

Background: Small nuclear RNA (snRNA) 3′ end processing is carried out by the poorly understood integrator complex.
Results: An essential microdomain within IntS12 binds IntS1 and is required for integrator complex activity.
Conclusion: A small binding interface between the largest and smallest integrator subunits is critical for snRNA processing.
Significance: These data uncover the unexpected findings that the IntS12 PHD finger is nearly dispensable for snRNA processing, whereas a microdomain is required.
Keywords: Drosophila, Protein-Protein Interactions, RNA, RNA Processing, Transcription Termination, IntS12 Microdomain, Integrator Complex, snRNA Biosynthesis
Abstract
The Drosophila integrator complex consists of 14 subunits that associate with the C terminus of Rpb1 and catalyze the endonucleolytic cleavage of nascent snRNAs near their 3′ ends. Although disruption of almost any integrator subunit causes snRNA misprocessing, very little is known about the role of the individual subunits or the network of structural and functional interactions that exist within the complex. Here we developed an RNAi rescue assay in Drosophila S2 cells to identify functional domains within integrator subunit 12 (IntS12) required for snRNA 3′ end formation. Surprisingly, the defining feature of the Ints12 protein, a highly conserved and centrally located plant homeodomain finger domain, is not required for reporter snRNA 3′ end cleavage. Rather, we find a small, 45-amino acid N-terminal microdomain to be both necessary and nearly sufficient for snRNA biogenesis in cells depleted of endogenous IntS12 protein. This IntS12 microdomain can function autonomously, restoring full integrator processing activity when introduced into a heterologous protein. Moreover, mutations within the microdomain not only disrupt IntS12 function but also abolish binding to other integrator subunits. Finally, the IntS12 microdomain is sufficient to interact and stabilize the putative scaffold integrator subunit, IntS1. Collectively, these results identify an unexpected interaction between the largest and smallest integrator subunits that is essential for the 3′ end formation of Drosophila snRNA.
Introduction
The accurate production of snRNAs is an important bioprocess needed for efficient downstream RNA processing events, including pre-mRNA intron removal and histone mRNA 3′ end formation (reviewed in Refs. 1, 2). With the exception of the U6 snRNA, spliceosomal snRNAs are transcribed by RNA polymerase II (RNAPII)2, and their 3′ ends are processed by the integrator complex. Both the snRNA promoter and the 3′ box sequence element located downstream of the cleavage site have been established as features required for snRNA 3′ end formation in metazoans (3–5). Unlike the rigid requirements of the poly(A) signal in protein coding genes or the histone downstream element in histone genes, the snRNA 3′ box can be removed with only minor perturbation to snRNA biosynthesis (6, 7). This demonstrates that the snRNA processing machinery is moderately tolerant to mutations within demarcating cis elements and that specificity of the RNA cleavage event is in part brought about through additional means.
Precise snRNA 3′ end cleavage is predicated on transcription being initiated from an snRNA promoter, with distinct RNAPII C-terminal domain protein modifications found on transcription complexes active at snRNA loci. Phosphorylations within the C-terminal domain heptad repeat at serines 2 and 7 have been shown to be essential for integrator recruitment and subsequent snRNA 3′ end formation (8, 9). Replacement of native snRNA promoters with RNAPII promoters from protein-coding genes prevents proper snRNA 3′ end formation, consistent with the notion that the integrator complex is assembled onto the RNAPII complex early in the transcription cycle (3, 5).
The initial biochemical identification of the integrator complex, subsequent analyses of immunoprecipitates, and a recent genome-wide RNAi screen have identified 14 members of the complex to date (IntS1 through IntS12, Asun/IntS13 and IntS14) (10–13). Each member of the human integrator complex is conserved in Drosophila, and RNAi-mediated depletion of nearly any integrator subunit in S2 cells causes snRNA misprocessing (7). This latter result suggests the existence of a network of interactions within the integrator complex that is highly sensitive to disruption.
The only well established protein-protein interaction among integrator subunits is between IntS9 and IntS11 (14, 15). These two proteins contain highly conserved metallo-β-lactamase and β-CASP (CPSF, Artemis, SNM1/PSO2) domains and likely represent the catalytic core of the complex (reviewed in Ref. 16). They form a highly stable heterodimer in vivo, and their association is mediated through conserved C-terminal domains on both proteins. Formation of this heterodimer is required for snRNA 3′ end formation and likely is important to activate the endonuclease activity of IntS11 (14). The role of the remaining subunits in snRNA 3′ end formation has yet to be determined, and functional domains within other subunits have yet to be identified experimentally.
