

Background: rRNA synthesis by Pol-I is a key and rate-limiting stage of ribosome biogenesis.
Results: Ellipticines selectively inhibit Pol-I transcription both in vitro and in cells.
Conclusion: Interactions of essential transcription factor SL1 with the promoter is the primary target of the drugs.
Significance: This study reveals a novel class of Pol-I inhibitors and analyses their mechanism of action.
Keywords: Cancer, Drug Design, Ribosomal RNA (rRNA), RNA Polymerase I, Transcription, Drug Action, Drug Development, Ellipticines
Abstract
Transcription by RNA polymerase I (Pol-I) is the main driving force behind ribosome biogenesis, a fundamental cellular process that requires the coordinated transcription of all three nuclear polymerases. Increased Pol-I transcription and the concurrent increase in ribosome biogenesis has been linked to the high rates of proliferation in cancers. The ellipticine family contains a number of potent anticancer therapeutic agents, some having progressed to stage I and II clinical trials; however, the mechanism by which many of the compounds work remains unclear. It has long been thought that inhibition of Top2 is the main reason behind the drugs antiproliferative effects. Here we report that a number of the ellipticines, including 9-hydroxyellipticine, are potent and specific inhibitors of Pol-I transcription, with IC50 in vitro and in cells in the nanomolar range. Essentially, the drugs did not affect Pol-II and Pol-III transcription, demonstrating a high selectivity. We have shown that Pol-I inhibition occurs by a p53-, ATM/ATR-, and Top2-independent mechanism. We discovered that the drug influences the assembly and stability of preinitiation complexes by targeting the interaction between promoter recognition factor SL1 and the rRNA promoter. Our findings will have an impact on the design and development of novel therapeutic agents specifically targeting ribosome biogenesis.
Introduction
Ribosome biogenesis is a fundamental cellular process that requires the coordinated transcription of all three nuclear polymerases (Pols).4 Pol-I transcribes the ribosomal RNAs (rRNAs) that form the backbone of the ribosomal subunits, Pol-II transcribes the messenger RNAs (mRNAs), and Pol-III transcribes the transfer RNAs (tRNAs), the 5 S rRNA, and other small RNAs. Disruptions to ribosome biogenesis that lead to increased levels of ribosome production and consequently to increased rates of protein synthesis and cell growth are commonly found in many chemotherapy-resistant and hard to treat cancers (1).
Distributed over the short arms of chromosomes 13, 14, 15, 21, and 22 are ∼400 copies of the ribosomal DNA (rDNA) repeat; however, only 50% of these are active at any time (2). Pol-I transcription of the rDNA yields the 47 S pre-rRNA, which after extensive processing yields the 5.8, 18, and 28 S rRNAs. These rRNAs then associate with the ribosomal proteins to yield the large and small ribosomal subunits. Pol-I transcription is responsible for about 80% of all the transcription carried out by the cell (3). Several transcription factors are essential for rRNA transcription including Pol-I itself, RRN3, SL1, and UBF (for the latest reviews see Refs. 4 and 5).
Despite the essential role that rDNA transcription plays with regard to cell growth and proliferation, the Pol-I transcription machinery has only recently become a target for anticancer therapy (4). The compounds CX-3543 and CX-5461, both developed by Cylene Pharmaceuticals, have entered clinical trials (stages II and later) and have been shown to target rRNA synthesis through disruption of G-quadruplex formation, which leads to decreased accessibility to the DNA for proteins required in transcription (6) and SL1-promoter binding (7), respectively. Other research examining various known chemotherapeutic agents has shown that a diverse range of compounds target ribosome biogenesis at different stages (8).
The ellipticine family are planar alkaloid compounds capable of entering the cell by diffusion (9), and many of the ellipticines display a wide range of cellular effects due to the diverse number of targets (10–12). Several derivatives of the parent compound, such as hydroxyl-methyl-ellipticine, have been validated for use in clinical trials (13); however, these have not progressed beyond stage one or two due to adverse side effects of the treatments (14). 9-Hydroxyellipticine (9HE), a water soluble derivative of ellipticine, was discovered by screening of ellipticine derivatives that were capable of intercalating into DNA with an affinity higher than of the parent compound ellipticine (15). 9HE has a diverse range of cellular targets including Top2 (16), telomerase (17), cytochrome P450 (18), and p53 (19). One of the effects of 9HE treatment in cells is decreased p53 phosphorylation, and prolonged treatment with 9HE leads to a G0/G1 block of the cell cycle followed by p53-mediated apoptosis (19, 20).
In this study we have shown that 9HE and two other ellipticines are efficient inhibitors of eukaryotic Pol-I transcription in vitro. We have demonstrated that 9HE efficiently inhibits rRNA transcription in cells and that this inhibition occurs independently of p53, ATM/ATR, or Top2. Importantly, the drug selectively inhibits Pol-I without affecting transcription by Pols II and III. We have shown that 9HE rapidly localizes to the nucleus of the cell and functions primarily through disrupting the interactions of Pol-I transcriptional machinery and the promoter, with SL1 appearing as the drug main target.
EXPERIMENTAL PROCEDURES
Tissue Culture
MCF7, MCF10A, U2OS, HCT116, and H1299 cells were obtained from ATCC and maintained according to the supplier's instructions. HTETOP cells were a kind gift from Dr. A. Porter (Imperial College London) and were maintained in DMEM (high glucose) in a 37 °C incubator with 5% CO2. All media were supplemented with 10% FBS (PAA) and 100 units/ml penicillin and 100 μg/ml streptomycin (Invitrogen). HTETOP media was additionally supplemented with 1 mm sodium pyruvate (PAA) and 4× MEM (PAA). For Top2α depletion, HTETOP medium was supplemented with 1 μg/ml tetracycline for 48 h.
Real-time Cell Growth Measurements
Cell growth over a 48-h period was monitored using an IncuCyte live-cell imaging system (Essen BioScience, Ann Arbor, MI) housed in a 37 °C incubator at 5% CO2 with readings taken every hour. Cells were seeded onto 24-well plates at 50,000 cells per well, and drugs (etoposide (50 μm), 9HE (5 μm), caffeine (125 mm), or a combination of 9HE and caffeine) were added after overnight incubation. The data analysis was performed using the IncuCyte FLR software. S.D. were calculated from three independent experiments.
In Vitro Transcription Assays
Nonspecific transcription assays were performed as described (21). Specific transcription reactions (either for S1 nuclease protection assay or for run-off assay) were performed essentially as described (22, 23) using supercoiled plasmid DNA or immobilized rDNA fragments containing the human rRNA gene promoter. Reactions were supplemented either with HeLa nuclear extract (NE) or with appropriate purified factors (22). Run-off reactions were terminated by the addition of RLT buffer (Qiagen), and RNA transcripts (purified using RNeasy mini kits (Qiagen)) were electrophoresed on denaturing 4% acrylamide, 8 m urea gels visualized by phosphorimaging using a FLA-7000 scanner (Fuji) and analyzed using Aida software. S1 nuclease protection was performed as described (22) with a 5′-end-labeled oligonucleotide identical to the region between −20 and +40 of the template strand in the human rRNA gene promoter. The amount of RNA produced by in vitro transcription reactions was quantified with the aid of phosphorimaging, FLA-7000 (Fuji), and Aida software. S.D. was calculated from three independent experiments.
9HE Fluorescence Detection
U2OS cells were grown on sterilized coverslips until 70% confluent, then treated with 10 μm 9HE for 10 min, cross-linked with 4% paraformaldehyde for 10 min, washed with PBS, and mounted onto slides with 90% glycerol. Excitation was at 405 nm, and emission was recorded within the range 410–620 nm using a Leica TCS SP5 scanning laser confocal microscope with a ×63 objective.
Immunoblotting and Immunoprecipitation
Antibodies used for immunoblotting and immunoprecipitations are listed in supplemental Table S1. Anti-FLAG M2 magnetic beads (Sigma) were used for immunoprecipitation (2 h at 4 °C) from nuclear extracts, prepared as described (24) from U2OS or HeLa cells transfected (FuGENE® HD reagent, Roche Applied Science) with one of the following expression vectors: FLAG-CAST expression vector pcFCAST (25), FLAG-RRN3 expression vector pcDNA3-FLAGhRRN3 (21), FLAG-TAFI110 expression vector pcfTAF110 and FLAG-UBF expression vector pcfUBF. The beads were washed 3 times in TM10 buffer (50 mm Tris-HCl, pH 7.9, 12.5 mm MgCl2, 1 mm EDTA, 10% glycerol, 1 mm sodium metabisulfite, and 1 mm dithiothreitol), 0.15 m KCl. Washed precipitates were split in half, and one-half was incubated with 50 μm 9HE and the other half with TM10, 0.15 m KCl buffer for 30 min on ice. Immunoprecipitated complexes were eluted from washed beads using FLAG-peptide (Sigma) according to the manufacturer's instruction. All buffers were supplemented with EDTA-free complete protease inhibitor mixture (Roche Applied Science) and Phosphatase Inhibitor Mixture 2 (Calbiochem).
