

Background: NSD proteins are histone H3K36 methyltransferases linked to multiple human diseases.
Results: Crystal structures of chromatin-binding motifs, PHD5-C5HCH, of NSD3 bound to the H3 N-terminal peptide were solved.
Conclusion: PHD5-C5HCHs of the NSD family are conserved in their overall structure yet variant in H3 recognition.
Significance: Their variant recognitions may play a role in the localization of NSD proteins to different genomic sites.
Keywords: Chromatin Histone Modification, Crystal Structure, Epigenetics, Histone Methylation, NMR, H3K36, H3K4, NSD Family, PHD Finger, WHSC1
Abstract
The NSD (nuclear receptor SET domain-containing) family members, consisting of NSD1, NSD2 (MMSET/WHSC1), and NSD3 (WHSC1L1), are SET domain-containing methyltransferases and aberrant expression of each member has been implicated in multiple diseases. They have specific mono- and dimethylase activities for H3K36, whereas play nonredundant roles during development. Aside from the well characterized catalytic SET domain, NSD proteins have multiple potential chromatin-binding motifs that are clinically relevant, including the fifth plant homeodomain (PHD5) and the adjacent Cys-His-rich domain (C5HCH) located at the C terminus. Herein, we report the crystal structures of the PHD5-C5HCH module of NSD3, in the free state and in complex with H31–7 (H3 residues 1–7), H31–15 (H3 residues 1–15), and H31–15K9me3 (H3 residues 1–15 with trimethylation on K9) peptides. These structures reveal that the PHD5 and C5HCH domains fold into a novel integrated PHD-PHD-like structural module with H3 peptide bound only on the surface of PHD5 and provide the molecular basis for the recognition of unmodified H3K4 and trimethylated H3K9 by NSD3 PHD5. Structural studies and binding assays show that differences exist in histone binding specificity of the PHD5 domain between three members of the NSD family. For NSD2, the PHD5-C5HCH:H3 N terminus interaction is largely conserved, although with a stronger preference for unmethylated H3K9 (H3K9me0) than trimethylated H3K9 (H3K9me3), and NSD1 PHD5-C5HCH does not bind to H3 peptides. Our results shed light on how NSD proteins that mediate H3K36 methylation are localized to specific genomic sites and provide implications for the mechanism of functional diversity of NSD proteins.
Introduction
Histone post-translational modifications (PTMs)4 have an important role in regulating chromatin dynamics and functions. The PTMs act either sequentially or in combination to form “histone codes”, which are recognized by protein modules, referred to as effectors or “readers” (1). Recognitions of readers to histone codes recruit or stabilize various chromatin-associated proteins or complexes (2). In addition, many reader-containing proteins or complexes also possess “writer” or “eraser” modules that further modify the epigenetic landscape by writing or erasing PTMs.
The NSD family of SET domain-containing histone methyltransferases includes NSD1, NSD2 (also known as WHSC1, Wolf-Hirschhorn syndrome candidate 1 or MMSET, multiple myeloma SET), and NSD3 (also known as WHSC1L1, WHSC1 like 1). All three NSD proteins have been directly linked to multiple human diseases. Haploinsufficiency and point mutations of NSD1 cause human Sotos syndrome, a childhood developmental disease (3, 4). NSD2 haploinsufficiency is implicated in the pathogenesis of Wolf-Hirschhorn syndrome, a disease characterized by severe growth retardation, mental as well as cardiac defects (5). Furthermore, each NSD family member behaves as an oncogene. Translocations resulting in NUP98 fusion to NSD1 and NSD3 lead to the development of acute myeloid leukemia (6, 7). The t(4;14)(p16.3;q32) translocation, which results in overexpression of NSD2, is responsible for about 15% of multiple myeloma (8, 9). Moreover, overexpression of NSD3 via gene amplification occurs in about 15% of breast cancer cell lines (10).
Despite the physiologic importance of NSD family proteins, their mechanisms of action are only beginning to become elucidated (11). A striking feature of the three NSD proteins is that they are highly similar within a block of about 700 amino acids, which contains a catalytic SET domain with its pre- (associated with SET) and post-domains, a PWWP (named after the conserved motif Pro-Trp-Trp-Pro in NSD2) domain, five PHD (plant homeodomain) fingers, and an NSD-specific Cys-His rich domain (C5HCH) (Fig. 1A) (10). However, the similar domain architecture of the three NSD family members does not indicate a functional redundancy (10, 12). For example, NSD1 defective mice exhibit embryonic lethality, but mice defective in NSD2 die shortly after birth and exhibit a spectrum of defects that are consistent with Wolf-Hirschhorn syndrome (13, 14). It has been suspected that functional diversity of the three NSD proteins might originate from different substrate specificities of their SET domains (15). Although there have been several conflicting reports on the catalytic activities of NSD proteins, it has become clear that all three members are highly specific H3K36 mono- and dimethylases when nucleosomes are used as their substrates (16–18). The rationale for this functional diversity between the three NSD members remains a mystery.
FIGURE 1.

PHD5-C5HCHNSD3 prefers to bind H3 N-terminal tail containing unmodified K4 and trimethylated K9. A, schematic representation of the domain architecture of human NSD1, NSD2, NSD3, and Drosophila melanogaster dmMes-4 (maternal-effect sterile 4). B, interactions of GST-tagged PHD5-C5HCHs of the human NSD family with whole calf thymus histones visualized by Coomassie Blue staining. C, the raw ITC titration data of PHD5-C5HCHNSD3 with histone peptides are shown (top for H31–15K4me2 and bottom for H31–15K9me3, respectively). D, ITC titration fitting curves for several histone H3 N-terminal peptides (residues 1–15). The peptide modification states and the measured binding constants are listed. N.D., no detectable binding. E–G, six superimposed 1H-15N HSQC spectra of PHD5-C5HCHNSD3 (0.25 mm) collected during titration of H31–15, H31–15K9me3, and H31–15K4me2, respectively. The spectra are color-coded according to the PHD5-C5HCHNSD3:peptide molar ratio.
H3K36 methylation has been implicated in multiple biological processes, and the modifications biological consequences are largely determined by where in the gene the methylated H3K36 mark is placed (12). Because the NSD proteins also contain several histone readers, one attractive possibility is that these readers target the NSD H3K36 methyltransferases to their specific gene sites, and thus lead to distinct biological outcomes. Several reports have revealed that NSD family members are possibly recruited to specific gene loci. NSD1 binds upstream of the bone morphogenetic protein 4 promoter, enforces H3K36 methylation levels within this region, and thus promotes bone morphogenetic protein 4 transcription (19). NSD3, LSD2 (an H3K4-specific demethylase, lysine demethylase 2, also known as KDM1B), and G9a (an H3K9-specific methyltransferase, also known as EHMT2) form complexes in vivo, predominantly localize to the gene bodies of actively transcribed genes and may coordinate modifications at H3K4, H3K9, and H3K36 during elongation for optimal transcription (20). So far, great efforts have been made to understand the substrate specificities of the enzymes, but little structural and functional information is known about the reader domains.
It has been reported that the PHD5-C5HCH module of NSD1 (PHD5-C5HCHNSD1) was the sole region required for tight binding of the NUP98-NSD1 fusion protein to the HoxA9 gene promoter, implicating that PHD5-C5HCHNSD1 might have chromatin targeting ability. In this study, we demonstrate that the PHD5-C5HCH module of NSD3 (PHD5-C5HCHNSD3) recognizes the H3 N-terminal peptide containing unmodified K4 and trimethylated K9. We have solved high resolution crystal structures of PHD5-C5HCHNSD3 in its free state and in complex with unmodified H3 residues 1–7 (H31–7), residues 1–15 (H31–15), and residues 1–15 with trimethylation on lysine 9 (H31–15K9me3), respectively. These structures reveal an integrated tandem PHD-PHD-like fold with H3 peptide bound only on the surface of PHD5 and provide the structural basis for peptide recognition by PHD5 of NSD3. Further mutagenesis and peptide binding assays show that PHD5-C5HCHNSD1 does not possess binding ability to H3 and the PHD5-C5HCH module of NSD2 (PHD5-C5HCHNSD2) prefers to bind the H3 N-terminal peptide containing unmodified K4 and K9. This is likely due to a minor sequence change at the H3 binding surface. Our findings suggest that PHD5-C5HCHs of the NSD family are conserved in the overall structure but vary in H3 recognition. These variant recognitions may play a role in localization of these H3K36 methyltransferases to different genome sites and are consistent with the distinct and nonredundant functions of NSD proteins.
