

Background: Cytochrome P450 46A1 is important for normal brain function.
Results: The x-ray structures of P450 46A1 were determined in complex with the R and S isomers of the anticancer drug bicalutamide.
Conclusion: The cyano group of the folded bicalutamide conformer coordinates the heme iron via a water molecule.
Significance: We demonstrate how conformational polymorphism affects drug entry and binding in the P450 active site.
Keywords: Androgen Receptor, Anticancer Drug, Brain Metabolism, Cholesterol Metabolism, Crystal Structure, Cytochrome P450, Drug Design, Enzyme Inhibitors, Enzyme Structure, Lipid Metabolism
Abstract
Cytochrome P450 46A1 (CYP46A1) is the cholesterol 24-hydroxylase initiating the major pathways of cholesterol removal from the brain, and bicalutamide (BIC) is a drug of choice for the treatment of progressive androgen-dependent prostate cancer. We evaluated the interactions of BIC with CYP46A1 by x-ray crystallography and by conducting solution and mutagenesis studies. Because BIC is administered to patients as a racemic mixture of the S and R isomers, we studied all three, racemic BIC as well as the S and R isomers. Co-crystallization of CYP46A1 with racemic BIC led to structure determinations at 2.1 Å resolution with the drug complexes exhibiting novel properties. Both BIC isomers bind to the CYP46A1 active site in very similar single orientation and adopt an energetically unfavorable folded conformation. This folded BIC conformation is correlated with drug-induced localized shifts of amino acid side chains in CYP46A1 and unusual interactions in the CYP46A1-BIC complex. One of these interactions involves a water molecule that is coordinated to the P450 heme iron and also hydrogen-bonded to the BIC nitrile. Due to polarization of the water in this environment, the heme elicits previously unreported types of P450 spectral responses. We also observed that access to the P450 active site was affected by differential recognition of S versus R isomers at the CYP46A1 surface arising from BIC conformational polymorphism.
Introduction
In humans ∼75% of the marketed drugs are metabolized by cytochrome P450 enzymes (CYP3 or P450) whereby the drugs not only serve as P450 substrates but also as inhibitors (1). In many cases inhibition of P450s is an unwanted side effect leading to adverse drug reactions. Currently, only a small fraction of therapeutic agents have been co-crystallized with human P450s, thus limiting our understanding of the structural basis of drug binding and precluding, in part, the development of more specific pharmaceuticals. Among the structurally characterized P450 drug complexes are those of CYP46A1, which catalyzes cholesterol 24-hydroxylation and controls cholesterol elimination from the brain (2–6). CYP46A1 is involved in memory and cognition through the effect on cerebral cholesterol turnover and could be a risk factor for Alzheimer disease (for review, see Ref. 6). A number of medications were found to bind tightly to this P450 in vitro, and the antifungal azole voriconazole was also shown to inhibit cholesterol 24-hydroxylation in vivo in mice (7–10). Five tight binding pharmaceuticals were co-crystallized with CYP46A1 (8, 10) revealing the structural basis of undesired drug binding to a P450 metabolizing an important endogenous compound, cholesterol.
The visible spectrum of CYP46A1 as well as of all P450s reflects the protein environment of the P450 chromophore (the heme group) and axial ligation of the heme iron. In P450s, the iron proximal ligation is always the covalent bond with the protein cysteine residue, whereas the distal ligation varies because of the reversible nature of ligand binding (for review, see Ref. 11). In the resting substrate-free, low spin ferric P450, the distal (sixth) ligand is usually a water molecule, which can be displaced upon substrate or inhibitor binding (12–15). If water displacement leaves the sixth axial position unoccupied, the P450 becomes high spin, and its Soret maximum (or γ band) shifts from 416–418 nm toward the shorter wavelength (the so-called blue shift; type I spectral response). Accordingly, in the difference spectrum, a peak appears at 380–390 nm with a trough at 415–420 nm (16, 17). In contrast, in a type II spectral response, the P450 remains low spin and the direction of the Soret peak shift is the opposite, toward the longer wavelength (the so called red shift). Consequently, the difference spectrum develops a trough at 390–410 nm and a peak at ∼421–435 nm (16–18). The type II spectral response often reflects the coordination of the heme iron with a nitrogen atom (15), which usually stabilizes the low reduction potential of P450s. Therefore, type II ligands are more often inhibitors rather than substrates for P450s (19). Yet, there are exceptions; some type II ligands could also be P450 substrates (20, 21), and some type II inhibitors do not coordinate the heme iron directly but through a bridging water molecule (22). A third type of P450 response was also described, reverse type I, in which P450 spectral properties become very similar or identical to those of substrate-free, water-coordinated enzyme. Although this type of response has not yet been characterized structurally, it is believed to reflect the formation of the Fe-O bond where the oxygen donor atom is either from a water molecule or the added exogenous compound (18, 23). The spectral signature of reverse type I response is a trough at 380–390 nm and peak at 415–420 nm in the difference spectrum, the mirror reflection of type I difference spectrum (18).
The present study was initiated by the observation that the anti-cancer drug and cyano-containing compound bicalutamide (BIC) elicits spectral changes in the CYP46A1 difference spectrum that do not correspond to any of the previously described P450 spectral responses: type I, type II, reverse type I, or the known variants of these responses (18, 23). BIC-induced spectral perturbations were also different from those induced by cyano-containing compounds (18, 24). We, therefore, decided to elucidate the structural basis for this unusual spectral response and determined the crystal structure of CYP46A1 in complex with BIC. This structure, the first of a P450 with a cyano-containing drug, revealed interesting features of BIC binding to CYP46A1.
EXPERIMENTAL PROCEDURES
Chemicals
Racemic BIC (racBIC) was from Sigma and contained S and R isomers of an unknown ratio. Individual BIC isomers were from Toronto Research Chemicals. Most other chemicals were from Sigma.
CYP46A1 Expression and Purification
All studies were carried out on truncated, Δ(2–50), human CYP46A1 that was heterologously expressed in Escherichia coli as described (25). The P450 was then purified either as a substrate-free resting enzyme (7) or as BIC-bound. The latter was obtained by saturating CYP46A1 with the drug during the last purification step as described (8).
Spectroscopic Characterizations
All spectra were recorded using a double beam Shimadzu UV-2450 spectrophotometer equipped with the temperature-controlled cell holder. The buffer was 50 mm potassium phosphate (KPi), pH 7.2, containing 100 mm NaCl. The CYP46A1 concentration was 0.4 μm. Difference spectra were generated by recording the CYP46A1 absorbance in a sample cuvette containing CYP46A1 and increasing amounts of compound under study versus the CYP46A1 absorbance in a reference cuvette containing CYP46A1 and increasing amounts of the solvent in which the compound under study was dissolved. Peak to trough absorption in the difference spectrum was measured and plotted against the concentration of the compound under study. Apparent binding constants (Kd) were calculated using either Equations 1 or 2,
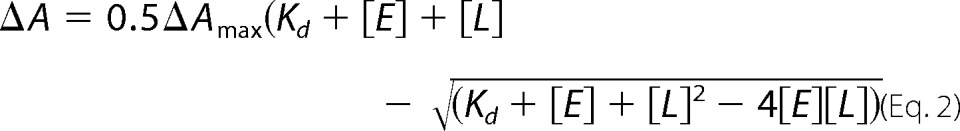 |
in which ΔA is the spectral response at different ligand (compound) concentrations [L], and ΔAmax is the maximal amplitude of the spectral response. Equation 1 was used when Kd was higher than the enzyme concentration, whereas Equation 2 was applied when Kd was lower than the enzyme concentration assuming 1:1 stoichiometry.
