

Background: Peroxisome proliferator-activated receptor γ coactivator 1β (PGC-1β) is a key regulator of biological processes.
Results: Overexpression of PGC-1β inhibits neointima formation and vascular smooth muscle cell proliferation.
Conclusion: PGC-1β inhibits proliferation through impaired minichromosome maintenance (MCM) complex loading onto chromatin.
Significance: PGC-1β may emerge as an important therapeutic target relevant for the treatment of proliferative vascular disorders.
Keywords: Cell Cycle, Diabetes, Gene Regulation, Vascular Biology, Vascular Smooth Muscle Cells, PGC-1β
Abstract
Proliferation of vascular smooth muscle cells (VSMCs) in response to vascular injury plays a critical role in vascular lesion formation. Emerging data suggest that peroxisome proliferator-activated receptor γ coactivator 1 (PGC-1) is a key regulator of energy metabolism and other biological processes. However, the physiological role of PGC-1β in VSMCs remains unknown. A decrease in PGC-1β expression was observed in balloon-injured rat carotid arteries. PGC-1β overexpression substantially inhibited neointima formation in vivo and markedly inhibited VSMC proliferation and induced cell cycle arrest at the G1/S transition phase in vitro. Accordingly, overexpression of PGC-1β decreased the expression of minichromosome maintenance 4 (MCM4), which leads to a decreased loading of the MCM complex onto chromatin at the replication origins and decreased cyclin D1 levels, whereas PGC-1β loss of function by adenovirus containing PGC-1β shRNA resulted in the opposite effect. The transcription factor AP-1 was involved in the down-regulation of MCM4 expression. Furthermore, PGC-1β is up-regulated by metformin, and metformin-associated anti-proliferative activity in VSMCs is at least partially dependent on PGC-1β. Our data show that PGC-1β is a critical component in regulating DNA replication, VSMC proliferation, and vascular lesion formation, suggesting that PGC-1β may emerge as a novel therapeutic target for control of proliferative vascular diseases.
Introduction
Vascular smooth muscle cells (VSMCs)3 play a key role in many physiological and pathological events occurring in blood vessels, such as hypertension, atherosclerosis, and neointima formation following angioplasty and vein graft (1, 2). Proliferation and migration of VSMCs stimulated by growth factors and cytokines, which are released by platelets and VSMCs, are essential for neointimal development upon injury. Recently, evidence from others and our own studies has shown that transcription factors play a critical role in the regulation of VSMC proliferation and migration (3–8).
Many transcription factors repress and/or activate transcription of their target genes through the recruitment of corepressors and coactivator complexes, respectively (9–12). These cofactor complexes transcriptionally regulate numerous signaling pathways that are essential for development, metabolism, and tissue repair as well as normal tissue homeostasis (9, 11). In particular conditions, differential gene expression is regulated by recruitment of specific transcriptional regulators (10, 13, 14). Consequently, we hypothesized that distinct transcriptional cofactors may be deregulated under VSMC proliferative conditions, contributing to VSMC proliferation and neointima formation. In this study, we report the effects of up- and down-regulation of peroxisome proliferator-activated receptor γ coactivator 1β (PGC-1β) and its biological consequences in proliferative vascular diseases.
The PGC-1 family of transcriptional coactivators consists of three members, PGC-1α, PGC-1β, and PGC-1-related coactivator (PRC) (15–17). The first member of the PGC-1 family was identified as a peroxisome proliferator-activated receptor γ (PPARγ)-interacting protein and named PGC-1α (18). PGC-1β is the closest homolog of PGC-1α (19, 20), whereas PRC has a limited homology to PGC-1α and PGC-1β (21). PGC-1α and PGC-1β have the ability to increase the transcriptional activity of many nuclear receptors, including PPARs (18, 22), estrogen receptors (23), glucocorticoid receptor (24), retinoid X receptor (25), liver X receptor (26), hepatic nuclear factor 4α (27), estrogen-related receptor α (28), and also members of the vitamin D and thyroid hormone receptor families (29). The PGC-1 family of coactivators also plays a key role in the regulation of mitochondrial biogenesis and the maintenance of energy homeostasis by regulating glucose and lipid metabolism (22, 27). PGC-1α inhibits high glucose-induced VSMC proliferation and migration and attenuates neointima formation by up-regulation of the expression of the mitochondrial antioxidant enzyme superoxide dismutase 2 (30, 31). However, the biological and, in particular, vascular-specific functions of PGC-1β in proliferative vascular diseases remain unknown.
DNA replication is a critical event in cell proliferation, and it is precisely controlled by the initiator proteins of DNA replication, such as the origin recognition complex, cyclin-dependent kinases, and the minichromosome maintenance (MCM) complex. The activated MCM complex serves as a DNA helicase that unwinds the helix at the replication origin, and chromatin becomes licensed for replication. The MCM protein family has six highly conserved subunits, MCM2 to MCM7. Among them, MCM6 and MCM7 are up-regulated by PDGF in smooth muscle cells (32).
We found that PGC-1β expression is down-regulated in VSMC under proliferative conditions and predicted that preserving its expression may reduce neointima formation. Accordingly, overexpression of PGC-1β inhibited neointima formation in a balloon injury carotid artery model. PGC-1β was able to reduce SMC proliferation by blocking the accumulation of MCM proteins and their loading onto chromatin via specific down-regulation of MCM4. AP-1 plays a role in the down-regulation of MCM4 expression by PGC-1β. Furthermore, metformin up-regulated PGC-1β, and metformin-associated antiproliferative activity in VSMCs is at least partially dependent on PGC-1β. Our data provide new insights into the mechanisms controlling VSMC proliferation and identify that PGC-1β, because of its role as a mediator of metformin protective effects, is emerging as a feasible therapeutic target for vascular proliferative disorders, especially diabetic vascular complications.
EXPERIMENTAL PROCEDURES
Cell Culture and Infection
The culture of rat aortic smooth muscle cells was performed as described previously (8, 33). VSMCs were grown overnight in growth medium and then rendered quiescent by changing to medium containing 0.2% FBS for 24 h. Adenoviral vectors containing PGC-1β (AdPGC-1β), GFP (AdGFP), shRNA specific for PGC-1β (AdshPGC-1β), or LacZ (AdshLacZ) were used in this study (34–36). The recombinant adenoviruses were purified by CsCl2 density gradient ultracentrifugation, and adenovirus titration was performed using the Adeno-Xtm quantitative PCR titration kit (Clontech). Cells were infected with adenovirus at a multiplicity of infection of 50 for 2 h and then changed to fresh medium containing 0.2% FBS for 24 h prior to stimulation with PDGF-BB (10 ng/ml). When no viral infection was needed, cells were treated in medium containing 0.2% FBS for 48 h prior to PDGF-BB treatment.
Cell Proliferation Assays
Cell proliferation was determined by cell counting, DNA synthesis assays, and cell cycle analysis in vitro (33). DNA synthesis assays were performed by BrdU incorporation according to the instructions of the cell proliferation ELISA kit (Roche). Cell cycle distribution was analyzed using a FACSCaliburTM system (BD Biosciences).
