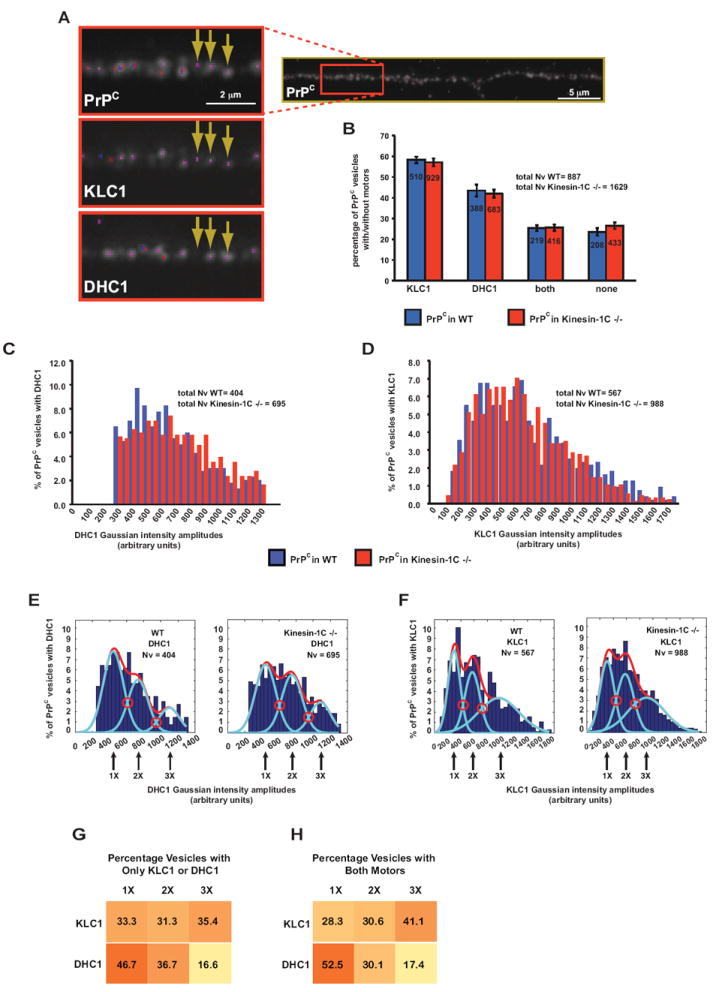
(A) Representative immunofluorescence image of a hippocampal axon stained with antibodies against PrPC, KLC1, and DHC1. Insets show enlargement with arrows pointing to three and two point sources in KLC1 and DHC1 channels, respectively, that associate with PrPC vesicles. Dots represent the location of fitted Gaussian functions.
(B) Percentage of PrPC vesicles that have only KLC1, only DHC1, both, or no motor subunits associated with them. Inside bars are the numbers of vesicles for each category. All values are shown as mean ± SEM.
(C-D) Gaussian intensity amplitude distributions comparing the frequency of PrPC vesicles associated with (C) DHC1 and (D) KLC1 intensities, in wild-type and Kinesin-1C -/- axons.
(E-F) Histograms of the same Gaussian intensity amplitude distributions shown in (C, D), depicting percent of PrPC vesicles associated with (E) DHC1 and (F) KLC1. Red and light blue curves represent the overall and predicted Gaussian modes, respectively. Red open circles point to intersections between modes
(G-H) Distribution of PrPC vesicles with (G) one or (H) both associated motor subunits. Numbers in boxes are percentages of PrPC vesicles in each category. Color gradient represents higher to lower percentage of PrPC vesicles in each category. Nv = # vesicles. See also Figure S5.
We designated four PrPC vesicle categories, those that had only DHC1, only KLC1, both motor subunits, or no motor subunits associated with them (Figure 5B). We found that in wild-type axons, 43% of PrPC vesicles colocalized with DHC1 (± 2.9% SEM), 57% of PrPC vesicles associated with KLC1 (± 1.5% SEM), and 25% of PrPC vesicles had both motor subunits (± 1.5% SEM). We did not detect motor subunits on 23% of PrPC vesicles (± 1.8% SEM). Removing Kinesin-1C resulted in almost identical DHC1- and KLC1-associated PrPC vesicle pools (Figure 5B). In addition, relative amounts of PrPC vesicle-associated DHC1 and KLC1, as measured by intensity distributions, were very similar between wild-type and Kinesin-1C -/- axons (permutation t-test p = 0.04, and p = 0.0681 for DHC1 and KLC1 comparisons, respectively; Figures 5C and 5D). Thus, while the DHC1 intensity distribution was borderline significantly different in Kinesin-1C-/- axons as compared to wildtype, DHC1 certainly did not appear to dissociate from PrPC vesicles. We conclude that Kinesin-1C is not required for DHC1 or KLC1 association with PrPC vesicles. Thus, impairment of retrograde movement observed after removing Kinesin-1C is likely a result of coordination between Kinesin-1C and DHC1 activities, rather than by altering DHC1-vesicle associations. Furthermore, because our tracking data show that retrograde movement is reduced after removal of KLC1 or Kinesin-1C, but removal of Kinesin-1C did not change the association of KLC1 with vesicles, we conclude that KLC1 is necessary but not sufficient to activate normal retrograde transport of PrPC vesicles. Thus, in the absence of KLC1 or Kinesin-1C, retrograde movement is impaired suggesting that maximal activation of retrograde motion requires the formation and association of a complete KLC1-Kinesin-1C holoenzyme with PrPC vesicles. It is possible that redundancy with KLC2 and Kinesin-1A or Kinesin-1B might stimulate residual retrograde motion still observed in KLC1 and Kinesin-1C mutant axons.