Enzyme Assay Using Isolated Bovine Brain Microsomes
The isolation of bovine brain microsomes and measurements of CYP46A1 activity were as described (10). Briefly, 1 mg of total microsomal protein was used for reconstitution with 1 μm cytochrome P450 oxidoreductase, varying concentrations of the racBIC and NADPH regenerating system as a source of reducing equivalents in a total volume of 1 ml. The enzymatic reaction was carried out for 30 min at 37 °C and terminated by the addition two times of 3 ml of Folch reagent (chloroform-methanol, 2:1, v/v). The CYP46A1 product 24S-hydroxycholesterol was quantified in the chloroform extract by gas chromatography mass spectrometry as described (26) using deuterated 24S-hydroxycholesterol as the internal standard.
Crystallization, Data Collection, and Structure Determination
BIC-bound CYP46A1 was crystallized at 18 °C in sitting drops after mixing 1 μl of an ∼40 mg/ml P450 preparation with 1 μl of precipitant solution (12% PEG 8000, 50 mm KPi, pH 5.8, and 20% glycerol). The crystals obtained were then used for microseeding with a cat whisker. X-ray diffraction data were collected from a single crystal at Stanford Synchrotron Radiation Lightsource beam line 11-1 at 100 K (27). The diffraction limit was 2.1 Å. The data set was processed with XDS (28) and CCP4 programs (29). The structure of BIC-bound CYP46A1 was determined by molecular replacement using cholesterol 3-sulfate-bound structure as the search model with cholesterol 3-sulfate omitted (Protein Data Bank code 2Q9F) and Phaser (30). The model was refined using Refmac (31) and Coot for electron density fitting (32). The BIC complex crystallized in space group I4122 with one molecule per asymmetric unit. Data collection and refinement statistics are given in Table 1; coordinates and structure factors have been deposited in the PDB (code 4FIA).
TABLE 1.
Crystallographic statistics
| CYP46A1 complex | R,S-BIC |
|---|---|
| PDB code | 4FIA |
| Space group | I4122 |
| Unit cell dimensions (Å) | 121.1 121.1 142.3 |
| Molecules per asymmetric unit | 1 |
| Solvent content | 52.9% |
| Data | |
| Total observations > 0σF | 197,160 |
| Unique reflections > 0σF | 30,829 |
| Redundancy | 6.4 |
| Completeness | 99.3% |
| Resolution (last shell) (Å) | 2.21–2.10 |
| <I/ σI> all data (last shell) | 10.2 (1.3) |
| Rmerge all data (last shell) | 0.052 (0.584) |
| Refinement | |
| R-factor | 0.208 |
| Rfree | 0.253 |
| Reflections used | 29,263 |
| Test set | 1,557 (5.1%) |
| Root mean square deviation from ideality | |
| Bond lengths (Å) | 0.012 |
| Bond angles (deg.) | 1.25 |
| Ramachandran plot | |
| Favored regions | 98.1% |
| Allowed regions | 100.0% |
| Model | Residues/average B (Å2) |
| Protein | 426 (48.4) |
| Heme | 1 (30.0) |
| R-BIC | 1 (64.0) |
| S-BIC | 1 (62.2) |
| H2O molecules | 135 (54.3) |
Site-directed Mutagenesis
Mutations in the CYP46A1 expression construct were introduced using an in vitro QuikChange site-directed mutagenesis kit (Stratagene) according to the instructions. The correct generation of desired mutations and the absence of undesired mutations were confirmed by DNA sequencing of the entire CYP46A1 coding region.
RESULTS
Spectral Changes in CYP46A1 Induced by racBIC
The addition of the drug led to only very small changes in the absolute spectrum of CYP46A1 as compared with that of the P450 in the resting substrate-free state (Fig. 1A). The Soret and δ bands blue-shifted by 1 and 2 nm, respectively, and their intensities were slightly altered; the absorbance of the Soret band increased by <1%, and that of the δ band decreased by ∼5%. These spectral perturbations were similar but not identical to those observed in our previous studies when CYP46A1 was titrated with 24S-hydroxycholesterol (Fig. 1B), a product and simultaneously substrate for CYP46A1 that could be further metabolized by the enzyme to 24,25 and 24,27-dihydroxysterols (33). 24S-Hydroxycholesterol also induced a blue shift (by 0.5 nm) of the CYP46A1 Soret band and slightly changed its intensity but did not affect the position and intensity of the δ band. To confirm that the alterations of the CYP46A1 absolute spectrum upon the addition of racBIC are real, we used the approach described in the earlier work with P450cam (12, 34). Substrate-free, low spin CYP46A1 was first supplemented with a type II ligand imidazole to induce an 8-nm red shift of the Soret band and alter the intensity of the δ band (Fig. 1C), and then racBIC was added. Changes elicited by racBIC in CYP46A1-imidazole adduct were identical to those in substrate-free CYP46A1; the Soret and δ bands blue-shifted and their intensities increased and decreased, respectively, with the peak maxima at 417 and 358 nm corresponding to those in racBIC-complex obtained from substrate-free CYP46A1. Small changes in the absolute spectrum of racBIC-CYP46A1 complex as compared with the substrate-free P450 suggested that in the former the heme iron remains hexa-coordinated and the sixth iron ligand is still the oxygen atom. The electronic properties of this oxygen donor atom are, however, not the same as in substrate-free CYP46A1.
FIGURE 1.
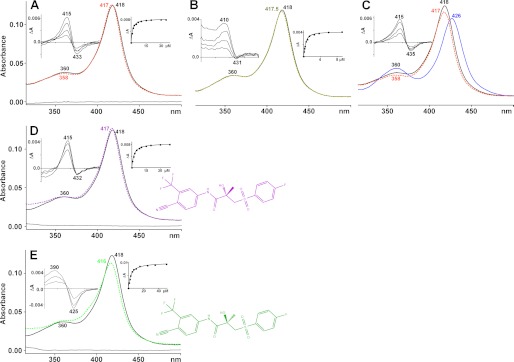
Absolute and difference (left side insets) spectra of CYP46A1 in the absence (black line) and presence (dashed colored lines) of different ligands. Right side insets show the fitting of the binding curves. Shown are P450 spectra with racBIC (orange dashed line, A), 24S-hydroxycholesterol (olive dashed line, B), imidazole (solid blue line) and racBIC (orange dashed line) (C), S-BIC (dashed purple line, D), and R-BIC (dashed green line, E). The concentration of CYP46A1 is 1 μm, and that of the ligand is 20 μm. The gray solid lines are the absorbance of BIC. Chemical structures of S-BIC (in purple) and R-BIC (in green) are also shown.