There are several evolutionarily conserved motifs identifiable within integrator subunits in addition to the conserved β-CASP/β-lactamase domains of IntS9/11 (reviewed in Ref. 17). These include the HEAT (Huntingtin, Elongation factor 3, protein phosphatase 2A, TOR1) repeats within IntS4, a von Willebrand factor type A domain within IntS6, and in IntS12 a plant homeodomain (PHD) finger. PHD fingers typically consist of ∼60 amino acid motifs, comprising a C4HC3 signature, that coordinate two zinc ions (reviewed in Refs. 18, 19). Proteins containing PHD fingers are almost exclusively found in the nucleus and are commonly present in protein complexes that govern transcriptional regulation. The PHD finger itself robustly interacts with N-terminal tails of histones, most commonly histone H3. Typically, PHD fingers possess specificity toward unique chemical modifications of amino acids in histone H3 with a particular preference toward lysine methylation. These attributes make analysis of the role of the IntS12 PHD finger in snRNA 3′ end formation an attractive entry point to further our understanding of integrator subunit function.
Here we investigate the role of Drosophila IntS12 in snRNA 3′ end formation using snRNA-specific GFP reporters to assess integrator complex activity. To identify regions of IntS12 required for integrator activity, we devised an RNAi rescue strategy to re-express RNAi-resistant forms of IntS12 mRNA in cells depleted for endogenous IntS12 protein. We unexpectedly determined the PHD finger to be dispensable for IntS12 activity and instead identified a small microdomain at the N terminus of IntS12 that is essential for activity. Surprisingly, the IntS12 microdomain is fully sufficient to rescue the snRNA processing defect from our reporters and nearly sufficient to rescue misprocessing of endogenous snRNAs observed in IntS12-depleted cells. Moreover, the microdomain is sufficient to mediate interaction between integrator subunits and a heterologous protein. We further show that the IntS12 microdomain interacts with IntS1 in the absence of other integrator subunits and that the stability of these two subunits requires their interaction. Collectively, these results suggest that a critical regulatory function for IntS1 and IntS12 is required for integrator activity in snRNA 3′ end formation.
EXPERIMENTAL PROCEDURES
Cloning and Plasmids
The U4-GFP construct was constructed similar to the previously described U7-GFP reporter (7), except that the U4:39B snRNA gene (CR31625), including the promoter, coding region, and 3′ untranslated region, was cloned upstream of the enhanced GFP open reading frame. The RNAi-resistant IntS12 cDNA (IntS12*) was designed using the online tool described by Schulz et al. (20) to avoid introducing rare codons or potential splice sites. A full-length IntS12* cDNA, encoding the entire IntS12 protein, including 188 silent mutations between bases 1 and 418, was synthesized by Integrated DNA Technologies (Coralville, IA) and cloned into pUB-Myc or pUB-FLAG. The pUB vectors were created using pUC19 as a backbone where we introduced the ubi-p63E promoter as well as the OpIE2 poly(A) site. Coding fragments used to complement the double-stranded RNA-mediated depletion of endogenous IntS12 were amplified from this IntS12* cDNA. Alanine-scanning mutants were generated from IntS12* by site-directed mutagenesis of groups of five amino acids. All oligonucleotide sequences used for cloning and PCR are available upon request. IntS12 microdomain secondary structure predictions were performed using the protein secondary structure prediction tool PSIPRED provided at the University College London Department of Computer Science Bioinformatics Group.