PrPC Vesicles Associate with Heterogeneous but Stable Motor Subunit Assemblies
To further confirm the presence of stable motor assemblies on PrPC vesicles, and characterize the nature of this motor composition, we asked whether motor subunits of both polarities were distributed evenly and stably on these vesicles. The non-normal distributions of wild-type KLC1 and DHC1 intensity amplitudes detected on PrPC vesicles were mode-fitted and selected using MCLUST and BIC (Figures 5E and 5F). The predicted modes on each KLC1 and DHC1 distributions showed three peaks corresponding to 1X, 2X, and 3X increments of intensities. Because the fluorescent intensity distribution of single molecules has a single Gaussian peak (Sugiyama et al., 2005), and the intensity of fluorescently labeled proteins has been shown to increase with increasing molecule concentration (Dixit et al., 2008), the multiple predicted modes in our data suggest the presence of a heterogeneous population of PrPC vesicles associated with 1X, 2X and 3X multiples of KLC1 or DHC1 motor subunits. We also tested that the KLC1 and DHC1 antibody signals scaled linearly with copy number, and were in linear range (Figures S5A-E; Supplemental Experimental Procedures). The KLC1 and DHC1 quantal intensity modes did not change after removal of Kinesin-1C (Figures 5E and 5F), indicating that a stable motor subunit population on vesicles is not affected by the presence or absence of other motors.
To further assess the distribution of motor subunits on vesicles, we divided PrPC vesicles into those associated with a single motor subunit (either KLC1 or DHC1), or with both, and quantified their intensity distributions and the percentage of vesicles within each predicted mode. When PrPC vesicles were associated only with KLC1, vesicles were distributed evenly in each of the 1X, 2X, and 3X motor subunit number modes (Figure 5G), while the majority of PrPC vesicles associated only with 1X DHC1. No changes to these distributions were observed when both KLC1 and DHC1 were associated simultaneously with vesicles or when Kinesin-1C was removed, suggesting that presence of KLC1 on the vesicle did not influence the association of DHC1 and vice versa (Figures 5H and 5SF-H). We conclude that the population of endogenous PrPC vesicles in axons is heterogeneous, having vesicles with overall associated motor subunit amounts in quantal multiples of 1X, 2X, 3X. However, this composition remained stable, and the presence of motor subunits on vesicles did not affect the binding of other subunits.
Motor Subunits are Associated with Stationary and Moving PrPC Vesicles
Many cargo-bound proteins and organelles stop and function at specified axonal micro-domains. Thus, mitochondria are largely stationary in axons at sites where there is a high demand for ATP (Kang et al., 2008). While stoppage can be achieved via changes in external factors such as Ca2+ levels or via interaction with docking adaptors (Kang et al., 2008), it is unknown whether a motionless state is achieved via dissociation of motors from cargo. Our system afforded us the opportunity to test this hypothesis because while PrPC vesicles move robustly in both directions, the majority are stationary (~70%; Figure 1B). To ask whether motor subunits are differently associated with individual stationary versus moving vesicles in vivo, we developed a ‘vesicle mapping’ technique to characterize relative amounts of KLC1 and DHC1 on YFP-PrPC vesicles following recording of their individual live motion (Supplemental Experimental Procedures). Hippocampal cells were plated in microfluidic chambers to promote the growth of straight axons, and were transfected with YFP-PrPC (Taylor et al., 2005; Figure 6A). Movement of vesicles was imaged and cells were fixed with paraformaldehyde for subsequent staining with KLC1 and DHC1 antibodies, but the trajectories of vesicles pre- and post-fixation were recorded (Figure 6B). Fixed images were superimposed to live movement kymographs to map (colocalize) fixed vesicles to their trajectories, after obtaining their precise Gaussian position coordinates and intensity amplitudes (Figure 6B). The advantage of this method is that we are able to assess motor subunit composition on vesicles for which we have recorded individual vesicular trajectories (anterograde, retrograde, or stationary).
Figure 6. Association of Motor Subunits with Stationary and Moving YFP-PrPC Vesicles.