Preparation of Total Cell Extracts, Nuclear Extracts, and Chromatin Fractionations
Whole cell lysate extracts were prepared by using the mammalian protein extraction reagent (Thermo Scientific), and nuclear extracts were prepared with NE-PER nuclear and cytoplasmic extraction reagents (Thermo Scientific) according to the instructions of the manufacturer. Chromatin fractionations were prepared according to previous studies (37). Proteins were subjected to immunoblotting using the following antibodies against PGC-1β (Santa Cruz Biotechnology, Inc., 1:500): PGC-1α (Abcam, 1:1000), MCM4 (Santa Cruz Biotechnology, Inc., 1:500), MCM2 (Santa Cruz Biotechnology, Inc., 1:500), cyclin D1 (Santa Cruz Biotechnology, Inc., 1:500), proliferating cell nuclear antigen (PCNA) (Santa Cruz Biotechnology, Inc., 1:500), Lamin B (Santa Cruz Biotechnology, Inc., 1:1000), and tubulin (Sigma, 1:2000). Membranes were incubated with a donkey anti-rabbit, goat, or mouse IRDye-conjugated IgG (Li-Cor Odyssey) secondary antibody. Blots were scanned, and the image was displayed in grayscale.
Coimmunoprecipitation
VSMCs were lysed in lysis buffer (50 mm Tris-HCl (pH 7.8), 137 mm NaCl, 1 mm EDTA) containing 0.1% Triton-X-100 and a protease inhibitor mixture (Roche). Protein-protein interaction was detected by coimmunoprecipitation assays as described previously (38). Cellular extracts were precleared with protein A/G-agarose for 1 h at 4 °C, incubated with an anti-PGC-1β polyclonal antibody for 1 h, and then protein A/G-agarose was added and incubated overnight at 4 °C. Normal IgG was used for a negative control. The samples were separated by SDS-PAGE and analyzed by immunoblotting using an anti-c-Jun antibody (Santa Cruz Biotechnology, Inc., 1:500).
Microarray Analysis
RNA from AdGFP- or AdPGC-1β-infected human aortic smooth muscle cells (Lonza) were extracted by use of a Qiagen RNAeasy kit (Qiagen, Valencia, CA). Labeling, hybridization, washing, scanning, and initial analysis were performed by the Microarray Core Facility at the University of Michigan using standard Affymetrix protocols, and this analysis was performed on the basis of human U-133 Plus 2.0 microarrays.
Construction of Plasmids and Transfections
For transient transfections measuring promoter activity, desired DNA fragments encoding different lengths of the rat MCM4 promoter region were PCR-amplified from rat genomic DNA and inserted into the pGL4.10 luciferase reporter vector (Promega, Madison, WI). The inserts were positioned between the KpnI and XhoI sites relative to the luciferase coding sequence. Proper insertion was verified by direct DNA sequencing. The mutation of the putative AP-1 binding site in the MCM4 promoter region was conducted using the QuikChange site-directed mutagenesis kit (Agilent Technologies). The reporter plasmids were transfected into cells using Lipofectamine LTX (Invitrogen). Twenty-four hours later, cells were infected with AdGFP or AdPGC-1β for 2 h and then stimulated with PDGF-BB for 24 h. Luciferase activity was measured with the dual luciferase reporter assay system (Promega) with a luminescence counter (PerkinElmer Life Sciences, Waltham, CA). The AP-1 luciferase reporter plasmid was purchased from Stratagene. Thymidine kinase-driven Renilla luciferase served as the internal control.
ChIP
ChIP assays were performed using the EZ ChIP kit (Millipore) with minor modifications (38). In brief, VSMCs were treated for 10 min with 1% formaldehyde at room temperature for cross-linking. Cells were lysed, and chromatin extracts were sonicated for obtaining DNA fragments between 500–1000 bp. The sonicated extracts were incubated overnight at 4 °C with 5 μg of anti-c-Jun antibody (Santa Cruz Biotechnology, Inc.) or normal-rabbit IgG. The immunoprecipitated DNA-protein complex was incubated with protein G-agarose for 1 h at 4 °C. After centrifugation the complexes were washed, and the protein-chromatin cross-linking in the immunoprecipitated complexes was reversed at 65 °C overnight. Proteins were eliminated using proteinase K for 30 min at 45 °C. Purified DNA was used as a template for real-time PCR. The PCR primers used for the analysis of MCM4 promoters are listed in supplemental Table 1.
RNA Isolation and Real Time Quantitative RT-PCR
Total RNA was extracted by RNeasy mini kit (Qiagen, Valencia, CA). cDNA was synthesized and subjected to PCR amplification with primers specific for proliferation-related rat genes. mRNA levels were analyzed in triplicate and normalized to 18S RNA, using the comparative CT method. PCR primers are described in supplemental Table 1.
Angiotensin II Infusion Protocol in Mice
C57BL/6J male mice homozygous for PGC-1β gene disruption (PGC-1β−/−) and littermate WT control animals, age 8–10 weeks, were utilized. Under anesthesia with ketamine/xylazine (80/5 mg/kg intraperitoneally), mice were implanted subcutaneously with osmotic minipumps (Alzet Corp., Palo Alto, CA) set to infuse AngII (Sigma) at 500 ng/kg/min. Mice were treated for 4 weeks. Aortas and mesenteric arteries were isolated from WT or PGC-1β−/− mice and frozen in liquid N2 for histological experiments. Cross-sections from aortas and mesenteric arteries were stained with hematoxylin and eosin for vascular remodeling analysis.
Rat Carotid Artery Balloon Injury Model and Adenovirus-mediated Gene Transfer
Rat carotid artery balloon injury and adenovirus-mediated gene transfer were performed in male Sprague-Dawley rats (250–300 g) as described in our previous studies (5, 33). Briefly, rats were anesthetized with ketamine/xylazine. A 2F Fogarty catheter (Baxter Edwards Life Sciences) was inserted into the common carotid artery via the external carotid artery, and the balloon was inflated for 1 min three times. After balloon injury, 50 μl of AdPGC-1β (4 × 1010 plaque-forming units/ml) or AdGFP (4 × 1010 plaque-forming units/ml) were infused into the ligated segment of the common carotid artery and incubated for 15 min. The external carotid artery was then permanently ligated with a 6-0 silk suture, and blood flow in the common carotid artery was restored. All animal work was performed in accordance with the University of Michigan Animal Care and Use Committee.
Immunohistochemistry and Lesion Assessment
The animals were sacrificed, and carotid arteries were perfused and fixed with 4% (v/v) paraformaldehyde in PBS, harvested, and embedded in paraffin. Six sections (5 μm thick) of injured carotid artery cut at equally spaced intervals were used. Cross-sections were stained with hematoxylin and eosin for lesion assessment. For immunohistochemistry, cross-sections were stained using mouse monoclonal anti-PCNA antibody (Santa Cruz Biotechnology, Inc., 1:200) and avidin-biotin-horseradish peroxidase for detection (Vectastain Elite ABC kit, Vector Laboratories). The intimal/medial cross-sectional area was quantified with ImagePro Plus software (University of Michigan). Total PCNA-positive cells were counted in a blinded manner manually at high magnification. The total cell number was counted after DAPI staining in the adjacent sections. The proliferation index was calculated as the mean number of PCNA-positive cells divided by the mean total number of cells and expressed as a percentage.
Statistical Analysis
Data are expressed as mean ± S.E. Statistical significance was analyzed by Student's t test comparing one group with the control group. To compare two or more groups with the control group, one-way analysis of variance followed by a Student-Newman-Keuls multiple comparison test was performed using GraphPad PRISMTM software version 5. p < 0.05 was considered statistically significant. All experiments were performed independently at least three times.