S2 Cell Culture and RNAi Treatment
Oligonucleotides (Sigma) containing T7 RNA polymerase promoter sequences at their 5′ ends were designed to amplify bases 419–740 (IntS12#1) or bases 1–418 (IntS12#2) of full-length IntS12 cDNA (Drosophila Genomics Resource Centre, Indiana University). dsRNA was generated by in vitro transcription of these amplicons with T7 RNA polymerase by standard methods. Gene silencing was initiated (day 0) by adding 1 μg of dsRNA into each well of a 96-well plate followed by 100 μl of Sf-900 II media (Invitrogen) containing 5 × 104 of S2 cells. Additional aliquots of 1 μg of dsRNA were added to each well on day 1 and again on day 2. To assess snRNA processing activity in dsRNA-treated S2 cells, 50 ng of reporter plasmids (U7-GFP or U4-GFP constructs) were transfected per well per 96-well plate using the Effectene transfection reagent (Qiagen, Hilden, Germany) on day 3. Cells were harvested for biochemical analysis on day 5 using radioimmunoprecipitation assay buffer (50 mm Tris-HCl (pH 8), 150 mm NaCl, 1% Nonidet P-40, 0.5% Na deoxycholate, 0.1% SDS) for protein expression studies or TRIzol for total RNA extraction. For the RNAi rescue/reporter experiments, RNAi-resistant rescue and GFP reporter plasmids were cotransfected into S2 cells on day 2, an additional 1 μg of dsRNA was added to cells on day 3, and cells were visualized by fluorescence microscopy on day 5 before being harvested for Western blot analysis. To generate S2 stable cell lines, FLAG-tagged cDNA clones were cotransfected with a blastocidin-expressing plasmid in a ratio 19:1 (w/w) using Effectene transfection reagent (Qiagen). Selection was performed in Drosophila media (Invitrogen) containing 10% FBS and 25 μg/ml blastocidin. Nuclear extracts from S2 stable cells were prepared as described previously (21).
Real-time PCR
Total RNA was subject to reverse transcription using Moloney Murine Leukemia Virus Reverse Transcriptase (MMLV-RT) (Invitrogen) to generate cDNA using standard methods. Quantitative real-time PCR was performed using primers specific for endogenous premature snRNAs (7) in SYBR Green master mix (Thermo Fisher Scientific, Waltham, MA). Data were analyzed using the ΔΔCt method with Rps17 as the reference gene and LacZ dsRNA-treated cells as control, as described previously (7).
Immunoprecipitation
For immunoprecipitation of FLAG-tagged proteins, 1 mg of S2 cell nuclear extracts were incubated with 20 μl of anti-FLAG M2 affinity gel (Sigma-Aldrich) with constant rotation for 2 h at 4 °C in buffer D (20 mm HEPES (pH7.9), 20% glycerol, 0.1 m KCl, 0.2 mm EDTA, 0.5 mm PMSF) plus 0.1% Triton X-100. Beads were washed twice with 1× TBS (50 mm Tris HCl (pH 7.4), 150 mm NaCl) and then with buffer D plus 0.1% Triton X-100. Samples were transferred to a new tube, and the buffer D plus 0.1% Triton X-100 was repeated. Finally, washed beads were suspended in 50 μl of 1× Laemmli buffer, boiled at 95 °C for 3 min, and then supernatants were resolved on a 12.5% SDS-PAGE gel. Electrophoresis and Western blot analysis were carried out using standard procedures. The generation of antibodies for IntS1, IntS9, and IntS12 were described previously (7). Anti-Myc (Santa Cruz Biotechnology, Santa Cruz, CA), Anti-FLAG (Sigma, catalog no. F3165), anti-HA (Covance, Princeton, NJ), and anti-GFP (Clontech, Palo Alto, CA) antibodies were obtained commercially.
Yeast Two-hybrid Assay
Full-length coding regions of the Drosophila integrator subunits were obtained from the Drosophila Genomics Resource Center and cloned in-frame into the pGBKT7 vector (BD). IntS12 full-length, first 45 amino acid microdomain (N45), N-terminal truncation (ΔN), and functionally deficient alanine-scanning mutants Mt3 and Mt5 were cloned into pGADT7 (AD) using standard methods. Pairwise cotransformations of the AD and BD constructs into yeast strain AH109 were performed according to the instructions of the manufacturer (Matchmaker 3 system, Clontech). Empty vectors were cotransformed to control for construct autoactivation. Positive interactions were analyzed using nutritional selection by spotting serial 10-fold dilutions on Synthetic Complete plates lacking leucine/tryptophan (vector control) or, additionally, histidine (medium stringency). Images of colony growth were taken after 3 or 5 days of incubation at 30 °C. To determine the HA-tagged protein expression, yeast transformants were harvested at ∼0.8 A600 in SC-Leu medium, lysed using the glass beads method, and tagged proteins were detected by Western blotting using anti-HA antibody (Covance, Princeton NJ).