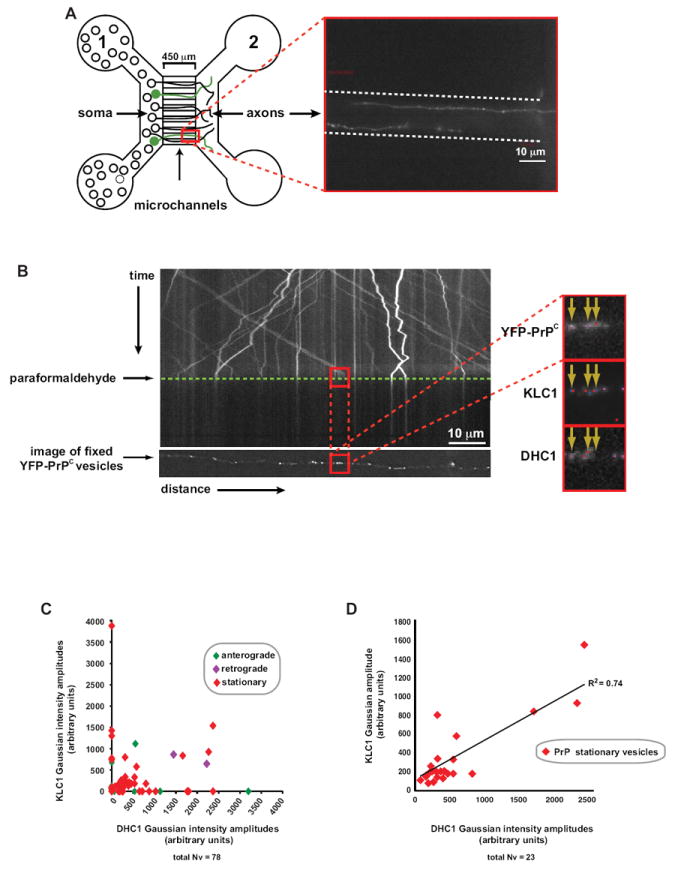
(A) Left panel: schematic diagram of a microfluidic chamber device. Transfected neurons are shown in green. Right panel: inset of two axons transfected with YFP-PrPC growing through a single microchannel (outlined with dotted lines)
(B) Kymograph of YFP-PrPC movement in the wild-type hippocampal axon shown in (A). Time of paraformaldehyde application is indicated by green dotted line. The panel below the kymograph is of the deconvolved image of the same fixed axon showing the YFP-PrPC channel. Red squares correspond to the same anterograde-moving vesicles in the kymograph that have been mapped to those in the fixed deconvolved image. Deconvolved images (inset) were taken of all three fixed/stained channels. Point sources were fitted with Gaussian functions (colored dots).
(C, D) Scatter plots of KLC1 versus DHC1 Gaussian intensity amplitudes of (C) all moving and stationary mapped vesicles from n = 7 axons (with and without associated motor subunits), and of (D) stationary vesicles with detected motor subunits.
Our data revealed that both KLC1 and DHC1 associated with stationary as well as moving vesicles (Figure 6C). Interestingly, we observed a correlation between KLC1 and DHC1 motor association on stationary vesicles, suggesting simultaneous increasing association of KLC1 and DHC1 (Figure 6D). Our data show motor subunits associate to vesicles regardless of whether they were moving or not, indicating that motor subunit association is necessary but not sufficient for active translocation along microtubules.
DISCUSSION
We identified Kinesin-1C and DHC1 as major anterograde and retrograde motors required to transport PrPC vesicles in mammalian axons, and showed that they reciprocally promote their activity independently of motor-association mechanisms. We developed an assay to robustly assess the relative amounts of motors on vesicular cargo, and thus provided the characterization of motor composition in vivo on a single type of vesicle. We report that PrPC vesicles have a stable complement of motor subunits regardless of changes in motor activity suggesting that regulation of activity, and not motor-vesicle attachment, determines directionality.
Differential Requirements of Kinesin-1 Subunits in PrPC Vesicle Transport
Previous studies showed that PrPC moved in anterograde and retrograde directions in nerves, but the mechanism of intracellular movement was unknown (Borchelt et al., 1994; Butowt et al., 2006; Moya et al., 2004; Rodolfo et al., 1999). Using a combination of genetic, live imaging, biochemical, and immunofluorescence approaches, we dissected the requirements of each Kinesin-1 subunit and of dynein, and identified Kinesin-1C and DHC1 as the main plus- and minus-end motors, respectively, with a minor role attributed to Kinesin-1A and Kinesin-1B. Reduction of any one KHC did not result in complete disruption of anterograde transport, suggesting either some redundancy, or the requirement of another as yet unidentified motor. Interestingly, Kinesin-1C mRNA expression was upregulated in Kinesin-1B null extraembryonic membranes, pointing to a possible redundant function between these KHCs (Tanaka et al., 1998). Whether higher transcript levels result in increased vesicle association of Kinesin-1C in Kinesin-1B -/- cells is unknown.
Our data also provide evidence for a role of KLC1 and KLC2 in PrPC vesicle transport. KLCs have been implicated in cargo-binding and transport regulation through their binding to KHCs, but are not always required. Mitochondria for example, are transported by KHC and a complex formed by Milton and Miro, but lack KLC association (Glater et al., 2006). For PrPC vesicles, redundancy between KLC1 and KLC2 is likely, as disruption of either KLC did not result in complete blockage of transport. We tested this by combining velocity distributions after reducing each KLC, and observing the partial reconstitution of the wild-type anterograde distribution but not the retrograde one (Figure 3G; Table S1). Our data further point toward the pairing of neuronally enriched KLC1 and Kinesin-1C as a primary PrPC vesicle transport complex: vesicle immunoisolations with KLC1 brought down Kinesin-1C, and disrupting either of these motor subunits resulted in almost identical bidirectional phenotypes, more severe than seen with other subunits, including the decrease in bidirectional velocity distributions (Table S1). Thus, our work establishes KLC1, KLC2, Kinesin-1C and DHC1 as main motor subunits for PrPC vesicle transport. These requirements appear specific, as movement of synaptophysin vesicles is not inhibited following their disruption. Differential use of Kinesin-1 components might be important for the selective targeting of different types of PrPC vesicles to distinct axonal domains. Whether the subunits are used in different cellular contexts is unknown, and functional experiments to address this issue for PrPC vesicles still need to be performed.