EXPERIMENTAL PROCEDURES
Cloning, Expression, and Purification
Human PHD5-C5HCHNSD1(2115–2220), PHD5-C5HCHNSD2(1232–1331), and PHD5-C5HCHNSD3(1310–1413) were amplified from the human brain cDNA library by PCR, and cloned into modified pET-28a(+) (Novagen) plasmids (for NMR, ITC, and crystallization) or pGEX-4T1 vectors (for GST pulldown and peptide pulldown assays). The modified pET-28a(+) plasmid contains an N-terminal His6 tag, and a tobacco etch virus protease cleavage site. All of the mutants were generated by the Mutant Best kit (Takara), and verified by DNA sequencing. The recombinant proteins were produced in Escherichia coli BL21(DE3) cells (Novagen). Cells were cultured in LeMaster and Richards minimal medium (LR medium), supplemented with 100 μm ZnSO4. For stable isotope labeling, LR medium was supplemented with 15NH4Cl and [13C6]glucose as the sole nitrogen and carbon sources, respectively. The protein expression was induced when A600 reached around 0.8 with 0.1 mm isopropyl 1-thio-β-d-galactopyranoside at 16 °C for 24 h. The expressed His-tagged proteins were purified using a nickel-chelating column (Qiagen), followed by tobacco etch virus cleavage, size-exclusion chromatography on a Hiload 16/60 Superdex 75 column (GE Healthcare), and anion exchange chromatography on a MonoQ HR 5/5 column (GE Healthcare). The GST fusion proteins were purified by glutathione-Sepharose (GE Healthcare), and further purified by size exclusion chromatography on a Hiload 16/60 Superdex 200 column (GE Healthcare). The purified proteins were dialyzed against Buffer A (20 mm BisTris-HCl, pH 7.0, 150 mm NaCl), and concentrated for subsequent analysis.
Peptide Synthesis
All of the peptides used in the study were synthesized by GL Biochem (Shanghai). The lyophilized synthesized peptides (purity >98%, verified by Mass Spectrometry) were carefully weighted and dissolved in Buffer A, and the pH of each sample was adjusted to a value of 7.0.
Peptide Pulldown Assays
16 μg of purified GST fusion proteins were incubated with biotinylated histone peptides (2 μg each) for 2 h at 10 °C in Buffer A, and the fusion proteins were also incubated without peptides as negative controls. Then 50 μl of 50% streptavidin beads (Thermo Scientific) were added and incubated for 1 h. After incubation, the beads were washed 3 times using Buffer A supplemented with 0.1% Triton X-100, and then the bound materials were subjected to SDS-PAGE and Coomassie Blue staining.
Calf Thymus Histone Binding Assays
The binding assays were performed as previously described (21). GST fusion proteins were bound to glutathione-Sepharose beads (GE Healthcare), and the beads were subsequently incubated with calf thymus histones (Worthington) in buffer containing 50 mm Tris-HCl, pH 7.5, 1 m NaCl, 1% Nonidet P-40. After a rigorous wash, the bound materials were subjected to SDS-PAGE and Coomassie Blue staining.
Peptide Arrays
A peptide array comprising 384 peptide spots purchased from Active Motif (catalog number 13001) was utilized and the detailed information can be obtained from the company website. The array was blocked overnight in 1× TBS-T (0.1%) containing 5% BSA. The array was then washed 3 times in 1× TBS, and incubated with 50 nm purified GST-PHD5-C5HCHNSD3 in Buffer A for 1 h at room temperature. After the reaction, the membrane was washed 3 times in 1× TBS-T, and incubated with goat anti-GST antibody (Sigma) for 1 h at room temperature. The unbound antibody was then washed three times with 1× TBS-T, and the array was incubated with horseradish peroxidase-conjugated anti-goat IgG antibody for 1 h at room temperature. After several washes in 1× TBS-T, the membrane was submerged in an ECL Western blotting reagent kit (Thermo Scientific) and the resulting image was captured with ImageQuant LAS 4000 mini-biomolecular imager (GE Healthcare).
Isothermal Titration Calorimetry
ITC experiments were carried out on a MicroCal iTC200 calorimeter (GE Healthcare) at 25 °C. The protein concentrations of the untagged PHD5-C5HCHNSD3 and its mutants were determined photometrically and diluted to 200 μm. The concentration of each peptide was 6 mm. A reference measurement (peptide injected into buffer) was carried out to compensate for the heat of dilution of each peptide. Curve fitting to a one-binding site model was performed by ITC data analysis module of Origin 7.5 (Origin Lab Corporation) provided with the iTC200 calorimeter.
NMR Experiments
The purified 13C,15N-labeled PHD5-C5HCHNSD3 was dissolved to a final concentration of 0.6 mm in 500 μl of Buffer A containing 10% D2O. All the NMR experiments were carried out on a Bruker DMX600 spectrometer or Varian 700 MHz spectrometer at 298 K.
The following spectra were acquired to obtain backbone resonance assignments: two-dimensional, 1H-15N HSQC; three-dimensional sets: HNCA, HNCOCA, CBCA(CO)NH, CBCANH. All of the NMR spectra were processed by NMRPipe (22) and the resonances were assigned with Sparky (T. D. Goddard and D. G. Kneller, University of California, San Francisco, CA).
For NMR titration experiments, 0.25 mm 15N-labeled PHD5-C5HCHNSD3 was prepared in Buffer A containing 10% D2O. Lyophilized H3 peptide was dissolved in the NMR buffer of identical composition, and added stepwise with a sample dilution of less than 10%. 1H-15N HSQC spectra were recorded to follow chemical shift perturbations at each titration point. Combined chemical shift perturbations and dissociation constants were estimated as previously described (23).
A sample containing 0.6 mm 15N-labeled PHD5-C5HCHNSD3 was prepared for 15N relaxation measurements performed on a Bruker DMX 600 spectrometer as described (24). Data analysis and exponential curve fitting were performed utilizing NMRPipe and Sparky.
Crystallization Conditions, Data Collection, and Structure Determination
All the crystals were grown using the hanging drop vapor diffusion method at 16 °C. Crystals of PHD5-C5HCHNSD3 in the free form were grown by mixing equal volumes of 15 mg ml−1 of protein in Buffer A and crystallization buffer (0.1 m Hepes, pH 7.0, 1.1 m sodium malonate, pH 7.0). Crystals of the PHD5-C5HCHNSD3·H31–7 complex were obtained by incubating 15 mg ml−1 of protein with peptide at a 1:3 ratio, followed by mixing an equal volume of reservoir solution containing 0.1 m Tris-HCl, pH 7.0, 30% (w/v) PEG-3000, and 0.2 m NaCl. For crystallization of the H31–15 complex, the peptide was incubated with PHD5-C5HCHNSD3 at a 2:1 ratio to a complex concentration of about 12 mg ml−1, and then the complex was mixed with an equal volume of crystallization buffer (0.1 m Hepes, pH 7.5, 70% (v/v) MPD). Crystals of the H31–15K9me3 complex were prepared by mixing the peptide with 12 mg ml−1 of proteins at a 2:1 ratio, followed by mixing with an equal volume of crystallization buffer (0.1 m Mes, pH 6.5, 20% PEG3350). Reservoir solutions with the addition of 25% glycerol were used as cryoprotectants for all four crystals mentioned above.
Data sets for all crystals mentioned above were collected on beam line 17U at Shanghai Synchrotron Radiation Facility (SSRF). A zinc SAD data set was collected from a single crystal of free PHD5-C5HCHNSD3 at 1.278 Å. The data set was integrated with iMosflm (25) and scaled with SCALA (26) from the CCP4 program suite (27). Twinning was performed utilizing Ctruncate and revealed a perfect merohedral twinning of the free PHD5-C5HCHNSD3 data set, and the twinning hampered the successful zinc SAD phasing. A data set of PHD5-C5HCHNSD3 complex with H31–7 was collected at 0.979 Å without twinning. Despite the fact of the wavelength being remote from the zinc edge, by using SHELX C/D/E (28), all four expected endogenous zinc atom positions were unambiguously determined, and the resulting experimental phases could be calculated. An initial model was automatically built by ARP/wARP (29), and was further built and refined using Coot (30) and Refmac (31), respectively. The other two complex structures and the free PHD5-C5HCHNSD3 were solved by molecular replacement using the program MOLREP (32) employing the PHD5-C5HCHNSD3·H31–7 structure as the search model. The free PHD5-C5HCHNSD3 structure was refined with twinning-based refinement implemented in Refmac. This twinning-based refinement gave good electron density (supplemental Fig. S6). For the high resolution data sets of PHD5-C5HCHNSD3·H31–7 (1.47 Å) and PHD5-C5HCHNSD3·H31–15 (1.55 Å), anisotropic B-factor refinement of protein/ligand heavy atoms (C/N/O or heavier), together with isotropic B-factor refinement of waters were applied in Refmac during the later stages of the refinement process. Procheck (33) was utilized for the validation of protein model geometry and the Ramachandran plots show that all the residues of the four structures are within the allowed regions. Crystal diffraction data and refinement statistics are displayed in Table 1.