RESULTS
PGC-1β Expression Is Significantly Decreased in PDGF-treated VSMCs and in the Balloon-injured Arteries
To investigate whether nuclear receptor coregulators might be involved in modulation of VSMC proliferation, we first selectively assessed expression levels of well known nuclear receptor coregulators in primary rat aortic SMCs. mRNA expression levels of most coregulators, including PGC-1α, P300, small heterodimer partner, PPAR-binding protein, PPARγ-interacting protein, silencing mediator of retinoid and thyroid hormone receptor, and dosage-sensitive sex reversal adrenal hypoplasia congenital critical region on X chromosome, gene 1 (DAX1), remained unchanged between PDGF-treated VSMCs and quiescent VSMCs. In contrast, VSMC mRNA of PGC-1β was significantly repressed in PDGF-treated VSMCs (Fig. 1A). PGC-1β mRNA was decreased by 80% 4 h after PDGF-BB treatment. A similar reduction in PGC-1β expression was also observed in angiotensin II-treated VSMCs (Fig. 1B). Time course experiments demonstrated that PGC-1β showed an early (2 h) significant decrease in mRNA levels in proliferating VSMCs compared with quiescent VSMCs that was sustained during the 24 h documented (Fig. 1C), whereas mRNA levels increased for PGC-1α and PRC in the first 1 and 2 h post-treatment, respectively, and then returned to basal levels (data not shown). Consistently, PGC-1β protein levels were significantly reduced in nuclear extracts after PDGF-BB treatment, whereas the levels of PCNA, a cell proliferation-related marker, were increased dramatically (Fig. 1D).
FIGURE 1.

PGC-1β is down-regulated by growth factors and during neointima formation. A and B, rat VSMCs grown to 60–70% confluence underwent mitogenic quiescence by serum deprivation for 48 h and were subsequently stimulated with 10 ng/ml PDGF-BB or angiotensin II (Ang II) (10−7 m) for 6 h. qRT-PCR for PGC-1β expression was performed, and 18 S RNA was used for normalization of cDNA. n = 3. C, qRT-PCR for PGC-1β expression was performed in cDNA from VSMCs treated with 10 ng/ml PDGF-BB at the indicated time points. n = 3. D, nuclear proteins extracted from VSMCs treated with 10 ng/ml PDGF-BB for the specified times were immunoblotted for the indicated proteins. E, expression of PGC-1β was determined by qRT-PCR in samples from balloon-injured carotid arteries harvested at the indicated times post-injury. n = 3 rats at each time point. *, p < 0.05; **, p < 0.01.
To test whether PGC-1β is involved in proliferative vascular disease, we examined the expression of PGC-1β in the balloon-injured carotid artery model, which is characterized by VSMC proliferation and neointima formation. PGC-1β mRNA expression was markedly down-regulated in arteries after balloon injury compared with sham controls during a 28-day time course (Fig. 1E). These data support our hypothesis that the expression of the nuclear receptor coregulator PGC-1β is impaired in proliferative disorders, suggesting that PGC-1β may be an antiproliferative factor in VSMCs.
PGC-1β Deficiency Aggravates Angiotensin II-induced Artery Remodeling
We analyzed the effect of PGC-1β on angiotensin II-induced vascular remodeling upon loss of function in PGC-1β knockout mice. After 4 weeks infusion with angiotensin II, the artery wall thickness of aortas increased markedly in PGC-1β−/− mice compared with WT mice (Fig. 2, A and B). The lumen areas were not different between the groups (data not shown). We observed mesenteric resistance artery remodeling and found that the thickness of third-order mesenteric arteries was also significantly increased in PGC-1β-deficient mice compared with control mice (Fig. 2, C and D). These observations suggest that PGC-1β plays a critical role in vascular remodeling.
FIGURE 2.

Effect of PGC-1β loss on artery remodeling. PGC-1β-deficient mice (PGC-1β−/−) and littermate WT mice were implanted subcutaneously with osmotic minipumps to infuse angiotensin II at 500 ng/kg/min. Shown is the histological analysis with hematoxylin and eosin staining of aorta (A) and the third-order mesenteric artery (C) from wild-type and PGC-1β−/− mice, respectively, after 4 weeks of infusion. Also shown is the quantification of the aortic wall thickness (B) and third-order mesenteric artery wall thickness (D) from WT and PGC-1β−/− mice. n = 5 in each animal group. Scale bars = 20 μm. **, p < 0.01.
Overexpression of PGC-1β Prevents Neointima Formation in Balloon-injured Carotid Arteries in Rats
To determine whether PGC-1β modulates the development of neointima formation, PGC-1β was overexpressed in the common carotid artery immediately after balloon injury by adenovirus-mediated gene delivery (5, 33). As depicted in Fig. 3, A–C, overexpression of PGC-1β was capable of attenuating neointimal lesion formation significantly without affecting the thickness of the media, thus resulting in a reduced intimal/medial ratio in the AdPGC-1β-treated group compared with the AdGFP-treated group.
FIGURE 3.

PGC-1β overexpression inhibits neointimal hyperplasia and cell proliferation. A, representative sections from balloon-injured carotid arteries treated with AdGFP or AdPGC-1β harvested at day 21 after injury and stained with hematoxylin and eosin. Scale bars = 100 μm. NI, neointima; M, media. B and C, decreased intima area and intima/media ratio in the AdPGC-1β-treated group. n = 9 rats in each group. **, p < 0.01. D, representative sections of balloon injured-arteries in AdGFP and AdPGC-1β group were immunostained for PCNA at day 7 after injury. Scale bars = 100 μm. Arrows show PCNA-positive cells. E, percentage of the total cells in the media of carotid arteries showing PCNA-positive nuclei. n = 5 rats in each group. **, p < 0.01. F, the sections adjacent to those used for PCNA-staining were labeled with DAPI and total cells in neointima were counted manually at high magnification in a fluorescence microscope. **, p < 0.01.
Immunohistochemical staining for PCNA (Fig. 3D) was performed to examine the effect of PGC-1β on VSMC proliferation in vivo. There is a 60% reduction in PCNA-positive cells in the media of AdPGC-1β-treated animals (Fig. 3E) and a 75% reduction in the number of cells in the neointima (F).
PGC-1β Inhibits PDGF-BB-induced VSMC Proliferation by Arresting Cells in the G0/G1 Phase
In cultured rat aortic vascular smooth muscle cells, PGC-1β overexpression caused cell cycle arrest with a 23% increase in the G0/G1 population and an 83% reduction in cells in the S phase compared with AdGFP-treated VSMCs (Fig. 4, A and B). Overexpression of PGC-1β significantly suppressed VSMC proliferation and reduced DNA synthesis detected by cell counting and BrdU incorporation, respectively (Fig. 4, C and D). Furthermore, overexpression of PGC-1β in VSMCs caused a marked inhibition of cyclin D1 and PCNA protein levels, two proliferation markers, as shown by Western blot analysis (Fig. 4E).
FIGURE 4.