Uncoupling Motor-Association from Motor-Activation Mechanisms to Drive Bidirectional Transport
To characterize transport mechanisms we assessed PrPC vesicle movement by live imaging, as well as motor-vesicle associations using immunofluorescence assays. Our data support the hypothesis that the total composition of motor subunits on PrPC vesicles is constant even when pronounced changes in motor activity are detected. Activity changes were revealed after impairing Kinesin-1 and DHC1, which resulted in PrPC vesicles that traveled shorter distances, paused more frequently, and had altered velocity distributions (Figures 3, 4). The lower distribution of velocities observed here are in contrast to at least one in vivo study on Drosophila embryo lipid droplets that showed that average velocities were slightly higher following reductions in anterograde motor copy number (Shubeita et al., 2008). While inherent differences between Drosophila lipid droplets versus axonal transport systems might explain the contrasting observations, it is also possible that mean velocities might not reveal differences that can be detected only in analyses of velocity distributions. Indeed, mean velocities were generally unaffected after reduction of motors in our system (Figure 4D; Figure S3A).
Are the changes in observed motor activity directly correlated to the amount and composition of motors associated to those vesicles? Previous in vitro studies estimated the number of cargo-bound motors and suggested that these directly correlate to level of movement activity during TOW (Hendricks et al., 2010; Shubeita et al., 2008; Soppina et al., 2009). However, during in vivo axonal transport of cargoes undergoing regulatory coordination, although cargoes can be moved by multiple motors, it is unclear whether all motors associated with cargo are active. Our data show that in axons in vivo, removing or reducing a motor reduces parameters of opposite-polarity transport, thus providing support for regulatory motor coordination, and against simple TOW scenarios. In this regulatory coordination setting, we show that a stable motor subunit complement associates with PrPC vesicles regardless of their activity level or directionality. A combination of motor-interactions and regulatory coordination could be at play in vivo, as motors of opposite-polarities can mechanically interact to influence their motility (Ally et al., 2009). Thus, the mechanism of motor activity regulation in axons in vivo appears to be uncoupled from the one that regulates motor-vesicle associations. Consistent with this stable motor association model, previous work showed that Kinesin-2 and dynein levels from purified melanosomes did not change during directionality switches, although these were bulk estimates and therefore it was unclear whether motor levels were unchanged on a per cargo basis (Gross et al., 2002).
We thus propose a coordination model in which a stable population of Kinesin-1 subunits and DHC1 associate with PrPC vesicles and a subset of these activate bidirectional movement, while the rest remain vesicle-bound but inactive (Figure 7A). Stationary states are likely achieved by regulatory inhibition of bound motors. Thus, this model does not support cargo-binding as the sole mechanism of motor activation (Akhmanova and Hammer, 2010). Indeed, in vitro work has shown that inactive motors can bind cargo and diffuse along the MT lattice (Lu et al., 2009). The ability of inactive motors to remain vesicle-bound could allow them to be activated “on the spot” according to cues specific to the cargo being transported.
Figure 7. A Stable Motor Association and Coordination Model of PrPC Vesicle Transport.
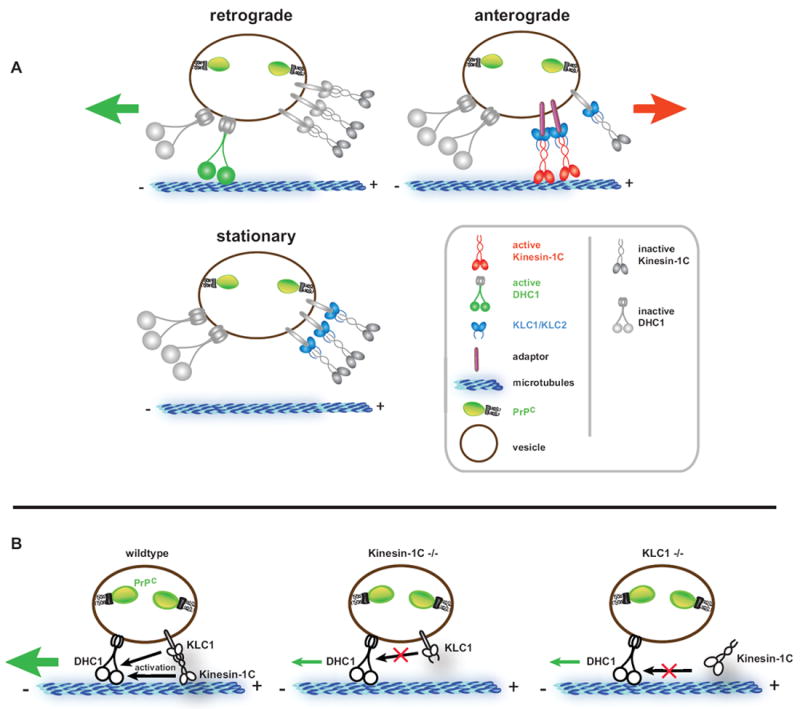
(A) A stable motor subunit composition on anterograde, retrograde, or stationary vesicles is depicted, but only a subset of those are active to drive transport in either direction. Our data suggests a coordination model whereby Kinesin-1 and dynein act as alternating activators of opposite polarity transport. Number of motors depicted is arbitrary. See Discussion for details.