The difference spectrum of the substrate-free CYP46A1 titrated with racBIC had the peak at 415 nm and trough at 433 nm (Fig. 1A, left side inset). This spectrum was similar to that induced in the imidazole-bound CYP46A1 (Fig. 1C, inset) and represented a type of a spectral shift that was not reported previously. This shift, however, was similar to that elicited by 24S-hydroxycholesterol, which had the peak at 410 nm and a trough at 431 nm (Fig. 1B, left side inset). With each, racBIC and 24S-hydroxycholesterol, the difference spectra induced in the substrate-free CYP46A1 lacked isosbestic points, suggesting that multiple spectral species are present. Nevertheless, the magnitude of the spectral responses was a function of ligand concentration and showed the hyperbolic dependence on ligand concentration (Fig. 1, A and B, right side insets). This enabled determination of the apparent Kd values, 1.2 μm for racBIC and 0.2 μm for 24S-hydroxycholesterol. Unusual spectral response, relatively tight binding to CYP46A1, pharmacological relevance (i.e. wide use for the treatment of prostate cancer), and the presence of a cyano group served as a rationale for crystallization of CYP46A1 in complex with racBIC after the evaluation of how racBIC inhibits CYP46A1-mediated cholesterol 24-hydroxylation in bovine brain microsomes.
Enzyme Assay Using Isolated Brain Microsomes
RacBIC was originally identified as a binder and inhibitor of CYP46A1 in our previous work assessing different Food and Drug Administration-approved pharmaceuticals for the effect on activity of CYP46A1 (10). In this investigation we screened drugs in the in vitro enzyme assay containing purified recombinant CYP46A, cytochrome P450 redox partner cytochrome P450 oxidoreductase, and a NADPH-regenerating system with the concentrations of cholesterol and tested drug being equal to 2.7 and 43 μm, respectively, or 0.5 Km (2.7 μm) and 8 Km (43 μm) of cholesterol for CYP46A1. Under these conditions, the extent of CYP46A1 inhibition by racBIC was 65% (10). In the present work we decided to further characterize racBIC and used isolated bovine brain microsomes as a more physiologically relevant system. In brain microsomes, CYP46A1 is associated with the native membrane and, when supplemented with cytochrome P450 oxidoreductase and NADPH, metabolizes endogenous cholesterol present in the endoplasmic reticulum. Similar to the assay with recombinant CYP46A1, brain microsomes also showed inhibition of CYP46A1-mediated cholesterol 24-hydroxylation by racBIC (Fig. 2). The extent of the inhibition was dependent on the drug concentration and was 30% at 100 μm BIC, the maximal concentration that we tested. The data obtained indicate that racBIC reaches the active site of CYP46A, buried, as indicated by our previous studies (35), in the membrane. However, at a relatively high 100 μm concentration, the inhibition is moderate suggesting that additional studies are required to clarify whether CYP46A1 could be inhibited by racBIC in vivo. In particular, it would be important to clarify whether there is BIC accumulation in cellular membranes upon long term treatments (as in cancer patients) leading to high drug concentrations in the endoplasmic reticulum and thus inhibition of CYP46A1.
FIGURE 2.

Inhibition of CYP46A1-mediated cholesterol hydroxylation by racBIC in isolated bovine brain microsomes. Assay conditions are described under “Experimental Procedures.” The results represent the average of triplicate measurements ± S.D.
Binding of racBIC to CYP46A1
Determination of the crystal structure of the CYP46A1-racBIC co-complex revealed that for most of the drug molecule, the electron density was well defined, except the region around the asymmetric carbon containing the methyl and hydroxyl groups, whose position determines the chirality of BIC (Fig. 3). We hypothesized that this ambiguity in the electron density likely reflects binding of both BIC isomers to CYP46A1 as racBIC was used for co-crystallization. Therefore, CYP46A1 was titrated with individual S and R isomers. The S-BIC produced the same unusual type of the difference spectrum as racBIC (Fig. 1D, left side inset), whereas the signal in response to the addition of R-BIC was different and of type I (Fig. 1E, left side inset). The apparent Kd values were determined (1.8 μm for the S-BIC and 2.9 μm for the R-BIC), and the occupancy of the active site with each of the isomers was calculated based on the ratio of the isomer reciprocal Kd values. The isomer geometry was then optimized, and the models obtained were used for geometric restraints in the refinement. In the final structure, the S- and R-BIC are bound identically to CYP46A1 except that the methyl and hydroxyl groups at the chiral center are interchanged. After refinement of the S- and R-BIC with occupancy 0.58 and 0.42, respectively, the Fo − Fc map showed no significant electron density for either isomer, whereas the density in the 2Fo − Fc map enclosed both isomers (Fig. 3; refined occupancies for ligands with comparable B factors, as in this case, are accurate to no more than ±10%). Hence, the crystallographic refinement is consistent with a mixture of the two isomers in the crystals.
FIGURE 3.

Unbiased σA weighted |Fo| − |Fc| electron density for the R,S-BIC complex of CYP46A1 at 2.1 Å resolution contoured from 3σ to 5σ in intervals of 1σ. BIC carbon atoms are white, oxygen atoms are red, nitrogen atoms are blue, fluorine atoms are purple, and sulfur atoms are cyan; heme carbon atoms are orange; Cα atoms of the I-helix are yellow. Both BIC isomers are shown.
The S-BIC hydroxyl is in a polar environment (Fig. 4A) formed by Wat-725 at 3.4 Å, the side chain of Arg-226 at 3.4 Å, and Ala-474 carbonyl at 2.9 Å and is hydrogen-bonded to this carbonyl, consistent with the lower Kd of the S isomer as compared with the R isomer. Conversely, the chiral methyl group of the S isomer is flanked by the hydrophobic side chains of Phe-80 at 3.2 Å and Ile-222 at 3.6 Å. In the R-BIC, the chiral functionalities are rotated 180 °C relative to those in the S-BIC and are also shifted by 0.6–0.7 Å (Fig. 4B). This rotation and shift eliminate the hydroxyl bonding to the Ala-474 carbonyl and likely underlie the weaker isomer binding. A lack of the anchoring bonding to the Ala-474 carbonyl is also probably the reason why the R-BIC elicits a type I spectral response in CYP46A1, as this isomer is more mobile and can move closer (by up 0.8 Å) to the heme iron and displace the coordinating water while keeping other interactions essentially the same. Perhaps the spectral properties of the racBIC-bound CYP46A1 are similar to those of the S-BIC-CYP46A1 complex because the S isomer is the predominant isomer in the racemic mixture and in addition has a 1.6-fold lower Kd for CYP46A1 than the R isomer.
