

Background: Calmodulin regulation of Ca2+ channels mediates short term synaptic plasticity.
Results: Binding of CaMKII to CaV2.1 channels induces Ca2+-independent kinase activity.
Conclusion: A complex of CaMKII and CaV2.1 channels is required for short term synaptic plasticity.
Significance: CaMKII bound to CaV2.1 may regulate synaptic plasticity.
Keywords: Calcium, Calcium Channels, Neurotransmitters, Protein Kinases, Protein Phosphorylation, Synapses, Synaptic Plasticity, Nerve Terminal, Signaling Complex
Abstract
Ca2+/calmodulin-dependent protein kinase II (CaMKII) forms a major component of the postsynaptic density where its functions in synaptic plasticity are well established, but its presynaptic actions are poorly defined. Here we show that CaMKII binds directly to the C-terminal domain of CaV2.1 channels. Binding is enhanced by autophosphorylation, and the kinase-channel signaling complex persists after dephosphorylation and removal of the Ca2+/CaM stimulus. Autophosphorylated CaMKII can bind the CaV2.1 channel and synapsin-1 simultaneously. CaMKII binding to CaV2.1 channels induces Ca2+-independent activity of the kinase, which phosphorylates the enzyme itself as well as the neuronal substrate synapsin-1. Facilitation and inactivation of CaV2.1 channels by binding of Ca2+/CaM mediates short term synaptic plasticity in transfected superior cervical ganglion neurons, and these regulatory effects are prevented by a competing peptide and the endogenous brain inhibitor CaMKIIN, which blocks binding of CaMKII to CaV2.1 channels. These results define the functional properties of a signaling complex of CaMKII and CaV2.1 channels in which both binding partners are persistently activated by their association, and they further suggest that this complex is important in presynaptic terminals in regulating protein phosphorylation and short term synaptic plasticity.
Introduction
P/Q-type calcium currents (1) are conducted by voltage-gated Ca2+ channel Cav2.1 (2), which is localized in high density in presynaptic nerve terminals (3, 4) and initiates synaptic transmission at most synapses in the central nervous system (1, 5, 6). The Cav2.1 channel protein consists of a pore-forming α1 subunit associated with auxiliary β, α2δ, and possibly γ subunits (7, 8). The function of Cav2.1 channels is dynamically regulated by interaction with SNARE proteins, G-protein βγ subunits, RIM and other SNARE-interacting proteins, calmodulin (CaM),4 and related calcium sensor proteins (8, 9). Cav2.1 channels form a presynaptic complex that is co-localized with nearly 100 interacting proteins at the active zone, which serves to dock neurotransmitter vesicles, modulate Cav2.1 channel function, and mediate exocytosis (9, 10).
Ca2+/CaM-dependent protein kinase II (CaMKII) is a ubiquitous, multifunctional serine/threonine kinase (11–14). It mediates Ca2+-dependent phosphorylation of a wide range of neuronal targets (15, 16). CaMKII in the brain is composed of dodecamers of 52-kDa α and 60-kDa β subunits (17, 18). Under basal conditions, an autoinhibitory domain binds to the catalytic domain, rendering the kinase inactive. Upon Ca2+ influx, Ca2+/CaM binds to the autoinhibitory domain, relieves the block of kinase activity, and stimulates autophosphorylation of Thr-286 and phosphorylation of other substrates. After phosphorylation of Thr-286, block of catalytic activity is re-established slowly even after the Ca2+ level falls and Ca2+/CaM dissociates from the kinase, which allows CaMKII to integrate signals from trains of Ca2+ transients (19–22). On the postsynaptic side of the synapse, CaMKII phosphorylation regulates glutamate receptors in long term potentiation and has several other well established functions (15, 16).
CaMKII is also present in presynaptic terminals (23, 24), but its presynaptic functions are not well established. Previous studies implicate presynaptic CaMKII in different forms of synaptic plasticity (25–29), including modulation of paired-pulse facilitation (30–32). CaMKII forms a complex with CaV2.1 channels in transfected cells via a site in the proximal C-terminal domain (33). Interaction with CaMKII increases CaV2.1 channel activity and enhances CaV2.1 channel facilitation by slowing inactivation and shifting the voltage dependence of inactivation to more positive membrane potentials (33). These effects of CaMKII did not require the catalytic activity of the kinase, suggesting that binding per se was sufficient for channel regulation (33). Here we report that CaMKII binds directly to a site in the C-terminal domain of CaV2.1 channels and that autophosphorylation of CaMKII stimulates binding to this site. Autophosphorylated CaMKII can bind to the CaV2.1 channel and synapsin-1 simultaneously. Binding of CaV2.1 to CaMKII induces Ca2+-independent kinase activity, which mediates both autophosphorylation and phosphorylation of synapsin-1 at Ser-603. Block of this interaction with competing peptides or the endogenous brain-specific CaMKII inhibitor, CaMKIIN, prevents short term synaptic facilitation and depression. We present a molecular model in which the pore-forming α1 subunit of the Cav2.1 channel serves as a platform for association of CaMKII at the site of influx of Ca2+, where it is persistently activated and poised to phosphorylate synapsin-1 and other nearby substrates to regulate synaptic vesicle dynamics and synaptic plasticity.
EXPERIMENTAL PROCEDURES
Plasmids and Antibodies
CaV2.1(1848–1964)pGEX-4T-2, encoding the C-terminal domain of CaV2.1 channel containing the IQ-like motif in pGEX-4T-2, was amplified via PCR using CaV2.1(1766–2212) as template. The PCR product was cloned into the BamHI/EcoR1 sites of pGEX-4T-2. CaV2.1(1959–2035)pGEX-4T-2, encoding the C-terminal domain of CaV2.1 channel containing the CaM binding domain in pGEX-4T-2, was amplified via PCR using CaV2.1(1766–2212) as template. The PCR product was cloned into the BamHI/EcoR1 sites of pGEX-4T-2. CaV2.1(1848–1964)/EEDAAA, encoding the C-terminal domain of CaV2.1 containing the IQ-like motif, was cloned into BamHI/EcoR1 sites of pGEX-4T-2. The CaMKII tethering site TVGKIY, located upstream of IQ-like containing domain, was mutated to EEDAAA. N-terminal MGALCC lipid anchor + CaV2.1(1766–2211) was amplified via PCR using the CaV2.1 α1 subunit as template. The PCR product was cloned into the BamHI/EcoR1 sites of pCDNA3.1/myc-HisA. Myc-CaV2.1(1764–2211) was generated by subcloning α12.1 C-terminal fragment into CS2 + Myc with six consecutive myc tags (33).
cDNA encoding full-length CaMKII was amplified by PCR, and the product was cloned into the BamH1/EcoR1 sites of the pMALc2X vector. Synapsin-1/pEGFP c-1 encodes full-length synapsin-1 in pEGFP c-1. Syntaxin-1A pGEX-4T-2 encodes syntaxin-1A in pGEX-4T-2 (34). NaV1.2(1848–1964)pGEX-4T-2, encoding a segment of the C-terminal domain of the NaV1.2 channel, was amplified via PCR using full-length NaV1.2 as template. The PCR product was cloned into the BamHI/Xho1 sites of pGEX-4T-2.
The following antibodies were diluted and used as described below: anti-myc, 1:5,000 (monoclonal, Sigma); anti-CaMKII, 1:5,000 (monoclonal, BD Transduction Laboratories); anti-GST, 1:10,000 (monoclonal, Sigma); anti-phospho-CaMKII(Thr-286), 1:10,000 (polyclonal, PhosphoSolutions); anti-CaM, 1:5,000 (monoclonal, Millipore); anti-Hsp 90, 1:10,000 (monoclonal, BD Transduction Laboratories); anti-phosphoSynapsin-1(Ser-603), 1:10,000 (polyclonal, PhosphoSolutions); anti-Synapsin-1, 1:10,000 (polyclonal, PhosphoSolutions).
Culture and Transfection of tsA-201 Cells
TsA-201 cells were plated and grown at the density of 9 × 105 cells/100 mm dish in Hyclone DMEM/F medium and afterward used in experiments (33). The cells were transfected using transit-LT1 (Mirus) and 8 μg of total DNA.
Protein Extraction and Immunoblotting
At least 24 h post transfection, tsA-201 cells were processed to extract recombinant protein. The Petri dishes were placed on ice, and cells were harvested with a rubber scraper and sedimented at 3500 rpm at 4 °C for 20 min. Cells were washed once with 20 ml of ice-cold PBS to remove serum proteins. The cell pellet was resuspended in 500 μl of lysis buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 1 mm EGTA, 1 mm EDTA, 50 mm NaF, 20 mm β-glycerophosphate, 320 mm sucrose, complete protease inhibitor mixture (Roche Diagnostics), HaltTM phosphatase inhibitor mixture (Thermo Scientific), and pipetted up and down 10 times. The preparation was sedimented at 2500 rpm at 4 °C for 5 min to remove the nuclear fraction. The supernatant was collected and sedimented at 14,000 rpm at 4 °C for 30 min. The pellet was washed with 500 μl of lysis buffer and sedimented again at 14,000 rpm at 4 °C for 30 min to obtain pure pellet devoid of contaminants from the previous supernatant fraction. The pellet was resuspended in 20 μl of 2× sample buffer and heated at 50 °C for 5 min. Proteins were resolved on 4–20% SDS-PAGE gels, and immunoblotting was performed with the indicated antibodies.
Co-immunoprecipitation
TsA-201 cells were cultured and transfected as described above. For co-immunoprecipitation studies, the cells were processed as described previously (33).
Recombinant Protein Production
Purified recombinant CaMKII was a kind gift of Dr. Thomas Soderling (Oregon Health and Science University, Portland, OR (35)). GST-tagged CaV2.1(1848–1964), CaV2.1(1959–2035), or GST alone were expressed in Escherichia coli (BL21) in baffled shaker flasks. Isolated single colonies were inoculated and grown overnight in 50 ml of LB medium containing 100 μl of ampicillin (100 mg/ml) at 37 °C and 220 rpm to obtain precultures. Large-scale cultures were prepared using 400 ml of LB containing 800 μl of ampicillin with the addition of 10 ml of overnight precultures. The cells were grown at 37 °C and 220 rpm until the absorbance increased to 1.0 at 600 nm. Protein expression was induced by the addition of 400 μl of 100 mm isopropyl 1-thio-β-d-galactopyranoside for 14 h at 11 °C. Cells were harvested by centrifugation at 3500 rpm, 4 °C for 30 min, and washed 3 times by resuspension in 50 ml of cold PBS. The 15-ml cell suspension (in PBS) was incubated with 500 μl of lysozyme (10 mg/ml) for 1 h at 4 °C with shaking and subsequently sonicated four times at 20-s intervals. Cell-free supernatant was obtained by 2 rounds of centrifugation at 13,000 rpm at 4 °C for 1 h. Glutathione-Sepharose beads (100 μl) were washed 3 times in 50 ml of cold PBS and sedimented at 1500 rpm at 4 °C for 1 min before use. Cell-free supernatant from the earlier step was incubated overnight with glutathione-Sepharose beads. Nonspecific binding was reduced by washing the bound protein six times with cold PBS. The washes were carried out for 30 min at 4 °C on a shaker, and beads/resin were sedimented at 1500 rpm for 1 min at 4 °C. The yield of fusion protein was estimated by Coomassie Blue staining after SDS-PAGE using a calibration curve with bovine serum albumin. These proteins are pure by SDS-PAGE analysis and are native with respect to binding of CaM as expected (Fig. 1).