PGC-1β inhibits VSMC proliferation in vitro. VSMCs were infected with adenoviruses encoding GFP or PGC-1β. A and B, results from flow cytometry analysis to measure the DNA content in AdGFP- or AdPGC-1β-treated VSMCs after serum starvation for 24 h followed by stimulation with 10 ng/ml PDGF-BB for 18 h. Data are expressed as the percentage of the total cells. n = 3. C, VSMCs were seeded in 96-well plates and infected with the indicated adenovirus. Cell proliferation was measured by BrdU incorporation. n = 10. D, cell proliferation was measured by counting cell numbers at each time point. VSMCs were infected with adenoviruses encoding GFP or PGC-1β and changed to growth medium containing 10% FBS. n = 4. E, AdGFP- or AdPGC-1β-treated VSMCs were serum-starved for 24 h and stimulated with PDGF-BB at 10 ng/ml for 12 and 24 h. Total cell lysates were analyzed by Western blot analysis using the indicated antibodies. Tubulin served as the internal loading control. The blot shown is one representative of three independent experiments. * p < 0.05; **, p < 0.01 versus AdGFP group.
To confirm the antiproliferative role of PGC-1β in VSMCs, an adenovirus containing an shRNA designed to target PGC-1β (AdshPGC-1β) was used (35). Infection with AdshPGC-1β decreased the content of PGC-1β mRNA by 73%, without significantly affecting PGC-1α and PRC mRNA (Fig. 5, A–C). Treatment of VSMCs with AdshPGC-1β significantly promoted PDGF-BB-induced cell proliferation and increased cyclin D1 and PCNA (Fig. 5, D–E).
FIGURE 5.
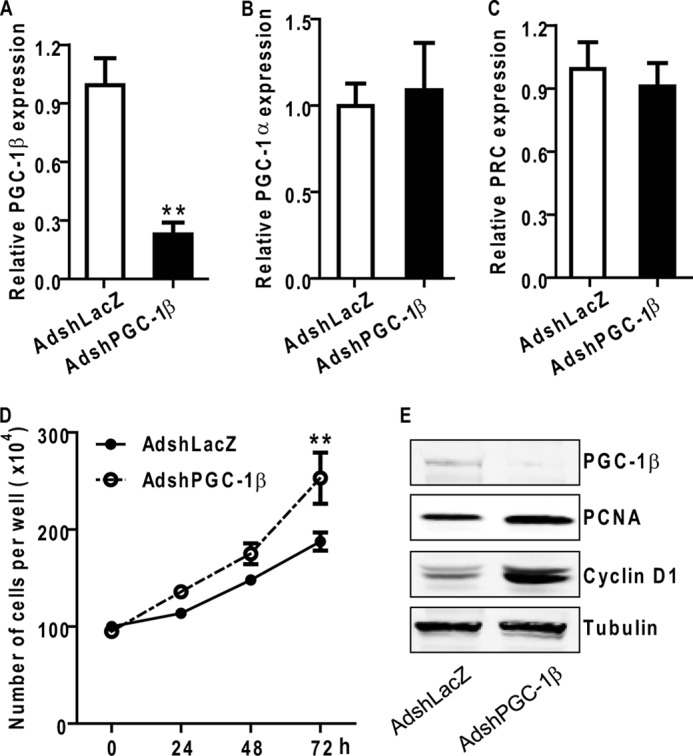
Knockdown of PGC-1β increases VSMC proliferation. A–C, knockdown efficiency of adenovirus-mediated shRNA against PGC-1β on PGC members in VSMCs. VSMCs were infected with adenoviruses AdshLacZ or AdshPGC-1β and maintained in 10% FBS medium for 48 h. PGC-1β (A), PGC-1α (B), and PRC (C) mRNA expression levels were analyzed by qRT-PCR. D, VSMCs were seeded in 6-well plates and infected with the indicated adenovirus. Cell proliferation was measured by cell counting at the specified time points. n = 4. E, AdshLacZ- or AdshPGC-1β-treated VSMCs were serum-starved for 24 h and stimulated with PDGF-BB at 10 ng/ml for 12 h. Total cell lysates were analyzed by Western blot analysis using the indicated antibodies. Tubulin served as the internal control. **, p < 0.01 versus AdshLacZ-treated cells.
PGC-1β Blocks Accumulation of MCM Proteins and Their Loading onto Chromatin
To begin addressing the molecular mechanisms underlying the inhibitory effect of PGC-1β on VSMC proliferation, we evaluated global gene expression profiles to determine the effect of PGC-1β on target genes by cDNA microarray analysis in total mRNA from human aortic smooth muscle cells infected with AdGFP or AdPGC-1β. Of the genes involved in cell cycle/DNA replication, six genes showed elevated expression levels, whereas two genes were reduced in expression, as further verified by qRT-PCR.
MCM proteins, including six highly conserved subunits, MCM2 to MCM7, are essential regulators of initiation and elongation of DNA replication in all eukaryotes (39, 40). The mammalian MCM protein family exists in two forms, chromatin-bound and unbound, in the nuclei. The former is generally believed to function as a helicase during DNA replication (39, 40). Cyclin D1 is a key regulator of the G1 phase progression, and it is expressed to a maximum in the mid-G1 phase of the cell cycle when cells are stimulated with growth factors (41). We show that MCM2, MCM4, and MCM7 mRNA levels are increased in VSMCs during G1 phase, as indicated by increased Cyclin D1 levels (Fig. 6, A–D). In AdPGC-1β-treated VSMCs, we observed selective down-regulation of MCM4 mRNA and protein levels (Fig. 7, B and I) concomitant with reduced cyclin D1 expression (A), whereas the levels of MCM2 and MCM7 remained unaltered (C, D, and I). Conversely, down-regulation of endogenous PGC-1β with AdshPGC-1β resulted in increased mRNA levels of cyclin D1 and MCM4 with no effect on MCM2 or MCM7 expression (Fig. 7, E–I). We also determined chromatin-bound MCM2 and MCM4 levels in VSMCs by Western blot analysis. As shown in Fig. 7J, both chromatin-bound MCM2 and MCM4 are decreased in PGC-1β-treated VSMCs, suggesting that down-regulation of MCM4 by PGC-1β affects MCM complex loading on to chromatin.
FIGURE 6.

Cyclin D1, MCM2, MCM4, and MCM7 expression are increased during the S phase in VSMCs. Rat VSMCs grown to 60–70% confluence underwent mitogenic quiescence by serum deprivation for 48 h and were subsequently stimulated with 10 ng/ml PDGF-BB for the indicated time points. The expression of cyclin D1 (A), MCM4 (B), MCM2 (C), and MCM7 (D) were analyzed by qRT-PCR. 18 S RNA served as the internal control. n = 3. *, p < 0.05; **, p < 0.01.
FIGURE 7.

PGC-1β inhibits PDGF-induced MCM4 expression and MCM complex chromatin loading in VSMCs. A–F, VSMCs were infected with AdPGC-1β, AdGFP, AdshPGC-1β, or AdshLacZ, maintained in 0.2% FBS medium for 24 h, and then stimulated with 10 ng/ml PDGF-BB for 8 h or 18 h. Cyclin D1 (Ccnd1), MCM4, and MCM2 mRNA levels were analyzed by qRT-PCR. 18 S RNA served as internal control. n = 3. G and H, VSMCs were infected with AdPGC-1β, AdGFP, AdshPGC-1β, or AdshLacZ, maintained in 0.2% FBS medium for 24 h, and then stimulated with 10 ng/ml PDGF-BB for 12 h. VSMCs were collected and separated into total cell lysates or chromatin-bound extracts. Immunoblotting with the indicated antibodies was performed on lysates from equal cell numbers loaded onto each lane. *, p < 0.05; **, p < 0.01 versus AdGFP-treated group; #, p < 0.05; ##, p < 0.01 versus AdshLacZ-treated group.