RESULTS
Development of an RNAi Rescue Assay to Study IntS12 Function
We previously developed a GFP-based reporter system for in vivo monitoring of U7 snRNA 3′ end formation in Drosophila cells (7, 13). The advantage of the U7-GFP reporter as a method of monitoring integrator complex activity is that loss of activity can be readily detected in vivo with high sensitivity via GFP fluorescence. A potential limitation of this reporter is that it uses the U7 snRNA gene, which is somewhat atypical from the spliceosomal snRNA genes because of unique variations within its core promoter elements and a non-consensus Sm binding site (22, 23). To control for potential experimental bias, we created a second analogous spliceosomal snRNA reporter on the basis of the Drosophila U4:39B snRNA gene. To test the functionality of this new reporter (U4-GFP), we treated S2 cells with two separate non-overlapping dsRNAs (IntS12#1 or IntS12#2) to induce RNAi-mediated depletion of IntS12. Cells were then transfected with either the U7-GFP or U4-GFP reporter to assess integrator complex functionality (Fig. 1A). Depletion of IntS12 resulted in robust levels of GFP expression from both reporters relative to control dsRNA (LacZ)-treated cells. Western blot analysis of lysates prepared from treated cells confirmed both the loss of endogenous IntS12 expression and a consequential increase in GFP production from both reporters (Fig. 1B). Although the overall sensitivity of the U4-GFP reporter is similar to that of the U7-GFP reporter, the U4-GFP reporter displayed greater specificity, as seen by the lower background GFP in the control-treated cells. Finally, to confirm that dsRNA depletion of IntS12 led to misprocessing of endogenous snRNAs, we isolated total RNA and performed quantitative real-time PCR analysis using primers that specifically detect the misprocessed forms of the U1, U2, U4, U5, and U6 snRNAs (Fig. 1C). Both dsRNAs directed against IntS12 were found to increase the levels of misprocessed U1, U2, U4, and U5 snRNA ∼2–7-fold, consistent with our previous observations (7). Moreover, as expected, depletion of IntS12 did not affect the RNAPIII-transcribed U6 snRNA 3′ end formation.
FIGURE 1.

Sensitive dual GFP reporters reveal snRNA misprocessing following IntS12 knockdown in Drosophila S2 cells. A, fluorescence and bright field (BF) images of dsRNA-treated S2 cells (upper panel) and schematic representations of the snRNA reporters (bottom panel). EGFP, enhanced GFP. B, Western blot analysis of cell lysates from A using anti-GFP or anti-IntS12 antibodies. A nonspecific band that cross-reacts with the IntS12 antibody was used as a loading control (Con.). C, graphical representation of quantitative real-time PCR of endogenous snRNA misprocessing. Shown are amplicons specific for non-processed snRNAs isolated from S2 cells treated with either IntS12#1 or IntS12#2 dsRNAs. Data are the average of triplicate independent experiments normalized to Rps17 mRNA and plotted as fold increase relative to LacZ-treated control cells.
Using the dual U7 and U4 reporter system, we undertook an RNAi rescue strategy to ultimately investigate the relative contribution of different coding domains within IntS12 to its activity. As attempts to target the endogenous IntS12 5′ or 3′ UTRs using dsRNA failed to generate adequate depletion (not shown), we developed an RNAi-resistant IntS12 expression construct containing a series of 188 silent mutations within the region (cDNA bases 1–418) targeted by the IntS12 dsRNA#2 (Fig. 2A; a complete sequence alignment between wild-type IntS12 and RNAi-resistant IntS12* is shown in supplemental Fig. S1). Expression of this RNAi-resistant IntS12 cDNA (IntS12*) from a ubiquitin promoter yielded an ∼50-kDa doublet of IntS12 protein, albeit at slightly reduced levels relative to the wild-type IntS12 cDNA expressed under the same conditions (Fig. 2B, left panel). Despite the reduced accumulation of IntS12 protein from the IntS12* cDNA-transfected cells, we could readily detect overexpressed IntS12 protein in cells treated with IntS12 dsRNA#2, demonstrating its ability to evade the RNAi response (Fig. 2B, right panel). Next we tested the ability of IntS12* to restore snRNA 3′ end processing in cells where endogenous IntS12 has been depleted. To this end, we cotransfected either the U4-GFP or U7-GFP reporters with decreasing amounts of plasmid DNA encoding IntS12* into S2 cells depleted of endogenous IntS12 after treatment with dsRNA#2. Both fluorescence microscopy and Western blot analysis of lysates from transfected cells revealed a clear dose-dependent response between the amount of transfected IntS12* and the level of snRNA 3′ end processing as measured by both reporters (Fig. 2, C and D). As 10 ng of transfected IntS12* cDNA was found to be the minimal amount required to achieve full rescue of IntS12 knockdown, this amount was used in all further experiments. Collectively, these results demonstrate that depletion of endogenous IntS12 leads to measureable and reproducible GFP expression from both U4-GFP and U7-GFP reporters and that dsRNA mediated knockdown of endogenous IntS12 can be fully rescued through the expression of the RNAi resistant IntS12* cDNA.