FIGURE 1.
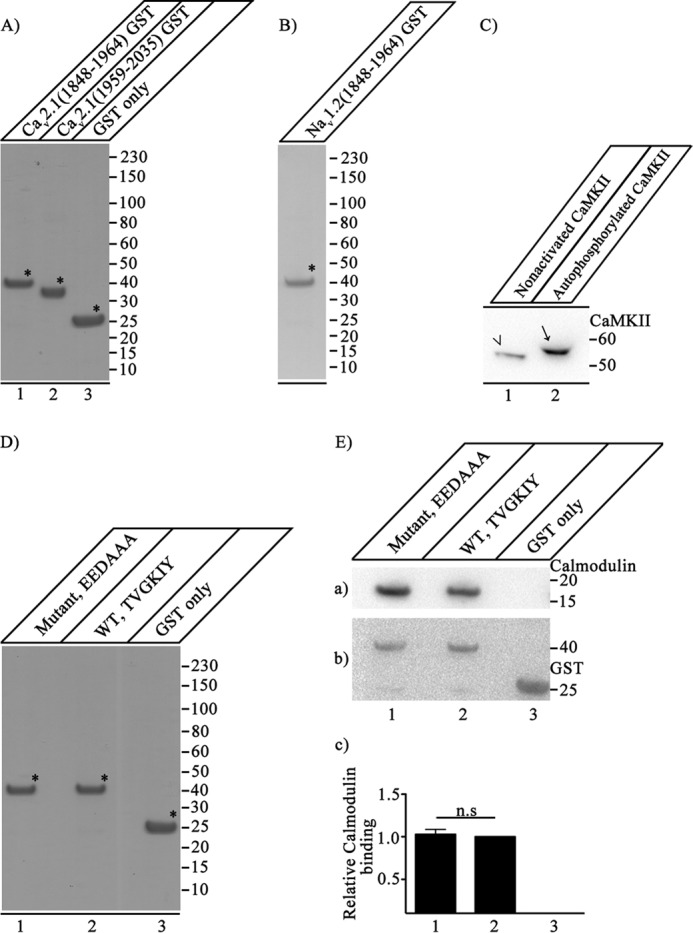
Expression, purification, and function of CaV2.1 and NaV1.2 proteins. A, CaV2.1(1848–1964)-GST (lane 1), CaV2.1(1959–2035)-GST (lane 2), and GST (lane 3) were expressed and purified as described under “Experimental Procedures.” Samples (7.5 μg of purified protein) were denatured, loaded, and resolved on 10% SDS-PAGE. The gel was stained with Coomassie Brilliant Blue to show the purity of the expressed proteins, which were used for in vitro binding assays. B, expression and purification of NaV1.2(1848–1964)-GST. NaV1.2(1848–1964)-GST was expressed and purified as described under “Experimental Procedures.” Samples (7.5 μg of purified protein) were denatured, loaded, and resolved on 10% SDS-PAGE. The gel was stained with Coomassie Brilliant Blue to show the quality of the expressed proteins. C, extent of CaMKII phosphorylation. Nonactivated CaMKII was analyzed side by side with autophosphorylated CaMKII to estimate the extent of CaMKII autophosphorylation by comparison of their migration positions in SDS-PAGE. Autophosphorylation of CaMKII causes an upward shift in the migration of CaMKII (lane 2, arrow) as compared with the nonactivated CaMKII (lane 1, arrowhead) when separated on 4–20% SDS-PAGE. The blot was developed using anti-CaMKII that detects both nonactivated and autophosphorylated CaMKII. Lack of any band corresponding to the nonactivated CaMKII in lane 2 (compared with lane 1) suggests that CaMKII is autophosphorylated to near completion under our experimental conditions. D, CaV2.1(1848–1964)-EEDAAA-GST (lane 1), CaV2.1(1848–1964)-GST (lane 2), and GST (lane 3) were expressed and purified as described under “Experimental Procedures.” Samples (7.5 μg of purified protein) were denatured, loaded, and resolved on 10% SDS-PAGE. The gel was stained with Coomassie Brilliant Blue to show the purity of the expressed proteins, which were used for in vitro binding assays. E, binding of CaV2.1(1848–1964)-EEDAAA, CaV2.1(1848–1964)-GST, and GST alone to CaM. a, the blot was probed with anti-CaM. b, the same blot after stripping and re-probing with anti-GST to show loading of CaV2.1(1848–1964)-EEDAAA (lane 1), CaV2.1(1848–1964)-GST (lane 2), and GST (lane 3). c, quantitation of relative CaM binding under the indicated conditions (mean ± S.E.; n.s., not significant by Student's t test; n = 5). Asterisks mark the primary protein band corresponding to the expressed protein of interest in panels A, B, and D.
For some experiments, CaMKII was expressed with a maltose-binding protein (MBP) epitope tag on its N terminus (36). MBP-CaMKII or MBP alone was expressed in E. coli (BL21) in baffled shaker flasks. Isolated single colonies were inoculated, grown overnight in 50 ml of LB medium containing 100 μl ampicillin (100 mg/ml) at 37 °C and 220 rpm to obtain precultures. Large-scale cultures were prepared using 400 ml of LB containing 800 μl of ampicillin with the addition of 10 ml overnight precultures. The cells were grown at 37 °C and 220 rpm until the absorbance increased to 1.0 at 600 nm. Protein expression was induced by the addition of 400 μl of 100 mm isopropyl 1-thio-β-d-galactopyranoside for 14 h at 11 °C. Cells were harvested by centrifugation at 3500 rpm and 4 °C for 30 min and washed 3 times by resuspension in 50 ml of cold PBS. The 15-ml cell suspension (in PBS) was incubated with 500 μl of lysozyme (10 mg/ml) for 1 h at 4 °C with shaking and subsequently sonicated 4 times at 20-s intervals. Cell-free supernatant was obtained by 2 rounds of centrifugation at 13,000 rpm and 4 °C for 1 h. Amylose resin (100 μl) was washed 3 times in 50 ml of cold PBS and sedimented at 1500 rpm and 4 °C for 1 min before use. Cell-free supernatant from the earlier step was incubated overnight with amylose resin beads. Nonspecific binding was reduced by washing the bound protein six times with cold PBS. The washes were carried out for 30 min at 4 °C on a shaker, and beads/resin were sedimented at 1500 rpm for 1 min at 4 °C. The yield of fusion protein was estimated by Coomassie Blue staining after SDS-PAGE using a calibration curve with bovine serum albumin.
Binding Experiments
Binding of CaV2.1-GST fusion proteins and CaMKII was analyzed by GST pulldown assays using Tris binding buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 0.1% Tween 20, and 0.1% BSA). Autophosphorylation of Thr-286 in purified CaMKII was carried out by incubation on ice for 5 min in Tris binding buffer containing 0.025 mm CaCl2,0.125 μm CaM, 0.025 mm ATP, and 0.125 mm MgCl2 (37). This procedure resulted in essentially complete phosphorylation of CaMKII as judged by its change in mobility in SDS-PAGE (Fig. 1C). The binding reaction was carried out for 1 h at 4 °C. After incubation, washing was performed in binding or washing buffer with the addition of 5 mm EGTA where indicated. The proteins were washed 3 times for 5 min at 4 °C by sedimentation at 500 rpm. The beads were boiled at 95 °C in 10 μl of 4× sample buffer. Proteins were resolved on 10% SDS-PAGE, and immunoblotting was performed. Binding of CaMKII was detected using anti-CaMKII (monoclonal, BD Biosciences). CaMKII phosphorylation at Thr-286 was detected using anti-phospho-CaMKII(Thr-286) (polyclonal, PhosphoSolutions). For re-probing of immunoblots, nitrocellulose membranes were stripped as described (38). Equal bait loading was confirmed by blotting with anti-GST (Sigma). Immunoblots (ECL detection) were documented with Chemidoc XRS (Bio-Rad). Binding was quantified using densitometric measurement of band intensity using NIH ImageJ software.
Expression and Electrophysiological Recording in Cultured Neurons
Superior cervical ganglion (SCG) neurons were cultured as described to allow synapse formation (39). cDNAs encoding α12.1 subunit, eGFP, and (where indicated) CaMKIIN was microinjected into the nuclei of SCG neurons through glass micropipettes with 5% fast-green dye (Sigma). Entry of the injected reagents into the cell nucleus was monitored by the intensity of green dye in the nucleus. The cells were maintained at 37 °C in a 95% air, 5% CO2-humidified incubator for 2–3 days.
Excitatory postsynaptic potentials (EPSPs) were recorded from SCG neurons cultured for 6 weeks as described (39). Injected neurons were identified with an inverted microscope equipped with an epifluorescence unit. Conventional intracellular membrane potential recordings were made from two neighboring neurons using microelectrodes filled with 1 m KAc (70–90 mΩ). Paired action potentials were generated in the injected, presynaptic neuron expressing the α12.1 channels and eGFP by passing 1–2 nA of current for 5 ms through the intracellular recording electrode. EPSPs were recorded from a neighboring non-injected neuron. CaMKIIN was co-expressed with CaV2.1 channels or CaV2.1(1897–1912) was injected 30 min before recordings as indicated. Endogenous synaptic transmission was blocked by bath application of 3 μm ω-conotoxin GVIA in a modified Krebs solution consisting of 136 mm NaCl, 5.9 mm KCl, 1 mm CaCl2, 1.2 mm MgCl2, 11 mm glucose, and 3 mm Na-HEPES, pH 7.4. For recording sub-threshold EPSPs, the membrane potential of the postsynaptic cell was held at −70 or −80 mV by passing current (0.2–0.4 nA) through the recording electrode. Each 30-s recording protocol was repeated 4 times for each inter-stimulus interval, and the ratio of the second EPSP to the first EPSP was averaged for each synapse. Data values with associated error shown in the text and figures represent the mean ± S.E.
RESULTS
CaMKII Binds Directly to the C-terminal Domain of Cav2.1 Channels
As shown in previous studies, we confirmed that CaMKII binds to the C-terminal domain of CaV2.1 channels in transfected cells, as assessed by co-immunoprecipitation (Fig. 2A) (33). However, it was not known whether this binding interaction was direct or required other intermediary proteins. To measure direct interactions between CaMKII and CaV2.1, we investigated binding of CaMKII in vitro to adjacent segments of the C-terminal domain that contain the two components of the calcium sensor protein interaction site: the IQ-like motif (CaV2.1(1848–1964)) and the CaM binding domain (CaV2.1(1959–2035)) (40, 41). We prepared autophosphorylated CaMKII in which Thr-286 was essentially completely phosphorylated (Fig. 1C). We incubated purified preparations of nonactivated, Ca2+/CaM-treated, and autophosphorylated CaMKII with purified GST-tagged CaV2.1(1848–1964), CaV2.1(1959–2035), or GST alone in vitro and detected the complex by GST pulldown assay. Binding of autophosphorylated CaMKII to GST-CaV2.1(1848–1964) (Fig. 2B, lane a3) was substantially greater than binding of nonactivated CaMKII or CaMKII incubated with Ca2+/CaM but without ATP (Fig. 2B, lanes a1 and a2). In contrast, GST-tagged CaV2.1(1959–2035) did not bind any of the three forms of CaMKII (Fig. 2B, lanes a4–a6), and GST itself was also unable to bind CaMKII (Fig. 2B, lanes a7–a9). Quantification of the results revealed that binding of the autophosphorylated kinase to GST-CaV2.1(1848–1964) was ∼8-fold greater than binding to the non-activated or Ca2+/CaM-treated kinase (Fig. 2C). These results demonstrate direct and specific binding of CaMKII to CaV2.1(1848–1964) containing the IQ-like motif.