AP-1 Is Required for Transcriptional Regulation of MCM4 by PGC-1β
To further establish whether PGC-1β regulates MCM4 expression at the transcriptional level, we performed transient transcriptional assays in vitro using rat MCM4 gene luciferase reporter constructs that contain DNA fragments in varying lengths corresponding to the MCM4 promoter (MCM4-Luc). Treatment of VSMCs with PDGF-BB resulted in increased activation of the MCM4 promoter (-736/+131 and −377/+131), an effect readily inhibited by PGC-1β (Fig. 8A). Because PGC-1β is a transcriptional cofactor lacking a DNA binding domain, at least one other transcription factor should be involved in the regulation of MCM4 transcriptional activation in VSMCs. Analysis of binding sites for transcription factors with the Genomatix software uncovered a highly conserved AP-1 site in both human and rat MCM4 promoters. The putative AP-1 binding site (-274 to −281) was mutated to determine whether this site is a functional target element. The MCM4-Luc promoter mutation could not be activated by PDGF-BB (Fig. 8B), whereas PGC-1β overexpression inhibited PDGF-BB activation to basal levels comparable with those of the mutant reporter, suggesting that the AP-1 site within the MCM4 promoter region is functional and critical for PGC-1β to control MCM4 expression.
FIGURE 8.

PGC-1β inhibits PDGF-induced AP-1 transcriptional activity. A, serial deletion constructs of the rat MCM4 promoter were transfected into VSMCs. After 12 h, cells were infected with AdPGC-1β or AdGFP and stimulated with PDGF-BB for an additional 24 h. Luciferase activity was measured with the dual luciferase reporter assay system. Cotransfected thymidine kinase-driven Renilla luciferase served as the internal control. The numbers indicate the distance in nucleotides from the transcription start site (+1) of the rat MCM4 gene. B, VSMCs were transfected with the MCM4 promoter (-377/+161) or MCM4 (-377/+161) mut-luciferase constructs, and luciferase activity was measured as above. C, VSMCs were transiently transfected with AP-1-Luc and infected with AdGFP or AdPGC-1β. Cells were stimulated with PDGF-BB (10 ng/ml) for 24 h, and luciferase activity was measured as above. D, VSMCs were infected with AdGFP or AdPGC-1β and treated with 10 ng/ml PDGF-BB for 8 h. ChIP assays were performed using antibodies against c-Jun and normal rabbit IgG. Data shown are from three independent experiments. E, Coimmunoprecipitation was performed with an antibody against PGC-1β in VSMCs. Normal rabbit IgG served as a negative control. Input samples were loaded as 10% of total cell extracts. Blots were probed with antibodies as indicated in each panel. IP, immunoprecipitation. F, coimmunoprecipitation was performed with an antibody against c-Jun in VSMCs treated with AdGFP or AdPGC-1β. Normal rabbit IgG served as a negative control. Input samples were loaded as 10% of total cell extracts used for the coimmunoprecipitation. Blots were probed with antibodies as indicated in each panel. *, p < 0.05; **, p < 0.01.
The effect of PGC-1β on AP-1 transcriptional activity was also confirmed by a reporter plasmid containing isolated AP-1 response elements (pAP1-Luc) from Stratagene. SMCs were transfected with pAP1-Luc and subsequently treated with PDGF-BB. AP-1 transcriptional activity increased 2.2-fold in response to PDGF-BB treatment and was reduced to basal levels when infected with AdPGC-1β (Fig. 8C).
Next, ChIP assays were performed to further determine the effect of PGC-1β on AP-1 binding to the MCM4 promoter region. Consistent with the data shown above, we demonstrated that c-Jun can directly bind to the putative AP-1 binding site (-274 to −281) in the MCM4 promoter upon PDGF-BB treatment and that PGC-1β potently inhibits the binding of c-Jun to this AP-1 binding site in VSMCs infected with Ad PGC-1β in the same conditions (Fig. 8D).
To determine whether PGC-1β interacts with AP-1, we performed a coimmunoprecipitation assay and found that PGC-1β physically interacts with c-Jun in VSMCs (Fig. 8E). To further establish whether PGC-1β affects the AP-1 complex formation, we determined c-Fos levels in a c-Jun pull-down complex and found decreased c-Fos levels in AdPGC-1β-treated VSMCs (Fig. 8F), indicating that PGC-1β could suppress the binding of AP-1 to the MCM4 promoter by interaction with c-Jun, a component of the AP-1 complex resulting in reduced c-Fos/c-Jun interaction. These findings suggest that the inhibitory effect of PGC-1β on VSMC proliferation is at least partially carried out by inhibition of AP-1 transcriptional activity.
PGC-1β Is Required for the Antiproliferative Effect of Metformin in VSMCs
In addition to our discovery that PGC-1β is an effective regulator of VSMC proliferation, we determined that the expression of PGC-1β is up-regulated by metformin, one agent used for treatment of the metabolic syndrome or type 2 diabetes (42, 43). Metformin significantly up-regulated PGC-1β mRNA levels ∼3-fold in VSMCs (Fig. 9A). Metformin induces activation of AMP-activated protein kinase (42). Consistently, we also determined that 5-aminoimidazole-4-carboxamide-1-b-4-ribofuranoside, another AMP-activated protein kinase activator, resulted in up-regulation of PGC-1β (Fig. 9B). Conversely, compound C, a selective inhibitor of AMP-activated protein kinase, completely prevented the up-regulation of PGC-1β mRNA by metformin and 5-aminoimidazole-4-carboxamide-1-b-4-ribofuranoside in smooth muscle cells (Fig. 9, A and B), suggesting that PGC-1β may be a mediator of the metformin inhibitory effects in VSMC proliferation.
FIGURE 9.

PGC-1β is required for the inhibitory effect of metformin on VSMC proliferation. A and B, VSMCs were maintained in 0.2% FBS medium for 24 h and then treated with metformin (10 ng/ml), 5-aminoimidazole-4-carboxamide-1-b-4-ribofuranoside (200 μm), or vehicle for 24 h with dimethyl sulfoxide (DMSO) or compound C (10 μm). PGC-1β mRNA expression levels were determined by qRT-PCR. C, VSMCs were infected with AdshLacZ or AdshPGC-1β and maintained in 0.2% FBS medium for 24 h and then stimulated with PDGF-BB (10 ng/ml) for 24 h in the presence or absence of metformin at 10 ng/ml. Cell proliferation was measured by BrdU incorporation. n = 10. *, p < 0.05 versus vehicle-treated group. D, proposed anti-proliferative mechanism of PGC-1β by inhibition of expression of MCM4 and MCM complex loading onto chromatin.
Metformin has demonstrated antiproliferative effect in VSMCs and some cancer cells by causing G0/G1 cell cycle arrest (43–47). Treatment of VSMCs with metformin resulted in decreased DNA replication and enhanced the antiproliferative effect of PGC-1β as measured by BrdU incorporation (Fig. 9C). Importantly, knockdown of PGC-1β reversed the inhibitory effect of metformin on VSMC proliferation (Fig. 9C). These data suggest that the metformin-associated potent antiproliferative activity in VSMCs is at least partially dependent on PGC-1β. The antiproliferative mechanisms of PGC-1β by inhibition of the expression of MCM4 and MCM complex loading onto chromatin in VSMCs is summarized in Fig. 9D.