FIGURE 2.

RNAi-resistant IntS12* rescues dsRNA-induced U4 and U7 snRNA misprocessing. A, schematic of the features encoded in the Drosophila IntS12 (CG5491) gene showing the relative location of dsRNA#2 and the 188 silent site changes used to generate the IntS12* cDNA. B, Western blot analysis of lysates from cells transfected with myc-tagged IntS12 (wild-type) cDNA or IntS12* cDNA using anti-myc antibodies (left panel) or from cells transfected with myc vector or myc-tagged IntS12* followed by IntS12 dsRNA#2 treatment using anti-IntS12 antibodies (right panel). The asterisk represents a degradation product of the overexpressed IntS12*. Unt., untreated; Resc., rescue plasmid; VA, vector alone. C, Western blot analysis demonstrating dose-dependent rescue of the U7-GFP and U4-GFP misprocessing phenotype using the IntS12* cDNA following RNAi-mediated depletion. Lanes 2–7 are from S2 cells treated with IntS12 dsRNA#2 subsequently cotransfected with reporter plasmid and rescue plasmid DNAs at doses of rescue plasmid indicated in nanograms. D, representative fluorescence images of S2 cells treated as described in C. In all panels, an ∼60-kDa cross-reacting band with anti-IntS1 polyclonal antibodies is used as the loading control (Con.).
The N Terminus Is Both Necessary and Sufficient for IntS12 Function
Alignment of IntS12 protein sequences from multiple diverse species identified two regions with significant homology. The largest region includes the amino acids within the PHD finger known to be important for zinc coordination as well as several other residues known to be essential for maintaining PHD structure (18). A smaller conserved region within the first 50 amino acids of the N terminus bears no resemblance to other known motifs. Lastly, the C-terminal region consists of a poorly conserved serine-rich region (supplemental Fig. S2). To investigate the functional role of these conserved features, we generated a series of RNAi-resistant deletion constructs derived from the IntS12* cDNA and tested their ability to rescue reporter snRNA misprocessing in cells depleted of endogenous IntS12 (Fig. 3A). Constructs lacking various combinations of the N terminus, centrally located PHD finger domain, or serine-rich C terminus were myc-tagged and transfected into S2 cells. Western blot analysis using anti-myc antibodies confirmed protein expression from all constructs (Fig. 3B). To determine the relative ability of these deletion mutants to restore snRNA processing after knockdown of endogenous IntS12, we cotransfected each mutant construct with either the U7-GFP reporter or the U4-GFP into S2 cells pretreated with IntS12 dsRNA#2. Data were obtained using GFP fluorescence imaging (Fig. 3C), and Western blot analysis with anti-GFP antibodies were congruent (D). The results gathered using both GFP reporters were remarkably consistent and, to our surprise, demonstrated that the conserved PHD finger is not required for IntS12 to promote reporter snRNA 3′ end processing. We found that constructs containing N-terminal amino acids (ΔC, NP, N, and ΔP) were as active as full-length (FL) protein in restoring reporter snRNA 3′ end processing after endogenous IntS12 knockdown (Fig. 3D). Conversely, all mutants lacking the N terminus (C, PC, ΔN, and P) were incapable of restoring integrator activity. These results indicate that the N-terminal 45 amino acids are required for reporter snRNA 3′ end processing activity and the first 129 amino acids are sufficient, whereas the PHD domain is dispensable.