(B) Activation of retrograde motility requires the vesicle association of a complete Kinesin-1 holoenzyme comprised of both KLC1 and Kinesin-1C. Removal of either subunit downregulates activation of DHC1, but does not dissociate DHC1 from PrPC vesicles. Thus, Kinesin-1C can activate DHC1 via interaction with KLC1.
Coordination of Bidirectional Transport in Axons
Our data show that coordination of retrograde activity by Kinesin-1C is independent of DHC1-vesicle association, but involves the simultaneous attachment of both types of motors to vesicles (Figures 5B). Kinesin-1C thus appears to perform a dual function as a mediator of anterograde movement, and as an activator of retrograde transport, but is not required for cargo binding by dynein. How might this retrograde activation occur? Our data suggest that it does so by the formation and vesicle association of an intact Kinesin-1C/KLC1 complex, which is required for proper retrograde activity. The absence of either Kinesin-1C or KLC1 precludes normal retrograde activation (Figure 7B). However, KLC1 remains vesicle associated in Kinesin-1C mutants, so its presence alone is not sufficient to activate normal retrograde motion. Likewise, Kinesin-1C is decreased but still present in KLC1 -/- cells (Rhiannon Killian, personal communication), suggesting that this KHC subunit alone cannot induce normal retrograde activity. It is possible that Kinesin-1C can bind to KLC2 in KLC1 -/- axons, as our data shows KLC2 is also required for normal levels of bidirectional movement, and is likely responsible for the residual retrograde motion observed. However, such a putative interaction is clearly not sufficient to rescue normal retrograde transport. A possible outcome of requiring a complete vesicle-bound Kinesin-1 complex is that it could facilitate rapid activation and/or autoinhibition, which has been shown to occur via KLC with KHC interactions (Cai et al., 2007; Verhey et al., 1998). Changes in Kinesin-1 autoregulation could translate to changes in dynein activity. Thus, our data is consistent with a model for bidirectional coordination in which activities of Kinesin-1 and dynein might be linked via physical contacts (Martin et al., 1999), and points to a role of KLCs in this linkage. Whether contact occurs via KLC interactions with dynein accessory subunits, dynactin, and/or other unidentified components has been suggested but is unclear (Ligon et al., 2004; Martin et al., 1999). Alternatively, direct motor-motor interactions could coordinate movement, as mechanical pulling of plus- and minus-end motors against each other has been suggested to be necessary and sufficient to activate opposing motor activity (Ally et al., 2009). In the case of PrPC vesicles, coordination of a stable population of simultaneously bound motors is likely to be important for their efficient transport and delivery to various axonal regions or to the cell surface, where PrPC has been implicated in signal transduction and cell adhesion (Malaga-Trillo et al., 2009; Mouillet-Richard et al., 2000). This mechanism might also be a strategy for efficient distribution of many other cargoes along axons, with stable complexes of motors loading onto vesicles presumably at or near the cell body, where motors are produced. Stable associations would allow differential regulation and coordination of subsets of motors, either by themselves or by factors specific to the vesicular cargo.
EXPERIMENTAL PROCEDURES
Mice and Cell Culture
Mice used throughout this study were in the C57/Bl6 background. KLC1, Kinesin-1A -/-, and conditional Kinesin-1B mice were described previously (Cui et al., 2010; Rahman et al., 1999; Xia et al., 2003). Generation of Kinesin-1C -/- mice is detailed in Supplemental Experimental Procedures. Hippocampal cultures were plated from either E15-E18 or 1 day old pups (Falzone et al., 2009).
Transfection and Adenovirus Cre-Recombinase Transduction
Transfections of hippocampal neurons were done 10 days post-plating following a standard Lipofectamine 2000 protocol (Invitrogen). Cells were imaged or fixed 18-24 hours later. Plated hippocampal cells from conditional homozygous Kinesin-1B II/II mice were treated 10 days post-plating with 0, 100 or 400 MOI adenovirus cre-recombinase (Ad5CMVCre from the University of Iowa, Gene Transfer Vector Core), corresponding to 0, 1.1e-7, and 4.4e-7 plaque forming units (PFU), respectively.
Vesicle Immunoisolation
Vesicle immunoisolations were performed with antibodies against KLC1 or GFP to pull-down vesicular membrane components obtained from floated membrane fractions (Supplemental Experimental Procedures).
Immunofluorescence and Microscopy
Hippocampal neurons and N2a cells were fixed with 4% paraformaldehyde, permeabilized, and stained with antibodies against KLC1 and DHC1. Fixed immunofluorescence images were taken on a Deltavision RT deconvolution system, and live images were taken with a Nikon Eclipse TE2000-U inverted microscope (Supplemental Experimental Procedures).
Data and Statistical Analysis
Trajectories of individual YFP-PrPC or synaptophysin-mCherry vesicles (i.e. tracks) were detected using a custom-made semi-automated particle tracking software written in MATLAB (Mathworks) and C++ (Reis et al. unpublished data). Definitions and calculations for each parameter are detailed in Supplemental Experimental Procedures. ‘Motor colocalization’ and ‘vesicle mapping’ analyses are detailed in Supplemental Experimental Procedures. All tracking parameters reported herein were first tested for normality using the Lilliefors test implemented in the nortest package of R. Most parameters were not normally distributed so a non-parametric permutation t-test was used for comparison between genotypes (Moore and McCabe, 2005). Differences in medians were also compared between genotypes for all parameters using the Wilcoxon-Mann-Whitney rank-sum test.