Immunocytochemistry
U2OS cells were grown until 70% confluent on sterilized coverslips then treated for 10 min with 5 μm 9HE, fixed for 10 min with 4% paraformaldehyde, permeabilized for 10 min with 1% Triton X-100, and blocked for 10 min with 1% donkey serum (Jackson Immuno Research) in PBS. Cells were then incubated with one of the antibodies listed in supplemental Table S1 in blocking buffer (containing 1% donkey serum) for 1 h followed by three 10-min washes with PBS and then incubated with α-mouse, α-sheep, and α-rabbit Cy3-labeled secondary antibodies (Jackson Immuno Research) for 1 h. Coverslips were then washed 3 × 10 min with PBS and mounted onto glass slides using Vectashield with DAPI and visualized using a Leica TCS SP5 confocal microscope with a ×40 objective.
BrU Incorporation
Cells were grown until 70% confluent on sterilized coverslips. Cells were then washed 2× in buffer PB (100 mm KOAc, 30 mm KCl, 10 mm Na2PO4, 1 mm MgCl2, 1 mm Na2ATP, 1 mm DTT, 0.2 mm PMSF, 5 units/ml RNasin) followed by permeabilization on ice with PB + 0.05% Triton X-100 for 2 min. The cells were then washed 2× in PB on ice and then incubated at 33 °C in buffer PB. Transcription was carried out by the addition of transcription mix (100 mm CTP, 100 mm ATP, 100 mm GTP, 100 mm BrUTP, 1 mm MgCl2, 1 mg/ml α-amanitin as indicated, 2.5 μm 9HE as indicated in buffer PB) for 15 min at 33 °C. Cells were then washed 2× on ice with buffer PB and fixed at −20 °C by the addition of acetone:methanol mix for 4 min followed by aspiration of the fixation buffer, air drying of the slides, and rehydration of the cells by 2× PBS washes. Cells were then incubated with α-BrdU antibody (supplemental Table S1) for 1 h followed by three 5-min washes with PBS and incubation with α-mouse Cy3-labeled secondary antibody. Coverslips were then washed 3× with PBS and mounted onto glass slides using Vectashield with DAPI and visualized using a Leica TCS SP5 confocal microscope with ×40 objective.
Analysis of Pol-II and Pol-III Transcription
Cells were grown until 70% confluent and then treated with 25 μm 9HE for 1 h. RNA was purified using the RNeasy purification kit (Qiagen) following the manufacturer's instructions, and the RNA concentration was determined spectroscopically. 1 μg of RNA was converted to cDNA using the High Capacity RNA-to-cDNA reverse transcriptase kit (Applied Biosystems). Pol-II transcripts were analyzed on the LightCycler 480 thermocycler (Roche Applied Science) using the Human Housekeeper reference gene plate (Roche Applied Science) according to manufacturer's instructions. Pol-III transcripts were analyzed on the LightCycler 480 thermocycler (Roche Applied Science) using primers and conditions described previously (for tRNA-Arg (26) and for 5S rRNA and 7SL RNA (27)). S.D. were calculated from three independent experiments.
Analysis of Pol-I Pre-initiation Complexes (PICs)
Purified Pol-I, UBF, and SL1 were incubated with the immobilized DNA template (22) in TM10, 50 mm KCl for 20 min on ice. After PIC formation, beads were washed by the same buffer twice, and half of them were used for in vitro transcription assay, the other half used for Western blot analysis. Proteins were eluted with 8 m urea, and samples were analyzed by Western blotting with antibodies specific to human Pol-I subunits A135 and PAF53, and UBF, RRN3, and SL1 subunits TAFI110, TAFI63, and TBP (see supplemental Table S1). The TM buffer was supplemented with EDTA-free complete protease inhibitor mixture (Roche Applied Science) and Phosphatase Inhibitor Mixture 2 (Calbiochem).
Analysis of Pre-rRNA Level in Cells
Total RNA was isolated from 9HE-treated and untreated cells using the RNeasy kit (Qiagen) according to the manufacturer's instructions. Pre-rRNA levels were determined by S1 nuclease protection assay as described (22) using 5 and 10 μg of RNA per reaction. S.D. were calculated from three independent experiments.
In Vivo RNA Labeling and rRNA Analysis
In vivo labeling of RNA from cells (∼70% confluent) was performed essentially as described (28) using 10 μCi of [3H]uridine for ∼0.2–0.4 × 105 cells per well of a 6-well plate. In pulse-chase labeling, cells were incubated for 2 h with [3H]uridine, washed, and incubated in unlabeled medium containing 0.5 mm uridine (±Pol-I inhibitors). RNA was extracted (RNeasy Mini Kit (Qiagen)). 2 μg of 3H-labeled total RNA was run on a 1% formaldehyde agarose gel at 120 V for 90 min in 1× MOPS, blotted onto a Hybond-N membrane (Amersham Biosciences), cross-linked using UV crosslinker (UVP), and analyzed by tritium imaging using a Fuji Tritium image plate (or after PerkinElmer Life Sciences En3Hance spray, exposed to Kodak Biomax XAR film at −80 °C), then quantified using Aida software. S.D. were calculated from three independent experiments.
Chromatin Immunoprecipitation (ChIP) Assay
Cells were grown until 70% confluent and cross-linked with formaldehyde (final concentration 1%) for 10 min, and the cross-linking was stopped by the addition of glycine (final concentration 0.125 m) for 5 min. Cross-linked chromatin was shared using Biorupter (Diagenode) to a 300-base pair average size. Immunoprecipitation was carried out using chromatin isolated from 1 × 106 cells (antibodies are listed in Supplemental Table S1) or appropriate control IgGs and 30 μl of Protein A/Protein G magnetic beads (Invitrogen). Purified immunoprecipitated DNA was analyzed by two tetraplex quantitative PCR panels designed for eight regions of the rDNA repeat; Promoter, IGS4, 18 S, 5.8 S, 28 S, Terminator, IGS1, and 5′ETS (see supplemental Table S2 for primers and probes sequences). Reactions were carried out on a LightCycler 480 with a reaction volume of 10 μl per well in triplicate. Results were expressed as the percentage of input chromatin and normalized to control IgG levels, with S.D. calculated from three independent ChIP experiments.
Binding Constant Determination
Experiments were carried essentially as described previously (29) with minor modifications. 5–20 nm Pol-I promoter containing DNA sequences or nonspecific DNA of similar lengths were incubated with 0.5 nm ethidium bromide and 9HE (0.5–10 μm) on ice for 15 min. Experiments were performed in triplicate with 100-μl samples added to black 96-well plates, and ethidium bromide fluorescence was monitored by excitation at 510 nm and emission at 590 nm. Plates were incubated at 30 °C during each experiment. KD value determination and analysis of 9HEs mode of DNA binding (single or multiple DNA binding site analysis) was carried out using Graphpad Prism software (Version 2). Dissociation constant determination was carried out using Lineweaver-Burke analysis. One site binding was chosen as the predominant binding mode, and two-site binding was only accepted if KD values were found to be significantly different between each model based on F-test analysis; to ensure data validity, residual plots were plotted for all data points. S.D. were calculated from at least three independent experiments.
Molecular Modeling
The pharmacophore search of 9HE, GQC-Qi, and CX-5461 was conducted using PHASE (Version 3.1 Schrodinger, LLC, New York, NY, 2009) with default settings. The crystal structure of the DNA-9HE complex with the PDB code 1Z3F (30) was used to dock 9HE, GQC-Qi, and CX-5461. The docking was done manually using the results of the pharmacophore search where pharmacophoric features were defined using ellipticine in Maestro (Version 9.2, Schrödinger, LLC, New York, NY, 2011) and optimized with MacroModel (Version 9.7, Schrödinger, LLC, New York, NY, 2009) with the default settings. All pictures were prepared in the PyMOL Molecular Graphics System.
RESULTS
9HE, Ellipticine, and 9-Methoxy-2-methylellipticinium Acetate Inhibit Pol-I Transcription in Vitro
In the course of a separate study we found that 9HE (Fig. 1A, structure 1), a member of the family of ellipticines and potent Top2α inhibitor (IC50 = 3.3 μm), efficiently represses Pol-I transcription in reconstituted reactions in a dose-dependent manner (Fig. 1B) with IC50 = 585 nm (Fig. 1C). Importantly, other Top2 inhibitors (Merbarone, ICRF-193, etoposide, and genistein) have no effect on Pol-I transcription in this assay (Fig. 1D), suggesting that 9HE represses Pol-I by a mechanism independent from its Top2 inhibitory activity. We next tested if other members of the ellipticine family are inhibitors of Pol-I transcription or if this is a unique property of 9HE. Two commercially available compounds (ellipticine and 9-methoxy-2-methylellipticinium acetate) (Fig. 1A, structures 2 and 3, respectively) were tested in the reactions supplemented with HeLa nuclear extract. Both compounds repress specific transcription (Fig. 1E), suggesting that the ability to inhibit Pol-I is a common feature of ellipticine family. Using a nonspecific activity assay (promoter independent transcription assay) we have shown that ellipticines do not inhibit the catalytic activity of Pol-I (Fig. 1F). Interestingly, 9HE is also capable of inhibiting specific Pol-I transcription in a yeast-reconstituted transcription system without affecting the activity of the RNA polymerase III (Fig. 2).
FIGURE 1.