TABLE 1.
Data collection and refinement statistics
| FREE | PHD5-C5HCHNSD3·H31–7 | PHD5-C5HCHNSD3·H31–15 | PHD5-C5HCHNSD3·H31–15K9me3 | |
|---|---|---|---|---|
| Wavelength (Å) | 1.278 | 0.979 | 0.978 | 0.978 |
| Space group | P65 | P 65 2 2 | I 2 2 2 | P 21 21 21 |
| Cell parameters | ||||
| a, b, c (Å) | 58.5, 58.5, 108.6 | 57.2, 57.2, 107.4 | 60.0, 62.7, 62.8 | 56.2, 59.9, 95.2 |
| α, β, γ (°) | 90, 90, 120 | 90, 90, 120 | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 50.67–2.27 | 36.42–1.479 | 31.37–1.55 | 48.40–1.73 |
| (2.39–2.27)a | (1.54–1.47) | (1.63–1.54) | (1.82–1.73) | |
| Rmerge (%) | 9.1 (30.8) | 8.5 (28.1) | 6.4 (42.3) | 10.8 (61.6) |
| I/σI | 28.0 (10.5) | 36.1 (15.4) | 13.2 (3.4) | 10.8 (3.3) |
| Completeness (%) | 100.0 (100.0) | 99.5 (98.5) | 99.8 (100.0) | 100.0 (100.0) |
| Average redundancy | 19.0 (17.9) | 39.7 (37.4) | 6.8 (6.9) | 5.8 (5.4) |
| Refinement (F>0) | ||||
| No. reflections (overall) | 9,050 | 17,507 | 16,643 | 32,556 |
| No. reflections (test set) | 706 | 948 | 890 | 1,733 |
| Twinning fraction | 0. 443 | |||
| Rwork/Rfree (%) | 17.16/21.90 | 14.79/17.85 | 16.02/20.46 | 18.15/21.37 |
| No. non-H atoms | ||||
| Protein | 1596 | 810 | 758 | 1524 |
| Peptide/zinc ion | -/8 | 48/4 | 73/4 | 164/8 |
| Water | 25 | 136 | 98 | 294 |
| B factors (Å2) | ||||
| Protein | 26.51 | 17.18 | 28.88 | 21.69 |
| Peptide/zinc ion | −/20.18 | 24.54/12.96 | 42.26/27.92 | 28.20/19.02 |
| Water | 18.55 | 30.12 | 38.23 | 33.36 |
| Root mean square deviations | ||||
| Bond lengths (Å) | 0.0074 | 0.0074 | 0.0083 | 0.0095 |
| Bond angles (°) | 1.195 | 1.297 | 1.314 | 1.314 |
| Ramachandran plot % residues | ||||
| Most favored | 84.5 | 88.5 | 89.8 | 91.0 |
| Additional allowed | 15.5 | 11.5 | 10.2 | 9.0 |
| Generously allowed | 0 | 0 | 0 | 0 |
| Disallowed | 0 | 0 | 0 | 0 |
a Values in parentheses are for highest-resolution shell.
RESULTS
PHD5-C5HCHNSD3 Recognizes Unmodified K4 and Trimethylated K9 in Histone H3
To identify whether the PHD5-C5HCH region could bind to histones, we performed GST pulldown assays with calf thymus histones using recombinant GST fusion proteins corresponding to the PHD5-C5HCH modules of human NSD1, NSD2, and NSD3, respectively. We detected substantial binding of PHD5-C5HCHNSD2 and PHD5-C5HCHNSD3 with histone H3, whereas almost no binding could be detected for PHD5-C5HCHNSD1 (Fig. 1B). A small quantity of H4 appeared to also be pulled down, which may be caused by the formation of the heterotetramer with H3 and being simultaneously pulled down (Fig. 1B).
To further investigate the potential interaction of PHD5-C5HCHNSD3 with modified histone tails, we applied peptide arrays comprising 384 histone tail peptides, which contain 59 PTMs in different combinations. Binding of PHD5-C5HCHNSD3 to the histone peptide arrays significantly occurred at unmodified H3 (residues 1–19), methylated H3R2 (H3R2me2a, H3R2me2s), methylated H3R8 (H3R8me2a, H3R8me2s), methylated H3K9 (H3K9me1, H3K9me2, and H3K9me3), but not methylated or acetylated H3K4 (supplemental Fig. S1). Moreover, an enhancement of the interaction with trimethylated H3K9 peptides was observed (supplemental Fig. S1). PHD5-C5HCHNSD3 showed no or very weak binding to other histone peptides such as H3 (7–26), H3 (16–35), H3 (26–45), H4 (1–19), H2A (1–19), and H2B (1–19) irrespective of their modification states (Fig. 1C). These results suggest that PHD5-C5HCHNSD3 interacts with the N-terminal tail of histone H3 containing unmodified K4 and senses modification states of H3K9.
The interactions of PHD5-C5HCHNSD3 with H3 tail peptides were further quantitatively analyzed by ITC as well as nuclear magnetic resonance (NMR) spectroscopy (Fig. 1, C–G). The ITC measurements showed that PHD5-C5HCHNSD3 binds the unmodified H31–12 and H31–15 peptides, with similar binding affinity (supplemental Table S1). Shortening of the H3 tail to residues 1–7 significantly reduced this interaction, indicating that the H38–12 region is directly involved in the interaction (supplemental Table S1). Moreover, the binding of PHD5-C5HCHNSD3 to histone H31–15 was almost completely abolished by mono-, dimethylation, or acetylation at H3K4 (Fig. 1, C and D, and supplemental Table S1). By contrast, di- and trimethylation at H3K9 slightly improved the binding affinity by a factor of ∼1.2 and 1.6, respectively (Fig. 1, C and D, and supplemental Table S1).
Peptide binding was monitored by NMR chemical shift perturbation experiments (Fig. 1, E–G). We observed no major changes in the HSQC spectrum of PHD5-C5HCHNSD3 upon titration of the H31–15K4me2 peptide (Fig. 1G). However, noticeable changes in chemical shift or reduction in signal intensity occurred upon addition of the H31–15 or H31–15K9me3 peptides (Fig. 1, E and F). Analysis of the ligand-concentration dependence of the changes induced in the spectrum gave dissociation constants of 0.28 and 0.48 mm for the interactions of H31–15K9me3 and H31–15, respectively (supplemental Fig. S3). Thus, the NMR data are consistent with the ITC as well as the peptide array results, and further confirm that PHD5-C5HCHNSD3 specifically recognizes H3 N-terminal peptides containing unmodified K4 with a preference for trimethylated K9.
PHD5-C5HCHNSD3 Folds as an Integrated Structural Unit, and Only PHD5 Is Responsible for H3 Tail Peptide Recognition
In NMR chemical shift perturbations, large perturbations or disappearances of signals were observed only for residues of PHD5, which suggested that the PHD5 but not C5HCH domain might be involved in the binding with H3 tail (supplemental Fig. S4A). In addition, the 15N relaxation data implied that these two domains might fold as an integrated structural unit (supplemental Fig. S5).