FIGURE 2.
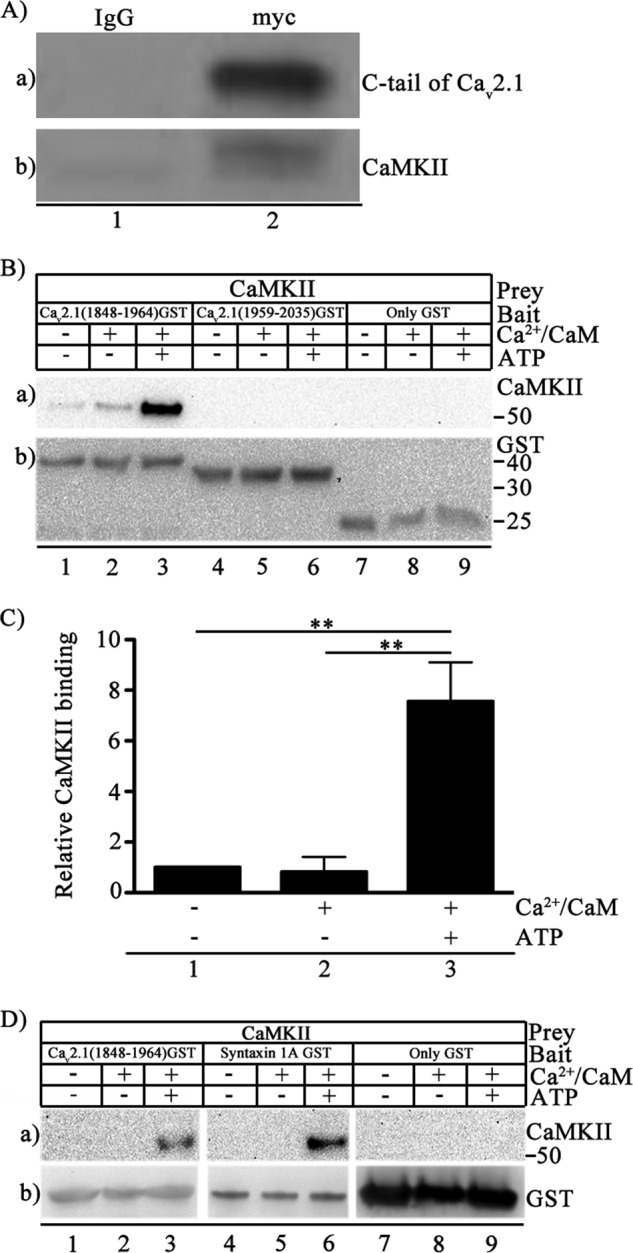
Binding of CaMKII to CaV2.1 channels. A, Myc-tagged Cav2.1(1764–2211) was expressed in tsA-201 cells, and immunoprecipitation was carried out with control IgG (lane 1) or anti-myc antibody (lane 2). a, immunoblotting with anti-myc. b, immunoblotting with anti-CaMKII antibody. B, binding of autophosphorylated CaMKII to Cav2.1 channel. Left to right, CaV2.1(1848–1964), CaV2.1(1959–2035), or GST alone were incubated with Mg2+ (5 mm) and CaMKII (20 nm) in the presence/absence of Ca2+/CaM (5 μm) and ATP (1 mm) as indicated. a, binding of CaMKII was probed using anti-CaMKII. b, the same blot after stripping and re-probing using anti-GST antibody to show equal loading of CaV2.1(1848–1964), CaV2.1(1959–2035), or GST alone. C, quantitation of relative CaMKII binding to CaV2.1(1848–1964) using anti-CaMKII under the indicated conditions (means ± S.E.; **, p < 0.01 by Student's t test; n = 4). D, left to right, CaV2.1(1848–1964) (lanes 1–3), syntaxin 1A (lanes 4–6), or GST (lanes 7–9) were incubated with CaMKII (20 nm) and Mg2+ (5 mm) in the presence/absence of Ca2+/CaM (5 μm) and ATP (1 mm) as indicated. a, binding of CaMKII probed using anti-CaMKII. b, the same blot after stripping and re-probing using anti-GST to show loading of CaV2.1(1848–1964), syntaxin 1A, or GST alone. The section of each blot containing GST-labeled proteins or GST itself was aligned for presentation even though the molecular weights are different.
In similar experiments we used GST-tagged syntaxin 1A as a positive control ligand for specific binding to autophosphorylated CaMKII. Syntaxin 1A is a key component of the SNARE complex that initiates regulated exocytosis, and it binds only autophosphorylated CaMKII (42). We observed CaMKII-syntaxin 1A interaction only when CaMKII was autophosphorylated (Fig. 2D, lanes a4–a6). Autophosphorylated CaMKII also bound specifically to CaV2.1(1848–1964) (Fig. 2D, lanes a1–a3) in a parallel experiment, demonstrating preferential binding of autophosphorylated CaMKII to CaV2.1(1848–1964) as compared with nonactivated or Ca2+/CaM-treated CaMKII. GST alone did not bind non-activated, Ca2+/CaM-treated, or autophosphorylated kinase (Fig. 2D, lanes a7–a9).
Location of the CaMKII Binding Site
CaV2.1(1848–1964) contains an IQ-like motif as part of a bipartite regulatory site that is important for CaM-induced facilitation and inactivation of CaV2.1 channels (9). IQ-like motifs bind CaM, which could potentially serve as a docking site for CaMKII (43). On the other hand, CaM-independent docking of CaMKII to cardiac CaV1.2 channels is well described, suggesting that CaM is not required for CaMKII binding to Ca2+ channels (37). Sequence alignment of the C-terminal domains of Cav2.1 and CaV1.2 channels revealed a conserved amino acid sequence motif in CaV2.1(1848–1964): TVGKFY in CaV1.2 channels versus TVGKIY in CaV2.1 channels (37). Peptides containing this motif prevented CaMKII regulation of CaV2.1 channels, suggesting an important role in CaMKII binding (33); however, intact CaV2.1 channels with this sequence mutated were inactive. To ascertain the role of the TVGKIY motif in binding to CaV2.1(1848–1964), we substituted the sequence EEDAAA used previously (37) for TVGKIY and tested CaMKII binding. Autophosphorylated CaMKII bound specifically to WT CaV2.1(1848–1964) (Fig. 3A, lane a4), but its binding to CaV2.1(1848–1964)/EEDAAA was significantly reduced to 0.42 ± 0.02 of WT when quantified using phospho-CaMKII(Thr-286)-specific antibody (Fig. 3A, lane a5; Fig. 3B, p < 0.01). Autophosphorylated CaMKII did not bind to the GST control (Fig. 3A, lane a1), and non-activated CaMKII did not bind to either CaV2.1(1848–1964)/TGVKIY or CaV2.1(1848–1964)/EEDAAA (Fig. 3A, lanes a2 and a3), confirming the specificity of binding to phospho-CaMKII(Thr-286). These results indicate that the TGYKIY motif forms an important part of the binding site for CaMKII, but other nearby sequence elements must also contribute substantially to kinase binding when this sequence is mutated.
FIGURE 3.
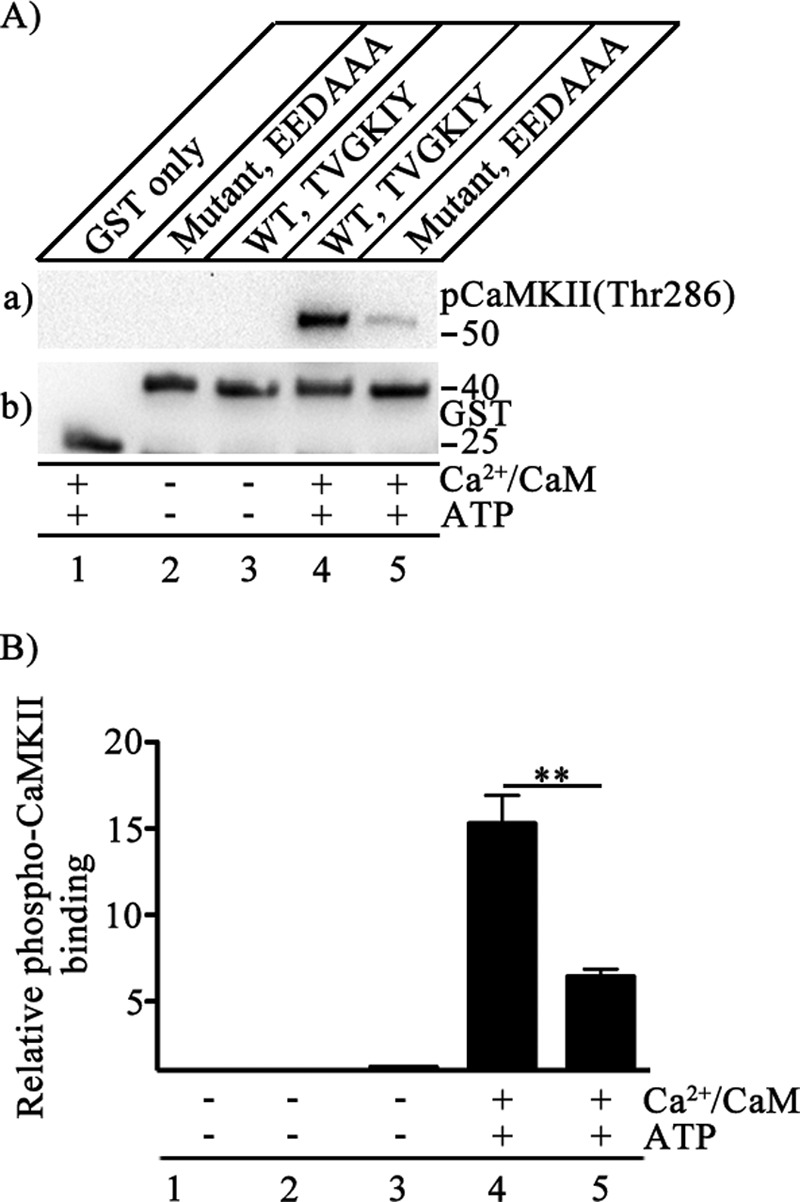
Binding site for CaMKII on CaV2.1 channels. A, binding of CaMKII was detected by GST pulldown assay. CaMKII (20 nm) was used as prey in the presence/absence of Ca2+/CaM (5 μm) and ATP (1 mm) as indicated. GST alone or GST-tagged CaV2.1(1848–1964) was used as bait as indicated: WT, 1903TVGKIY1908; mutant, 1903EEDAAA1908. a, the blot was probed with anti-phospho-CaMKII(Thr-286). b, the same blot after stripping and re-probing using anti-GST to show equal loading of CaV2.1(1848–1964). B, quantitation of relative CaMKII autophosphorylation using anti-phospho CaMKII(Thr-286) under the indicated conditions is shown (mean ± S.E.; **, p < 0.01 by Student's t test; n = 6).