Supplementary Material
HIGHLIGHTS.
Prion protein (PrPC) vesicles are newly identified Kinesin-1 and dynein cargoes
Kinesin-1 activates normal retrograde motion independent of dynein vesicle association
A complete Kinesin-1 holoenzyme is required for normal retrograde transport activity
Motor composition on PrPC vesicles does not determine directionality of cargo
Acknowledgments
We thank Ge Yang, Gaudenz Danuser, Khuloud Jaqaman, and Daniel Whisler for assistance with the development of software; Liz Roberts, Eileen Westerman for technical assistance; Jennifer Meerloo for assistance with imaging (UCSD Neuroscience Microscopy Shared Facility Grant P30 NS047101); Anne-Marie Craig (University of British Columbia) for the synaptophysin-mCherry construct; Anthony Williamson and Laura Solforosi (The Scripps Research Institute) for the HuM-D13 anti-PrPC antibody; Scott T. Brady for the KLC1/2 (63-90) antibody; Nancy Jenkins (Institute of Molecular and Cell Biology, Singapore), and Jiandong Huang (University of Hong Kong) for the Kinesin-1B mice; Tony Orth at the Genomics Institute of the Novartis Foundation for help with design and construction of shRNA lentiviral constructs; and Jeff W. Kelly (The Scripps Research Institute) for critical feedback on the manuscript. This work was supported in part by NIH-NIA grant AG032180 to L.S.B.G, who is an Investigator of the Howard Hughes Medical Institute. L.S. was supported by NIH Bioinformatics Training Grant T32 GM008806, and S.E.E was supported by a Damon Runyon Cancer Research Foundation Postdoctoral Fellowship and NIH Neuroplasticity Training Grant AG000216.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, five figures, and a table and can be found with this article online at
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akhmanova A, Hammer JA., 3rd Linking molecular motors to membrane cargo. Curr Opin Cell Biol. 2010;22:479–487. doi: 10.1016/j.ceb.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ally S, Larson AG, Barlan K, Rice SE, Gelfand VI. Opposite-polarity motors activate one another to trigger cargo transport in live cells. J Cell Biol. 2009;187:1071–1082. doi: 10.1083/jcb.200908075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baas PW, Deitch JS, Black MM, Banker GA. Polarity orientation of microtubules in hippocampal neurons: uniformity in the axon and nonuniformity in the dendrite. Proc Natl Acad Sci U S A. 1988;85:8335–8339. doi: 10.1073/pnas.85.21.8335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchelt DR, Davis J, Fischer M, Lee MK, Slunt HH, Ratovitsky T, Regard J, Copeland NG, Jenkins NA, Sisodia SS, et al. A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genet Anal. 1996;13:159–163. doi: 10.1016/s1050-3862(96)00167-2. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Koliatsos VE, Guarnieri M, Pardo CA, Sisodia SS, Price DL. Rapid anterograde axonal transport of the cellular prion glycoprotein in the peripheral and central nervous systems. J Biol Chem. 1994;269:14711–14714. [PubMed] [Google Scholar]
- Butowt R, Abdelraheim S, Brown DR, von Bartheld CS. Anterograde axonal transport of the exogenous cellular isoform of prion protein in the chick visual system. Mol Cell Neurosci. 2006;31:97–108. doi: 10.1016/j.mcn.2005.09.004. [DOI] [PubMed] [Google Scholar]
- Butowt R, Davies P, Brown DR. Anterograde axonal transport of chicken cellular prion protein (PrPc) in vivo requires its N-terminal part. J Neurosci Res. 2007;85:2567–2579. doi: 10.1002/jnr.21229. [DOI] [PubMed] [Google Scholar]
- Cai D, Hoppe AD, Swanson JA, Verhey KJ. Kinesin-1 structural organization and conformational changes revealed by FRET stoichiometry in live cells. J Cell Biol. 2007;176:51–63. doi: 10.1083/jcb.200605097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caughey B, Baron GS, Chesebro B, Jeffrey M. Getting a grip on prions: oligomers, amyloids, and pathological membrane interactions. Annu Rev Biochem. 2009;78:177–204. doi: 10.1146/annurev.biochem.78.082907.145410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Wang Z, Cheng Q, Lin R, Xin-Mei Z, Leung PS, Copeland NG, Jenkins NA, Yao KM, Huang JD. Targeted Inactivation of Kinesin-1 in Pancreatic {beta}-Cells in vivo Leads to Insulin Secretory Deficiency. Diabetes. 2010 doi: 10.2337/db09-1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBoer SR, You Y, Szodorai A, Kaminska A, Pigino G, Nwabuisi E, Wang B, Estrada-Hernandez T, Kins S, Brady ST, et al. Conventional kinesin holoenzymes are composed of heavy and light chain homodimers. Biochemistry (Mosc) 2008;47:4535–4543. doi: 10.1021/bi702445j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit R, Ross JL, Goldman YE, Holzbaur EL. Differential regulation of dynein and kinesin motor proteins by tau. Science. 2008;319:1086–1089. doi: 10.1126/science.1152993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falzone TL, Stokin GB, Lillo C, Rodrigues EM, Westerman EL, Williams DS, Goldstein LS. Axonal stress kinase activation and tau misbehavior induced by kinesin-1 transport defects. J Neurosci. 2009;29:5758–5767. doi: 10.1523/JNEUROSCI.0780-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraley C. MCLUST: Software for Model-Based Cluster Analysis. Journal of Classification. 1999;16:297–306. [Google Scholar]