Ellipticines inhibit Pol-I transcription in vitro. A, chemical structures of 9HE (1), ellipticine (2), and 9-methoxy-N2-methylellipticinium acetate (3) are shown. B, specific transcription reactions containing 2 μl of HeLa NE and 200 ng of supercoiled plasmid DNA template were supplemented with various amounts of 9HE as indicated. To ensure an equal loading of the DNA template and NE, the reaction mixture containing all components (excluding drugs) was first prepared, then aliquoted into individual reactions, and then the drug or buffer were added. Transcripts were analyzed by S1 nuclease protection; a representative image is shown. Lane 1 is the control reaction containing no drug. C, the results were quantified with the aid of phosphorimaging. The data are expressed as a percentage of the highest value (set at 100%) and were used for IC50 calculation. The data represent an average from three independent experiments. S.D. is shown. D, specific transcription reactions containing 2 μl of HeLa NE and 200 ng of supercoiled plasmid DNA template were supplemented with different Top2 inhibitors (merbarone, etoposide, and genistein were used at 100 μm final concentration; ICRF-193 at 50 μm, and AMP-PNP at 500 μm). To ensure equal loading of the DNA template and NE, the reaction mixture containing all components (excluding drugs) was first prepared, then aliquoted into individual reactions, and then the drug or buffer were added. Transcripts were analyzed as in B, and representative image is shown. Lane 2 is the control reaction containing no drug. E, specific transcription reactions (as in B) were supplemented with various amounts of ellipticine or 9-methoxy-2-methylellipticinium acetate (MMEA) as indicated. Transcripts were analyzed as in B and quantified as in C. F, nonspecific transcription reactions were supplemented with 1 μl of HeLa nuclear extract and various amounts of 9HE as indicated. The data expressed as a percentage of the highest value (set at 100%) represent an average from three independent experiments. S.D. and statistical significance (*, p < 0.05) are shown. p values have been calculated using one and two-way analysis of variance on R software.
FIGURE 2.

9HE efficiently inhibits Pol-I transcription in the yeast-reconstituted transcription system without affecting transcription by Pol-III. A, Pol-I in vitro transcription was assayed with increasing concentrations of 9HE (0.1–1 μm). In each lane, a constant concentration (0.63 μg/μl) of yeast transcription extract prepared as described earlier (60, 61) was incubated with transcription matrix pSIRT or pSIRTΔ (62). Both plasmids contained RNA Pol-I minigene that led, respectively, to Pol-I transcripts of 681 (35 S long) and 516 (35 S short) nucleotides and carry RNA Pol-III transcribe gene RDN5.3 (5 S). nt, nucleotides. Transcription reactions were performed as described previously (61). 9HE was diluted at the required concentration and preincubated together with transcription extract for 30 min before starting the reaction by the addition of NTP mix and radiolabeled UTP. Transcription products were analyzed on 6% urea-acrylamide gel and revealed by phosphorimaging. B, quantification of transcription products is shown. The histogram shows the percentage of specific activity of RNA Pol-I (gray bars) and RNA Pol-III (white bars) as a function of 9HE concentration using pSIRTΔ (35 S short) as transcription matrix. For each 9HE concentration, the specific activity is defined as the ratio of RNA Pol-I activity or RNA Pol-III activity versus total activity (RNA Pol-I+RNA Pol-III). Results are normalized, respectively, against the maximum RNA Pol-I and RNA Pol-III activity. S.D. is shown.
9HE Inhibits Pol-I Transcription in Cells by a Mechanism That Is Not Linked to Other Known Activities of the Drug
We next tested the effect of 9HE on rDNA transcription on a range of human-derived cell lines including U2OS, HeLa, H1299, and MCF10A (Fig. 3A). 9HE efficiently repressed Pol-I transcription (with IC50 ∼ 120–800 nm depending on cell line, supplemental Table S3). Notably, the drug acts very rapidly, and we observed significant repression of rRNA synthesis within 5 min of treatment (Fig. 3B). Interestingly, that non-malignant MCF10A cell line displays highest resistance to 9HE treatment among all cell lines tested (supplemental Table S3). However, at the moment the reasons for lower sensitivity of rDNA transcription to the drug in this particular cell line are unknown.
FIGURE 3.

Ellipticines rapidly inhibit rDNA transcription in cells, and inhibition mechanism is not linked to known activities of the drugs. A, HeLa, H1299, U2OS, and MCF10A cell lines were incubated for 60 min with various concentrations of 9HE as indicated. The relative level of pre-rRNA was determined by S1 nuclease protection assay. The results were quantified with aid of phosphorimaging and expressed as a percentage of the highest value (set at 100%). The data represent an average from three independent experiments. S.D. is shown. B, U2OS cells were incubated for various times with two concentrations of 9HE as indicated. The relative level of pre-rRNA was determined as in A. C, HTETOP cells were incubated for 48 h with 1 μg/ml (+Tet) or without (no Tet) tetracycline and treated with different 9HE concentrations for 30 min. The relative level of pre-rRNA was determined as in A. D, p53 null H1299 cells were incubated for 2 h with 125 μm caffeine (+caffeine) or without (control) and treated with different 9HE concentrations for 30 min. The relative level of pre-rRNA was determined as in A. E, the level of confluence of growing H1299 (p53−/−) cells was measured by IncuCyte (Essen) each hour for 43 h in total and expressed as %. Mean values for three independent experiments are represented on the graph, and S.D. are shown. Cells were grown untreated (control) and in the presence of 125 μm caffeine, 50 μm etoposide, 5 μm 9HE and 125 μm caffeine, and 5 μm 9HE combined (9HE + caffeine). Growth media were replaced each 12 h. AU, arbitrary units.
9HE is a potent Top2 inhibitor, and in this capacity it can affect rRNA transcription in cells by inducing DNA damage and activating DNA damage response pathways that can cause repression of Pol-I transcription in cells (31, 32). We determined the effect of 9HE on rDNA transcription in cells lacking Top2α, p53 null cells and in cells treated with the ATM/ATR inhibitor caffeine. In HTETOP cells >99.5% Top2α expression can be silenced in all cells by 48 h of treatment by tetracycline (33) (supplemental Fig. S1). We found that 9HE inhibits rRNA synthesis with the same efficiency regardless of the presence or absence of Top2α (Fig. 3C and supplemental Table S3). Furthermore, 9HE represses rRNA synthesis in p53 null and in p53 null cells pretreated with ATM/ATR inhibitor caffeine (Fig. 3D). Similarly, cell proliferation assays showed that the effect of 9HE on cell proliferation is different from the effect of the Top2 poison and DNA-damaging agent etoposide and does not depend on p53 and ATM/ATR pathways (Fig. 3E). Together these data strongly suggest that 9HE affects Pol-I transcription by a mechanism unrelated to its topoisomerase inhibitory activity and independent from p53 and ATM/ATR pathways.
9HE Selectively Inhibits Pol-I Transcription and Has No Effect on rRNA Processing
The three nuclear polymerases (Pol-I, Pol-II, and Pol-III) exhibit similar catalytic properties and share the same structural layout. Our results from the nonspecific assay suggest that 9HE does not affect the catalysis; however, 9HE may interfere with other stages of the transcription cycle that are similar for all three enzymes (e.g. transcription bubble formation, first phosphodiester bond synthesis). Moreover, in cells, 9HE may inhibit transcription indirectly by affecting other factors/pathways. To explore such possibilities and to determine the selectivity of 9HE, we measured the effect of 9HE treatment on the transcription by Pol-II and Pol-III genes in cells (Fig. 4A and Supplemental Table S4) and by Pol-II in a reconstituted transcription assay (supplemental Fig. S2). We determined relative expression levels of 18 housekeeping genes (GenExp Panel, Roche Applied Science) and of three Pol-III dependent genes in 9HE-treated and -untreated cells. We found no effect of 9HE on transcription of these genes under conditions when Pol-I transcription was significantly repressed (Fig. 4A). In vitro transcription assay also shows no effect of 9HE on Pol-II driven transcription (supplemental Fig. 2). All these results together reveal that the transcription of Pol-II and Pol-III targets is not affected by the drug, demonstrating a high selectivity of 9HE.
FIGURE 4.

9HE selectively inhibits Pol-I transcription in cells without affecting Pol-II and Pol-III transcription. A, actively growing U2OS cells (∼70% confluent) were treated with 25 μm 9HE for 1 h followed by cell lysis, RNA purification, and first strand cDNA synthesis. The cDNA generated was analyzed by quantitative PCR using the Human Reference Gene panel (Roche Applied Science) for Pol-II genes on a LightCycler 480 (Roche Applied Science) following the manufacturer's instructions and cycling conditions. Pol-III genes were analyzed as described previously (26, 27). RNA samples were analyzed for Pol-I transcription by S1 nuclease protection assay as described under “Experimental Procedures.” Pol-II and Pol-III transcripts were analyzed by relative quantification comparing untreated to 9HE-treated samples, and error bars represent S.D. for triplicate reactions. TBP, TATA-box binding protein; ALAS, aminolevulinate synthase. B, actively growing U2OS cells (confluency ∼70%) were treated either with 1 mg/ml α-amanitin or with 2.5 μm 9HE or with a combination of both for 1 h, and BrU incorporation was determined by immunostaining using a Leica SP5 scanning laser confocal microscope with ×40 objective (scale bar is 10 μm). The representative image is shown; two other images are shown in supplemental Fig. S3. C, the results were quantified and expressed as a percentage of the BrU incorporation level in untreated cells (set at 100%) The data represent an average from three independent experiments; S.D. and statistical significance (***, p < 0.001) are shown. p values have been calculated using one and two-way analysis of variance on R software.