DISCUSSION
This study sheds light on the functional importance of PGC-1β, a transcriptional cofactor, in modulating SMC proliferation and neointima formation. First, we found that PGC-1β is expressed in VSMCs and is down-regulated by mitogens, including PDGF-BB and angiotensin II. PGC-1β deficiency aggravates angiotensin II-induced artery remodeling. Additional in vivo data show that PGC-1β is down-regulated in the arteries after balloon injury and overexpression of PGC-1β inhibited neointimal lesion formation. Second, after performing a microarray analysis to identify the effect of PGC-1β on target genes, we demonstrated that PGC-1β inhibits VSMC proliferation by down-regulating MCM4 expression and reducing MCM complex loading to chromatin. We also identified that PGC-1β inhibits AP-1 transcriptional activity in the rat MCM4 promoter in VSMCs. Furthermore, we identified that metformin up-regulates PGC-1β expression and established PGC-1β as a key mediator in the metformin-mediated inhibitory effect on VSMC proliferation. Our findings provide a novel mechanism of controlling SMC proliferation and proliferative vascular diseases.
It is well known that SMC proliferation in response to vascular injury is central to neointima formation. In the eukaryotic cell cycle, DNA replication is precisely controlled and is initiated at specific sites on chromosomes (48–50). MCM proteins are loaded onto the chromatin by forming a stable ring structure around the DNA and provide the helicase activity necessary for unwinding double-stranded DNA and loading of DNA polymerase (40, 51, 52). Several studies have reported that MCM proteins are highly expressed in human malignant cancer cells, indicating that MCM proteins are essential for DNA replication and may serve as cell proliferation markers in cycling cells (39, 40, 53). Data in this study demonstrate that expression of MCM4 is induced in response to PDGF-BB stimulation in VSMCs. Bruemmer et al. (32) also found that the expression of MCM6 and MCM7 was up-regulated by PDGF-BB via the ERK/MAPK signal transduction pathway in smooth muscle cells. Overexpression of PGC-1β specifically down-regulates the expression of MCM4 but not MCM2 or MCM7, and lack of MCM4 at the origins abolishes MCM complex loading onto chromatin, resulting in the inhibition of DNA synthesis and cell proliferation. Kikuchi et al. (53) reported that knockdown of MCM4 reduces proliferation in lung cancer cells. The exact underlying the effects of PGC-1β on proliferative vascular disease may be pleiotropic and involve different molecular mechanisms. Here we focused on cell proliferation and found that PGC-1β inhibited DNA replication in VSMCs, indicating that the PGC-1β/MCM pathway plays a key role in controlling cell proliferation.
The PGC cofactors, including PGC-1α, PGC-1β, and PRC, are important components of the transcriptional machinery (16, 17, 47). PGC-1α and PGC-1β are recruited by a large number of activated transcription factors such as PPAR, estrogen receptor, mineralocorticoid receptor, and other transcription factors (18, 23, 24, 26). In transiently transfected cells, transcriptional activity of these transcription factors was significantly increased upon overexpression of PGC-1α and PGC-1β (18, 23, 24, 26). PGC-1α also serves as a corepressor by inhibiting tumor necrosis factor α-induced NF-κB transcriptional activity in human aortic smooth and human aortic endothelial cells (55, 56). The inhibitory effect of PGC-1β on NF-κB transcriptional activity was also confirmed by a reporter plasmid containing isolated NF-κB response elements in our study (data not shown). Our studies identified MCM4 as a putative novel target for PGC-1β inhibitory effects as evidenced by reduced endogenous mRNA levels in response to up-regulation of PGC-1β. Genomatix software analysis identified a highly conserved AP-1 binding site position (-274 to −281) in human and rat MCM4 promoters. Our data demonstrated that PGC-1β negatively regulates PDGF-BB-induced transcription through this functional AP-1 binding site in the MCM4 promoter region by the use of a reporter plasmid containing isolated AP-1 response elements and site-directed mutagenesis as well as coimmunoprecipitation with c-Jun. The inhibition of AP-1 activity by PGC-1β might constitute the basis for new treatment rationales.
In this study, we found that metformin up-regulates the expression of PGC-1β via the AMP-activated protein kinase pathway. It has been well known for decades that metformin can inhibit vascular complications in type 2 diabetic patients (46, 47, 57). Many studies also have shown that metformin has significant and diverse anticancer activity and that the administration of metformin prolongs survival (44, 54). The antiproliferative mechanisms of metformin action are not well understood. Here we identified that the inhibitory effect of metformin on VSMC proliferation is dependent on PGC-1β. Knockdown of PGC-1β totally blocked the suppressive effect of metformin in VSMCs. These findings suggest that the metformin-associated potent antiproliferative activity in VSMCs is at least partially mediated by PGC-1β.
In summary, we have shown that the transcription cofactor PGC-1β is decreased in response to pro-proliferative stimulation and that PGC-1β specifically down-regulates the expression of MCM4 and blocks DNA replication (Fig. 8D). Knockout of PGC-1β enhanced angiotensin II-mediated vascular remodeling, whereas increased PGC-1β expression attenuated neointima formation after vascular injury. This coregulator factor thus may emerge as an important therapeutic target relevant for the treatment of proliferative vascular disorders.
Acknowledgments
We thank Dr. Minerva T. Garcia-Barrio at the Morehouse School of Medicine for critical review of the manuscript. We also thank the University of Michigan DNA Sequencing Core Affymetrix and Microarray Core Group for global gene expression profiles.
This work was supported, in whole or in part, by National Institutes of Health Grants HL068878, HL105114, and HL089544 (to Y. E. C.). This work was also supported by American Heart Association Grants 10POST3270008 (to Y. F.), 09SDG2230270 (to L. C.), and 0835237N (to J. Z.).

This article contains supplemental Table 1.
- VSMC
- vascular smooth muscle cell
- PGC-1β
- peroxisome proliferator-activated receptor γ coactivator 1β
- PRC
- peroxisome proliferator-activated receptor γ coactivator 1-related coactivator
- PPARγ
- peroxisome proliferator-activated receptor γ
- MCM
- minichromosome maintenance
- PCNA
- proliferating cell nuclear antigen
- qRT-PCR
- quantitative real-time PCR.