FIGURE 4.

Views of the CYP46A1 active site illustrating interactions with BIC isomers (A and C, S-BIC; B and D, R-BIC). A and B, interactions with the BIC chiral hydroxyl, amide nitrogen, sulfonyl oxygen, and cyano group are shown. C and D, interactions with the BIC fluorophenyl and cyanotrifluoromethylphenyl rings are shown. S- and R-BIC are shown as sticks and balls in magenta and gray, respectively, and amino acid residues in CYP46A1 are depicted as sticks in light magenta. Water molecules are shown as green spheres. Dashed black lines indicate hydrogen bonds. The heme group is in red. The nitrogen, oxygen, fluorine, sulfur, and iron atoms are in blue, red, pale cyan, yellow, and orange, respectively.
The positioning and interactions of the chiral functionalities are the only differences between the binding of the S- and R-BIC to CYP46A1. In both isomers the BIC amide nitrogen forms a hydrogen bond with the Ala-474 carbonyl (2.9 Å), and the O22 atom of the sulfonyl group is hydrogen-bonded to the side chain of Trp-368 (Fig. 4, A and B). The Glu-472 carboxylate is positioned 3.2 Å from this sulfonyl oxygen, participates in a water network with Wat-725 and Wat-723, and is also hydrogen-bonded to the Trp-368 amide. The fluorophenyl moiety of BIC is in the hydrophobic side pocket (Fig. 4, C and D) formed by the side chains of Trp-368, Phe-371, and Tyr-109; consequently the fluorine atom and the Gly-369 carbonyl are only 2.7 Å apart. The cyanotrifluoromethylphenyl occupies the center of the active site and is confined by the BIC fluorophenyl on one side and the side chains of Leu-112, Phe-121, Val-126, Ile-301, and Thr-475 on the other. The key interaction stabilizing the position of this moiety is a hydrogen bond between the nitrogen of the cyano group and Wat-705 oxygen (3.0 Å) located orthogonally to the heme iron and serving as its distal ligand (the Fe3+-O bond is 2.4 Å) (Fig. 4, A and B).
The cyano group in BIC is a strong hydrogen bond acceptor; hence, the hydrogen bond in the -CN····(H)OH−-Fe3+ adduct is likely polarized to the cyano nitrogen making Wat-705 more like the hydroxyl anion, OH−, rather than a neutral water coordinating the heme iron in the resting state. In addition, the Ala-302 carbonyl and Thr-306 hydroxyl at 3.2 and 3.4 Å, respectively, from Wat-705 (Fig. 4, C and D) may also affect the electronic configuration of the water oxygen and tune the strength and polarization in the -CN····(H)OH−-Fe3+ complex. We suggest that it is the partial or complete deprotonation of the aqua sixth heme ligand due to its interactions in the active site that leads to the small blue shifts in the racBIC-induced CYP46A1 absolute spectrum and unusual type of response in the difference spectrum. To test this interpretation, we attempted to obtain the Fe3+-OH− adduct by placing CYP46A1 in 100 mm glycine/KOH buffer, pH 10.0, at 4 °C as described in previous studies of P450cam (24). Similar to P450cam, we were unable to obtain a stable CYP46A1-OH− complex because the P450 quickly denatured (within minutes) at high pH as assessed by the reduced CO difference spectrum (36).
Different Conformations of BIC in the Active Site of CYP46A1 and Androgen Receptor (AR)
The crystal structure of the BIC-CYP46A1 complex is the second structure of BIC bound to a protein. Previously, BIC was co-crystallized with the ligand binding domain of the AR, a therapeutic target for the treatment of prostate cancer (37). This structure (PDB 1Z95) was determined in complex with the R-BIC having a ∼30-fold higher binding affinity to the AR than the S-BIC as indicated by the R-BIC Ki of 11 versus 364 nm Ki of the S-BIC (38). Like BIC isomers in CYP46A1, R-BIC in the AR resides in the hydrophobic cavity, and its functionalities (the chiral hydroxyl, amide nitrogen, one of the sulfonyl oxygens, fluorophenyl fluorine as well as the cyano group) are involved in hydrogen-bonding with the protein side chains, backbone atoms, or water molecules (Fig. 5). Consistent with the cyano group being a strong hydrogen bond acceptor, it interacts with Arg-752 (at 3.0 Å; -N+H····NC-) as well as a water molecule (Wat-108, at 3.0 Å) and possibly Gln-711 (at 3.7 Å). The interaction of the fluorophenyl fluorine is, however, different; a hydrogen bond is formed to Wat-101 in the AR structure versus contact with a glycine carbonyl in the CYP46A1 structure. The most notable difference between BIC binding to the AR and CYP46A1 is the BIC conformation. In the AR structure, R-BIC is in a bent conformation in which its amide nitrogen, one of the sulfonyl oxygens, and the chiral hydroxyl form a triangle and at distances for intramolecular hydrogen bonds. These intramolecular interactions, which are precluded in the CYP46A1 complex due to the folded conformation of BIC, orient the chiral hydroxyl group for specific recognition in the AR. Hence, the folded conformation of BIC appears to afford greater potential for interactions with the protein, consistent with lack of chiral recognition of BIC in CYP46A1. The different BIC conformations in the two protein co-complexes prompted us to examine BIC structures when the drug was crystallized alone or the structure that was calculated theoretically (39–42) (Fig. 6, C--G). We found that in one of the experimentally determined structures (Fig. 6D) and the computed structure (Fig. 6G), BIC is in the folded conformation. Thus, in the active site of CYP46A1, BIC is in one of the possible conformations. However, in such a conformation BIC cannot enter the P450 because this conformer is wider than the substrate access channel in a substrate-free enzyme (PDB 2Q9G). This suggests that BIC enters CYP46A1 in the extended conformation and once inside the active site becomes folded. To gain insight into the mechanism of BIC folding, we compared crystal structures of the BIC-bound and substrate-free CYP46A1.
FIGURE 5.

Views of the AR binding pocket illustrating interactions with R-BIC (in dark gray). Carbon atoms of AR are shown in purple. Water molecules are shown as green spheres. Dashed black lines indicate hydrogen bonds. The nitrogen, oxygen, fluorine and sulfur atoms are in blue, red, pale cyan, and yellow, respectively.
FIGURE 6.
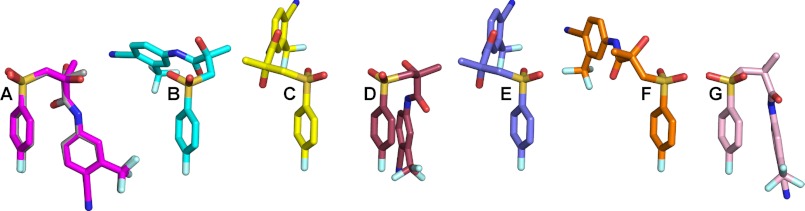
Conformations of BIC. A, in co-complex with CYP46A1 (both isomers are shown, S- is in magenta, and R- is in gray). B, in co-complex with the AR (37). C–F, in structures of BIC alone (39–41), and G, in the computed structure (42). The nitrogen, oxygen, fluorine and sulfur atoms are in blue, red, pale cyan, and yellow, respectively.