As a complimentary approach, we analyzed the effect of 9HE treatment on de novo RNA synthesis in cells treated with the Pol-II/Pol-III inhibitor α-amanitin (Fig. 4B and supplemental Fig. S3). BrU incorporation was drastically reduced after 45 min when cells were treated with 9HE and α-amanitin together, suggesting inhibition of all three RNA polymerases (Fig. 4B, panel 2). In contrast, when cells were treated either by α-amanitin (panel 3) or 9HE (panel 4), BrU incorporation (and, therefore, RNA synthesis) was still detected (compare panels 2, 3, and 4). The quantification of BrU incorporation (Fig. 4C) shows that combined α-amanitin/9HE treatment reduces BrU incorporation to ∼10%. α-Amanitin alone (Pol-II/Pol-III-inhibited) or 9HE alone (Pol-I-inhibited) led to an ∼30 and 50% percent reduction, respectively (Fig. 4C). After deduction of 10% of the drug-insensitive signal (that most likely represents a nonspecific background), the estimated level of Pol-I transcription was ∼60%, and Pol-II/Pol-III was ∼40% of the total transcription.
Transcription of rDNA and rRNA processing in eukaryotes are two coordinated events (34, 35). To test the effect of 9HE on pre-RNA processing we used [3H]uridine pulse-chase labeling (as outlined in Fig. 5A). Our results suggested that 9HE has no detectable effect on rRNA processing in concentrations that fully repress rRNA synthesis (Fig. 5, B and C). All these results together clearly suggest that 9HE selectively inhibits Pol-I transcription without affecting rRNA processing and transcription by Pol-II and Pol-III.
FIGURE 5.

9HE has no detectable effect on rRNA processing in actively growing cells. A, shown is a schematic representation of the pulse-labeling cold-chase of cells with [3H]uridine to determine the effects 9HE on pre-rRNA processing in cells. HCT116 (p53−/−) cells were grown to 70% confluence and incubated for 2 h with 10 μCi of [3H]uridine to label newly synthesized pre-rRNA (47/45 S). At time point 0 the cells were washed and incubated for 4 and 12 h in unlabeled medium without (no drug) or with 5 μm 9HE. B, total RNA was analyzed as described under “Experimental Procedures.” C, to determine the relative efficiencies of pre-rRNA processing, the data were quantitated (bar graphs: −, no 9HE, gray bars; +9HE, white bars). Transcript levels for 47S/45S pre-rRNA are shown on the left, and the 28S/18S rRNA ratio is shown on the right.
9HE Rapidly Accumulates in the Cell Nucleus and Displaces Pol-I Transcription Machinery from rDNA Repeat
Analysis of the cellular localization of 9HE by direct fluorescence microscopy (excitation at 405 nm, emission at 470 nm) and cellular distribution of Pol-I factors by immunostaining show that the drug rapidly accumulates in the nucleus (Fig. 6A) and causes changes in the localization of Pol-I and SL1, whereas the localization of UBF and RRN3 remains unchanged (Fig. 6B). This suggests that 9HE might affect early stages of PIC formation.
FIGURE 6.

9HE rapidly localizes in the nucleus and affects localization and occupancy of various components of the Pol-I transcription apparatus. A, actively growing U2OS cells (confluency ∼70%) were either treated with 10 μm 9HE for 10 min or untreated, fixed, washed, and visualized using a Leica TCS SP5 scanning laser confocal microscope. B, actively growing U2OS cells (confluency ∼70%) were either treated with 5 μm 9HE for 10 min or untreated, fixed, and analyzed by indirect immunofluorescence using antibodies specific to human SL1 subunit TAFI110 (panels 1 and 2), human UBF (panels 3 and 4), human Pol-I largest subunit A194 (panels 5 and 6), and human Pol-I transcription factor RRN3 (panels 7 and 8). Nuclear DNA was stained by DAPI (blue). Images were obtained by laser scanning confocal microscopy (scale bar is 15 μm). C, a diagram of the human rDNA repeat is shown indicating the positions of the PCR primer/probes used in ChIP analysis. The primer and probe sets used in the quantitative real-time PCR for the rDNA repeat were from the following regions: the rDNA promoter, 5′ETS, 18 S, 5.8 S, and 28 S rRNA gene-transcribed sequences (included in the 47 S pre-rRNA) and the intergenic spacers IGS1 and IGS2. kb, kilobases. D, ChIP assays were performed on chromatin from cells treated with 5 μm 9HE for the indicated periods of time using antibodies specific to human SL1 subunit TAFI110, human Pol-I second largest subunit A135, and human UBF and normalized to control IgG samples. The signal representing the transcribed region (TrR) is the average of the combined signal from 5′ETS, 18 S, 5.8 S, and 28 S rRNA. The signal representing the non-transcribed region (nTrR) is average of the combined signal from IGS1 and IGS2. Prom, a promoter region. Bar graphs are the combined data of three independent ChIP experiments. S.D. is shown.
To test this hypothesis we performed time course ChIP experiments harvesting cells at different time points after 9HE treatment and analyzing the association of Pol-I, UBF, and SL1 with rDNA (Fig. 6, C and D). Drug treatment led to a rapid decrease in the promoter occupancy of SL1 (Fig. 6D, top panel) followed by a decrease in Pol-I occupancy (Fig. 6D, middle panel). Interestingly, UBF occupancy was decreased at the promoter but not at the other regions of rDNA (Fig. 6D, bottom panel). Together these data suggest that the drug initially affects SL1 and then UBF and Pol-I occupancy at the rRNA promoter and consequently Pol-I occupancy across the transcribed region. SL1 is the factor responsible for promoter recognition, and interactions of other factors with the rRNA promoter are dependent upon the presence of SL1 (36, 37). Therefore, our results suggest that the drug targets the interaction of SL1 and the rRNA promoter.
9HE Interferes with SL1 Binding to the Promoter
The results of in vivo analysis suggest that 9HE treatment led to a decrease of SL1 and Pol-I occupancies at the rDNA, whereas UBF was largely unaffected. Potentially the drug can affect SL1-promoter interactions and Pol-I-SL1 interactions either directly (by interacting with particular factors or the promoter) or indirectly (the drug interacts with other factors that determine the ability of factors to bind to the promoter or interact with each other (e.g. via posttranslational modifications). To assess the mechanism of the drugs actions, we employed in vitro based assays where we can rule out the indirect effects of the dug.
We used an immobilized template containing the Pol-I promoter (23) and purified factors (Pol-I, SL1, and UBF) to examine the effect of 9HE on PIC formation. As outlined in Fig. 7A, we analyzed transcription efficiency and proteins bound to the template when the drug was added either before or simultaneously or after the factors. It is evident that the drug affects transcription (Fig. 7B) and binding of SL1 and Pol-I (Fig. 7C) when added during or after PIC formation, suggesting that the drug can displace already-bound factors (Fig. 7C, compare lane 1 and lanes 3 and 4). As in our in vivo experiments, binding of UBF was largely unaffected.
FIGURE 7.

9HE interferes with preinitiation complex assembly by affecting SL1 interactions with the rRNA promoter. A, the effect of 9HE on formation of Pol-I PICs was analyzed when the drug was added before (I), during (II), or after (III) PIC formation. Immobilized ribosomal promoter templates (IT) were initially incubated either with 10 μm 9HE (I) or with 10 μm 9HE and purified Pol-I, UBF, and SL1 (TF) (II) or with Pol-I factors only (III) for 15 min on ice. The beads were washed and incubated for an additional 15 min on ice either with Pol-I factors (I) or with buffer (II) or with 10 μm 9HE (III). The beads were washed and split in half and used either for in vitro transcription assay or for Western blot (WB) analysis. B, Pol-I PICs were assayed for transcriptional activity, and transcripts were detected by S1 nuclease protection and quantified. The data expressed as a percentage of the highest value (set at 100%) represent an average from three independent experiments. S.D. is shown. C, for immunoblotting, proteins were eluted from the IT-DNA with 8 m urea, subjected to SDS-polyacrylamide gel electrophoresis, and blotted onto PVDF membranes that were probed with antibodies against A135, PAF53, UBF, TAFI110, TAFI63, and TBP. D, the effect of 9HE on SL1 binding to the ribosomal promoter was analyzed when the drug was added either after (I) or during (II) binding. Immobilized ribosomal promoter templates were initially incubated either with purified SL1 (I) or with various amounts of 9HE (as indicated) and purified SL1 (II) for 15 min on ice. The beads were washed and incubated for an additional 15 min on ice either with various amounts of 9HE (I) or with buffer (II). The beads were washed and used for Western blot analysis. E, for immunoblotting, proteins were eluted from the IT-DNA with 8 m urea, subjected to SDS-polyacrylamide gel electrophoresis, and blotted onto PVDF membranes that were probed with antibodies against SL1 subunits TAFI110 and TAFI63.