Binding of Autophosphorylated CaMKII Persists after Ca2+/CaM Dissociation
Binding of CaMKII to CaV1.2 and CaV2.1 channels modulates their function (33, 37). The local Ca2+ concentration at the active zone is tightly regulated to create a Ca2+ nanodomain (9). Because of its slowly reversible autophosphorylation, activation of CaMKII can integrate repetitive Ca2+ signals and serve as a molecular memory of synaptic activity (15). Therefore, it is important to test the persistence of the kinase-channel interaction as the level of Ca2+ declines. To address this question, we chelated Ca2+ using 5 mm EGTA in the washing buffers. CaMKII was isolated by binding to CaV2.1(1848–1964) immobilized to glutathione-Sepharose beads, and the binding of CaMKII was analyzed in the absence or presence of 5 mm EGTA in the washing buffer (Fig. 4A). CaMKII is highly phosphorylated in the presence of Ca2+/CaM and ATP. Autophosphorylated CaMKII bound more effectively to CaV2.1(1848–1964) (Fig. 4A, lane a3) than nonactivated or Ca2+/CaM-treated CaMKII (Fig. 4A, lanes a1 and a2). Both Ca2+/CaM and ATP are essential components in triggering the autophosphorylation reaction, and omission of ATP followed by chelation of Ca2+ using EGTA (5 mm) in the washing reaction reduced binding significantly (Fig. 4A, lane a4). However, binding of the autophosphorylated kinase persisted even after Ca2+/CaM was removed from the kinase-channel complex by washes with EGTA-containing washing buffer (Fig. 4A, lane a5), suggesting that Ca2+/CaM removal does not rapidly reverse binding of autophosphorylated CaMKII. The efficacy of chelation of Ca2+/CaM by EGTA treatment was probed using anti-CaM. No bound CaM was detected after EGTA treatment (Fig. 4A, lanes c4–c5). We quantified the relative CaMKII binding in each of these conditions and found significant retention of bound CaMKII after EGTA treatment (Fig. 4B). Collectively, these results show that binding of CaMKII to CaV2.1(1848–1964) is greatly enhanced by Ca2+/CaM-dependent autophosphorylation of CaMKII, but once formed, the complex persists even after removal of Ca2+/CaM.
FIGURE 4.
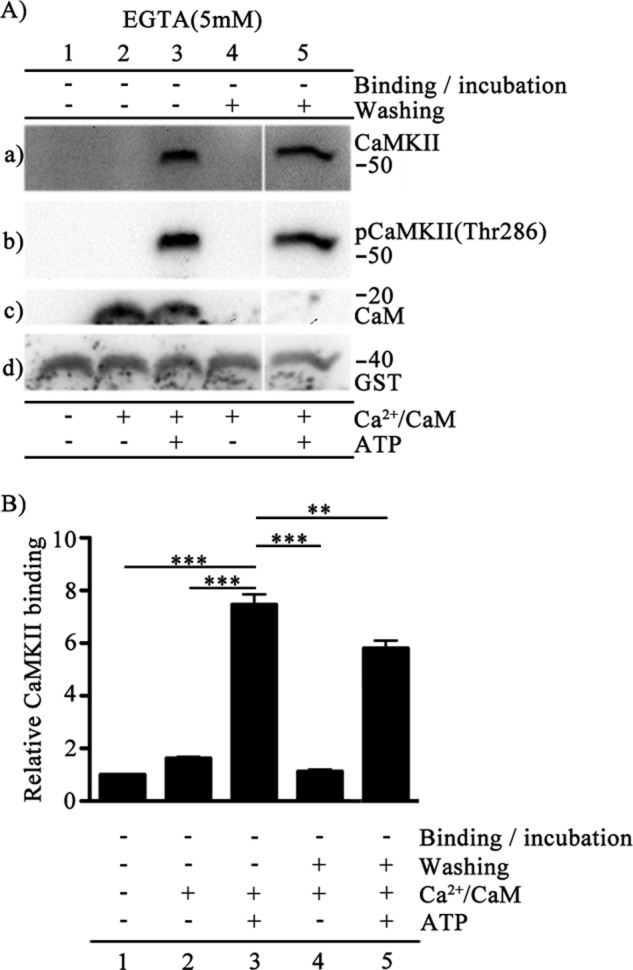
Persistence of binding of autophosphorylated CaMKII to CaV2.1(1848–1964). A, binding of CaMKII was detected by GST pulldown assay. CaV2.1(1848–1964) was incubated with CaMKII (20 nm) and Mg2+(5 mm) in the presence/absence of Ca/CaM (5 μm) and ATP (1 mm) as indicated. Samples were washed in the absence or presence of 5 mm EGTA as indicated. a, the blot was probed with anti-CaMKII antibody. b, the same blot after stripping and re-probing using anti-phospho-CaMKII(Thr-286). c, the same blot after stripping and reprobing using anti-CaM. d, the same blot after stripping and re-probing using anti-GST to show equal loading of CaV2.1(1848–1964). B, quantitation of relative CaMKII binding using anti-CaMKII under the indicated conditions (mean ± S.E.; **, p < 0.01; ***, p < 0.001 by Student's t test; n = 4).
Dephosphorylation of CaMKII Does Not Reverse Binding
To test whether autophosphorylation is required to maintain binding of CaMKII to CaV2.1(1848–1964), we dephosphorylated the channel-bound CaMKII and tested the fate of kinase-channel interaction. Protein phosphatases PP1, PP2A, PP2B, and PP2C represent the majority of serine/threonine phosphatase activity in brain and other tissues (44). CaMKII in isolated postsynaptic densities was mostly dephosphorylated by PP1 (44). Autophosphorylated CaMKII preferentially bound to CaV2.1(1848–1964) (Fig. 5A, lane a1). Dephosphorylation of autophosphorylated CaMKII by PP1 treatment before incubation of CaMKII with the channel peptide or both before and after incubation greatly reduced its binding (Fig. 5A, lane a2 and a3), confirming that autophosphorylation is prerequisite for CaMKII binding to the channel. The remaining faint signal in the absence of autophosphorylation is in accordance with our earlier finding of a low level of binding of nonactivated or Ca2+/CaM-treated CaMKII to the channel (Fig. 2B). Surprisingly, dephosphorylation of autophosphorylated CaMKII by post-PP1 treatment did not destabilize its binding (Fig. 5A, lane a4). Evidently, once kinase is bound to the channel, dephosphorylation does not reverse its binding, even when dephosphorylation is essentially complete (Fig. 5A, lanes b2–b5). Combining post-PP1 treatment and washing the kinase-channel complex with EGTA-containing buffer also did not reverse CaMKII binding (Fig. 5A, lane a5). Together these results show that once the autophosphorylated CaMKII is bound to CaV2.1(1848–1964), the interaction cannot be rapidly reversed by dephosphorylation (Fig. 5A, lane a4), Ca2+/CaM removal (Fig. 5A, lane a6), or both processes carried out simultaneously in vitro (Fig. 5A, lane a5). GST alone does not have any effect (Fig. 5B), further strengthening evidence for the specificity of these interactions.
FIGURE 5.
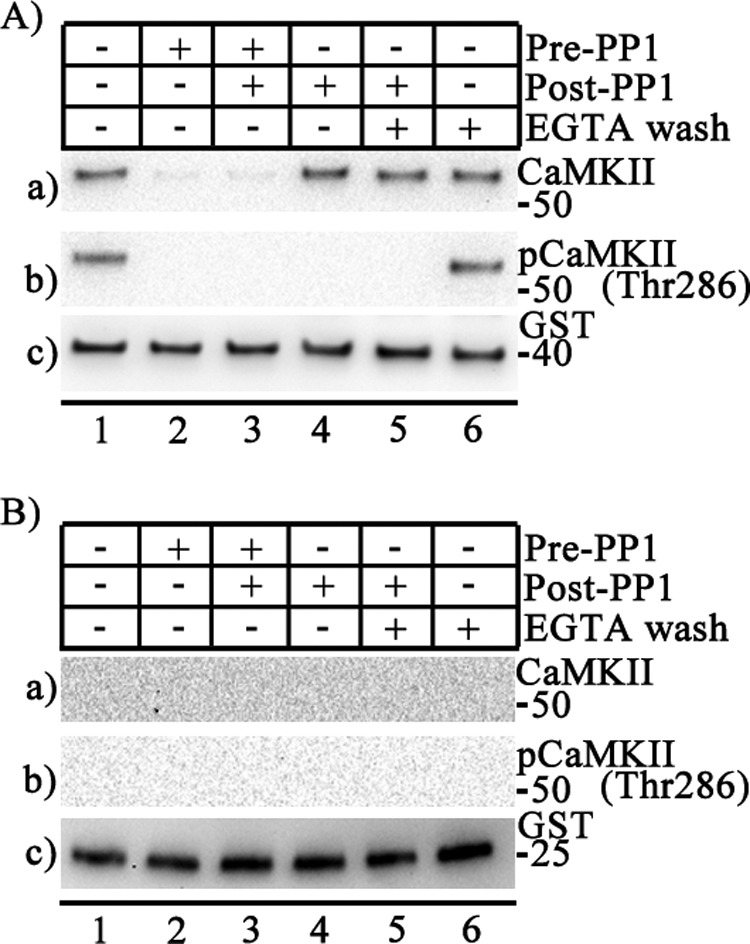
Persistence of binding of CaMKII after dephosphorylation. Purified recombinant protein phosphatase 1 (PP1) was added before (Pre-PP1) or after (Post-PP1) the kinase-channel complex was established as indicated. Washes were carried out in the presence or absence of 5 mm EGTA as indicated. A, binding of CaMKII to CaV2.1(1848–1964) GST was detected by GST pulldown assay. a, the blot was probed with anti-CaMKII. b, the same blot is shown after stripping and re-probing using anti-phospho-CaMKII(Thr-286). c, the same blot after stripping and re-probing using anti-GST to show equal loading of CaV2.1(1848–1964). B, binding of CaMKII to GST alone (control) was detected by GST pulldown assay. a, the blot was probed with anti-CaMKII. b, the same blot after stripping and re-probing using anti-phospho-CaMKII(Thr-286). c, the same blot after stripping and re-probing using anti-GST to show equal loading of GST.
Increased Autophosphorylation of CaMKII Bound to CaV2.1(1848–1964)
Binding of CaMKII to its target sites on NMDA-type glutamate receptors and KV11 channels causes Ca2+-independent activation of the kinase (45, 46). Although there is no detectable amino acid sequence similarity between CaV2.1 channels, KV11 channels, and glutamate receptors, expression of the C-terminal tail of CaV2.1 channels, CaV2.1(1766–2212), without the pore domain leads to enhancement of autophosphorylation of Thr-286 on endogenous CaMKII in human embryonic kidney tsA-201 cells (Fig. 6A, lane a2). These results suggest that binding of CaMKII to CaV2.1 channels per se may be sufficient to activate the kinase and increase its autophosphorylation substantially. The addition of ionomycin, which increases entry of Ca2+ into the cytosol, resulted in a comparable increase in CaMKII autophosphorylation on Thr-286 (Fig. 6A, lane a3). Increased cytosolic Ca2+ activates the Ca2+-dependent phosphoprotein phosphatase calcineurin (47), which might limit the level of autophosphorylation induced by ionomycin. However, in the presence of ionomycin (5 μm) and the calcineurin inhibitor cyclosporin A (5 μm), there was little increase in autophosphorylation compared with ionomycin alone (Fig. 6A, lane a4), suggesting that calcineurin does not play a major role in control of CaMKII phosphorylation in tsA-201 cells. Quantification of these results revealed an ∼6-fold increase in autophosphorylation upon transfection of CaV2.1(1766–2212) or treatment with ionomycin plus cyclosporin A (Fig. 6B).