REFERENCES
- 1. Dzau V. J., Braun-Dullaeus R. C., Sedding D. G. (2002) Vascular proliferation and atherosclerosis. New perspectives and therapeutic strategies. Nat. Med. 8, 1249–1256 [DOI] [PubMed] [Google Scholar]
- 2. Gibbons G. H., Dzau V. J. (1994) The emerging concept of vascular remodeling. N. Engl. J. Med. 330, 1431–1438 [DOI] [PubMed] [Google Scholar]
- 3. Blaschke F., Leppanen O., Takata Y., Caglayan E., Liu J., Fishbein M. C., Kappert K., Nakayama K. I., Collins A. R., Fleck E., Hsueh W. A., Law R. E., Bruemmer D. (2004) Liver X receptor agonists suppress vascular smooth muscle cell proliferation and inhibit neointima formation in balloon-injured rat carotid arteries. Circ. Res. 95, e110–123 [DOI] [PubMed] [Google Scholar]
- 4. Bonta P. I., Pols T. W., van Tiel C. M., Vos M., Arkenbout E. K., Rohlena J., Koch K. T., de Maat M. P., Tanck M. W., de Winter R. J., Pannekoek H., Biessen E. A., Bot I., de Vries C. J. (2010) Nuclear receptor Nurr1 is expressed in and is associated with human restenosis and inhibits vascular lesion formation in mice involving inhibition of smooth muscle cell proliferation and inflammation. Circulation 121, 2023–2032 [DOI] [PubMed] [Google Scholar]
- 5. Fu M., Zhang J., Zhu X., Myles D. E., Willson T. M., Liu X., Chen Y. E. (2001) Peroxisome proliferator-activated receptor γ inhibits transforming growth factor β-induced connective tissue growth factor expression in human aortic smooth muscle cells by interfering with Smad3. J. Biol. Chem. 276, 45888–45894 [DOI] [PubMed] [Google Scholar]
- 6. Gizard F., Amant C., Barbier O., Bellosta S., Robillard R., Percevault F., Sevestre H., Krimpenfort P., Corsini A., Rochette J., Glineur C., Fruchart J. C., Torpier G., Staels B. (2005) PPAR α inhibits vascular smooth muscle cell proliferation underlying intimal hyperplasia by inducing the tumor suppressor p16INK4a. J. Clin. Invest. 115, 3228–3238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nomiyama T., Nakamachi T., Gizard F., Heywood E. B., Jones K. L., Ohkura N., Kawamori R., Conneely O. M., Bruemmer D. (2006) The NR4A orphan nuclear receptor NOR1 is induced by platelet-derived growth factor and mediates vascular smooth muscle cell proliferation. J. Biol. Chem. 281, 33467–33476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang J., Fu M., Zhu X., Xiao Y., Mou Y., Zheng H., Akinbami M. A., Wang Q., Chen Y. E. (2002) Peroxisome proliferator-activated receptor δ is up-regulated during vascular lesion formation and promotes post-confluent cell proliferation in vascular smooth muscle cells. J. Biol. Chem. 277, 11505–11512 [DOI] [PubMed] [Google Scholar]
- 9. Smith C. L., O'Malley B. W. (2004) Coregulator function. A key to understanding tissue specificity of selective receptor modulators. Endocr. Rev. 25, 45–71 [DOI] [PubMed] [Google Scholar]
- 10. Yu S., Reddy J. K. (2007) Transcription coactivators for peroxisome proliferator-activated receptors. Biochim. Biophys. Acta 1771, 936–951 [DOI] [PubMed] [Google Scholar]
- 11. Lee J. W., Lee Y. C., Na S. Y., Jung D. J., Lee S. K. (2001) Transcriptional coregulators of the nuclear receptor superfamily. Coactivators and corepressors. Cell Mol. Life Sci. 58, 289–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Horwitz K. B., Jackson T. A., Bain D. L., Richer J. K., Takimoto G. S., Tung L. (1996) Nuclear receptor coactivators and corepressors. Mol. Endocrinol. 10, 1167–1177 [DOI] [PubMed] [Google Scholar]
- 13. Yu C., Markan K., Temple K. A., Deplewski D., Brady M. J., Cohen R. N. (2005) The nuclear receptor corepressors NCoR and SMRT decrease peroxisome proliferator-activated receptor γ transcriptional activity and repress 3T3-L1 adipogenesis. J. Biol. Chem. 280, 13600–13605 [DOI] [PubMed] [Google Scholar]
- 14. Guan H. P., Ishizuka T., Chui P. C., Lehrke M., Lazar M. A. (2005) Corepressors selectively control the transcriptional activity of PPARγ in adipocytes. Genes Dev. 19, 453–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu C., Lin J. D. (2011) PGC-1 coactivators in the control of energy metabolism. Acta Biochim. Biophys. Sin. 43, 248–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rowe G. C., Jiang A., Arany Z. (2010) PGC-1 coactivators in cardiac development and disease. Circ. Res. 107, 825–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Arany Z. (2008) PGC-1 coactivators and skeletal muscle adaptations in health and disease. Curr. Opin. Genet. Dev. 18, 426–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Puigserver P., Wu Z., Park C. W., Graves R., Wright M., Spiegelman B. M. (1998) A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 92, 829–839 [DOI] [PubMed] [Google Scholar]
- 19. Lin J., Puigserver P., Donovan J., Tarr P., Spiegelman B. M. (2002) Peroxisome proliferator-activated receptor γ coactivator 1β (PGC-1β), a novel PGC-1-related transcription coactivator associated with host cell factor. J. Biol. Chem. 277, 1645–1648 [DOI] [PubMed] [Google Scholar]
- 20. Kressler D., Schreiber S. N., Knutti D., Kralli A. (2002) The PGC-1-related protein PERC is a selective coactivator of estrogen receptor α. J. Biol. Chem. 277, 13918–13925 [DOI] [PubMed] [Google Scholar]
- 21. Andersson U., Scarpulla R. C. (2001) Pgc-1-related coactivator, a novel, serum-inducible coactivator of nuclear respiratory factor 1-dependent transcription in mammalian cells. Mol. Cell. Biol. 21, 3738–3749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vega R. B., Huss J. M., Kelly D. P. (2000) The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor α in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol. Cell Biol. 20, 1868–1876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tcherepanova I., Puigserver P., Norris J. D., Spiegelman B. M., McDonnell D. P. (2000) Modulation of estrogen receptor-α transcriptional activity by the coactivator PGC-1. J. Biol. Chem. 275, 16302–16308 [DOI] [PubMed] [Google Scholar]
- 24. Knutti D., Kaul A., Kralli A. (2000) A tissue-specific coactivator of steroid receptors, identified in a functional genetic screen. Mol. Cell Biol. 20, 2411–2422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Delerive P., Wu Y., Burris T. P., Chin W. W., Suen C. S. (2002) PGC-1 functions as a transcriptional coactivator for the retinoid X receptors. J. Biol. Chem. 277, 3913–3917 [DOI] [PubMed] [Google Scholar]
- 26. Oberkofler H., Schraml E., Krempler F., Patsch W. (2003) Potentiation of liver X receptor transcriptional activity by peroxisome-proliferator-activated receptor γ co-activator 1α. Biochem. J. 371, 89–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yoon J. C., Puigserver P., Chen G., Donovan J., Wu Z., Rhee J., Adelmant G., Stafford J., Kahn C. R., Granner D. K., Newgard C. B., Spiegelman B. M. (2001) Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 413, 131–138 [DOI] [PubMed] [Google Scholar]