However, when the drug was first added to the template and the template was washed and the factors added, we observed no effect on transcription or binding (Fig. 7, B and C, compare lanes 1 and 2). This suggests that the interactions of 9HE with the DNA template are not strong enough to survive the washes.
Taken together, both in vivo and in vitro results suggest that interaction of SL1 with the rRNA promoter is the 9HE target. To verify this hypothesis we analyzed the ability of 9HE to displace prebound SL1 and prevent SL1 from binding to the rDNA template as outlined in Fig. 7D. The results show that 9HE can displace and prevent SL1 binding with similar efficiency (Fig. 7E). Therefore, we demonstrated that 9HE inhibits Pol-I transcription directly by targeting essential interactions between transcription factor SL1 and the rRNA promoter.
We also performed “order of addition” experiments using HeLa nuclear extract (that supports multiround transcription) and purified factors (that support only single round transcription) (Fig. 8). As outlined in Fig. 8, A and D, the drug was added either before PIC formation, or PIC was allowed to form for 15 min before the addition of the drug. Then transcription was initiated by the addition of NTP, and Pol-I specific activity was determined by an S1 nuclease protection assay (Fig. 8, B and E) and quantified (Fig. 8, C and F). The results demonstrate that 9HE inhibits transcription with the same efficiency when added before or after PIC formation, and it represses both multi- and single-round transcription.
FIGURE 8.

9HE inhibits multi- and single- round transcription with similar efficiency when added before or after PIC formation. A, the effect of 9HE on multi-round transcription was analyzed when various concentrations of the drug were added either before or after PIC formation. 2 μl of HeLa NE and 200 ng of supercoiled plasmid DNA template were incubated for 15 min on ice either with (before) or without (after) 9HE. 9HE was added to “after” reactions, and transcription was initiated by the addition of NTP. B, transcripts were analyzed by S1 nuclease protection, and a representative image is shown. C, the results were quantified with the aid of phosphorimaging. The data are expressed as a percentage of the highest value (set at 100%) and represent an average from three independent experiments. S.D. is shown. D, the effect of 9HE on single-round transcription was analyzed when various concentrations of the drug were added either before or after PIC formation. 4 μl of purified Pol-I, 1 μl of purified SL1, 0.1 μl of recombinant UBF, and 200 ng of supercoiled plasmid DNA template were incubated for 15 min on ice either with (before) or without (after) 9HE. 9HE was added to “after” reactions, and transcription was initiated by the addition of NTP. E, transcripts were analyzed by S1 nuclease protection, and a representative image is shown. The black triangle indicates a specific signal, and an asterisk indicates a nonspecific band. F, the results were quantified with the aid of phosphorimaging. The data are expressed as a percentage of the highest value (set at 100%) and represent an average from three independent experiments. S.D. is shown.
9HE Binds to DNA with Relaxed Sequence Specificity
It has been shown previously that ellipticine inhibits the activity of Top2 via formation of a ternary DNA-enzyme-drug complex; however, it can bind to DNA as well as to Top2 (29).
The human rRNA promoter is composed of three elements: the upstream binding element, the Core element, and the linker (38). The Core element is the binding site for promoter recognition factor SL1, upstream binding element is the binding site for transcriptional activator UBF, and the length of the linker is essential for topological organization of the promoter during the preinitiation complex formation. We generated DNA fragments containing the rRNA promoter, fragments representing different elements of the promoter, and DNA fragments of similar sizes derived from non-human sequences (see supplemental sequences for all DNA fragments). Using an approach described earlier (29), we measured KD values for these DNA fragments. The drug binds to all DNA fragments regardless of the source, with similar KD values (Fig. 9 and supplemental Table S5), thus exhibiting relaxed sequence specificity.
FIGURE 9.

9HE does not bind preferentially to the rRNA promoter. A, shown is a schematic outline of different DNA fragments containing either whole promoter or its regions. B, KD values of 9HE for various DNA fragments were determined from at least three independent experiments. Gray bars, nonspecific DNA fragments; white bars, rDNA-derived DNA fragments. The approximate length of the fragments and S.D. are shown. kb, kilobases.
9HE Binds to SL1 but Not to Other Components of the Pol-I Transcription Apparatus
We have previously shown that the promoter-initiating form of Pol-I contains Top2α (39). 9HE can bind to Top2 and, therefore, to Pol-Iβ. As a result, the drug could be delivered to the rRNA promoter by Pol-Iβ, where it can interfere with SL1 binding. It is not technically feasible to measure the strength of interaction between 9HE and Pol-Iβ directly, but using HTETOP cells we compared the efficiency of inhibition of Pol-I transcription by 9HE in wild type cells and in cells lacking Top2α (Fig. 10A). We found that cells lacking Top2α are slightly less sensitive to 9HE treatment than cells expressing normal levels of Top2α (Fig. 10A). However, this difference disappears at higher drug concentrations or longer treatments (Fig. 10A), suggesting that Top2α - 9HE interaction plays a limited role in drug targeting.
FIGURE 10.

9HE binds to SL1 but not to other components of the Pol-I machinery. A, HTETOP cells were incubated for 48 h with 1 μg/ml (+Tet) or without (no Tet) tetracycline and treated with low 9HE concentrations (0.1 μm) for various times as indicated. The relative level of pre-rRNA was determined by S1 nuclease protection assay. The results were quantified with aid of a phosphorimaging and are expressed as a percentage of the highest value (set at 100%) The data represent an average from three independent experiments, S.D. and statistical significance (***, p < 0.001) are shown. B, nuclear extract of cells transiently expressing FLAG-tagged components of the Pol-I transcription machinery (RRN3, Pol-I, SL1) or purified recombinant FLAG-UBF were incubated with α-FLAG beads for 2 h on ice and washed with TM10, 0.15 m KCl buffer, and beads were split in half. One-half was incubated with 10 μm 9HE; the other with buffer for 30 min on ice. After washing, proteins were eluted by FLAG-peptide, and activity of specific factor was assayed in a reconstituted transcription reaction. C, activity of immunopurified Pol-I factors, which were preincubated with or without 9HE, was measured by run-off assay. Top panel: Pol-I (lanes 5 and 6), RRN3 (lanes 7 and 8), and SL1 (lanes 9 and 10). Bottom panel: UBF (lanes 1–6). IP, immunoprecipitation. Transcription reactions in lanes 3–8 (top panel) were supplemented with purified SL1 and UBF, lanes 9–12 (top panel) were supplemented with Pol-Iβ and UBF, and lanes 1–9 (bottom panel) were supplemented with purified Pol-I and SL1. D, transcripts from C were quantified with aid of phosphorimaging. The data are expressed as a percentage of the highest value (set at 100%) and represent an average from three independent experiments. S.D. and statistical significance (***, p < 0.001; *, p < 0.05) are shown. p values have been calculated using one and two-way analysis of variance on R software.
We next examined if 9HE directly interacts with other components of the Pol-I transcription machinery (RRN3, Pol-I, SL1, and UBF) in the absence of DNA. As for Pol-Iβ, it is not technically feasible to directly measure the strength of interaction between 9HE and the components of the Pol-I transcription machinery; thus, we employed the approach schematically outlined in Fig. 10B. We have immunoprecipitated an appropriate factor either from nuclear extracts of cells transiently expressing the FLAG-tagged subunit (Pol-I and SL1) or from a solution of purified recombinant protein (UBF). Immunoprecipitated material was split; one-half was incubated with 9HE and another without. Proteins were eluted by FLAG peptide, and the activity of each factor was determined in reconstituted transcription reactions (Fig. 10, C and D). If 9HE interacts with the immunopurified factor, it will be carried to the test tube and subsequently repress transcription. No inhibition was detected in a reaction containing total Pol-I (fCAST IP), Pol-Iβ, RRN3 (fRRN3 IP) and UBF (Fig. 10, C, top panel, lanes 5–8; bottom panel, lanes 1–6, and D). However, immunopurified SL1 (fTAF1110 IP) treated with 9HE exhibits ∼2-fold less activity compare with untreated (Fig. 10, C, top panel, lanes 9–10, and D). These results suggest that 9HE binds to SL1 but not to Pol-I, RRN3, or UBF, and these interactions may play a limited role in the selectivity and specificity of the drug.
DISCUSSION
Transcription of rDNA by Pol-I is a very promising target for anti-cancer chemotherapeutics (4, 40). However, until 2009, the list of small molecules specifically inhibiting rRNA production included only actinomycin D. Recently, however, two new compounds were added to the list, CX-3543 and CX-5461 (6, 7), and we also learned that many known therapeutic agents target ribosome biogenesis at different stages, including transcription of rDNA (8). In this study we have shown for the first time that ellipticines, well known anti-proliferative agents affecting a large variety of cellular processes by various mechanisms (10–12, 41–44), are selective and efficient inhibitors of Pol-I transcription in eukaryotes. Importantly, the drugs inhibit Pol-I specific transcription in vitro (Figs. 1, 2, and Fig. 8), suggesting a direct mechanism of action.