To better understand the structural basis for H3 binding by PHD5-C5HCHNSD3, we solved the crystal structures of PHD5-C5HCHNSD3 in its free state (2.27 Å) and in complex with unmodified H31–7 (1.47 Å), H31–15 (1.55 Å), and H31–15K9me3 (1.73 Å) with the structural statistics listed in Table 1. The crystal structures demonstrate that the PHD5 and C5HCH domains of NSD3 indeed fold dependently as a single structural unit, coordinating four zinc atoms in all, whereas only PHD5 is responsible for H3 peptide binding (Fig. 2, A–C). PHD5 adopts a canonical PHD finger fold, which consists of a two-strand anti-parallel β-sheet (β1 and β2) followed by two 310-helixs (Fig. 2, A and B), and is stabilized by two zinc atoms coordinated by the Cys4-His-Cys2-His motif in a cross-brace topology (Fig. 2A and supplemental Fig. S7). Interestingly, the C5HCH domain folds into a compact module containing two small β-hairpins: one positioned at its core (β3 and β4), and the other positioned (β5 and β6) close to the C terminus (Fig. 2, A and B). The C5HCH finger is also stabilized by two zinc atoms in a cross-brace topology, anchored by the novel Cys5-His-Cys-His motif (Fig. 2, A and B, and supplemental Fig. S7). A structure-based homology search using DALI (34) shows that the C5HCH finger structurally resembled both the PHD and FYVE fingers. Actually, at the protein fold level, the C5HCH finger is more similar to the FYVE domain due to having a second β-hairpin close to the C terminus. However, it lacks characteristic amino acids in the FYVE domain for lipid binding (supplemental Fig. S8A); conversely, the C5HCH finger has been implicated in chromatin-associated processes that most PHD domains function in, so we still termed the C5HCH domain a PHD-like finger.
FIGURE 2.
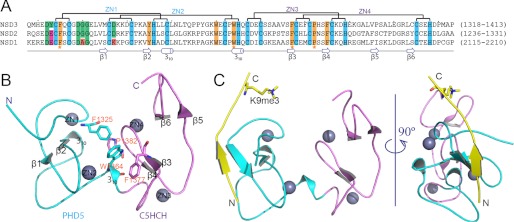
Crystal structures of PHD5-C5HCHNSD3 in the free and H31–15K9me3 peptide-bound states. A, sequence alignment of the PHD5-C5HCH regions of human NSD1, NSD2, and NSD3. Conserved residues are colored: cyan, zinc-coordinating; yellow, hydrophobic or aromatic. Residues for PHD5 and C5HCH inter-domain interactions are further emphasized by yellow star markers. Residues potentially responsible for interactions with H3 are highlighted in green. In comparison with PHD5-C5HCHNSD3, residues probably regulating and disrupting interactions with H3 are shown in magenta and red background, respectively. B, a ribbon representation of the 2.27-Å crystal structure of PHD5-C5HCHNSD3 in its free state. PHD5 (cyan) and C5HCH (violet) associate with each other through several hydrophobic residues, which are shown as sticks. Zinc ions are depicted as gray spheres. C, ribbon representations of the 1.73-Å crystal structure of PHD5-C5HCHNSD3 in complex with H31–15K9me3. The H3 peptide that binds to the surface of PHD5 finger is colored in yellow, with K9me3 shown as sticks.
The compact fold of PHD5-C5HCHNSD3 originates from extensive contacts, primarily hydrophobic, between the PHD5 and C5HCH finger. Specifically, the hydrophobic residue Trp1364, positioned at the interface of the two zinc fingers, bridges these two fingers through hydrophobic interactions with Phe1325 of PHD5 and Phe1377 and Pro1382 of C5HCH (Fig. 2, A and B). These four key residues form the tandem domain fold and are highly conserved in the NSD family (Fig. 2A). In addition, Tyr1345 and Trp1360 are characterized as the hydrophobic core residues of PHD5NSD3, and Phe1385 is characterized as the key residue responsible for the fold of C5HCHNSD3, and all are identical among the three NSD proteins (Fig. 2A). Moreover, the three PHD5-C5HCHs share about 65% sequence similarity with each other and have the same zinc chelating motifs. This suggests that the PHD5-C5HCH motifs of the NSD family fold as a similar integrated PHD-PHD-like structural module.
Molecular Basis for H3K4me0 and H3K9me3 Readout by PHD5-C5HCHNSD3
In our crystal structure of PHD5-C5HCHNSD3 bound to unmodified H31–7, we could trace residues 1–6 of the peptide. Residues 1–9 could be traced in the H31–15 complex and residues 1–10 in the H31–15K9me3 complex (Figs. 2C, 3A, and supplemental Fig. S9). Similar to the previously solved PHD:H3 structures, the histone H3 N-terminal peptide binds to the surface groove of PHD5NSD3 through formation of an antiparallel β-sheet, stabilized by several direct hydrogen bonds (Fig. 3B).
FIGURE 3.

Molecular basis for H3 N-terminal peptide recognition by PHD5-C5HCHNSD3. A, a ribbon representation of PHD5-C5HCHNSD3 in complex with H31–15K9me3 peptide. B, the hydrogen bonding network formed between the backbones of H31–15K9me3 peptide and PHD5-C5HCHNSD3. The intermolecular interactions are depicted as black dashed lines. C, an electrostatic surface view of PHD5-C5HCHNSD3 in complex with the H31–15K9me3 peptide. Red and blue colors denote negative and positive surface charge, respectively. The peptide is shown in stick representation. D, detailed intermolecular interactions between the side chains of H3A1, R2, T3, K4, R8, and K9me3 with PHD5-C5HCHNSD3. The hydrogen bonds are depicted as black dashed lines, and water molecules involved in intermolecular recognition are shown as red spheres. The intermolecular interaction between Asp1329 and the main chain amide of H3K9me3 is also shown. E, comparison of ITC plots for wild-type with D1322A, Y1323A, D1337A, and D1329A mutants and D1322A/D1329A/D1337A triple mutant of PHD5-C5HCHNSD3 bound to H31–15K9me3 peptide. The inset lists the measured dissociation constants. N.D., no detectable binding.
In addition to the backbone interactions, side chains of the H3 N-terminal residues establish a network of interactions with PHD5 residues. The side chains of H3A1 and H3T3 are accommodated within two separate shallow hydrophobic pockets, respectively (Fig. 3, C and D). The side chains of H3R2 and H3K4 are isolated to two shallow negatively charged channels separated by the side chain of Met1335 (Fig. 3, C and D). In the complex structure of PHD5-C5HCHNSD3 with H31–15K9me3 or H31–7, the density of the side chain of H3R2 can be clearly observed (supplemental Fig. S10), and its Nη proton forms a hydrogen bond to the side chain carbonyl oxygen of Asp1337 (Fig. 3D and supplemental Fig. S9, C and D). However, in the PHD5-C5HCHNSD3·H31–15 complex structure, the side chain of H3R2 is fully exposed to solvent, with relatively poor electron density, indicating a moderate positional variability (supplemental Figs. S9G). Notably, asymmetric dimethylation of H3R2 only causes a slight affinity reduction of less than 2-fold, whereas a remarkable affinity decrease of the H31–15K9me3 peptide was observed if Asp1337 of PHD5 was mutated to Ala (Fig. 3E and supplemental Table S1). A reasonable explanation for this is that the D1337A mutant not only disrupts hydrogen bonding with H3R2, but also destroys hydrogen bonding with the backbone amide of Trp1360 (Fig. 3D and supplemental Fig. S9, C and D), which might interfere with the H3A1 interactions.
Specific recognition of H3K4 in the unmodified state is secured by a canonical mode as observed in BHC80 (also known as PHF21A) (35). The ϵ-amino group of K4 forms hydrogen bonds to the side chain carbonyl oxygen of Asp1322, the backbone carbonyl oxygen of His1320 and Glu1321, as well as a water molecule (Fig. 3D and supplemental Fig. S9, C and G). Additional K4 recognition is achieved by hydrophobic contacts between the K4 alkyl chain and the side chain of Leu1333. Methylations at H3K4 reduce binding significantly due to steric clash and unfavorable polar-hydrophobic interactions.