- 28. Hentschke M., Süsens U., Borgmeyer U. (2002) PGC-1 and PERC, coactivators of the estrogen receptor-related receptor γ. Biochem. Biophys. Res. Commun. 299, 872–879 [DOI] [PubMed] [Google Scholar]
- 29. Wu Y., Delerive P., Chin W. W., Burris T. P. (2002) Requirement of helix 1 and the AF-2 domain of the thyroid hormone receptor for coactivation by PGC-1. J. Biol. Chem. 277, 8898–8905 [DOI] [PubMed] [Google Scholar]
- 30. Qu A., Jiang C., Xu M., Zhang Y., Zhu Y., Xu Q., Zhang C., Wang X. (2009) PGC-1α attenuates neointimal formation via inhibition of vascular smooth muscle cell migration in the injured rat carotid artery. Am. J. Physiol. Cell Physiol. 297, C645–653 [DOI] [PubMed] [Google Scholar]
- 31. Zhu L., Sun G., Zhang H., Zhang Y., Chen X., Jiang X., Jiang X., Krauss S., Zhang J., Xiang Y., Zhang C. Y. (2009) PGC-1α is a key regulator of glucose-induced proliferation and migration in vascular smooth muscle cells. PLoS ONE 4, e4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bruemmer D., Yin F., Liu J., Kiyono T., Fleck E., Van Herle A. J., Law R. E. (2003) Expression of minichromosome maintenance proteins in vascular smooth muscle cells is ERK/MAPK dependent. Exp. Cell Res. 290, 28–37 [DOI] [PubMed] [Google Scholar]
- 33. Chen K. H., Guo X., Ma D., Guo Y., Li Q., Yang D., Li P., Qiu X., Wen S., Xiao R. P., Tang J. (2004) Dysregulation of HSG triggers vascular proliferative disorders. Nat. Cell Biol. 6, 872–883 [DOI] [PubMed] [Google Scholar]
- 34. Lin J., Tarr P. T., Yang R., Rhee J., Puigserver P., Newgard C. B., Spiegelman B. M. (2003) PGC-1β in the regulation of hepatic glucose and energy metabolism. J. Biol. Chem. 278, 30843–30848 [DOI] [PubMed] [Google Scholar]
- 35. Hernandez C., Molusky M., Li Y., Li S., Lin J. D. (2010) Regulation of hepatic ApoC3 expression by PGC-1β mediates hypolipidemic effect of nicotinic acid. Cell Metab. 12, 411–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Uldry M., Yang W., St-Pierre J., Lin J., Seale P., Spiegelman B. M. (2006) Complementary action of the PGC-1 coactivators in mitochondrial biogenesis and brown fat differentiation. Cell Metab. 3, 333–341 [DOI] [PubMed] [Google Scholar]
- 37. Méndez J., Stillman B. (2000) Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle. Assembly of prereplication complexes in late mitosis. Mol. Cell Biol. 20, 8602–8612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fan Y., Guo Y., Hamblin M., Chang L., Zhang J., Chen Y. E. (2011) Inhibition of gluconeogenic genes by calcium-regulated heat-stable protein 1 via repression of peroxisome proliferator-activated receptor α. J. Biol. Chem. 286, 40584–40594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Giaginis C., Vgenopoulou S., Vielh P., Theocharis S. (2010) MCM proteins as diagnostic and prognostic tumor markers in the clinical setting. Histol. Histopathol. 25, 351–370 [DOI] [PubMed] [Google Scholar]
- 40. Lei M. (2005) The MCM complex. Its role in DNA replication and implications for cancer therapy. Curr. Cancer Drug Targets 5, 365–380 [DOI] [PubMed] [Google Scholar]
- 41. Barnes D. M., Gillett C. E. (1998) Cyclin D1 in breast cancer. Breast Cancer Res. Treat. 52, 1–15 [DOI] [PubMed] [Google Scholar]
- 42. Boyle J. G., Salt I. P., McKay G. A. (2010) Metformin action on AMP-activated protein kinase. A translational research approach to understanding a potential new therapeutic target. Diabet. Med. 27, 1097–1106 [DOI] [PubMed] [Google Scholar]
- 43. Ben Sahra I., Le Marchand-Brustel Y., Tanti J. F., Bost F. (2010) Metformin in cancer therapy. A new perspective for an old antidiabetic drug? Mol. Cancer Ther. 9, 1092–1099 [DOI] [PubMed] [Google Scholar]
- 44. Bost F., Sahra I. B., Le Marchand-Brustel Y., Tanti J. F. (2012) Metformin and cancer therapy. Curr. Opin. Oncol. 24, 103–108 [DOI] [PubMed] [Google Scholar]
- 45. Gallagher E. J., LeRoith D. (2011) Diabetes, cancer, and metformin. Connections of metabolism and cell proliferation. Ann. N.Y. Acad. Sci. 1243, 54–68 [DOI] [PubMed] [Google Scholar]
- 46. Mamputu J. C., Wiernsperger N. F., Renier G. (2003) Antiatherogenic properties of metformin. The experimental evidence. Diabetes Metab. 29, 6S71–76 [DOI] [PubMed] [Google Scholar]
- 47. Sharma R. V., Bhalla R. C. (1995) Metformin attenuates agonist-stimulated calcium transients in vascular smooth muscle cells. Clin. Exp. Hypertens. 17, 913–929 [DOI] [PubMed] [Google Scholar]
- 48. Bell S. P., Dutta A. (2002) DNA replication in eukaryotic cells. Annu. Rev. Biochem. 71, 333–374 [DOI] [PubMed] [Google Scholar]
- 49. Kelly T. J., Brown G. W. (2000) Regulation of chromosome replication. Annu. Rev. Biochem. 69, 829–880 [DOI] [PubMed] [Google Scholar]
- 50. Nishitani H., Lygerou Z. (2004) DNA replication licensing. Front. Biosci. 9, 2115–2132 [DOI] [PubMed] [Google Scholar]
- 51. Kearsey S. E., Labib K. (1998) MCM proteins. Evolution, properties, and role in DNA replication. Biochim. Biophys. Acta 1398, 113–136 [DOI] [PubMed] [Google Scholar]
- 52. Maiorano D., Lutzmann M., Méchali M. (2006) MCM proteins and DNA replication. Curr. Opin. Cell Biol. 18, 130–136 [DOI] [PubMed] [Google Scholar]
- 53. Kikuchi J., Kinoshita I., Shimizu Y., Kikuchi E., Takeda K., Aburatani H., Oizumi S., Konishi J., Kaga K., Matsuno Y., Birrer M. J., Nishimura M., Dosaka-Akita H. (2011) Minichromosome maintenance (MCM) protein 4 as a marker for proliferation and its clinical and clinicopathological significance in non-small cell lung cancer. Lung Cancer 72, 229–237 [DOI] [PubMed] [Google Scholar]
- 54. Liu B., Fan Z., Edgerton S. M., Yang X., Lind S. E., Thor A. D. (2011) Potent anti-proliferative effects of metformin on trastuzumab-resistant breast cancer cells via inhibition of erbB2/IGF-1 receptor interactions. Cell Cycle 10, 2959–2966 [DOI] [PubMed] [Google Scholar]
- 55. Kim H. J., Park K. G., Yoo E. K., Kim Y. H., Kim Y. N., Kim H. S., Kim H. T., Park J. Y., Lee K. U., Jang W. G., Kim J. G., Kim B. W., Lee I. K. (2007) Effects of PGC-1α on TNF-α-induced MCP-1 and VCAM-1 expression and NF-κB activation in human aortic smooth muscle and endothelial cells. Antioxid. Redox Signal. 9, 301–307 [DOI] [PubMed] [Google Scholar]
- 56. Xiong S., Salazar G., San Martin A., Ahmad M., Patrushev N., Hilenski L., Nazarewicz R. R., Ma M., Ushio-Fukai M., Alexander R. W. (2010) PGC-1 α serine 570 phosphorylation and GCN5-mediated acetylation by angiotensin II drive catalase down-regulation and vascular hypertrophy. J. Biol. Chem. 285, 2474–2487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bailey C. J. (2008) Metformin. Effects on micro and macrovascular complications in type 2 diabetes. Cardiovasc. Drugs Ther. 22, 215–224 [DOI] [PubMed] [Google Scholar]