It has been shown previously that ellipticines induce cell cycle arrest and apoptosis in cells (20, 42, 45–47). This effect was attributed to various activities of ellipticines including anti-topoisomerase activity (48), Fas ligand stimulatory activity (47), induction of reactive-oxygen species (42), and DNA adduct formation (49). Pol-I inhibition induces cell cycle arrest and apoptosis (6, 7, 40, 50, 51) and our findings that ellipticines inhibit rDNA transcription in cells, suggest that this is the main, underlying reason for the ellipticines anti-proliferative activity. Importantly, the anti-Pol-I activity of ellipticines is not linked to other known activities of the drugs (Figs. 1 and 3) and is specific to Pol-I (Fig. 1 and 4, supplemental Table S4 and Fig. S2). Results sown in Fig. 2 and supplemental Fig. S2 suggest that in reconstituted transcription system 9HE has high specificity toward Pol-I and it has no detectable effect on Pol-II and Pol-III driven transcription. Furthermore, in cells, 9HE also demonstrates a high level of selectivity, and our quantitative analysis of expression level of 18 Pol-II and 3 Pol-III targets (Fig. 4 and supplemental Table S4) clearly confirms this statement. Interestingly, we found that the level of Pol-I transcription in asynchronous population of U2OS cells is not very different from the level of Pol-II and Pol-III transcription combined (60 versus 40%) (Fig. 4, B and C), suggesting that a text book figure of 80% (Pol-I) versus 20% (Pol-II/III) might need a revision.
Localization experiments (Fig. 6B) demonstrate that SL1 and Pol-I are two components of Pol-I machinery displaced from the nucleolus by 9HE treatment. Importantly, this effect is confirmed by a ChIP assay (Fig. 6D), which also demonstrates orderly dissociation of factors. SL1 dissociates first, and Pol-I and UBF (only promoter bound fraction) follow (Fig. 6D). Overall these results lead us to conclude that SL1/promoter interaction is the primary drug target. This conclusion was further supported by the results of in vitro experiments (Fig. 7) which directly demonstrate that 9HE targets the Pol-I PIC and selectively affect the interactions of the basal Pol-I transcription factor SL1 with the promoter (Figs. 6D and 7E), thus preventing formation of PIC and initiation of transcription.
Intriguingly, we observed no specific interaction between the drug and the rRNA promoter (Fig. 9 and supplemental Table S5) as well as the rDNA in general (Fig. 6A). Overall our results suggested that ellipticines bind non-specifically to DNA (Fig. 9 and supplemental Table S5), which is supported by the absence of specific nucleoli staining (Fig. 6A). However, even in cells, ellipticines acted rapidly (Fig. 3D) and selectively (Fig. 4 and supplemental Table S4), indicating that they have a specific target(s) and/or a specific delivery mechanism.
Recently, ellipticine derivatives have been shown to bind and stabilize G-quadruplexes (G4) at the promoter region of the MYC gene (43). rDNA sequences are GC-rich and are known to form G4 structures that are involved in rDNA transcription (6, 52). Moreover, one of the first Pol-I inhibitors to enter clinical trials targets a G4-nucleolin interaction, thereby affecting the elongation of Pol-I (6). We have reasoned that ellipticines can stabilize (or induce formation) G4 structures, which then interfere with SL1 binding to the promoter. Using tools available on-line (53), we analyzed the human rRNA promoter sequence (Fig. 11). We found four potential G4 structures, but none was positioned within the SL1 binding site (the Core region). Therefore, this analysis suggests that the inhibitory effect of ellipticines is unlikely to be mediated through stabilization of G-quadruplexes.
FIGURE 11.
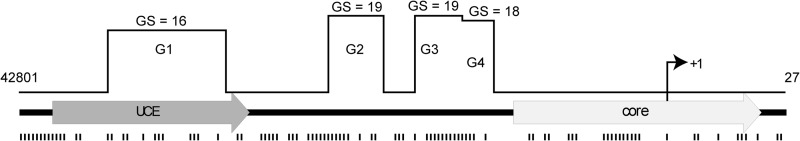
The rRNA promoter contains a putative quadruplex forming G-rich sequences and enriched by G/C sequences. The sequence of human rRNA promoter between positions 42801 and 27 (numbering corresponds to that of the complete human rDNA repeat, accession number U13369, GenBank) was analyzed by QGRC Mapper software (53) and positions of quadruplex forming sequences (G1–G4) and G-scores are indicated above schematic representation of the promoter. The same sequence was analyzed by Vector NTI (Invitrogen) and positions of GpC, GpG, CpG, and CpG motifs are shown below schematic representation of the promoter. UCE, upstream binding element.
Ellipticines are known DNA intercalators (54, 55) that have a strong preference to guanine/cytosine sites (e.g. GpC, CpG, CpC, and GpG) (30, 56). 9HE intercalation leads to DNA unwinding and lengthening, mainly at the intercalation sites, whereas the neighboring sites still adopt an almost canonical B-conformation (30). The human rRNA promoter contains a relatively large number of guanine/cytosine sites (Fig. 11 and supplemental Table S6) with an average frequency of 0.472 per base (compare with average frequency for the total rDNA repeat = 0.333 per base). Therefore, it is possible that binding of ellipticines within the rRNA promoter will significantly disrupt DNA structure, thus weakening SL1-DNA (by binding within the Core) and/or SL1-UBF-DNA interactions (by affecting the length of the linker). Notably, the aromatic core of CX-5461 compound, which selectively inhibits Pol-I transcription by interfering with SL1-rRNA promoter interactions (7), can be aligned to the structure of ellipticine using aromatic pharmacophoric features of the condensed rings (Fig. 12A). In addition, CX-5461 can be docked to the DNA structure (PDB code 1Z3F) (30) with a similar position of the condensed rings as ellipticine rings without causing steric hindrance with the DNA strand (Fig. 12B). This likely suggests a similar structural basis for Pol-I inhibiting activity for both drugs.
FIGURE 12.

Structural alignment of 9HE, GQC-Qi, and CX-5461 hydrophobic cores and their docking position in the DNA strand. A, shown is a three-dimensional superimposition of aromatic cores of 9HE (carbon atoms are in green) and CX-5461 (carbon atoms are in yellow). B, shown is a three-dimensional superimposition of 9HE (green) and CX-5461 (yellow) in the complex with d(GpC) (gray). Only aromatic cores are shown. Shown are the views from the major (left) and minor (right) grooves. C, shown is a three-dimensional superimposition of 9HE (green) and GQC-Qi (red) in complex with d(GpC) (gray). Only aromatic cores are shown. Shown are the views from the major (left) and minor (right) grooves.
Ellipicines are also structurally similar to quindolines (43), and using the same approach as above we found that an aromatic core of quindoline derivative GQC-Qi (57) can be also docked to the same DNA structure (PDB code 1Z3F) (Fig. 12C). There is no information on Pol-I inhibiting activity of quindolines, but we predict that quindolines will directly inhibit Pol-I transcription in vitro and in cells by a mechanism similar to that of the ellipticines.
Interestingly, some quindolines and ellipticines can down-regulate expression of the Myc oncogene (43, 57), which is a known activator of Pol-I transcription (58, 59). Therefore, drugs based on ellipticine or quindoline structural cores can potentially target ribosome biogenesis through two independent mechanisms (direct inhibition of rDNA transcription and down-regulation of expression of transcriptional activator), resulting in enhanced anti-proliferative activity of such drugs, especially in cancer cells expressing a high level of Myc protein.
It has been shown that ellipticines can bind to Top2α (29), which as we have shown recently is also a component of initiation competent Pol-Iβ (39). We initially hypothesized that the Pol-Iβ isoform can serve as a delivery vehicle for ellipticines that transports the drug precisely to the promoter where it displaces the essential factor SL1. However, experiments in cells lacking Top2α showed only a marginal effect of Top2α depletion (Fig. 10A), and we were unable to detect binding of 9HE to Pol-I (Fig. 10, C and D), suggesting a different model. Similarly, 9HE did not bind to UBF or RRN3 (Fig. 10, C and D) but did bind to SL1 (Fig. 10, C and D). Despite that the effect on transcription is modest (∼2-fold decrease), we envisage that these interactions might facilitate a precise targeting of the drug to the active rRNA genes.
Taken together, our results strongly suggest that ellipticines target the interactions between SL1 and the rRNA promoter most likely by affecting three-dimensional DNA structure. The drugs can specifically bind to the SL1 complex even in the absence of DNA, which might increase the efficiency and selectivity of the ellipticines. These findings revealed a new class of Pol-I inhibitors, and we propose that drugs based on an quindoline or ellipticine-like structural core (but displaying lower levels of DNA damaging activity) could be effective agents in the fight against cancer.
Acknowledgments
We thank Andrew Porter (Imperial College London) for HTETOP cells, Joost Zomerdijk (University of Dundee) for antibodies, Stefan Roberts (University of Bristol) for help with in vitro Pol-II transcription assay, S. Church (Queen's University Belfast) for help with microscopy, all members of the Panov laboratory for help, and D. McCance (Queen's University Belfast) and D. Timson (Queen's University Belfast) for critical reading of the manuscript and helpful suggestions.
This work was supported by Medical Research Council New Investigator Award Grant 89365 (to K. P.).

This article contains supplemental sequences, Tables S1–S6, and Figs. S1–S3.
- Pol
- polymerase
- 9HE
- 9-hydroxyellipticine
- NE
- nuclear extract
- PIC
- preinitiation complex
- AMP-PNP
- adenosine 5′-(β,γ-imino)triphosphate
- SL1
- Selectivity factor 1
- UBF
- Upstream Binding Factor
- TBP
- TATA-box Binding Protein.