Compared with the N-terminal first seven amino acids of His3, the Arg8 and Lys9 or K9me3 have relatively larger B factors for their side chain atoms (data not shown), which suggests a higher degree of flexibility and agrees with relatively little shape complementarity for these residues (Fig. 3C). The side chain carboxyl group of Asp1329 forms hydrogen bonds to the guanidinium group of H3R8 and the backbone amide of H3K9 or H3K9me3, undergoing an induced flipping coupled with the peptide binding (supplemental Figs. S11B and S12). Interestingly, in the complex structure of PHD5-C5HCHNSD3·H31–15, the carboxylate side chain of Asp1329 possesses alternative conformations: one similar to that in the PHD5-C5HCHNSD3·H31–15K9me3 complex and the other corresponding to that in the free state and PHD5-C5HCHNSD3·H31–7 complex (supplemental Figs. S9G), indicating Asp1329 might be involved in transient electrostatic interactions with unmodified K9 of the peptide. Moreover, the ammonium group of unmodified K9 is positioned close to the backbone carbonyl oxygen of Gly1328 (about 3.7 Å for side chain nitrogen of Lys9) in the structure involving unmodified K9 (supplemental Fig. S9G), whereas the K9me3 points its side chain near the aromatic ring of Tyr1323 and slightly away from Gly1328 (Fig. 3D), probably losing the electrostatic interaction with Gly1328. Consistently, the K9A mutation of the H3 peptide decreased the binding affinity to PHD5-C5HCHNSD3, thus confirming that the side chain of unmodified K9 plays a significant role in the interaction (supplemental Table S1). H3K9me1 was observed to induce a reduced affinity compared with H3K9me0 (supplemental Figs. S1 and S2). This can be explained, the enhanced cation-π interaction with the aromatic ring of Tyr1323 by monomethylation at H3K9 may not be sufficient to compensate for the loss of interaction with Gly1328. However, the increased cation-π interaction caused by H3K9me3 may have overcome this barrier and further provides a basis for the observed preference for K9me3 over K9me0. This phenomenon agrees well with the NMR chemical shift perturbation data. The majority of chemical shift changes in PHD5-C5HCHNSD3 induced by H31–15K9me3 were paralleled to those caused by unmodified H31–15, whereas the amide cross-peaks of Gly1328 and Asp1329 were shifted to a larger extent in the PHD5-C5HCHNSD3·H3K9me3 complex (supplemental Fig. S4).
The introduction of an Ala mutation in Asp1322, Asp1329, or Asp1337 caused a significant reduction in binding affinity of PHD5-C5HCHNSD3 to the H31–15K9me3 peptide, confirming the significance of these residues for H3 binding at K4 (Asp1322), R2 (Asp1337), R8 and K9 (Asp1329). The D1322A/D1329A/D1337A triple mutant led to a complete loss of binding (Fig. 3E and supplemental Table S1). Furthermore, the Ala mutation of Tyr1323 weakened the H31–15K9me3 binding of PHD5-C5HCHNSD3 to nearly the level of unmodified H31–15 peptide binding. This was accompanied with remarkably reduced binding enthalpy, hence confirming the importance of the Tyr side chain for this interaction (Fig. 3E and supplemental Table S1). Taken together, these results demonstrate how the sequence-specific histone H3 binding by PHD5-C5HCHNSD3 is modulated negatively by methylation at H3K4 and positively by trimethylation at H3K9.
A Conservative Sequence Motif Defines a Subgroup of PHD Fingers That Conducts H3K4me0 and H3K9 Cross-talk
Recent structural studies suggest that the N-terminal sequence motif (NTS motif) around the first and the second zinc coordinating cysteines of PHD fingers is often critical in histone H3 peptide recognition (36, 37). From sequence alignment (Fig. 4A) and structural comparison and analysis (Fig. 4, B–G), we extracted an NTS motif, which defines a subgroup of PHD fingers reading out H3K4me0 and H3K9me3 (or H3K9me0) simultaneously. This motif is “(D/E){(W/Y/F)/(D/E)}CXXCX(D/N)GG”, where “(/)” means alternative amino acids, “{/}” denotes alternative types of amino acid, and “X” represents any amino acid. We identify the “0” position of the NTS motif as the key Asp or Glu residue participating in the recognition of H3K4me0 (Fig. 4, A–G). The residue positioned at “+1” is responsible for K9 recognition, which has alternative roles: one is an Asp or a Glu forming hydrogen bonds with the ϵ-amino group of H3K9, such as in AIRE-PHD1 (the first PHD finger of autoimmune regulator) (Fig. 4B) (38); the other is an aromatic residue optimal for H3K9me3 recognition through a cation-π interaction, such as in CHD4-PHD2 (the second PHD of chromo domain helicase DNA-binding protein 4), TRIM33-PHD (the PHD finger of tripartite motif 33), and NSD3-PHD5 (Fig. 4, C–E) (39, 40). The Asp or Asn residues located at the “+7” position is another critical amino acid implicated in the formation of hydrogen bonds with the main chain amide of H3K9 and sometimes the side chain guanidinium group of H3R8 (Fig. 4, B–E) (38–40). When substituted for a Lys at this position, such as in BHC80-PHD, a disruption in the sensitivity to trimethylated K9 is observed even if the “+1” residue is a Phe (Fig. 4F) (35). In addition, the “+8 to +9” positions seem to be a conserved “G-G” motif, and bulky residues at these two positions might exclude the segment after H3A7 from the binding surface. BRPF2-PHD1 (the first PHD finger of Bromodomain-PHD finger protein 2) does not contain such an NTS motif and can only bind with the first six residues of H3 (Fig. 4G) (41). In brief, these key residues that we defined in the NTS motif cooperate to mediate the relatively long range cross-talk between H3K9me3 (or H3K9me0) and H3K4me0.
FIGURE 4.

The NTS motif in PHD fingers for H3K4me0 and H3K9 cross-talk. A, sequence alignment of PHD fingers that are known to conduct H3K4me0 and H3K9 cross-talk, including PHD5 of NSD3, PHD1 of AIRE, the PHDs of TRIM33 and TRIM24, and PHD2 of CHD4. The PHD finger of BHC80 and PHD1 of BRPF2, which are not involved in cross-talk, are also included in the alignment. Key residues involved in H3K4me0 and H3K9 recognition are grouped by green background. The conservative NTS motif dedicated to H3K4me0 and H3K9 dual readout is listed at the top of the alignment, with specific positions numbered above. B–G, ribbon representations of the structures of PHD·H3 complexes that are indicated in the alignment in A. The H3 peptides are colored yellow with K4, R8, and K9 or K9me3 side chains shown in stick representation. Key residues of the PHD fingers involved in the recognition of H3 are also shown as sticks.
We searched for this motif in a large sequence collection of human PHD domain containing proteins obtained from SMART (42) and Pfam (43) databases, and the results, listed in supplemental Table S2, suggested that the NTS motif is a common motif among PHD fingers in the human proteome. Interestingly, the NTS motif of PHD5NSD2 falls into the subset of H3K4me0 and H3K9me0 recognition.
Histone Binding Ability and Specificity of PHD5-C5HCH of Each Constituent NSD Protein
As mentioned above, a calf thymus histone pulldown assay revealed that PHD5-C5HCHNSD3 and PHD5-C5HCHNSD2 displayed substantial binding to histone H3, whereas PHD5-C5HCHNSD1 exhibited diminished binding. Moreover, the NTS motif search suggested a binding specificity of PHD5NSD2 for H3K4me0 and H3K9me0 (Fig. 2A and supplemental Table S2). Expectedly, a peptide pulldown assay clearly showed that PHD5-C5HCHNSD2 indeed prefers to bind the H3 tail containing unmodified K4 and K9 (Fig. 5A). To verify the significance of the “+1” position residue responsible for preference for the H3K9 methylation state (me0 or me3), Tyr1323 in PHD5-C5HCHNSD3 was mutated to Glu. The preference for H31–15 and H31–15K9me3 are reversed, as determined by ITC experiments, with a Kd of 0.22 and 0.35 mm to H31–15 and H31–15K9me3, respectively (Fig. 5B and supplemental Table S1).
FIGURE 5.

Differences exist in histone binding ability and preference of the three PHD5-C5HCHs. A, in vitro binding assays using biotinylated peptides: unmodified H31–18 and H31–18K9me3. The GST-tagged PHD5-C5HCHs used are as indicated. B, ITC titration fitting curves for the PHD5-C5HCHNSD3 mutant Y1323E with dissociation constant (Kd) values indicated. The mutant prefers unmodified H31–15 peptide in contrast to the WT, which prefers H31–15K9me3.
The Lys2134 residue in PHD5NSD1, corresponding to residue Asp1337 of NSD3, may interfere with the binding of H3 by disrupting hydrogen bonding interactions with the guanidinium group of H3R2 and destabilizing the H3A1 interaction loop (Fig. 2A). Besides, Ala2127 in NSD1, which corresponds to residue Gly1330 of NSD3, may have a negative effect on the binding to the backbones of H3A7 and H3R8 (Fig. 2A). Therefore, the completely blocked interaction of PHD5-C5HCHNSD1 with H3 peptides revealed by the peptide pulldown assay might be due to these two residues (Fig. 5A). To confirm this notion, we prepared a K2134D/A2127G double mutant of PHD5-C5HCHNSD1. Expectedly, this mutant exhibited binding ability with H3 N-terminal peptides, and showed a preference for unmethylated H3K9 over H3K9me3 as PHD5-C5HCHNSD2 (Fig. 5A).