Putative BIC Folding in the CYP46A1 Active Site
Interaction with BIC does not significantly affect the position of the secondary structural elements in CYP46A1; the major conformational changes are the shifts of the amino acid side chains (Fig. 7). His-81, Lys-82, and Thr-228 on the protein surface move because they are probably involved in the initial recognition of BIC by CYP46A1 (Fig. 7A). This initial recognition may be followed by BIC sliding along the substrate access channel toward the heme iron accompanied by the displacement of Phe-80, Arg-226, and Asn-227 lining the interior of this channel. In addition, the side chain of Arg-226 rotates and becomes hydrogen-bonded to the side chains of Asn-227 and Thr-223 and carbonyl of Gln-473 (Fig. 7B). Gln-473 in turn pulls the Ala-474 closer to BIC placing its carbonyl at a hydrogen bond distance to the drug amide, chiral hydroxyl of the S isomer, and Wat-725 (Fig. 7C). The altered position of Gln-473 affects the side chain of Glu-472, which becomes hydrogen-bonded to Wat-725 and Trp-368 and shifts the latter. As a result, the Trp-368 indole nitrogen forms a hydrogen bond with one of the BIC sulfonyl oxygens and likely triggers BIC folding in the CYP46A1 active site. Anchoring of BIC via hydrogen bonding to Ala-474 carbonyl, Glu-472 side chain, and possibly Gly-369 carbonyl likely favors the interaction of the BIC cyano group with Wat-705 ligating the heme iron. Once BIC is positioned in the active site, there are also adjustments of the side chains inside the binding cavity to improve the fit of the BIC molecule (Fig. 7D). These include conformational changes of Met-108, Tyr-109, Ala-111, and Leu-112 on the B′-helix, Val-115 and Phe-121 on the B′-C loop, Met-218, Ala-224, and Ser-225 on the F-helix, and Ile-301 and Thr-475 on the I helix and β4–1 sheet. The side chains on the secondary structural elements forming the entrance to the substrate access channel move as well (Fig. 7E). These are Lys-107 and Arg-110 on the B′-helix and Thr-114 and Phe-116 on the B′-C-loop. Collectively, the conformational changes improve the CYP46A1 complementarity fit to BIC and reduce the volume of the active site from 470 Å3 in substrate-free enzyme to 402 Å3 in BIC-bound P450. Despite all the adjustments and strong hydrogen-bond interactions, the binding affinity of BIC for CYP46A1 is only 1.2 μm, as assessed by the spectral binding Kd, >100-fold higher than that for the AR (38). A possible reason for this difference in apparent binding affinity could be a low solution concentration of the BIC conformer that can enter the CYP46A1 substrate access channel and reach the active site, thus reducing the effective drug concentration and the apparent Kd. To test this hypothesis, we generated a number of CYP46A1 mutants and characterized them for binding of BIC.
FIGURE 7.

Superimposed views of the active sites in BIC-bound (light magenta) and ligand-free (light blue) CYP46A1 showing the amino acid residues undergoing conformational changes upon BIC binding. The putative sequence of the conformational shifts is also shown. A and B, shown are the movements induced by the initial BIC recognition on the CYP46A1 surface and subsequent BIC sliding inside the substrate access channel. C, shown are key conformational changes in the active site. D, shown are secondary adjustments in the active site. E, shown are secondary adjustments on the CYP46A1 surface. The enclosed volumes of the active site in BIC-bound and ligand-free CYP46A1 are shown as a semitransparent pink surface and blue mesh. Water molecules are shown as green spheres. Dashed black lines indicate hydrogen bonds. The heme group in BIC-bound and ligand-free CYP46A1 are in red and salmon, respectively. The nitrogen, oxygen, fluorine, sulfur, and iron atoms are in blue, red, pale cyan, yellow, and orange, respectively.
BIC Binding Properties of the CYP46A1 Mutants
His-81 is a flexible amino acid residue on the CYP46A1 surface covering the entrance to the substrate access channel. Placing a smaller Ala at this position should increase BIC access to the active site. The H81A mutation could also weaken the histidine interaction with the neighboring F-G loop and favor drug access. Consistent with our predictions, the Kd of the S-BIC decreased >20-fold, whereas those of the R isomer and racBIC did not change significantly (Fig. 8). Perhaps His-81 is more restrictive to the access of the S-BIC than to the access of the R-BIC adopting in solution likely the extended conformation, in which it can enter the substrate access channel. This is in contrast to S-BIC, which cannot form intramolecular hydrogen bonds stabilizing the extended conformation and thus adopts the bulkier conformations in which it cannot enter the wild type CYP46A1. Although the Kd of the H81A mutant for the R-BIC decreased only slightly, its difference spectra upon the addition of this isomer had a blue shift in the position of the maxima when drug concentrations were increased, probably because of a somewhat different position of R-BIC inside the active site.
FIGURE 8.

Effect of replacements of amino acid residues outside the active site on difference spectra and apparent Kd values of the CYP46A1 mutants. Insets show the fitting of the binding curves. BIC binding properties of the CYP46A1 wild type (WT) are shown for comparison. Titrations were carried with racBIC, and S and R isomers as described under “Experimental Procedures.” The results represent the average of triplicate measurements ± S.D.
The surface Lys-82 at the entrance to the access channel is also flexible, and like His-81 controls drug access. In addition, Lys-82 may be involved in the initial recognition of the BIC nitrile. Hence, Lys-82 was mutated to a shorter but negatively charged (at neutral pH) Glu and a shorter and hydrophobic Met. Both mutants had a similar decrease in the Kd for S-BIC (∼4 fold) and also a similar change in the Kd for R-BIC (∼2 fold), which increased. Possibly the size of Lys-82 and His-81 differentially controls the isomer access to the active site, and the polarity of the amino acid residue at position 82 is not important for the isomer recognition by CYP46A1. The residue polarity, however, seems to affect isomer binding when it is mixed, as indicated by ∼2-fold difference in the Kd values of the K82E and K82M mutants for racBIC and a change of the difference spectrum of the K82E mutant from the “unusual” form to the reverse type I form.
Arg-110 is another conformationally flexible amino acid residue at the entrance to the access channel, yet it points away from the channel. Because Arg-110 is on the outside surface of the B′ helix, which defines the shape of the CYP46A1 active site, mutation of Arg-110 may affect BIC binding indirectly by altering the position of the active site Tyr-109 and Leu-112. Indeed, the R110L mutation changed BIC Kd values, and this effect was isomer-dependent; the Kd of the S-BIC decreased 18-fold, and that of the R-BIC was essentially unchanged. The Kd of racBIC increased 1.7-fold, supporting our previous observation with the K82E mutant that isomer interactions with BIC are not the same when they are added to CYP46A1 as a mixture. As in the case of the H81A mutant and R-BIC, the difference spectrum of the R110L mutant also had a blue shift in the position of the maximum upon increased binding of the R-BIC.