REFERENCES
- 1. Holland E. C., Sonenberg N., Pandolfi P. P., Thomas G. (2004) Signaling control of mRNA translation in cancer pathogenesis. Oncogene 23, 3138–3144 [DOI] [PubMed] [Google Scholar]
- 2. Sanij E., Poortinga G., Sharkey K., Hung S., Holloway T. P., Quin J., Robb E., Wong L. H., Thomas W. G., Stefanovsky V., Moss T., Rothblum L., Hannan K. M., McArthur G. A., Pearson R. B., Hannan R. D. (2008) UBF levels determine the number of active ribosomal RNA genes in mammals. J. Cell Biol. 183, 1259–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Warner J. R. (1999) The economics of ribosome biosynthesis in yeast. Trends Biochem. Sci. 24, 437–440 [DOI] [PubMed] [Google Scholar]
- 4. Drygin D., Rice W. G., Grummt I. (2010) The RNA polymerase I transcription machinery. An emerging target for the treatment of cancer. Annu. Rev. Pharmacol. Toxicol. 50, 131–156 [DOI] [PubMed] [Google Scholar]
- 5. Schneider D. A. (2012) RNA polymerase I activity is regulated at multiple steps in the transcription cycle. Recent insights into factors that influence transcription elongation. Gene 493, 176–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Drygin D., Siddiqui-Jain A., O'Brien S., Schwaebe M., Lin A., Bliesath J., Ho C. B., Proffitt C., Trent K., Whitten J. P., Lim J. K., Von Hoff D., Anderes K., Rice W. G. (2009) Anticancer activity of CX-3543. A direct inhibitor of rRNA biogenesis. Cancer Res. 69, 7653–7661 [DOI] [PubMed] [Google Scholar]
- 7. Drygin D., Lin A., Bliesath J., Ho C. B., O'Brien S. E., Proffitt C., Omori M., Haddach M., Schwaebe M. K., Siddiqui-Jain A., Streiner N., Quin J. E., Sanij E., Bywater M. J., Hannan R. D., Ryckman D., Anderes K., Rice W. G. (2011) Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 71, 1418–1430 [DOI] [PubMed] [Google Scholar]
- 8. Burger K., Mühl B., Harasim T., Rohrmoser M., Malamoussi A., Orban M., Kellner M., Gruber-Eber A., Kremmer E., Hölzel M., Eick D. (2010) Chemotherapeutic drugs inhibit ribosome biogenesis at various levels. J. Biol. Chem. 285, 12416–12425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stiborová M., Bieler C. A., Wiessler M., Frei E. (2001) The anticancer agent ellipticine on activation by cytochrome P450 forms covalent DNA adducts. Biochem. Pharmacol. 62, 1675–1684 [DOI] [PubMed] [Google Scholar]
- 10. Vistica D. T., Kenney S., Hursey M. L., Boyd M. R. (1994) Cellular uptake as a determinant of cytotoxicity of quaternized ellipticines to human brain tumor cells. Biochem. Biophys. Res. Commun. 200, 1762–1768 [DOI] [PubMed] [Google Scholar]
- 11. Garbett N. C., Graves D. E. (2004) Extending nature's leads. The anticancer agent ellipticine. Curr. Med. Chem. Anticancer Agents 4, 149–172 [DOI] [PubMed] [Google Scholar]
- 12. Stiborova M., Rupertova M., Schmeiser H. H., Frei E. (2006) Molecular mechanisms of antineoplastic action of an anticancer drug ellipticine. Biomed Pap. Med. Fac. Univ. Palacky Olomouc Czech. Repub. 150, 13–23 [DOI] [PubMed] [Google Scholar]
- 13. Mathé G., Triana K., Pontiggia P., Blanquet D., Hallard M., Morette C. (1998) Data of pre-clinical and early clinical trials of acriflavine and hydroxy-methyl-ellipticine reviewed, enriched by the experience of their use for 18 months to 6 years in combinations with other HIV1 virostatics. Biomed. Pharmacother. 52, 391–396 [DOI] [PubMed] [Google Scholar]
- 14. Kenney S., Vistica D. T., Linden H., Boyd M. R. (1995) Uptake and cytotoxicity of 9-methoxy-N2-methylellipticinium acetate in human brain and non-brain tumor cell lines. Biochem. Pharmacol. 49, 23–32 [DOI] [PubMed] [Google Scholar]
- 15. Le Pecq J. B., Gosse C., Nguyen-Dat-Xuong, Paoletti C. (1973) A new antitumor compound: hydroxy-9 ellipticin. Effect on mouse L 1210 leukemia]. C. R. Acad. Sci. Hebd. Seances Acad. Sci. D 277, 2289–2291 [PubMed] [Google Scholar]
- 16. Renault G., Malvy C., Venegas W., Larsen A. K. (1987) In vivo exposure to four ellipticine derivatives with topoisomerase inhibitory activity results in chromosome clumping and sister chromatid exchange in murine bone marrow cells. Toxicol. Appl. Pharmacol. 89, 281–286 [DOI] [PubMed] [Google Scholar]
- 17. Sato N., Mizumoto K., Kusumoto M., Niiyama H., Maehara N., Ogawa T., Tanaka M. (1998) 9-Hydroxyellipticine inhibits telomerase activity in human pancreatic cancer cells. FEBS Lett. 441, 318–321 [DOI] [PubMed] [Google Scholar]
- 18. Lesca P., Lecointe P., Paoletti C., Mansuy D. (1978) Ellipticines as potent inhibitors of aryl hydrocarbon hydroxylase. Their binding to microsomal cytochromes P450 and protective effect against benzo(a)pyrene mutagenicity. Biochem. Pharmacol. 27, 1203–1209 [DOI] [PubMed] [Google Scholar]
- 19. Ohashi M., Sugikawa E., Nakanishi N. (1995) Inhibition of p53 protein phosphorylation by 9-hydroxyellipticine. A possible anticancer mechanism. Jpn. J. Cancer Res. 86, 819–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sugikawa E., Hosoi T., Yazaki N., Gamanuma M., Nakanishi N., Ohashi M. (1999) Mutant p53 mediated induction of cell cycle arrest and apoptosis at G1 phase by 9-hydroxyellipticine. Anticancer Res. 19, 3099–3108 [PubMed] [Google Scholar]
- 21. Miller G., Panov K. I., Friedrich J. K., Trinkle-Mulcahy L., Lamond A. I., Zomerdijk J. C. (2001) hRRN3 is essential in the SL1-mediated recruitment of RNA polymerase I to rRNA gene promoters. EMBO J. 20, 1373–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Panov K. I., Friedrich J. K., Zomerdijk J. C. (2001) A step subsequent to preinitiation complex assembly at the ribosomal RNA gene promoter is rate-limiting for human RNA polymerase I-dependent transcription. Mol. Cell. Biol. 21, 2641–2649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Panov K. I., Friedrich J. K., Russell J., Zomerdijk J. C. (2006) UBF activates RNA polymerase I transcription by stimulating promoter escape. EMBO J. 25, 3310–3322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tanaka T., Grusby M. J., Kaisho T. (2007) PDLIM2-mediated termination of transcription factor NF-κB activation by intranuclear sequestration and degradation of the p65 subunit. Nat. Immunol. 8, 584–591 [DOI] [PubMed] [Google Scholar]
- 25. Panov K. I., Panova T. B., Gadal O., Nishiyama K., Saito T., Russell J., Zomerdijk J. C. (2006) RNA polymerase I-specific subunit CAST/hPAF49 has a role in the activation of transcription by upstream binding factor. Mol. Cell. Biol. 26, 5436–5448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Daly N. L., Arvanitis D. A., Fairley J. A., Gomez-Roman N., Morton J. P., Graham S. V., Spandidos D. A., White R. J. (2005) Deregulation of RNA polymerase III transcription in cervical epithelium in response to high-risk human papillomavirus. Oncogene 24, 880–888 [DOI] [PubMed] [Google Scholar]
- 27. Winter A. G., Sourvinos G., Allison S. J., Tosh K., Scott P. H., Spandidos D. A., White R. J. (2000) RNA polymerase III transcription factor TFIIIC2 is overexpressed in ovarian tumors. Proc. Natl. Acad. Sci. U. S. A. 97, 12619–12624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stefanovsky V., Langlois F., Gagnon-Kugler T., Rothblum L. I., Moss T. (2006) Growth factor signaling regulates elongation of RNA polymerase I transcription in mammals via UBF phosphorylation and r-chromatin remodeling. Mol. Cell 21, 629–639 [DOI] [PubMed] [Google Scholar]
- 29. Froelich-Ammon S. J., Patchan M. W., Osheroff N., Thompson R. B. (1995) Topoisomerase II binds to ellipticine in the absence or presence of DNA. Characterization of enzyme-drug interactions by fluorescence spectroscopy. J. Biol. Chem. 270, 14998–15004 [DOI] [PubMed] [Google Scholar]
- 30. Canals A., Purciolas M., Aymamí J., Coll M. (2005) Crystallization and preliminary X-ray analysis of the Pax6 paired domain bound to the Pax6 gene enhancer. Acta Crystallogr. D Biol. Crystallogr. 61, 1009–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Budde A., Grummt I. (1999) p53 represses ribosomal gene transcription. Oncogene 18, 1119–1124 [DOI] [PubMed] [Google Scholar]