FIGURE 6.
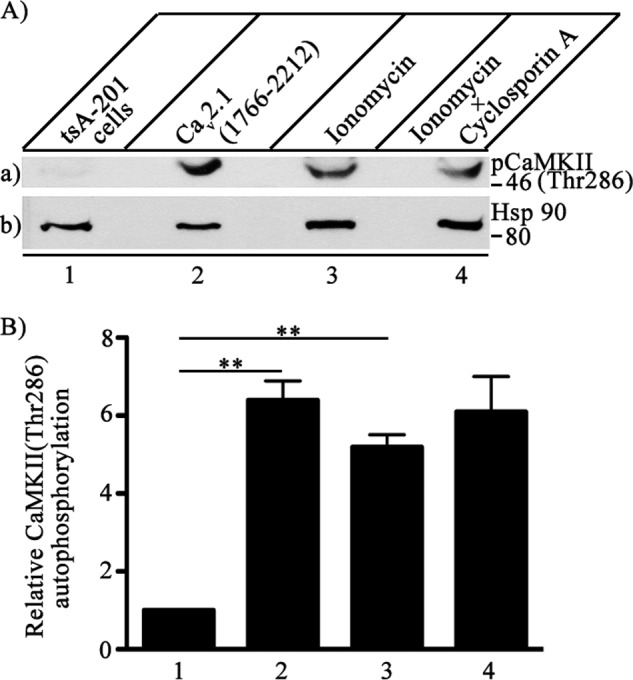
Activation of autophosphorylation of CaMKII by binding to CaV2.1 channels. CaV2.1(1766–2122) was expressed in tsA-201 cells, and autophosphorylation of endogenous CaMKII was measured with anti- phospho-CaMKII(Thr-286). A, left to right, untreated control tsA-201 (lane 1), cells transfected with C-terminal domain of Cav2.1 (1766–2212) (lane 2), cells treated with 5 μm ionomycin to allow Ca2+ entry for 15 min before lysis (lane 3), and cells treated with 5 μm ionomycin and additionally with cyclosporin A to inhibit the protein phosphatase calcineurin (lane 4). a, autophosphorylation of CaMKII was assayed by immunoblotting with anti- phospho-CaMKII(Thr-286). b, the same blot after stripping and re-probing with anti-Hsp90 to show equal loading of cell lysates. B, quantitation of relative CaMKII autophosphorylation using anti-phospho-CaMKII(Thr-286) under the indicated conditions (means ± S.E.; **, p < 0.01 by Student's t test; n = 3).
To extend these findings of increased autophosphorylation of CaMKII bound to Cav2.1 channels in transfected tsA-201 cells to pure proteins, we reconstituted the kinase-channel dimeric complex in vitro and measured autophosphorylation. We activated CaMKII in the presence of Ca2+/CaM, ATP, and Mg2+ and studied the autophosphorylation levels in the absence or presence of the CaV2.1(1848–1964) peptide (Fig. 7A). Autophosphorylation of CaMKII increased in the presence of increasing concentrations of the Cav2.1 channel peptide (Fig. 7Aa, lanes 1–4) and then decreased at higher concentrations of the peptide (Fig. 7Aa, lanes 4–6). Quantification of the levels of CaMKII autophosphorylation using anti-phospho-CaMKII(Thr-286) (Fig. 7B) showed that direct binding of CaV2.1(1848–1964) in vitro mimics the activation of kinase autophosphorylation observed in transfected tsA-201 cells. These results confirm that direct binding of CaV2.1(1848–1964) activates CaMKII autophosphorylation. As negative controls, we used a GST-tagged protein of the same size from the C-terminal of Nav1.2 channels (Fig. 7, C and D) or GST itself (data not shown), and we found no effect on CaMKII activity, further supporting the specificity of this interaction.
FIGURE 7.
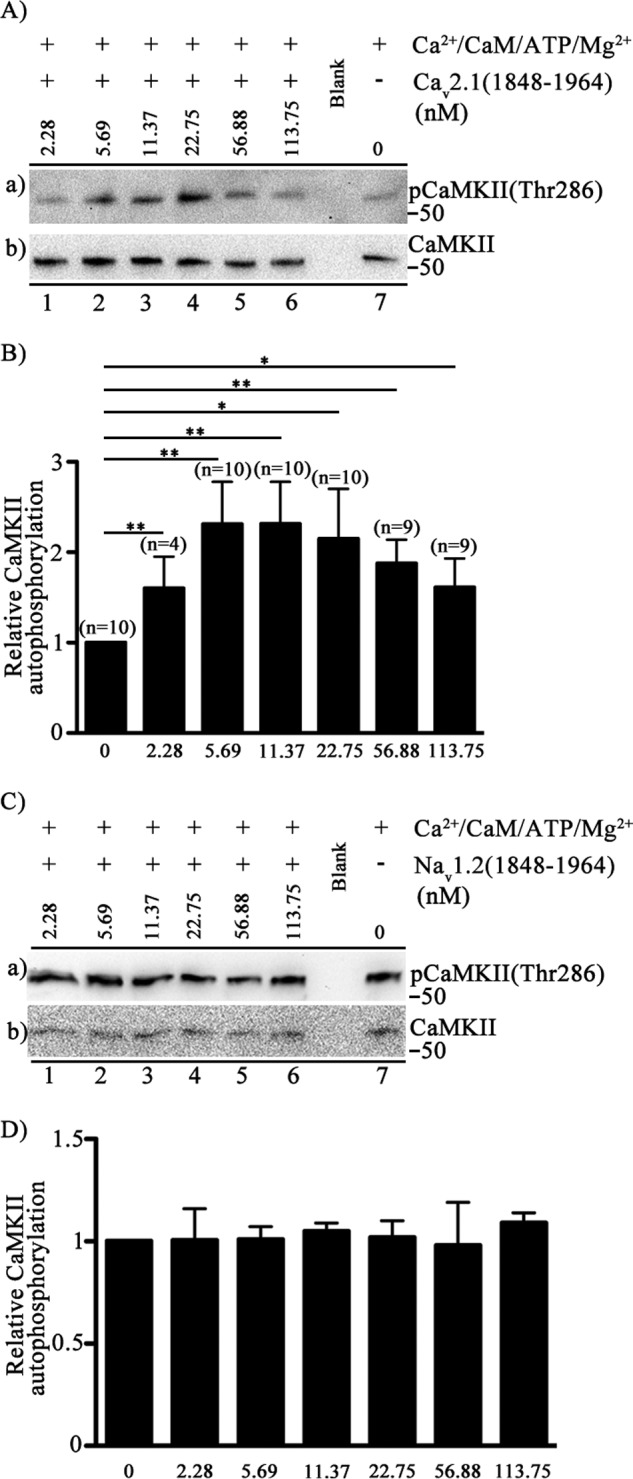
Activation of autophosphorylation of CaMKII by binding of CaV2.1(1848–1964). A, autophosphorylation of CaMKII was carried out by incubation in the presence of Ca2+ (0.6 μm), CaM (2.2 μm), ATP (32 μm), Mg2+(2.3 mm), CaMKII (20 nm) and the indicated concentrations (0–113.75 nm) of Cav2.1 (1848–1964). Free Ca2+ was precisely controlled. Left to right (lanes 1–6), the concentration of CaV2.1(1848–1964) channel peptide ranging from 2.28 nm to 113.75 nm. Extreme right (lane 7), the control in the absence of CaV2.1(1848–1964). a, the blot was probed with anti-phospho-CaMKII(Thr-286). b, the same blot after stripping and re-probing using anti-CaMKII. B, quantitation of relative CaMKII autophosphorylation using anti-phospho-CaMKII(Thr-286) under the indicated conditions (mean ± S.E.; *, p < 0.05; **, p < 0.01 by Student's t test). C, CaMKII autophosphorylation in the presence of NaV1.2(1848–1964)-GST is shown. Autophosphorylation of CaMKII was carried out by incubation in the presence of Ca2+ (0.6 μm), CaM (2.2 μm), ATP (32 μm), Mg2+(2.3 mm), CaMKII (20 nm), and the indicated concentrations (0–113.75 nm) of NaV1.2(1848–1964). Free Ca2+ was precisely controlled. Left to right (lanes 1–6), concentration of NaV1.2(1848–1964) channel peptide ranging from 0 to 113.75 nm. Extreme right (lane 7), control in the absence of NaV1.2(1848–1964). a, the blot was probed with anti- phospho-CaMKII(Thr-286). b, the same blot after stripping and re-probing using anti-CaMKII. D, the quantification of relative CaMKII autophosphorylation using anti-phospho- CaMKII(Thr-286) under the indicated conditions (mean ± S.E.; not significant by Student's t test; n = 3).
Phosphorylation of Synapsin-1 by CaMKII Bound to CaV2.1(1766–2212)
Synapsin-1 is a major presynaptic phosphoprotein that is a prominent substrate for CaMKII, and phosphorylation by CaMKII regulates the effects of synapsin-1 on synaptic vesicle trafficking (23). Phosphorylation of synapsin-1 by CaMKII substantially increases synaptic transmission at the squid giant synapse (28, 29). Expression of CaV2.1(1766–2212) with synapsin-1 in tsA-201 cells led to a substantial increase in synapsin-1 phosphorylation at Ser-603 (Fig. 8A, lane a3) compared with untransfected tsA-201 cells (Fig. 8A, lane a1) or cells expressing synapsin-1 alone (Fig. 8A, lane a2). Increasing the cytosolic Ca2+ concentration with ionomycin, which triggers CaMKII autophosphorylation, also led to a significant increase in synapsin-1 phosphorylation at Ser-603 (Fig. 8, lane a4), and these levels are comparable to those observed when synapsin-1 is coexpressed with CaV2.1(1766–2212). Ionomycin treatment of tsA-201 cells co-expressing CaV2.1(1766–2212), and synapsin-1 shows further enhancement in synapsin-1 phosphorylation (Fig. 8, lane a5). These results indicate that the C-terminal domain of CaV2.1 channels stimulates activation and autophosphorylation of CaMKII as effectively as Ca2+/CaM, and this leads to phosphorylation of synapsin-1 at Ser-603 and potentially to phosphorylation of other presynaptic substrates.
FIGURE 8.