Arg-226 is a residue lining the interior of the CYP46A1 substrate access channel. In addition to likely controlling BIC access to the CYP46A1 active site, Arg-226 is at a hydrogen-bond distance (at 3.4 Å) to the S-BIC chiral hydroxyl and is close to Glu-472 (at 4.0 Å), which is in the vicinity of the BIC sulfone of both isomers (at 3.2 Å). The R226E mutation deletes a potential hydrogen bond to S-BIC chiral hydroxyl, but the replacing Glu could also interact with either or both R- and S-BIC by accepting a hydrogen bond from the BIC hydroxyl. In contrast, the R226M replacement knocks out any possible interactions with BIC. Consistent with possible multiple interactions of the replacement Glu, S-BIC, R-BIC, and racBIC had reduced Kd values for the R226E mutant (by 6-, 3.7-, and 2.7-fold, respectively) (Fig. 9). The pattern of the Kd changes for the R226M mutant was different; the Kd of the S isomer was not affected, and those of the R isomer and racBIC increased by 1.5- and 3.3-fold, respectively, consistent with placement of the hydrophobic Met side chain near the BIC chiral methyl group (S-BIC) versus the chiral hydroxyl (R-BIC) (Fig. 4, A and B). Like the difference spectrum of the R110L mutant, the difference spectra of the R226E and R226M mutants had shifted maxima in response to the addition of the R-BIC. The Met mutation also affected the spectral response to racBIC, which showed the trough at 383 nm besides the peak at 416 nm and trough at 434 nm.
FIGURE 9.

Effect of replacements of amino acid residues inside the active site on difference spectra and apparent Kd values of the CYP46A1 mutants. Insets show the fitting of the binding curves. Titrations were carried with racBIC, S and R isomers as described under “Experimental Procedures.” The results represent the average of triplicate measurements ± S.D.
We also investigated the effect of the replacements of the amino acid residues inside the active site involved directly (Trp-368 and Ala-474) or indirectly (Thr-306) in the interactions with BIC. The W368F substitution decreased the Kd of both S- and R-BIC 2.6- and 2.9-fold, respectively, but did not affect the Kd of racBIC. Perhaps Phe-368 is able to stack more effectively on the fluorophenyl moiety of BIC. This mutant also showed the shifted maximum in the difference spectrum induced by R-BIC. The effect of the A474V substitution was mainly on the shape of the difference spectrum, which, as with the H81A, R110L, R226E, R226M, and W368F mutants, had a blue shift in the position of the maxima upon increased concentrations of the R-BIC. Similarly, the Kd values of the S-BIC, R-BIC, and racBIC for the T306A mutant were essentially unchanged. Because Wat-705 and Wat-712 have multiple interactions, they could remain bound in the T306A mutant, making its BIC binding properties and spectra very similar to that of the CYP46A1 wild type, e.g. Wat-705 could still hydrogen-bond to the carbonyl of Ala-302 while interacting with the BIC nitrile.
DISCUSSION
The present work provides novel important insights into areas of drug binding that are not yet sufficiently addressed. These insights pertain to 1) the effect of protein target on a conformation of a bound drug, 2) conformational flexibility or polymorphism of drugs, 3) selection of drug conformers at the entrance to the substrate access channel, 4) interactions of cyano groups in drugs, and 5) unusual drug-induced spectral shifts in a P450, which represents a major family of drug-metabolizing enzymes whose spectral responses are widely used for characterization of the drug-P450 interactions.
We first encountered the effect of protein target on a conformation of a bound drug in our previous studies investigating the interactions of CYP46A1 with the antidepressant medicine fluvoxamine (10). The structure of the CYP46A1-fluvoxamine co-complex suggested that fluvoxamine enters the P450 substrate access channel in one (compact) conformation, which changes (unfolds) when the drug reaches the active site. In contrast, in the present study the drug (BIC) likely folds rather than unfolds once in the CYP46A1 active site, and remarkably, this folding is accomplished without the large scale conformational changes in the P450. Quantum mechanical calculations (42) indicate that BIC has two energetic minima: one when it is in the extended conformation, similar to that observed in the AR structure and the second minimum when it is in the folded conformation, similar to those in some of the structures when BIC was crystallized alone (39–41) or in complex with CYP46A1. The present work shows that in CYP46A1, local movements of amino acid residues induce BIC folding in a conformation that is energetically less favorable, likely because it is stabilized by the hydrogen bonds between the BIC functionalities and the protein side chains and water. This finding is of importance for in silico screening for predicting P450-drug interactions. Apparently, energetically less favorable drug conformations could also be present in the enzyme active site and, therefore, should be considered during screening.
Conformational polymorphism of BIC, a drug that has rotatable bonds, was first shown by x-ray crystallography (39–41) and then confirmed by the investigation of the properties of the individual BIC conformers (40). BIC in the folded conformation was found to be less stable but more soluble than BIC in the extended conformation and had a different extinction coefficient in the UV region as compared with the bent isomer (40). Moreover, the recognition appeared to depend on how the isomers were added to the P450: as a racemic mixture or individually. Perhaps the preferred conformations of BIC isomers in solution are different; mostly extended for R-BIC because of the intramolecular hydrogen bonding and mostly folded for the S isomer, which cannot form these hydrogen bonds. It is also possible that when mixed in solution, there are interactions between BIC isomers that affect their conformations. This will be consistent with different spectral properties of racBIC and its isomers that we observed (Fig. 1, A, D, and E). Collectively, distinct effects of surface mutations on apparent binding affinities of racBIC and its isomers demonstrate that drug-protein interactions at the entrance to the substrate access channel could play a role in selecting a specific drug conformation and that the apparent Kd of a drug could reflect the concentration of a conformer that can enter the substrate access channel. The present work expands our knowledge of the diversity of drug-protein interactions.
The structural information on binding of racBIC also provides insight, in our opinion, into the nature of the CYP46A1 heme iron interactions with 24S-hydroxycholesterol, an abundant brain sterol. We suggest that the sterol 24S-hydroxyl ligates the iron through the bridging water, but since the oxygen in the 24S-hydroxyl is a weaker hydrogen bond acceptor than the nitrogen in the BIC cyano group, the 24S-hydroxycholesterol-induced spectral shifts are similar to those elicited by racBIC but do not match them exactly. If the sterol 24S-hydroxyl ligated the heme iron directly, we would probably observe a type I spectral response in CYP46A1, as we did in CYP11A1 when its heme iron was directly coordinated by the 22R-hydroxyl of 22R-hydroxycholesterol (43).