- 32. Kruhlak M., Crouch E. E., Orlov M., Montaño C., Gorski S. A., Nussenzweig A., Misteli T., Phair R. D., Casellas R. (2007) The ATM repair pathway inhibits RNA polymerase I transcription in response to chromosome breaks. Nature 447, 730–734 [DOI] [PubMed] [Google Scholar]
- 33. Carpenter A. J., Porter A. C. (2004) Construction, characterization, and complementation of a conditional-lethal DNA topoisomerase IIα mutant human cell line. Mol. Biol. Cell 15, 5700–5711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kopp K., Gasiorowski J. Z., Chen D., Gilmore R., Norton J. T., Wang C., Leary D. J., Chan E. K., Dean D. A., Huang S. (2007) Pol I transcription and pre-rRNA processing are coordinated in a transcription-dependent manner in mammalian cells. Mol. Biol. Cell 18, 394–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kos M., Tollervey D. (2010) Yeast pre-rRNA processing and modification occur cotranscriptionally. Mol. Cell 37, 809–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bell S. P., Learned R. M., Jantzen H. M., Tjian R. (1988) Functional cooperativity between transcription factors UBF1 and SL1 mediates human ribosomal RNA synthesis. Science 241, 1192–1197 [DOI] [PubMed] [Google Scholar]
- 37. Friedrich J. K., Panov K. I., Cabart P., Russell J., Zomerdijk J. C. (2005) TBP-TAF complex SL1 directs RNA polymerase I pre-initiation complex formation and stabilizes upstream binding factor at the rDNA promoter. J. Biol. Chem. 280, 29551–29558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Learned R. M., Smale S. T., Haltiner M. M., Tjian R. (1983) Regulation of human ribosomal RNA transcription. Proc. Natl. Acad. Sci. U. S. A. 80, 3558–3562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Panova T. B., Panov K. I., Russell J., Zomerdijk J. C. (2006) Casein kinase 2 associates with initiation-competent RNA polymerase I and has multiple roles in ribosomal DNA transcription. Mol. Cell. Biol. 26, 5957–5968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bywater M. J., Poortinga G., Sanij E., Hein N., Peck A., Cullinane C., Wall M., Cluse L., Drygin D., Anderes K., Huser N., Proffitt C., Bliesath J., Haddach M., Schwaebe M. K., Ryckman D. M., Rice W. G., Schmitt C., Lowe S. W., Johnstone R. W., Pearson R. B., McArthur G. A., Hannan R. D. (2012) Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer-specific activation of p53. Cancer Cell 22, 51–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stiborová M., Poljaková J., Martínková E., Boek-Dohalská L., Eckschlager T., Kizek R., Frei E. (2011) Ellipticine cytotoxicity to cancer cell lines. A comparative study. Interdiscip. Toxicol. 4, 98–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kim J. Y., Lee S. G., Chung J. Y., Kim Y. J., Park J. E., Koh H., Han M. S., Park Y. C., Yoo Y. H., Kim J. M. (2011) Ellipticine induces apoptosis in human endometrial cancer cells. The potential involvement of reactive oxygen species and mitogen-activated protein kinases. Toxicology 289, 91–102 [DOI] [PubMed] [Google Scholar]
- 43. Brown R. V., Danford F. L., Gokhale V., Hurley L. H., Brooks T. A. (2011) Demonstration that drug-targeted down-regulation of MYC in non-Hodgkins lymphoma is directly mediated through the promoter G-quadruplex. J. Biol. Chem. 286, 41018–41027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shahabuddin M. S., Nambiar M., Moorthy B. T., Naik P. L., Choudhary B., Advirao G. M., Raghavan S. C. (2011) A novel structural derivative of natural alkaloid ellipticine, MDPSQ, induces necrosis in leukemic cells. Invest. New Drugs 29, 523–533 [DOI] [PubMed] [Google Scholar]
- 45. Kuo Y. C., Kuo P. L., Hsu Y. L., Cho C. Y., Lin C. C. (2006) Ellipticine induces apoptosis through p53-dependent pathway in human hepatocellular carcinoma HepG2 cells. Life Sci. 78, 2550–2557 [DOI] [PubMed] [Google Scholar]
- 46. Kuo P. L., Hsu Y. L., Kuo Y. C., Chang C. H., Lin C. C. (2005) The anti-proliferative inhibition of ellipticine in human breast mda-mb-231 cancer cells is through cell cycle arrest and apoptosis induction. Anti-cancer Drugs 16, 789–795 [DOI] [PubMed] [Google Scholar]
- 47. Kuo P. L., Hsu Y. L., Chang C. H., Lin C. C. (2005) The mechanism of ellipticine-induced apoptosis and cell cycle arrest in human breast MCF-7 cancer cells. Cancer Lett. 223, 293–301 [DOI] [PubMed] [Google Scholar]
- 48. Godard T., Deslandes E., Sichel F., Poul J. M., Gauduchon P. (2002) Detection of topoisomerase inhibitor-induced DNA strand breaks and apoptosis by the alkaline comet assay. Mutat. Res. 520, 47–56 [DOI] [PubMed] [Google Scholar]
- 49. Stiborová M., Breuer A., Aimová D., Stiborová-Rupertová M., Wiessler M., Frei E. (2003) DNA adduct formation by the anticancer drug ellipticine in rats determined by 32P postlabeling. Int. J. Cancer 107, 885–890 [DOI] [PubMed] [Google Scholar]
- 50. Yuan X., Zhou Y., Casanova E., Chai M., Kiss E., Gröne H. J., Schütz G., Grummt I. (2005) Genetic inactivation of the transcription factor TIF-IA leads to nucleolar disruption, cell cycle arrest, and p53-mediated apoptosis. Mol. Cell 19, 77–87 [DOI] [PubMed] [Google Scholar]
- 51. Donati G., Brighenti E., Vici M., Mazzini G., Treré D., Montanaro L., Derenzini M. (2011) Selective inhibition of rRNA transcription down-regulates E2F-1. A new p53-independent mechanism linking cell growth to cell proliferation. J. Cell Sci. 124, 3017–3028 [DOI] [PubMed] [Google Scholar]
- 52. Hershman S. G., Chen Q., Lee J. Y., Kozak M. L., Yue P., Wang L. S., Johnson F. B. (2008) Genomic distribution and functional analyses of potential G-quadruplex-forming sequences in Saccharomyces cerevisiae. Nucleic Acids Res. 36, 144–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kikin O., D'Antonio L., Bagga P. S. (2006) QGRS Mapper. A web-based server for predicting G-quadruplexes in nucleotide sequences. Nucleic Acids Res. 34, W676–W682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ross W. E., Glaubiger D. L., Kohn K. W. (1978) Protein-associated DNA breaks in cells treated with adriamycin or ellipticine. Biochim. Biophys. Acta 519, 23–30 [DOI] [PubMed] [Google Scholar]
- 55. Bertrand J. R., Giacomoni P. U. (1985) Ellipticines. Correlation between in vitro DNA intercalation and physiological properties? Chemioterapia 4, 445–453 [PubMed] [Google Scholar]
- 56. Elcock A. H., Rodger A., Richards W. G. (1996) Theoretical studies of the intercalation of 9-hydroxyellipticine in DNA. Biopolymers 39, 309–326 [DOI] [PubMed] [Google Scholar]
- 57. Ou T. M., Lu Y. J., Zhang C., Huang Z. S., Wang X. D., Tan J. H., Chen Y., Ma D. L., Wong K. Y., Tang J. C., Chan A. S., Gu L. Q. (2007) Stabilization of G-quadruplex DNA and down-regulation of oncogene c-myc by quindoline derivatives. J. Med. Chem. 50, 1465–1474 [DOI] [PubMed] [Google Scholar]
- 58. Arabi A., Wu S., Ridderstråle K., Bierhoff H., Shiue C., Fatyol K., Fahlén S., Hydbring P., Söderberg O., Grummt I., Larsson L. G., Wright A. P. (2005) c-Myc associates with ribosomal DNA and activates RNA polymerase I transcription. Nat. Cell Biol. 7, 303–310 [DOI] [PubMed] [Google Scholar]
- 59. van Riggelen J., Yetil A., Felsher D. W. (2010) MYC as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer 10, 301–309 [DOI] [PubMed] [Google Scholar]
- 60. Buhler J. M., Sentenac A., Fromageot P. (1974) Isolation, structure, and general properties of yeast ribonucleic acid polymerase A (or I). J. Biol. Chem. 249, 5963–5970 [PubMed] [Google Scholar]
- 61. Milkereit P., Schultz P., Tschochner H. (1997) Resolution of RNA polymerase I into dimers and monomers and their function in transcription. Biol. Chem. 378, 1433–1443 [DOI] [PubMed] [Google Scholar]
- 62. Kulkens T., Riggs D. L., Heck J. D., Planta R. J., Nomura M. (1991) The yeast RNA polymerase I promoter. Ribosomal DNA sequences involved in transcription initiation and complex formation in vitro. Nucleic Acids Res. 19, 5363–5370 [DOI] [PMC free article] [PubMed] [Google Scholar]