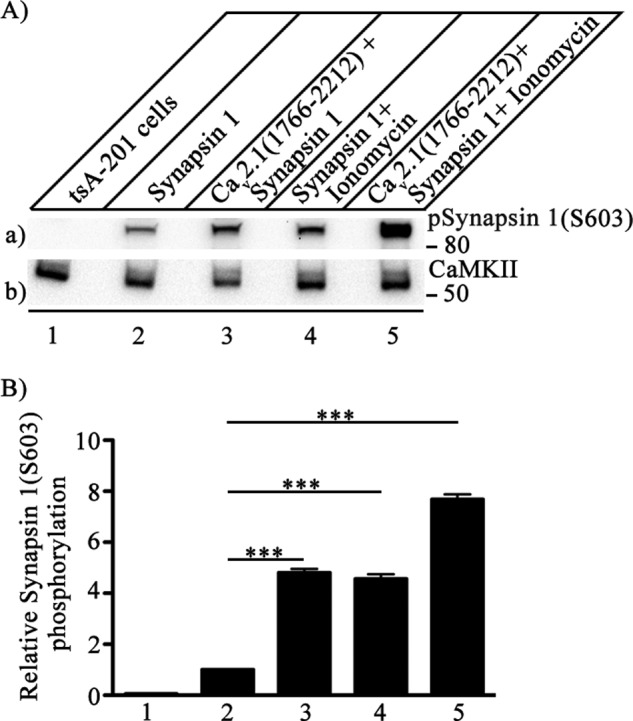
Phosphorylation of synapsin-1 by CaMKII bound to the C-terminal domain of Cav2.1 channels. CaV2.1(1766–2122) was co-expressed in tsA-201 cells along with synapsin-1, and phosphorylation of synapsin-1 was measured with anti-phosphosynapsin-1(Ser-603). A, left to right, untreated control tsA-201 cells (lane 1), cells single-transfected with synapsin-1 (lane 2), cells double-transfected with C-terminal domain of Cav2.1 (1766–2212) channel and synapsin-1 (lane 3), cells single-transfected with synapsin-1 and treated with 5 μm ionomycin to allow Ca2+ entry for 15 min before lysis (lane 4), and cells double-transfected with C-terminal domain of Cav2.1 (1766–2212) channel and synapsin-1 additionally treated with 5 μm ionomycin to allow Ca2+ entry for 15 min before lysis (lane 5). a, phosphorylation of synapsin-1 was assayed by immunoblotting with anti-phosphosynapsin-1(Ser-603) antibody. b, the same blot after stripping and re-probing with anti-CaMKII antibody to show equal loading of CaMKII. B, quantitation of relative synapsin-1(Ser-603) phosphorylation under the indicated conditions (mean ± S.E.; ***, p < 0.001 by Student's t test; n = 5).
If binding of CaV2.1 to CaMKII can lead to phosphorylation of synapsin-1, a stable ternary complex of CaV2.1 and synapsin-1 bound to CaMKII may be formed. To test this possibility we expressed CaMKII in bacteria with a MBP epitope tag and purified the resulting fusion protein. MBP-CaMKII attached to amylose resin was able to bind both CaV2.1(1848–1964) and synapsin-1 simultaneously (Fig. 9), whereas control experiments with MBP showed no binding (Fig. 9B). Formation of this ternary complex in presynaptic terminals would allow local phosphorylation of synapsin-1 by CaMKII bound to CaV2.1 channels to modulate the dynamics of synaptic vesicle function in active zones containing these proteins. As CaMKII is a dodecamer, this ternary complex may be formed within a single subunit or may reflect binding of CaV2.1 and synapsin-1 to different subunits in the CaMKII complex.
FIGURE 9.
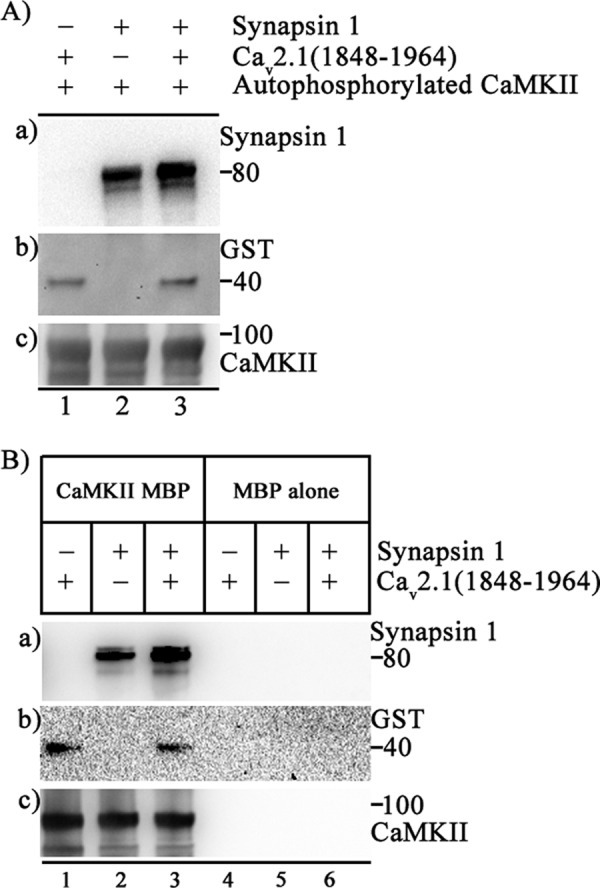
Formation of a ternary complex of CaV2.1(1848–1964), CaMKII, and synapsin-1. CaMKII was expressed as a MBP fusion protein and immobilized on amylose resin. A, incubations were carried out as depicted in the figure. a, binding of synapsin-1 was detected using anti-synapsin-1 antibody (PhosphoSolutions, polyclonal, 1;5000 TBST). b, shown is the same blot as a but after stripping and reprobing with anti-GST to show the binding of CaV2.1(1848–1964). c is the same blot as a and b but after stripping and re-probing with anti-CaMKII (Invitrogen, monoclonal, 1:1000 in TBST) to show the CaMKII bait. B, incubations were carried out as depicted in figure. a, binding of synapsin-1 was detected using anti-synapsin-1 antibody (PhosphoSolutions, polyclonal, 1;5000 TBST). b, shown is the same as a but after stripping and reprobing with anti-GST to show the binding of CaV2.1(1848–1964), and c is same blot as a and b but after stripping and reprobing with anti-CaMKII (Invitrogen, monoclonal, 1:1000 in TBST) to show the CaMKII bait.
Functional Role of Interaction of CaMKII with CaV2.1 Channels in Synaptic Plasticity
Binding of CaMKII to CaV2.1 channels enhances their functional activity by inhibiting their inactivation (33) and enhances the activity of CaMKII by increasing its autophosphorylation and its phosphorylation of other substrates as shown above. To critically test the potential effects of this specific interaction on synaptic transmission, it is necessary to manipulate the activity of CaMKII bound specifically to CaV2.1 channels in the presynaptic terminal without altering the functional activity of CaMKII in the postsynaptic compartment or CaMKII in other locations in the presynaptic terminal. Accordingly, we expressed CaV2.1 channels in SCG neurons in cell culture by microinjection of cDNA into the nucleus of a single cell using methods that were well defined in previous work (42, 48, 49). After 2–3 days, we impaled the injected cell and a nearby uninjected postsynaptic partner, and we measured synaptic transmission driven exclusively by the transfected CaV2.1 channels by blocking endogenous N-type Ca2+ currents with ω-conotoxin GVIA. We studied paired-pulse facilitation as a measure of short term synaptic plasticity. In this experimental paradigm, the synaptic response to the second stimulus in the pair is enhanced by residual Ca2+ remaining in the nerve terminal from the first stimulus (9). Paired-pulse facilitation of synaptic transmission in this transfected SCG neuron preparation is primarily caused by facilitation of CaV2.1 channel activity by Ca2+/CaM binding to the Ca2+ sensor protein binding site in the C-terminal domain (48). As illustrated in Fig. 10A, CaV2.1 channels expressed alone generate synaptic transmission in which the paired-pulse ratio is highly dependent on the inter-stimulus interval (ISI) between the paired pulses. At short ISI, synaptic depression is dominant, and paired-pulse ratio values are less than 1.0. At longer ISI, synaptic facilitation becomes dominant, peaks at ∼1.75 for an ISI of 80 ms, and declines to 1.0 at long ISI (Fig. 10A). Perfusion of a competing peptide that blocks the interaction of CaMKII with CaV2.1 channels (CaV2.1(1897–1912)) prevented both paired-pulse facilitation and paired-pulse depression at this model synapse (Fig. 10A), suggesting that binding of CaMKII to CaV2.1 channels is required for expression of this regulatory effect. Similarly, expression of the brain-specific CaMKII inhibitor CaMKIIN (50), which prevents CaMKII binding to CaV2.1 channels (33), also prevented paired-pulse facilitation and depression (Fig. 10A). This is consistent with previous results showing that facilitation of CaV2.1 channels expressed in tsA-201 cells also requires binding of CaMKII (33). It is unlikely that the basal release probability is affected by competing peptide injection or CaMKIIN expression because the mean amplitudes of the first EPSPs are unchanged (Fig. 10B). Because the competing peptide CaV2.1(1897–1912) applied acutely through the recording pipette and CaMKIIN expressed from cDNA both reduce paired-pulse facilitation of synaptic transmission, as expected from their inhibition of paired-pulse facilitation of CaV2.1 current, these results support the conclusion that these are specific effects. To further support the specificity of action of these agents, we applied CaV2.1(1897–1912) through the recording pipette and CaMKIIN by expression of injected cDNA and analyzed their effects on synaptic transmission initiated by endogenous CaV2.2 channels. We found no effect of either of these agents (Fig. 10, C and D), further supporting the specificity of their effects on facilitation of CaV2.1 currents and neurotransmission initiated by CaV2.1 channels. Evidently, binding of CaMKII by CaV2.1 channels is required for both up-regulation of channel activity in paired pulses and for Ca2+-independent activation of CaMKII by CaV2.1, and one or both of these effects is necessary for normal short term synaptic plasticity.
FIGURE 10.

Binding of CaMKII to CaV2.1 channels is required for short term synaptic plasticity. A, SCG neurons were cultured and injected with cDNA encoding CaV2.1 channels as described under “Experimental Procedures.” Where indicated, CaMKIIN was co-expressed with CaV2.1 channels, or CaV2.1(1897–1912) was injected through a whole-cell patch electrode 30 min before recording as described (39). Sharp microelectrode impalements were made in the previously transfected, presynaptic neuron and a neighboring, synaptically connected neuron. Action potentials were generated in the presynaptic cell, and EPSPs were recorded in the postsynaptic cell and analyzed as described under “Experimental Procedures.” The paired-pulse ratio (PPR) was plotted against inter-stimulus interval (mean ± S.E.; *, < 0.05, Student's t test, n = 6–9). WT, green; CaMKIIN, blue; CaV2.1(1897–1912), orange. B, amplitudes of the first EPSP in paired pulse experiments are shown. C, a similar experiment to that described in panel A was carried out with CaV2.1(1897–1912) injected through a whole-cell patch electrode in untransfected SCG neurons, and paired-pulse facilitation of neurotransmission initiated by the endogenous CaV2.2 channels was measured in the absence of ω-conotoxin GVIA. D, a similar experiment to that described in panel A was carried out with expression of CaMKIIN in untransfected SCG neurons, and paired-pulse facilitation of neurotransmission initiated by the endogenous CaV2.2 channels was measured in the absence of ω-conotoxin GVIA. Normalized paired-pulse ratios of control and CaMKIIN-expressing neurons are plotted.
DISCUSSION
CaMKII Binds Directly to CaV2.1 Channels
Previous studies showed that CaMKII can be co-immunoprecipitated with CaV2.1 channels from transfected cells and that binding of CaMKII per se was sufficient for up-regulation of CaV2.1 channel activity in transfected cells and neurons (33), but no evidence was provided for direct interaction of these two key Ca2+-signaling proteins. Our results show that CaMKII does indeed bind directly to the C-terminal domain of CaV2.1 channels at an interaction site located in CaV2.1(1848–1964). Autophosphorylation enhances binding of CaMKII, and autophosphorylated CaMKII remains bound to the C-terminal domain of CaV2.1 channels even after dephosphorylation and removal of the original Ca2+/CaM stimulus for binding. Thus, CaV2.1 channels with bound CaMKII are likely to serve as a signaling complex in the presynaptic active zone in neurons.