The CYP46A1-BIC complex is the first P450 structure with a bound cyano-containing drug. A comparison to the limited number of crystal structures of cyano-containing pharmaceuticals with proteins (for review, see Ref. 44) indicates that the interaction of the BIC cyano group with a water molecule in CYP46A1 is typical for this functionality that has a polarized triple bond and is a very strong hydrogen bond acceptor. However, this interaction is unusual for a P450, as the BIC cyano group accepts the hydrogen bond from the water serving as the sixth ligand to the heme iron. Moreover, this group induces a difference spectrum in CYP46A1 that was not described previously. Studies in the P450 field of the CYP121-fluconazole complex already showed that a drug nitrogen can accept a hydrogen bond from the P450 aqua ligand (22). Yet this nitrogen atom was from a triazole, not a cyano group as in our co-complex, and the CYP121 spectral peaks shifted in the opposite direction representing an incomplete type II spectral response. The latter suggests that in the BIC-CYP46A1 structure, the electronic properties of the ligating water (hydrogen bonded to the cyano group) are significantly different from those in the fluconazole-bound CYP121 (hydrogen-bonded to the triazole functionality). When combined with available data on spectral properties of P450s with different types of the heme iron ligands (12, 16, 18, 24, 34), our crystallographic and spectroscopic data suggest that it is possible that a hydroxyl anion serves as the sixth iron ligand in the BIC complex. If so, this would be the first P450 crystal structure in complex with such a type of ligand.
In summary, initiated by the observation of the unusual spectral response, studies of BIC-CYP46A1 interactions extended beyond the traditional crystallographic analysis and led to novel findings that enhance our understanding of drug binding to enzymes and protein receptors. Our findings could also lead to a design of more specific cyano-containing therapeutic agents.
Acknowledgments
We thank Dr. A. T. Hagler (Department of Chemistry, University of Massachusetts) for providing the coordinates of the computed BIC structure mentioned in Ref. (42) and Dr. M. Shimoji for generating the T306A CYP46A1 mutant. Portions of this research were carried out at Stanford Synchrotron Radiation Lightsource, a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the United States Department of Energy Office of Science by Stanford University. The Stanford Synchrotron Radiation Lightsource Structural Molecular Biology Program is supported by the Department of Energy Office of Biological and Environmental Research and by the NIGMS, National Institutes of Health (including Grant P41GM103393) and the National Center for Research Resources (Grant P41RR001209). We thank the staff at the Stanford Synchrotron Radiation Lightsource for assistance in data collection.
This work was supported in part by United States Public Health Service Grant GM62882 (to I. A. P.). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS, Center for Research Resources, or the National Institutes of Health.
The atomic coordinates and structure factors (code 4FIA) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- CYP
- cytochrome P450
- AR
- androgen receptor
- BIC
- bicalutamide
- racBIC
- racemic BIC
- Wat
- water.
REFERENCES
- 1. Guengerich F. P. (2008) Cytochrome P450 and chemical toxicology. Chem. Res. Toxicol. 21, 70–83 [DOI] [PubMed] [Google Scholar]
- 2. Lütjohann D., Breuer O., Ahlborg G., Nennesmo I., Sidén A., Diczfalusy U., Björkhem I. (1996) Cholesterol homeostasis in human brain. Evidence for an age-dependent flux of 24S-hydroxycholesterol from the brain into the circulation. Proc. Natl. Acad. Sci. U.S.A. 93, 9799–9804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Björkhem I., Lütjohann D., Diczfalusy U., Ståhle L., Ahlborg G., Wahren J. (1998) Cholesterol homeostasis in human brain. Turnover of 24S-hydroxycholesterol and evidence for a cerebral origin of most of this oxysterol in the circulation. J. Lipid Res. 39, 1594–1600 [PubMed] [Google Scholar]
- 4. Lund E. G., Guileyardo J. M., Russell D. W. (1999) cDNA cloning of cholesterol 24-hydroxylase, a mediator of cholesterol homeostasis in the brain. Proc. Natl. Acad. Sci. U.S.A. 96, 7238–7243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kotti T. J., Ramirez D. M., Pfeiffer B. E., Huber K. M., Russell D. W. (2006) Brain cholesterol turnover required for geranylgeraniol production and learning in mice. Proc. Natl. Acad. Sci. U.S.A. 103, 3869–3874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Russell D. W., Halford R. W., Ramirez D. M., Shah R., Kotti T. (2009) Cholesterol 24-hydroxylase. An enzyme of cholesterol turnover in the brain. Annu. Rev. Biochem. 78, 1017–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mast N., White M. A., Bjorkhem I., Johnson E. F., Stout C. D., Pikuleva I. A. (2008) Crystal structures of substrate-bound and substrate-free cytochrome P450 46A1, the principal cholesterol hydroxylase in the brain. Proc. Natl. Acad. Sci. U.S.A. 105, 9546–9551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mast N., Charvet C., Pikuleva I. A., Stout C. D. (2010) Structural basis of drug binding to CYP46A1, an enzyme that controls cholesterol turnover in the brain. J. Biol. Chem. 285, 31783–31795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shafaati M., Mast N., Beck O., Nayef R., Heo G. Y., Björkhem-Bergman L., Lütjohann D., Björkhem I., Pikuleva I. A. (2010) The antifungal drug voriconazole is an efficient inhibitor of brain cholesterol 24S-hydroxylase (CYP46A1) in vitro and in vivo. J. Lipid Res. 51, 318–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mast N., Linger M., Clark M., Wiseman J., Stout C. D., Pikuleva I. A. (2012) In silico and intuitive predictions of CYP46A1 inhibition by marketed drugs with subsequent enzyme crystallization in complex with fluvoxamine. Mol. Pharmacol. 82, 824–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Denisov I. G., Makris T. M., Sligar S. G., Schlichting I. (2005) Structure and chemistry of cytochrome P450. Chem. Rev. 105, 2253–2277 [DOI] [PubMed] [Google Scholar]
- 12. Dawson J. H., Andersson L. A., Sono M. (1982) Spectroscopic investigations of ferric cytochrome P-450-CAM ligand complexes. Identification of the ligand trans to cysteinate in the native enzyme. J. Biol. Chem. 257, 3606–3617 [PubMed] [Google Scholar]
- 13. Poulos T. L., Finzel B. C., Howard A. J. (1986) Crystal structure of substrate-free Pseudomonas putida cytochrome P-450. Biochemistry 25, 5314–5322 [DOI] [PubMed] [Google Scholar]