Taken together, the tandem PHD5-C5HCH modules of the three NSD proteins may have a similar structural fold, yet exhibit different binding abilities and specificities to the histone H3 tail as a consequence of a minor sequence variation located on the H3 binding surface: PHD5-C5HCHNSD1 loses the ability to bind the histone H3 N-terminal tail, PHD5-C5HCHNSD2 targets the H3 tail containing unmodified K4 and K9, whereas PHD5-C5HCHNSD3 prefers to bind the H3 peptide containing unmodified K4 and trimethylated K9. These differences associated with the PHD5-C5HCHs of the NSD proteins shed light on the functional diversity of this family.
DISCUSSION
The H3 Recognition Feature of Each NSD Family Member Is Conserved Across a Wide Variety of Species
The structural and biochemical data reveal that the PHD5 domains of the three human NSD proteins have distinct binding abilities and preferences toward the H3 N-terminal tail, which comes from minor sequence variations that are located on the H3 binding surface. Because our analysis was carried out predominantly using human NSD proteins, it would be interesting to determine whether this is a common phenomenon in other species.
There is only one NSD homolog called maternal-effect sterile 4 in worms or flies, which contains no PHD5-C5HCH fingers (Fig. 1A). All vertebrates, including species ranging from zebrafish to humans, have three NSD members and each member contains a PHD5-C5HCH region. A detailed sequence alignment of each NSD PHD5-C5HCH region along with homologues from different species demonstrate that the key residues for the three variant binding specificities of PHD5-C5HCHs to H3 are highly conserved during evolution. The lysine residue accounting for the loss of NSD1 binding to H3 is identical among NSD1 homologues, and the key residues that are responsible for unmodified H3K4 and K9 recognition of NSD2 are totally the same as with its homologous proteins (supplemental Fig. S13, A and B). For NSD3, the preference for H3K9me3 is also well conserved in mammals (supplemental Fig. S13C). Residues that are involved in hydrophobic core formation and zinc coordination for the tandem PHD-PHD-like module have been well maintained throughout evolution (supplemental Fig. S13). These imply that the overall fold and the disparity in the H3 tail target among the three PHD5-C5HCH members of the NSD family are both well conserved across a wide variety of species, which further indicates that the discrepancy of this region in binding with histones may be associated with the functional diversity associated with the NSD family.
The C5HCH Domain Acts as a Potential Protein-Protein Interaction Module and the Interaction Property May Also Be Divergent Among the Three NSD Members
The t(5;11)(q35;p15.5) translocation, which generates the NUP98-NSD1 fusion protein, gives rise to about 5% of human acute myeloid leukemia (7). NUP98-NSD1 induces acute myeloid leukemia by enforcing expression of the HoxA7, HoxA9, HoxA10, and Meis1 proto-oncogenes. In a mouse model, the PHD5-C5HCHNSD1 motif is essential for recruiting the fusion protein to the HoxA9 gene promoter, indicating that PHD5-C5HCHNSD1 harbors a specific chromatin-targeting ability (44). However, our data strongly suggests that PHD5-C5HCHNSD1 lacks a direct binding ability to histones. Therefore, the manner in which the motif contributes to the recruitment currently remains unclear. One possibility is that PHD5-C5HCHNSD1 targets the chromatin via an adapter protein, which binds to the chromatin and the PHD5-C5HCH motif simultaneously. The C5HCHNSD1 domain has been implicated to directly interact with the C2HR domain of Nizp1 (NSD1-interacting zinc finger protein 1) (45). Nizp1 contains four consensus C2H2-type zinc fingers and the C2H2 finger is widely identified as a sequence-specific DNA-binding motif. Thus, Nizp1 is likely characterized as an adapter protein that recruits PHD5-C5HCH of NSD1 to chromatin.
Our GST pulldown assay showed that PHD5-C5HCHNSD1 strongly binds to C2HR of Nizp1, whereas PHD5-C5HCHNSD3 does not (supplemental Fig. S14A), consistent with previous reports (45). Besides, the triple mutant of PHD5-C5HCHNSD3 or PHD5-C5HCHNSD2, in which the three key Asp residues were all mutated to alanine, was proven to demonstrate a significant loss of binding to calf thymus histones, which suggested that the histone binding ability of the PHD5-C5HCH module either in NSD3 or NSD2 only originates from the binding of PHD5 to H3 N-terminal tail but not from C5HCH (supplemental Fig. S14B). Taken together, these results imply that the C5HCH domain may act as a nonhistone protein interacting motif, and similar to PHD5 recognizing specific histone H3 modifications, each NSD C5HCH may have distinct protein interaction partners. All of these raise the possibility that the PHD5-C5HCH motif has dual roles in H3 and nonhistone protein interactions, which are carried out by PHD5 and C5HCH, respectively. These hybrid interactions may play important roles in targeting the NSD proteins that mediate H3K36 dimethylation to different genomic sites and thus lead to distinct physiological outcomes.
PHD5-C5HCH Is a New Type of Tandem PHD Fingers
Thus far, only two tandem PHD finger structures have been reported in the literature. These include the tandem PHD (PHD1-PHD2, PHD12) domains of MOZ (monocytic leukemia zinc finger protein, a histone acetyltransferase) and Dpf3b (an adaptor component of BAF chromatin remodeling complex), which share high sequence identities and structural similarities (46, 47). However, the structure of PHD5-C5HCHNSD3 is considerably different from the PHD12s of MOZ and Dpf3b despite them all being tandem PHD finger modules. First, PHD1 and PHD2 of MOZ or Dpf3b associate with each other mainly through polar interactions (supplemental Fig. S15, B and C) (46, 47), whereas PHD5 and C5HCH of NSD3 associate with each other through hydrophobic interactions. Second, PHD1 and PHD2 of MOZ or Dpf3b are pressed against one another in a “face to back” direction, whereas PHD5 and C5HCH of NSD3 in a “face to side” manner (supplemental Fig. S15, A–C) (46, 47). The third, MOZ or Dpf3b recognizes the unmodified N-terminal residues and acetylated K14 of H3 with PHD2 and PHD1, respectively (supplemental Fig. S15, B and C) (46, 47), whereas PHD5-C5HCHNSD3 forms a complex with the H3 N-terminal peptide only through the first PHD (PHD5) of the tandem module (Fig. 2C and supplemental Fig. S15A). Interestingly, the PHD finger and Bromo domain of TRIM33 also associate with each other mainly through hydrophobic interactions, and the PHD finger of TRIM33 shows a similar H3 binding preference with PHD5NSD3 (40). Therefore, to some extent, PHD5-C5HCHNSD3 shares certain similarity with PHD-Bromo of TRIM33 (supplemental Fig. S15D and note the direction of the bound peptide). In summary, PHD5-C5HCH of the NSD family defines a new type of tandem PHD finger motif.
Sotos Mutations
In Sotos syndrome, multiple missense mutations that occur in the catalytic SET, PWWP2, PHD1–5, and C5HCH domains produce the same phenotype as the elimination of the entire NSD1 gene (4). The crystal structure of the SET domain of NSD1 and the histone lysine methyltransferase (HKMT) activity assays reflect that the missense mutations within the SET domain are responsible for the loss of or reduction of the histone lysine methyltransferase activity of NSD1, which may induce a berrant H3K36 methylation pattern during development (18).
In our study, we show that PHD5-C5HCHNSD1 does not directly interact with histones, and its overall structure may resemble PHD5-C5HCHNSD3. Previous work has (4) identified at least 12 missense mutations within the PHD5-C5HCH motif, 10 of which are zinc coordinating residues, and the other two mutations are conserved hydrophobic core residues of PHD5 and C5HCH, respectively (supplemental Fig. S16). Thus, Sotos syndrome mutations within PHD5-C5HCHNSD1 seem to obstruct the normal function of NSD1 through disrupting the proper folding of the PHD5-C5HCH region.
The Histone Target Specificity of PHD5-C5HCHNSD3 and Its Implication for the Function of Human NSD3
The N-terminal tail of histone H3 is rich in PTMs, thereby providing a platform for histone PTM cross-talk and a wealth of regulatory potential. Among the PTMs in H3 N-terminal tail, H3K4me3 and H3K9me3 are the most studied modifications. H3K4me3 is a hallmark of active transcription start sites, whereas H3K9me3 usually demarcates heterochromatin. In this study, we define a subgroup of PHD fingers, which conducts the H3K4me0 and H3K9 cross-talk. This subgroup contains an NTS motif that is responsible for the cross-talk. In addition to this subgroup of PHD fingers, the ADD (ATRX-DNMT3-DNMT3L) domain of ATRX was recently confirmed to be an H3K4me0 and H3K9me3 dual reader, in which an atypical “polar pocket” is utilized for K9me3 readout (48, 49).