Binding to CaV2.1 Channels Induces Sustained Ca2+-independent Activity of CaMKII
CaMKII activation normally requires binding of Ca2+/CaM, but previous studies have demonstrated Ca2+/CaM-independent activation of CaMKII by interaction with NMDA-type glutamate receptors and KV11 channels (45, 46). Our results show that binding of CaMKII to CaV2.1 channels persistently activates its catalytic activity, as measured by autophosphorylation. CaMKII remains activated even after removal of the Ca2+/CaM stimulus. This is a provocative result, as it implies that CaMKII bound to CaV2.1 channels is poised to phosphorylate nearby substrates and thereby regulate synaptic transmission locally.
It is interesting to compare the regulatory effects of CaM and CaMKII on CaV2.1 channels and CaV1.2 channels. These two types of Ca2+ channels are less than 50% identical in amino acid sequence and serve different physiological roles: CaV2.1 in initiation of synaptic transmission versus CaV1.2 in initiation of excitation-contraction coupling in muscle and in postsynaptic regulation in neurons (8). Although both proteins bind CaM and CaMKII to nearby sites in their C-terminal domains, the regulatory consequences are quite different. Binding of Ca2+/CaM to CaV2.1 channels causes facilitation followed by inactivation, whereas binding to CaV1.2 channels causes only Ca2+-dependent inactivation (9, 51). Binding of CaMKII to CaV2.1 channels enhances their activity and their facilitation, whereas phosphorylation of CaV1.2 channels by CaMKII bound to the C-terminal domain and/or the CaVβ2 subunit is required for facilitation of their activity (9, 37, 52, 53). The structural and mechanistic basis for this differential regulation of CaV1.2 and CaV2.1 channels by CaM and CaMKII bound to nearby sites in their C-terminal domains is an interesting area for further research.
CaMKII Bound to CaV2.1 Channels Phosphorylates Synapsin-1
Synapsin-1 is abundant in presynaptic terminals, where it tethers synaptic vesicles to the actin cytoskeleton and is required for normal replenishment of synaptic vesicles during periods of high synaptic activity (54). Actin surrounds clusters of synaptic vesicles in presynaptic terminals and concentrates synapsin-1 at vesicle clusters (55). It also plays an important role in the dynamics of synaptic vesicle transfer to the readily releasable pool that is poised for rapid exocytosis, and both synapsin-1 and CaM are involved in those processes (55–57). Synapsin-1 is phosphorylated at Ser-603, which regulates trafficking of synaptic vesicles in vivo (54). Our results show that CaMKII bound to CaV2.1 channels is effective in phosphorylating Ser-603 in the absence of stimulation by Ca2+/CaM. In the nerve terminal, phosphorylation of Ser-603 detaches synapsin-1 from synaptic vesicles and renders the vesicles mobile (58, 59). Thus, CaMKII bound to CaV2.1 channels may phosphorylate synapsin-1 nearby and regulate synaptic vesicle dynamics in and near the active zones in the presynaptic terminal.
Binding of CaMKII to CaV2.1 Channels Is Required for Short Term Synaptic Plasticity
Recent results from studies of neurotransmission at the Calyx of Held, a large synapse in the auditory system, and in cultured SCG neurons show that Ca2+-dependent facilitation and inactivation of CaV2.1 channel activity contribute substantially to short term synaptic facilitation and depression (9). Our results show that binding of CaMKII to CaV2.1 channels is required for short term synaptic plasticity in SCG neurons. Block of CaMKII binding with a competing peptide from its CaV2.1 binding site inhibits short term facilitation and depression of synaptic transmission, as does binding of the brain-specific CaMKII inhibitor CaMKIIN. Evidently, CaMKII binding to CaV2.1 channels is a necessary prerequisite for short term synaptic plasticity mediated by CaV2.1 channels, as observed previously in studies of synaptic plasticity in genetically modified mouse strains (31). Binding of CaMKII to CaV2.1 channels may play a permissive role by enhancing the activation of CaV2.1 channels in response to trains of depolarizing stimuli and the resulting influx of Ca2+, because binding of the kinase does not activate or facilitate channel activity by itself (33). In addition to the role of CaMKII binding to CaV2.1 channels in short term plasticity demonstrated here, it is possible that phosphorylation by CaMKII bound to CaV2.1 channels may also be essential in the longer-term effects of synapsin-1 in regulating synaptic vesicle dynamics and synaptic transmission in the local environment of CaV2.1 channels at active zones.
Functional Roles of the Presynaptic CaV2.1 Signaling Complex
Previous studies show that CaV2.1 channels are regulated by binding of SNARE proteins, G proteins, CaM and CaM-like calcium sensor proteins, and CaMKII (9). Proteomic analysis revealed a complex of ∼100 proteins associated with CaV2.1 channels in isolated nerve terminals from the mouse brain (10). This large protein complex serves to bring the essential machinery for neurotransmitter release close to presynaptic CaV2.1 channels, which provide the trigger of Ca2+ entry to initiate rapid exocytosis. It also serves to regulate the activity of CaV2.1 channels in response to Ca2+ and other regulatory messengers. Prior binding of CaMKII to CaV2.1 channels is required for facilitation and inactivation of the CaV2.1 channel during trains of repetitive depolarizations or action potentials. In this way, CaMKII binding to CaV2.1 serves as a molecular switch to turn on or off the millisecond timescale modulation of channel activity by Ca2+-dependent facilitation and inactivation.
The Effector Checkpoint Model for Calcium Channel Regulation
Voltage-gated Ca2+ channels are regulated by their effectors such that the channels are more active when the effectors of their Ca2+ signal are bound. Examples include regulation of the skeletal muscle Ca2+ channel by the ryanodine-sensitive Ca2+ release channel (60), its effector in excitation-contraction coupling, and regulation of presynaptic Ca2+ channels by SNARE proteins, which are the effectors for Ca2+-dependent exocytosis (9). Regulation of Cav2.1 channels by CaMKII also fits this regulatory theme (33). Binding of CaMKII to Cav2.1 increases the activity of both binding partners, and their interaction is required for facilitation of synaptic transmission and perhaps for other aspects of presynaptic function. Enhancement of the activity of Ca2+ channels whose effectors are bound would focus Ca2+ entry and Ca2+-dependent protein phosphorylation in locations where it can effectively generate a cellular response via local Ca2+ signaling. This mechanism would enhance local signal transduction and reduce ineffective Ca2+ entry and protein phosphorylation at other sites.
Acknowledgment
We thank Dr. Thomas Soderling (Oregon Health and Science University, Portland, OR) for CaMKII protein, cDNAs encoding CaMKII and CaMKIIN, and valuable discussions.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 NS22625 (to W. A. C.) and National Institute on Drug Abuse Grant DA10044 (to A. C. N.).
- CaM
- calmodulin
- CaMKII
- Ca2+/CaM-dependent protein kinase II
- CaMKIIN
- CaM kinase inhibitor
- MBP
- maltose-binding protein
- SCG
- superior cervical ganglion
- EPSP
- excitatory postsynaptic potential
- ISI
- inter-stimulus interval
- RIM
- Rab3 interacting molecule
- eGFP
- enhanced green fluorescent protein.
REFERENCES
- 1. Llinás R., Sugimori M., Hillman D. E., Cherksey B. (1992) Distribution and functional significance of the P-type, voltage-dependent Ca2+ channels in the mammalian central nervous system. Trends Neurosci. 15, 351–355 [DOI] [PubMed] [Google Scholar]
- 2. Ertel E. A., Campbell K. P., Harpold M. M., Hofmann F., Mori Y., Perez-Reyes E., Schwartz A., Snutch T. P., Tanabe T., Birnbaumer L., Tsien R. W., Catterall W. A. (2000) Nomenclature of voltage-gated calcium channels. Neuron 25, 533–535 [DOI] [PubMed] [Google Scholar]
- 3. Wu L. G., Westenbroek R. E., Borst J. G., Catterall W. A., Sakmann B. (1999) Calcium channel types with distinct presynaptic localization couple differentially to transmitter release in single calyx-type synapses. J. Neurosci. 19, 726–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Westenbroek R. E., Sakurai T., Elliott E. M., Hell J. W., Starr T. V., Snutch T. P., Catterall W. A. (1995) Immunochemical identification and subcellular distribution of the α1A subunits of brain calcium channels. J. Neurosci. 15, 6403–6418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wheeler D. B., Randall A., Tsien R. W. (1994) Roles of N-type and Q-type Ca2+ channels in supporting hippocampal synaptic transmission. Science 264, 107–111 [DOI] [PubMed] [Google Scholar]
- 6. Dunlap K., Luebke J. I., Turner T. J. (1995) Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci. 18, 89–98 [PubMed] [Google Scholar]
- 7. Liu H., De Waard M., Scott V. E., Gurnett C. A., Lennon V. A., Campbell K. P. (1996) Identification of three subunits of the high affinity ω-conotoxin MVIIC-sensitive Ca2+ channel. J. Biol. Chem. 271, 13804–13810 [PubMed] [Google Scholar]
- 8. Catterall W. A. (2000) Structure and regulation of voltage-gated Ca2+ channels. Annu. Rev. Cell Dev. Biol. 16, 521–555 [DOI] [PubMed] [Google Scholar]
- 9. Catterall W. A., Few A. P. (2008) Calcium channel regulation and presynaptic plasticity. Neuron 59, 882–901 [DOI] [PubMed] [Google Scholar]
- 10. Müller C. S., Haupt A., Bildl W., Schindler J., Knaus H.-G., Meissner M., Rammner B., Striessnig J., Flockerzi V., Fakler B., Schulte U. (2010) Quantitative proteomics of the CaV2 channel nano-environments in the mammalian brain. Proc. Natl. Acad. Sci. U.S.A. 107, 14950–14957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Soderling T. R., Chang B., Brickey D. (2001) Cellular signaling through multifunctional Ca2+/calmodulin-dependent protein kinase II. J. Biol. Chem. 276, 3719–3722 [DOI] [PubMed] [Google Scholar]
- 12. Haribabu B., Hook S. S., Selbert M. A., Goldstein E. G., Tomhave E. D., Edelman A. M., Snyderman R., Means A. R. (1995) Human calcium-calmodulin dependent protein kinase I. cDNA cloning, domain structure and activation by phosphorylation at threonine-177 by calcium-calmodulin-dependent protein kinase I kinase. EMBO J. 14, 3679–3686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hemmings H. C., Jr., Nairn A. C., McGuinness T. L., Huganir R. L., Greengard P. (1989) Role of protein phosphorylation in neuronal signal transduction. FASEB J. 3, 1583–1592 [DOI] [PubMed] [Google Scholar]
- 14. Hudmon A., Schulman H. (2002) Neuronal Ca2+/calmodulin-dependent protein kinase II. The role of structure and autoregulation in cellular function. Annu. Rev. Biochem. 71, 473–510 [DOI] [PubMed] [Google Scholar]