- 14. Poulos T. L., Finzel B. C., Howard A. J. (1987) High resolution crystal structure of cytochrome P450cam. J. Mol. Biol. 195, 687–700 [DOI] [PubMed] [Google Scholar]
- 15. Poulos T. L., Howard A. J. (1987) Crystal structures of metyrapone- and phenylimidazole-inhibited complexes of cytochrome P-450cam. Biochemistry 26, 8165–8174 [DOI] [PubMed] [Google Scholar]
- 16. Remmer H., Schenkman J., Estabrook R. W., Sasame H., Gillette J., Narasimhulu S., Cooper D. Y., Rosenthal O. (1966) Drug interaction with hepatic microsomal cytochrome. Mol. Pharmacol. 2, 187–190 [PubMed] [Google Scholar]
- 17. Schenkman J. B., Remmer H., Estabrook R. W. (1967) Spectral studies of drug interaction with hepatic microsomal cytochrome. Mol. Pharmacol. 3, 113–123 [PubMed] [Google Scholar]
- 18. Mailman R. B., Kulkarni A. P., Baker R. C., Hodgson E. (1974) Cytochrome P-450 difference spectra. Effect of chemical structure on type II spectra in mouse hepatic microsomes. Drug. Metab. Dispos. 2, 301–308 [PubMed] [Google Scholar]
- 19. Gigon P. L., Gram T. E., Gillette J. R. (1968) Effect of drug substrates on the reduction of hepatic microsomal cytochrome P-450 by NADPH. Biochem. Biophys. Res. Commun. 31, 558–562 [DOI] [PubMed] [Google Scholar]
- 20. Pearson J., Dahal U. P., Rock D., Peng C. C., Schenk J. O., Joswig-Jones C., Jones J. P. (2011) The kinetic mechanism for cytochrome P450 metabolism of type II binding compounds. Evidence supporting direct reduction. Arch. Biochem. Biophys. 511, 69–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Isoherranen N., Kunze K. L., Allen K. E., Nelson W. L., Thummel K. E. (2004) Role of itraconazole metabolites in CYP3A4 inhibition. Drug. Metab. Dispos. 32, 1121–1131 [DOI] [PubMed] [Google Scholar]
- 22. Seward H. E., Roujeinikova A., McLean K. J., Munro A. W., Leys D. (2006) Crystal structure of the Mycobacterium tuberculosis P450 CYP121-fluconazole complex reveals new azole drug-P450 binding mode. J. Biol. Chem. 281, 39437–39443 [DOI] [PubMed] [Google Scholar]
- 23. Kumaki K., Sato M., Kon H., Nebert D. W. (1978) Correlation of type I, type II, and reverse type I difference spectra with absolute changes in spin state of hepatic microsomal cytochrome P-450 iron from five mammalian species. J. Biol. Chem. 253, 1048–1058 [PubMed] [Google Scholar]
- 24. Sono M., Dawson J. H. (1982) Formation of low spin complexes of ferric cytochrome P-450-CAM with anionic ligands. Spin state and ligand affinity comparison to myoglobin. J. Biol. Chem. 257, 5496–5502 [PubMed] [Google Scholar]
- 25. White M. A., Mast N., Bjorkhem I., Johnson E. F., Stout C. D., Pikuleva I. A. (2008) Use of complementary cation and anion heavy-atom salt derivatives to solve the structure of cytochrome P450 46A1. Acta Crystallogr. D Biol. Crystallogr. 64, 487–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mast N., Reem R., Bederman I., Huang S., DiPatre P. L., Bjorkhem I., Pikuleva I. A. (2011) Cholestenoic acid is an important elimination product of cholesterol in the retina. Comparison of retinal cholesterol metabolism to that in the brain. Invest. Ophthalmol. Vis. Sci. 52, 594–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Soltis S. M., Cohen A. E., Deacon A., Eriksson T., González A., McPhillips S., Chui H., Dunten P., Hollenbeck M., Mathews I., Miller M., Moorhead P., Phizackerley R. P., Smith C., Song J., van dem Bedem H., Ellis P., Kuhn P., McPhillips T., Sauter N., Sharp K., Tsyba I., Wolf G. (2008) New paradigm for macromolecular crystallography experiments at SSRL. Automated crystal screening and remote data collection. Acta Crystallogr. D Biol. Crystallogr. 64, 1210–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Collaborative Computational Project, Number 4 (1994) The CCP4 suite. Programs for protein crystallography. Acta Crystallogr. D 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 30. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 32. Emsley P., Cowtan K. (2004) Coot. Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 33. Mast N., Norcross R., Andersson U., Shou M., Nakayama K., Bjorkhem I., Pikuleva I. A. (2003) Broad substrate specificity of human cytochrome P450 46A1, which initiates cholesterol degradation in the brain. Biochemistry 42, 14284–14292 [DOI] [PubMed] [Google Scholar]
- 34. Andersson L. A., Dawson J. H. (1984) The influence of oxygen donor ligation on the spectroscopic properties of ferric cytochrome P-450. Ester, ether, and ketone co-ordination to the haem iron. Xenobiotica 14, 49–61 [DOI] [PubMed] [Google Scholar]
- 35. Mast N., Liao W. L., Pikuleva I. A., Turko I. V. (2009) Combined use of mass spectrometry and heterologous expression for identification of membrane-interacting peptides in cytochrome P450 46A1 and NADPH-cytochrome P450 oxidoreductase. Arch. Biochem. Biophys. 483, 81–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Omura R., Sato R. (1964) The carbon monoxide-binding pigment of liver microsomes. J. Biol. Chem. 239, 2370–2378 [PubMed] [Google Scholar]
- 37. Bohl C. E., Gao W., Miller D. D., Bell C. E., Dalton J. T. (2005) Structural basis for antagonism and resistance of bicalutamide in prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 102, 6201–6206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mukherjee A., Kirkovsky L., Yao X. T., Yates R. C., Miller D. D., Dalton J. T. (1996) Enantioselective binding of Casodex to the androgen receptor. Xenobiotica 26, 117–122 [DOI] [PubMed] [Google Scholar]
- 39. Hu X.-R., Gu J.-M. (2005) N-[4-Cyano-3-(trifluoromethyl)phenyl]-3-(4-fluorophenylsulfonyl)-2-hydroxy-2-methylpropionamide. Acta Crystallogr. Sect. E Struct. Rep. Online 61, o3897–o3898 [Google Scholar]
- 40. Vega D. R., Polla G., Martinez A., Mendioroz E., Reinoso M. (2007) Conformational polymorphism in bicalutamide. Int. J. Pharm. 328, 112–118 [DOI] [PubMed] [Google Scholar]
- 41. Bis J. A., Vishweshwar P., Weyna D., Zaworotko M. J. (2007) Hierarchy of supramolecular synthons. Persistent hydroxyl-pyridine hydrogen bonds in cocrystals that contain a cyano acceptor. Mol. Pharm. 4, 401–416 [DOI] [PubMed] [Google Scholar]
- 42. Osguthorpe D. J., Hagler A. T. (2011) Mechanism of androgen receptor antagonism by bicalutamide in the treatment of prostate cancer. Biochemistry 50, 4105–4113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mast N., Annalora A. J., Lodowski D. T., Palczewski K., Stout C. D., Pikuleva I. A. (2011) Structural basis for three-step sequential catalysis by the cholesterol side chain cleavage enzyme CYP11A1. J. Biol. Chem. 286, 5607–5613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fleming F. F., Yao L., Ravikumar P. C., Funk L., Shook B. C. (2010) Nitrile-containing pharmaceuticals. Efficacious roles of the nitrile pharmacophore. J. Med. Chem. 53, 7902–7917 [DOI] [PMC free article] [PubMed] [Google Scholar]