The preference for H3K9me3 of these fingers implicates a role associating with heterochromatin. ADDATRX was recruited to pericentromeric heterochromatin in an H3K9me3-dependent manner (48, 49). Similarly, the NuRD (nucleosome remodeling and histone deacetylase) complex, which contains CHD4, has been shown to localize to heterochromatin and display CHD4-dependent activity in heterochromatin maintenance and assembly (50). In contrast to ATRX and CHD4 associated with heterochromatin, the PHD-Bromo cassette of TRIM33 recognizes a poised chromatin platform that contains H3K9me3 and K18ac present in the promoter region of master regulators in embryonic stem cells (40).
The details concerning how PHD5-C5HCHNSD3 contributes to the recruitment of NSD3 to specific genomic regions in vivo is still largely unknown. Shi et al. (21) recently reported that NSD3 participates in transcriptional elongation. Their research indicates that NSD3 can form complexes with H3K4-specific demethylase LSD2 and H3K9-specific methyltransferase G9a. Strikingly, the preferable simultaneous binding of PHD5-C5HCHNSD3 to unmethylated H3K4 and methylated H3K9, which are just demethylation and methylation products of LSD2 and G9a, indicates that NSD3 might function downstream of LSD2 and G9a. On the other hand, the distinct distribution of LSD2, which shows binding preferentially at the 3′ end of actively transcribed genes, is similar with the pattern of H3K36 methylation deposition (20). Therefore, our results support a model: the LSD2 and G9a enzymes first mediate methylated H3K4 demethylation and H3K9 methylation at the 3′ end of actively transcribed genes, producing a histone modification pattern recognized by the PHD5 finger of NSD3, which helps NSD3 locate and then methylate H3K36. However, additional investigations are required to fully understand how LSD2, G9a, and NSD3 orchestrate appropriate histone modifications for optimal transcription elongation.
In summary, it is the first time a detailed analysis on the reader domains of the NSD family is presented. We identify the different histone binding specificities of PHD5-C5HCHs and provide the underlying molecular basis. Thus our findings offer insights into the role of this novel tandem module, suggesting that it may play a role in targeting the NSD enzymes to different genomic sites and thus lead to distinct biological outcomes. Our results are consistent with the nonredundancy of NSD proteins and may also imply a link between the deposition of H3K36 methylation and the recognition of H3K4 and H3K9. Another important question emerges concerning the chromatin targeting roles of other reader domains, including the PHD1–4 and the two PWWP domains. Further studies are required to establish an understanding of the interplay between the reader domains and the catalytic SET domain. Each new piece of information seeks to elucidate the underlying mechanisms of the NSD family action.
Acknowledgments
We thank Dr. Lorenzo Finci, Dr. Qingguo Gong, Dr. Ying Liu, and Dr. Hongda Huang for critical reading of the manuscript. We thank Prof. Mian Wu, Dr. Guohong Li, Fan Yang, Qian Zhao, Dr. Yu Qiu, Lei Liu, and Rongsheng Ma for helpful discussions about the experiments and manuscript. We thank Dr. Jianye Zang, Minhao Wu, Deshun Gong, and Zhenhua Shao for assistance with x-ray data collection and data processing. We thank the staff at BL17U of Shanghai Synchrotron Radiation Facilities (SSRF) for assistance with x-ray data collection.
This work was supported by National Basic Research Program of China 973 Program Grants 2011CB966302, 2012CB917201, and 2011CB911104 and Chinese National Natural Science Foundation Grants 30830031 and 31170693.

This article contains supplemental Tables S1 and S2 and Figs. S1–S16.
The atomic coordinates and structure factors (codes 4GND, 4GNE, 4GNF, and 4GNG) have been deposited in the Protein Data Bank (http://wwpdb.org/).
The resonance assignments of PHD5-C5HCHNSD3 have been deposited in the Biological Magnetic Resonance Data Bank (BMRB) under accession numbers 18664.
- PTM
- post-translational modification
- NSD
- nuclear receptor SET domain
- ITC
- isothermal titration calorimetry
- HSQC
- heteronuclear single quantum coherence
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
REFERENCES
- 1. Strahl B. D., Allis C. D. (2000) The language of covalent histone modifications. Nature 403, 41–45 [DOI] [PubMed] [Google Scholar]
- 2. Taverna S. D., Li H., Ruthenburg A. J., Allis C. D., Patel D. J. (2007) How chromatin-binding modules interpret histone modifications. Lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 14, 1025–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kurotaki N., Imaizumi K., Harada N., Masuno M., Kondoh T., Nagai T., Ohashi H., Naritomi K., Tsukahara M., Makita Y., Sugimoto T., Sonoda T., Hasegawa T., Chinen Y., Tomita Ha H. A., Kinoshita A., Mizuguchi T., Yoshiura Ki K., Ohta T., Kishino T., Fukushima Y., Niikawa N., Matsumoto N. (2002) Haploinsufficiency of NSD1 causes Sotos syndrome. Nat. Genet. 30, 365–366 [DOI] [PubMed] [Google Scholar]
- 4. Faravelli F. (2005) NSD1 mutations in Sotos syndrome. Am. J. Med. Genet. C 137, 24–31 [DOI] [PubMed] [Google Scholar]
- 5. Wright T. J., Ricke D. O., Denison K., Abmayr S., Cotter P. D., Hirschhorn K., Keinänen M., McDonald-McGinn D., Somer M., Spinner N., Yang-Feng T., Zackai E., Altherr M. R. (1997) A transcript map of the newly defined 165 kb Wolf-Hirschhorn syndrome critical region. Hum. Mol. Genet. 6, 317–324 [DOI] [PubMed] [Google Scholar]
- 6. Rosati R., La Starza R., Veronese A., Aventin A., Schwienbacher C., Vallespi T., Negrini M., Martelli M. F., Mecucci C. (2002) NUP98 is fused to the NSD3 gene in acute myeloid leukemia associated with t(8;11)(p11.2;p15). Blood 99, 3857–3860 [DOI] [PubMed] [Google Scholar]
- 7. Cerveira N., Correia C., Dória S., Bizarro S., Rocha P., Gomes P., Torres L., Norton L., Borges B. S., Castedo S., Teixeira M. R. (2003) Frequency of NUP98-NSD1 fusion transcript in childhood acute myeloid leukaemia. Leukemia 17, 2244–2247 [DOI] [PubMed] [Google Scholar]
- 8. Keats J. J., Reiman T., Maxwell C. A., Taylor B. J., Larratt L. M., Mant M. J., Belch A. R., Pilarski L. M. (2003) In multiple myeloma, t(4;14)(p16;q32) is an adverse prognostic factor irrespective of FGFR3 expression. Blood 101, 1520–1529 [DOI] [PubMed] [Google Scholar]
- 9. Keats J. J., Maxwell C. A., Taylor B. J., Hendzel M. J., Chesi M., Bergsagel P. L., Larratt L. M., Mant M. J., Reiman T., Belch A. R., Pilarski L. M. (2005) Overexpression of transcripts originating from the MMSET locus characterizes all t(4;14)(p16;q32)-positive multiple myeloma patients. Blood 105, 4060–4069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Angrand P. O., Apiou F., Stewart A. F., Dutrillaux B., Losson R., Chambon P. (2001) NSD3, a new SET domain-containing gene, maps to 8p12 and is amplified in human breast cancer cell lines. Genomics 74, 79–88 [DOI] [PubMed] [Google Scholar]
- 11. Lucio-Eterovic A. K., Carpenter P. B. (2011) An open and shut case for the role of NSD proteins as oncogenes. Transcription 2, 158–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wagner E. J., Carpenter P. B. (2012) Understanding the language of Lys36 methylation at histone H3. Nat. Rev. Mol. Cell Biol. 13, 115–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rayasam G. V., Wendling O., Angrand P. O., Mark M., Niederreither K., Song L., Lerouge T., Hager G. L., Chambon P., Losson R. (2003) NSD1 is essential for early post-implantation development and has a catalytically active SET domain. EMBO J. 22, 3153–3163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nimura K., Ura K., Shiratori H., Ikawa M., Okabe M., Schwartz R. J., Kaneda Y. (2009) A histone H3 lysine 36 trimethyltransferase links Nkx2–5 to Wolf-Hirschhorn syndrome. Nature 460, 287–291 [DOI] [PubMed] [Google Scholar]