- Glater EE, Megeath LJ, Stowers RS, Schwarz TL. Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J Cell Biol. 2006;173:545–557. doi: 10.1083/jcb.200601067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein AY, Wang X, Schwarz TL. Axonal transport and the delivery of pre-synaptic components. Curr Opin Neurobiol. 2008;18:495–503. doi: 10.1016/j.conb.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross SP. Hither and yon: a review of bi-directional microtubule-based transport. Phys Biol. 2004;1:R1–11. doi: 10.1088/1478-3967/1/2/R01. [DOI] [PubMed] [Google Scholar]
- Gross SP, Tuma MC, Deacon SW, Serpinskaya AS, Reilein AR, Gelfand VI. Interactions and regulation of molecular motors in Xenopus melanophores. J Cell Biol. 2002;156:855–865. doi: 10.1083/jcb.200105055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris DA. Trafficking, turnover and membrane topology of PrP. Br Med Bull. 2003;66:71–85. doi: 10.1093/bmb/66.1.71. [DOI] [PubMed] [Google Scholar]
- Hendricks AG, Perlson E, Ross JL, Schroeder HW, 3rd, Tokito M, Holzbaur EL. Motor Coordination via a Tug-of-War Mechanism Drives Bidirectional Vesicle Transport. Curr Biol. 2010 doi: 10.1016/j.cub.2010.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Takemura R. Molecular motors and mechanisms of directional transport in neurons. Nat Rev Neurosci. 2005;6:201–214. doi: 10.1038/nrn1624. [DOI] [PubMed] [Google Scholar]
- Howard J. Mechanics of motor proteins and the cytoskeleton. Sunderland, Mass: Sinauer Associates; 2001. [Google Scholar]
- Jaqaman K, Loerke D, Mettlen M, Kuwata H, Grinstein S, Schmid SL, Danuser G. Robust single-particle tracking in live-cell time-lapse sequences. Nat Methods. 2008;5:695–702. doi: 10.1038/nmeth.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal A, Stokin GB, Yang Z, Xia CH, Goldstein LS. Axonal transport of amyloid precursor protein is mediated by direct binding to the kinesin light chain subunit of kinesin-I. Neuron. 2000;28:449–459. doi: 10.1016/s0896-6273(00)00124-0. [DOI] [PubMed] [Google Scholar]
- Kang JS, Tian JH, Pan PY, Zald P, Li C, Deng C, Sheng ZH. Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell. 2008;132:137–148. doi: 10.1016/j.cell.2007.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kardon JR, Vale RD. Regulators of the cytoplasmic dynein motor. Nat Rev Mol Cell Biol. 2009;10:854–865. doi: 10.1038/nrm2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karki S, Holzbaur EL. Cytoplasmic dynein and dynactin in cell division and intracellular transport. Curr Opin Cell Biol. 1999;11:45–53. doi: 10.1016/s0955-0674(99)80006-4. [DOI] [PubMed] [Google Scholar]
- King SJ, Brown CL, Maier KC, Quintyne NJ, Schroer TA. Analysis of the dynein-dynactin interaction in vitro and in vivo. Mol Biol Cell. 2003;14:5089–5097. doi: 10.1091/mbc.E03-01-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kural C, Kim H, Syed S, Goshima G, Gelfand VI, Selvin PR. Kinesin and dynein move a peroxisome in vivo: a tug-of-war or coordinated movement? Science. 2005;308:1469–1472. doi: 10.1126/science.1108408. [DOI] [PubMed] [Google Scholar]
- Ligon LA, Tokito M, Finklestein JM, Grossman FE, Holzbaur EL. A direct interaction between cytoplasmic dynein and kinesin I may coordinate motor activity. J Biol Chem. 2004;279:19201–19208. doi: 10.1074/jbc.M313472200. [DOI] [PubMed] [Google Scholar]
- Lu H, Ali MY, Bookwalter CS, Warshaw DM, Trybus KM. Diffusive movement of processive kinesin-1 on microtubules. Traffic. 2009;10:1429–1438. doi: 10.1111/j.1600-0854.2009.00964.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyman MG, Enquist LW. Herpesvirus interactions with the host cytoskeleton. J Virol. 2009;83:2058–2066. doi: 10.1128/JVI.01718-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaga-Trillo E, Solis GP, Schrock Y, Geiss C, Luncz L, Thomanetz V, Stuermer CA. Regulation of embryonic cell adhesion by the prion protein. PLoS Biol. 2009;7:e55. doi: 10.1371/journal.pbio.1000055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M, Iyadurai SJ, Gassman A, Gindhart JG, Jr, Hays TS, Saxton WM. Cytoplasmic dynein, the dynactin complex, and kinesin are interdependent and essential for fast axonal transport. Mol Biol Cell. 1999;10:3717–3728. doi: 10.1091/mbc.10.11.3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazumdar M, Mikami A, Gee MA, Vallee RB. In vitro motility from recombinant dynein heavy chain. Proc Natl Acad Sci U S A. 1996;93:6552–6556. doi: 10.1073/pnas.93.13.6552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore DS, McCabe GP. Introduction to the Practice of Statistics. 5. W. H. Freeman; 2005. [Google Scholar]