- 15. Lisman J., Schulman H., Cline H. (2002) The molecular basis of CaMKII function in synaptic and behavioural memory. Nat. Rev. Neurosci. 3, 175–190 [DOI] [PubMed] [Google Scholar]
- 16. Lisman J., Yasuda R., Raghavachari S. (2012) Mechanisms of CaMKII action in long-term potentiation. Nat. Rev. Neurosci. 13, 169–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Miller S. G., Kennedy M. B. (1985) Distinct forebrain and cerebellar isozymes of type II Ca2+/calmodulin-dependent protein kinase associate differently with the postsynaptic density fraction. J. Biol. Chem. 260, 9039–9046 [PubMed] [Google Scholar]
- 18. Hoelz A., Nairn A. C., Kuriyan J. (2003) Crystal structure of a tetradecameric assembly of the association domain of Ca2+/calmodulin-dependent kinase II. Mol. Cell 11, 1241–1251 [DOI] [PubMed] [Google Scholar]
- 19. Schworer C. M., Colbran R. J., Soderling T. R. (1986) Reversible generation of a Ca2+-independent form of Ca2+(calmodulin)-dependent protein kinase II by an autophosphorylation mechanism. J. Biol. Chem. 261, 8581–8584 [PubMed] [Google Scholar]
- 20. Lai Y., Nairn A. C., Greengard P. (1986) Autophosphorylation reversibly regulates the Ca2+/calmodulin-dependence of Ca2+/calmodulin-dependent protein kinase II. Proc. Natl. Acad. Sci. U.S.A. 83, 4253–4257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Miller S. G., Kennedy M. B. (1986) Regulation of brain type II Ca2+/calmodulin-dependent protein kinase by autophosphorylation. A Ca2+-triggered molecular switch. Cell 44, 861–870 [DOI] [PubMed] [Google Scholar]
- 22. Schworer C. M., Colbran R. J., Keefer J. R., Soderling T. R. (1988) Ca2+/calmodulin-dependent protein kinase II. Identification of a regulatory autophosphorylation site adjacent to the inhibitory and calmodulin binding domains. J. Biol. Chem. 263, 13486–13489 [PubMed] [Google Scholar]
- 23. Benfenati F., Valtorta F., Chieregatti E., Greengard P. (1992) Interaction of free and synaptic vesicle-bound synapsin I with F-actin. Neuron 8, 377–386 [DOI] [PubMed] [Google Scholar]
- 24. Benfenati F., Valtorta F., Rubenstein J. L., Gorelick F. S., Greengard P., Czernik A. J. (1992) Synaptic vesicle-associated Ca2+/calmodulin-dependent protein kinase II is a binding protein for synapsin I. Nature 359, 417–420 [DOI] [PubMed] [Google Scholar]
- 25. Greengard P., Valtorta F., Czernik A. J., Benfenati F. (1993) Synaptic vesicle phosphoproteins and regulation of synaptic function. Science 259, 780–785 [DOI] [PubMed] [Google Scholar]
- 26. Luk C. C., Naruo H., Prince D., Hassan A., Doran S. A., Goldberg J. I., Syed N. I. (2011) A novel form of presynaptic CaMKII-dependent short term potentiation between Lymnaea neurons. Eur. J. Neurosci. 34, 569–577 [DOI] [PubMed] [Google Scholar]
- 27. Pang Z. P., Cao P., Xu W., Südhof T. C. (2010) Calmodulin controls synaptic strength via presynaptic activation of calmodulin kinase II. J. Neurosci. 30, 4132–4142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Llinás R., Gruner J. A., Sugimori M., McGuinness T. L., Greengard P. (1991) Regulation by synapsin I and Ca2+-calmodulin-dependent protein kinase II of the transmitter release in squid giant synapse. J. Physiol. 436, 257–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Llinás R., McGuinness T. L., Leonard C. S., Sugimori M., Greengard P. (1985) Intraterminal injection of synapsin I or calcium/calmodulin-dependent protein kinase II alters neurotransmitter release at the squid giant synapse. Proc. Natl. Acad. Sci. U.S.A. 82, 3035–3039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chapman P. F., Frenguelli B. G., Smith A., Chen C. M., Silva A. J. (1995) The α-Ca2+/calmodulin kinase II. A bidirectional modulator of presynaptic plasticity. Neuron 14, 591–597 [DOI] [PubMed] [Google Scholar]
- 31. Hojjati M. R., van Woerden G. M., Tyler W. J., Giese K. P., Silva A. J., Pozzo-Miller L., Elgersma Y. (2007) Kinase activity is not required for αCaMKII-dependent presynaptic plasticity at CA3-CA1 synapses. Nature Neurosci. 10, 1125–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lu F. M., Hawkins R. D. (2006) Presynaptic and postsynaptic Ca2+ and CamKII contribute to long-term potentiation at synapses between individual CA3 neurons. Proc. Natl. Acad. Sci. U.S.A. 103, 4264–4269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jiang X., Lautermilch N. J., Watari H., Westenbroek R. E., Scheuer T., Catterall W. A. (2008) Modulation of CaV2.1 channels by Ca2+/calmodulin-dependent protein kinase II bound to the C-terminal domain. Proc. Natl. Acad. Sci. U.S.A. 105, 341–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yokoyama C. T., Myers S. J., Fu J., Mockus S. M., Scheuer T., Catterall W. A. (2005) Mechanism of SNARE protein binding and regulation of CaV2 channels by phosphorylation of the synaptic protein interaction site. Mol. Cell. Neurosci. 28, 1–17 [DOI] [PubMed] [Google Scholar]
- 35. Brickey D. A., Colbran R. J., Fong Y. L., Soderling T. R. (1990) Expression and characterization of the α-subunit of Ca2+/calmodulin-dependent protein kinase II using the baculovirus expression system. Biochem. Biophys. Res. Commun. 173, 578–584 [DOI] [PubMed] [Google Scholar]
- 36. Chao L. H., Pellicena P., Deindl S., Barclay L. A., Schulman H., Kuriyan J. (2010) Intersubunit capture of regulatory segments is a component of cooperative CaMKII activation. Nat. Struct. Mol. Biol. 17, 264–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hudmon A., Schulman H., Kim J., Maltez J. M., Tsien R. W., Pitt G. S. (2005) CaMKII tethers to L-type Ca2+ channels, establishing a local and dedicated integrator of Ca2+ signals for facilitation. J. Cell Biol. 171, 537–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Magupalli V. G., Schwarz K., Alpadi K., Natarajan S., Seigel G. M., Schmitz F. (2008) Multiple RIBEYE-RIBEYE interactions create a dynamic scaffold for the formation of synaptic ribbons. J. Neurosci. 28, 7954–7967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mochida S., Sheng Z. H., Baker C., Kobayashi H., Catterall W. A. (1996) Inhibition of neurotransmission by peptides containing the synaptic protein interaction site of N-type Ca2+ channels. Neuron 17, 781–788 [DOI] [PubMed] [Google Scholar]
- 40. Lee A., Wong S. T., Gallagher D., Li B., Storm D. R., Scheuer T., Catterall W. A. (1999) Ca2+/calmodulin binds to and modulates P/Q-type calcium channels. Nature 399, 155–159 [DOI] [PubMed] [Google Scholar]
- 41. Lee A., Zhou H., Scheuer T., Catterall W. A. (2003) Molecular determinants of Ca2+/calmodulin-dependent regulation of CaV2.1 channels. Proc. Natl. Acad. Sci. U.S.A. 100, 16059–16064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ohyama A., Hosaka K., Komiya Y., Akagawa K., Yamauchi E., Taniguchi H., Sasagawa N., Kumakura K., Mochida S., Yamauchi T., Igarashi M. (2002) Regulation of exocytosis through Ca2+/ATP-dependent binding of autophosphorylated Ca2+/calmodulin-activated protein kinase II to syntaxin 1A. J. Neurosci. 22, 3342–3351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bähler M., Rhoads A. (2002) Calmodulin signaling via the IQ motif. FEBS Lett. 513, 107–113 [DOI] [PubMed] [Google Scholar]
- 44. Colbran R. J. (2004) Protein phosphatases and calcium/calmodulin-dependent protein kinase II-dependent synaptic plasticity. J. Neurosci. 24, 8404–8409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bayer K. U., De Koninck P., Leonard A. S., Hell J. W., Schulman H. (2001) Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature 411, 801–805 [DOI] [PubMed] [Google Scholar]
- 46. Sun X. X., Hodge J. J., Zhou Y., Nguyen M., Griffith L. C. (2004) The eag potassium channel binds and locally activates calcium/calmodulin-dependent protein kinase II. J. Biol. Chem. 279, 10206–10214 [DOI] [PubMed] [Google Scholar]
- 47. Rusnak F., Mertz P. (2000) Calcineurin. Form and function. Physiol. Rev. 80, 1483–1521 [DOI] [PubMed] [Google Scholar]
- 48. Mochida S., Few A. P., Scheuer T., Catterall W. A. (2008) Regulation of presynaptic CaV2.1 channels by Ca2+ sensor proteins mediates short term synaptic plasticity. Neuron 57, 210–216 [DOI] [PubMed] [Google Scholar]
- 49. Mochida S., Westenbroek R. E., Yokoyama C. T., Itoh K., Catterall W. A. (2003) Subtype-selective reconstitution of synaptic transmission in sympathetic ganglion neurons by expression of exogenous calcium channels. Proc. Natl. Acad. Sci. U.S.A. 100, 2813–2818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chang B. H., Mukherji S., Soderling T. R. (1998) Characterization of a calmodulin kinase II inhibitor protein in brain. Proc. Natl. Acad. Sci. U.S.A. 95, 10890–10895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dai S., Hall D. D., Hell J. W. (2009) Supramolecular assemblies and localized regulation of voltage-gated ion channels. Physiol. Rev. 89, 411–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Grueter C. E., Abiria S. A., Dzhura I., Wu Y., Ham A. J., Mohler P. J., Anderson M. E., Colbran R. J. (2006) L-type Ca2+ channel facilitation mediated by phosphorylation of the β subunit by CaMKII. Mol. Cell 23, 641–650 [DOI] [PubMed] [Google Scholar]
- 53. Grueter C. E., Abiria S. A., Wu Y., Anderson M. E., Colbran R. J. (2008) Differential regulated interactions of calcium/calmodulin-dependent protein kinase II with isoforms of voltage-gated calcium channel β subunits. Biochemistry 47, 1760–1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cesca F., Baldelli P., Valtorta F., Benfenati F. (2010) The synapsins. Key actors of synapse function and plasticity. Prog. Neurobiol. 91, 313–348 [DOI] [PubMed] [Google Scholar]
- 55. Sankaranarayanan S., Atluri P. P., Ryan T. A. (2003) Actin has a molecular scaffolding, not propulsive, role in presynaptic function. Nat. Neurosci. 6, 127–135 [DOI] [PubMed] [Google Scholar]
- 56. Lee J. S., Ho W. K., Lee S. H. (2012) Actin-dependent rapid recruitment of reluctant synaptic vesicles into a fast-releasing vesicle pool. Proc. Natl. Acad. Sci. U.S.A. 109, E765–E774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sakaba T., Neher E. (2003) Involvement of actin polymerization in vesicle recruitment at the calyx of Held synapse. J. Neurosci. 23, 837–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Stefani G., Onofri F., Valtorta F., Vaccaro P., Greengard P., Benfenati F. (1997) Kinetic analysis of the phosphorylation-dependent interactions of synapsin I with rat brain synaptic vesicles. J. Physiol. 504, 501–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bennett A. F., Baines A. J. (1992) Bundling of microtubules by synapsin 1. Characterization of bundling and interaction of distinct sites in synapsin 1 head and tail domains with different sites in tubulin. Eur. J. Biochem. 206, 783–792 [DOI] [PubMed] [Google Scholar]
- 60. Nakai J., Dirksen R. T., Nguyen H. T., Pessah I. N., Beam K. G., Allen P. D. (1996) Enhanced dihydropyridine receptor channel activity in the presence of ryanodine receptor. Nature 380, 72–75 [DOI] [PubMed] [Google Scholar]