- 15. Douglas J., Coleman K., Tatton-Brown K., Hughes H. E., Temple I. K., Cole T. R., Rahman N., and Childhood Overgrowth Collaboration (2005) Evaluation of NSD2 and NSD3 in overgrowth syndromes. Eur. J. Hum. Genet. 13, 150–153 [DOI] [PubMed] [Google Scholar]
- 16. Li Y., Trojer P., Xu C. F., Cheung P., Kuo A., Drury W. J., 3rd, Qiao Q., Neubert T. A., Xu R. M., Gozani O., Reinberg D. (2009) The target of the NSD family of histone lysine methyltransferases depends on the nature of the substrate. J. Biol. Chem. 284, 34283–34295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kuo A. J., Cheung P., Chen K., Zee B. M., Kioi M., Lauring J., Xi Y., Park B. H., Shi X., Garcia B. A., Li W., Gozani O. (2011) NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol. Cell 44, 609–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Qiao Q., Li Y., Chen Z., Wang M., Reinberg D., Xu R. M. (2011) The structure of NSD1 reveals an autoregulatory mechanism underlying histone H3K36 methylation. J. Biol. Chem. 286, 8361–8368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lucio-Eterovic A. K., Singh M. M., Gardner J. E., Veerappan C. S., Rice J. C., Carpenter P. B. (2010) Role for the nuclear receptor-binding SET domain protein 1 (NSD1) methyltransferase in coordinating lysine 36 methylation at histone 3 with RNA polymerase II function. Proc. Natl. Acad. Sci. U.S.A. 107, 16952–16957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fang R., Barbera A. J., Xu Y., Rutenberg M., Leonor T., Bi Q., Lan F., Mei P., Yuan G. C., Lian C., Peng J., Cheng D., Sui G., Kaiser U. B., Shi Y., Shi Y. G. (2010) Human LSD2/KDM1b/AOF1 regulates gene transcription by modulating intragenic H3K4me2 methylation. Mol. Cell 39, 222–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shi X., Hong T., Walter K. L., Ewalt M., Michishita E., Hung T., Carney D., Peña P., Lan F., Kaadige M. R., Lacoste N., Cayrou C., Davrazou F., Saha A., Cairns B. R., Ayer D. E., Kutateladze T. G., Shi Y., Côté J., Chua K. F., Gozani O. (2006) ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature 442, 96–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe. A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 23. Shen W., Xu C., Huang W., Zhang J., Carlson J. E., Tu X., Wu J., Shi Y. (2007) Solution structure of human Brg1 bromodomain and its specific binding to acetylated histone tails. Biochemistry 46, 2100–2110 [DOI] [PubMed] [Google Scholar]
- 24. Farrow N. A., Muhandiram R., Singer A. U., Pascal S. M., Kay C. M., Gish G., Shoelson S. E., Pawson T., Forman-Kay J. D., Kay L. E. (1994) Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 33, 5984–6003 [DOI] [PubMed] [Google Scholar]
- 25. Battye T. G., Kontogiannis L., Johnson O., Powell H. R., Leslie A. G. (2011) iMOSFLM. A new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 67, 271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 27. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sheldrick G. M. (2008) A short history of SHELX. Acta Crystallogr. A 64, 112–122 [DOI] [PubMed] [Google Scholar]
- 29. Langer G., Cohen S. X., Lamzin V. S., Perrakis A. (2008) Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 32. Vagin A., Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22–25 [DOI] [PubMed] [Google Scholar]
- 33. Laskowski R. A., Macarthur M. W., Moss D. S., Thornton J. M. (1993) Procheck. A program to check the sterochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 34. Holm L., Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lan F., Collins R. E., De Cegli R., Alpatov R., Horton J. R., Shi X., Gozani O., Cheng X., Shi Y. (2007) Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene repression. Nature 448, 718–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li Y., Li H. (2012) Many keys to push. Diversifying the ”readership“ of plant homeodomain fingers. Acta Biochim. Biophys. Sin. 44, 28–39 [DOI] [PubMed] [Google Scholar]
- 37. Slama P., Geman D. (2011) Identification of family-determining residues in PHD fingers. Nucleic Acids Res. 39, 1666–1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chignola F., Gaetani M., Rebane A., Org T., Mollica L., Zucchelli C., Spitaleri A., Mannella V., Peterson P., Musco G. (2009) The solution structure of the first PHD finger of autoimmune regulator in complex with non-modified histone H3 tail reveals the antagonistic role of H3R2 methylation. Nucleic Acids Res. 37, 2951–2961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mansfield R. E., Musselman C. A., Kwan A. H., Oliver S. S., Garske A. L., Davrazou F., Denu J. M., Kutateladze T. G., Mackay J. P. (2011) Plant homeodomain (PHD) fingers of CHD4 are histone H3-binding modules with preference for unmodified H3K4 and methylated H3K9. J. Biol. Chem. 286, 11779–11791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xi Q., Wang Z., Zaromytidou A. I., Zhang X. H., Chow-Tsang L. F., Liu J. X., Kim H., Barlas A., Manova-Todorova K., Kaartinen V., Studer L., Mark W., Patel D. J., Massagué J. (2011) A poised chromatin platform for TGF-β access to master regulators. Cell 147, 1511–1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Qin S., Jin L., Zhang J., Liu L., Ji P., Wu M., Wu J., Shi Y. (2011) Recognition of unmodified histone H3 by the first PHD finger of bromodomain-PHD finger protein 2 provides insights into the regulation of histone acetyltransferases monocytic leukemic zinc-finger protein (MOZ) and MOZ-related factor (MORF). J. Biol. Chem. 286, 36944–36955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Letunic I., Doerks T., Bork P. (2012) SMART 7. Recent updates to the protein domain annotation resource. Nucleic Acids Res. 40, D302–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Punta M., Coggill P. C., Eberhardt R. Y., Mistry J., Tate J., Boursnell C., Pang N., Forslund K., Ceric G., Clements J., Heger A., Holm L., Sonnhammer E. L., Eddy S. R., Bateman A., Finn R. D. (2012) The Pfam protein families database. Nucleic Acids Res. 40, D290–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang G. G., Cai L., Pasillas M. P., Kamps M. P. (2007) NUP98-NSD1 links H3K36 methylation to Hox-A gene activation and leukaemogenesis. Nat. Cell Biol. 9, 804–812 [DOI] [PubMed] [Google Scholar]
- 45. Nielsen A. L., Jørgensen P., Lerouge T., Cerviño M., Chambon P., Losson R. (2004) Nizp1, a novel multitype zinc finger protein that interacts with the NSD1 histone lysine methyltransferase through a unique C2HR motif. Mol. Cell. Biol. 24, 5184–5196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zeng L., Zhang Q., Li S., Plotnikov A. N., Walsh M. J., Zhou M. M. (2010) Mechanism and regulation of acetylated histone binding by the tandem PHD finger of DPF3b. Nature 466, 258–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Qiu Y., Liu L., Zhao C., Han C., Li F., Zhang J., Wang Y., Li G., Mei Y., Wu M., Wu J., Shi Y. (2012) Combinatorial readout of unmodified H3R2 and acetylated H3K14 by the tandem PHD finger of MOZ reveals a regulatory mechanism for HOXA9 transcription. Genes Dev. 26, 1376–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Iwase S., Xiang B., Ghosh S., Ren T., Lewis P. W., Cochrane J. C., Allis C. D., Picketts D. J., Patel D. J., Li H., Shi Y. (2011) ATRX ADD domain links an atypical histone methylation recognition mechanism to human mental-retardation syndrome. Nat. Struct. Mol. Biol. 18, 769–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Eustermann S., Yang J. C., Law M. J., Amos R., Chapman L. M., Jelinska C., Garrick D., Clynes D., Gibbons R. J., Rhodes D., Higgs D. R., Neuhaus D. (2011) Combinatorial readout of histone H3 modifications specifies localization of ATRX to heterochromatin. Nat. Struct. Mol. Biol. 18, 777–782 [DOI] [PubMed] [Google Scholar]
- 50. Musselman C. A., Ramírez J., Sims J. K., Mansfield R. E., Oliver S. S., Denu J. M., Mackay J. P., Wade P. A., Hagman J., Kutateladze T. G. (2012) Bivalent recognition of nucleosomes by the tandem PHD fingers of the CHD4 ATPase is required for CHD4-mediated repression. Proc. Natl. Acad. Sci. U.S.A. 109, 787–792 [DOI] [PMC free article] [PubMed] [Google Scholar]