- Mouillet-Richard S, Ermonval M, Chebassier C, Laplanche JL, Lehmann S, Launay JM, Kellermann O. Signal transduction through prion protein. Science. 2000;289:1925–1928. doi: 10.1126/science.289.5486.1925. [DOI] [PubMed] [Google Scholar]
- Moya KL, Hassig R, Creminon C, Laffont I, Di Giamberardino L. Enhanced detection and retrograde axonal transport of PrPc in peripheral nerve. J Neurochem. 2004;88:155–160. doi: 10.1046/j.1471-4159.2003.02150.x. [DOI] [PubMed] [Google Scholar]
- Okada Y, Yamazaki H, Sekine-Aizawa Y, Hirokawa N. The neuron-specific kinesin superfamily protein KIF1A is a unique monomeric motor for anterograde axonal transport of synaptic vesicle precursors. Cell. 1995;81:769–780. doi: 10.1016/0092-8674(95)90538-3. [DOI] [PubMed] [Google Scholar]
- Plitz T, Pfeffer K. Intact lysosome transport and phagosome function despite kinectin deficiency. Mol Cell Biol. 2001;21:6044–6055. doi: 10.1128/MCB.21.17.6044-6055.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman A, Friedman DS, Goldstein LS. Two kinesin light chain genes in mice. Identification and characterization of the encoded proteins. J Biol Chem. 1998;273:15395–15403. doi: 10.1074/jbc.273.25.15395. [DOI] [PubMed] [Google Scholar]
- Rahman A, Kamal A, Roberts EA, Goldstein LS. Defective kinesin heavy chain behavior in mouse kinesin light chain mutants. J Cell Biol. 1999;146:1277–1288. doi: 10.1083/jcb.146.6.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodolfo K, Hassig R, Moya KL, Frobert Y, Grassi J, Di Giamberardino L. A novel cellular prion protein isoform present in rapid anterograde axonal transport. Neuroreport. 1999;10:3639–3644. doi: 10.1097/00001756-199911260-00032. [DOI] [PubMed] [Google Scholar]
- Sato-Yoshitake R, Yorifuji H, Inagaki M, Hirokawa N. The phosphorylation of kinesin regulates its binding to synaptic vesicles. J Biol Chem. 1992;267:23930–23936. [PubMed] [Google Scholar]
- Shubeita GT, Tran SL, Xu J, Vershinin M, Cermelli S, Cotton SL, Welte MA, Gross SP. Consequences of motor copy number on the intracellular transport of kinesin-1-driven lipid droplets. Cell. 2008;135:1098–1107. doi: 10.1016/j.cell.2008.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soppina V, Rai AK, Ramaiya AJ, Barak P, Mallik R. Tug-of-war between dissimilar teams of microtubule motors regulates transport and fission of endosomes. Proc Natl Acad Sci U S A. 2009 doi: 10.1073/pnas.0906524106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama Y, Kawabata I, Sobue K, Okabe S. Determination of absolute protein numbers in single synapses by a GFP-based calibration technique. Nat Methods. 2005;2:677–684. doi: 10.1038/nmeth783. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Kanai Y, Okada Y, Nonaka S, Takeda S, Harada A, Hirokawa N. Targeted disruption of mouse conventional kinesin heavy chain, kif5B, results in abnormal perinuclear clustering of mitochondria. Cell. 1998;93:1147–1158. doi: 10.1016/s0092-8674(00)81459-2. [DOI] [PubMed] [Google Scholar]
- Taylor AM, Blurton-Jones M, Rhee SW, Cribbs DH, Cotman CW, Jeon NL. A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat Methods. 2005;2:599–605. doi: 10.1038/nmeth777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorn KS, Ubersax JA, Vale RD. Engineering the processive run length of the kinesin motor. J Cell Biol. 2000;151:1093–1100. doi: 10.1083/jcb.151.5.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhey KJ, Hammond JW. Traffic control: regulation of kinesin motors. Nat Rev Mol Cell Biol. 2009;10:765–777. doi: 10.1038/nrm2782. [DOI] [PubMed] [Google Scholar]
- Verhey KJ, Lizotte DL, Abramson T, Barenboim L, Schnapp BJ, Rapoport TA. Light chain-dependent regulation of Kinesin’s interaction with microtubules. J Cell Biol. 1998;143:1053–1066. doi: 10.1083/jcb.143.4.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welte MA. Bidirectional transport along microtubules. Curr Biol. 2004;14:R525–537. doi: 10.1016/j.cub.2004.06.045. [DOI] [PubMed] [Google Scholar]
- Xia C, Rahman A, Yang Z, Goldstein LS. Chromosomal localization reveals three kinesin heavy chain genes in mouse. Genomics. 1998;52:209–213. doi: 10.1006/geno.1998.5427. [DOI] [PubMed] [Google Scholar]
- Xia CH, Roberts EA, Her LS, Liu X, Williams DS, Cleveland DW, Goldstein LS. Abnormal neurofilament transport caused by targeted disruption of neuronal kinesin heavy chain KIF5A. J Cell Biol. 2003;161:55–66. doi: 10.1083/jcb.200301026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.