FIGURE 3.
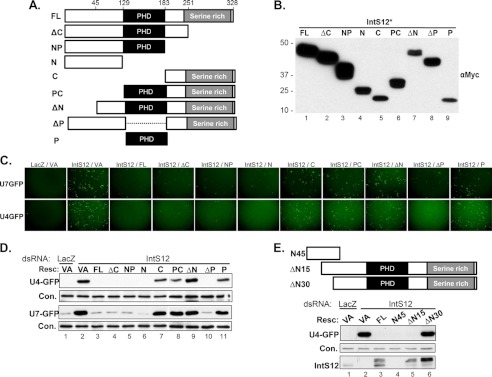
The N terminus of Drosophila IntS12 is required for snRNA 3′ end formation. A, schematic of IntS12* truncation and deletion constructs, which were designed on the basis of predicted domains. Relevant amino acid sequences are numbered. B, Western blot analysis of cell lysates isolated from S2 cells transiently transfected with plasmids encoding myc-tagged IntS12* proteins. C, representative fluorescence images of S2 cells treated with either control dsRNA or IntS12 dsRNA#2 followed by cotransfection of either the U4-GFP or U7-GFP reporters with the myc-tagged IntS12* cDNAs. D, Western blot analysis of cell lysates from C. Resc., rescue plasmid; VA, vector alone. E, schematic and Western blot analysis of three additional IntS12* deletion mutants cotransfected with the U4-GFP reporter. In all panels, an ∼60-kDa cross-reacting band with anti-IntS1 polyclonal antibodies is used as the loading control (Con.).
Mutations within an N-terminal Microdomain Disrupt IntS12 Activity
To further investigate the function of the N terminus, we generated three more IntS12 constructs derived from IntS12*. Two produced proteins harboring deletions of either the first 15 or first 30 amino acids, whereas a third generated a truncated protein consisting of only the first 45 amino acids (Fig. 3E, upper panel). The ΔN15 mutant was observed to function as well as full-length IntS12. However, deletion of the first 30 amino acids (ΔN30) abolished the ability of IntS12 to rescue misprocessing of the U4-GFP reporter (Fig. 3E, blot), despite both being expressed at levels comparable with the FL protein. In addition, expression of only the first 45 amino acids of IntS12 (N45) was sufficient to restore snRNA processing as effectively as the FL expression construct. These data demonstrate that amino acids 16–45 are required for IntS12 activity and that the first 45 amino acids are sufficient to restore reporter snRNA 3′ end processing after endogenous IntS12 knockdown.
Detailed examination of the evolutionary similarities within the N-terminal region of IntS12 (Fig. 4A) shows high conservation of the residues located within the region identified as required for IntS12 function. The combination of evolutionary conservation and requirement in snRNA 3′ end processing implicates this region as forming a functional microdomain critical for IntS12 activity. To achieve better resolution of the relative contribution of amino acids within this microdomain, we created a series of six further mutant constructs (Mt1 through Mt6) spanning amino acids 16–45, each comprising five contiguous alanine substitutions in the context of the full-length IntS12* (Fig. 4A). In instances where alanine was encoded in the wild-type protein at a specific residue, no change was introduced. Therefore, two of the mutants contained four amino acid changes (Mt2 and Mt6), and one (Mt4) contained only three substitutions. Western blot analysis confirmed each of these mutants to be expressed to similar levels at the wild-type IntS12 protein (Fig. 4B), yet they displayed strikingly different abilities to promote snRNA 3′ end processing. Mt1, Mt2, Mt4, and Mt6 were as capable as the wild-type IntS12 in restoring reporter snRNA processing. However, both Mt3 and Mt5 were completely ineffective in abrogating GFP reporter gene expression (Fig. 4B). When this same mutation series was retested in the context of the N-terminal region comprising the first 129 amino acids only, virtually identical results were obtained (not shown). These data define a highly conserved ∼15 amino acid “core” of a microdomain within the N terminus of IntS12 that is required for restoring snRNA 3′ end processing after endogenous IntS12 knockdown.
FIGURE 4.

Mapping critical residues within the N-terminal IntS12 microdomain required for snRNA 3′ end formation. A, upper panel, alignment of the IntS12 N termini of several species. Residues highlighted in blue represent similar amino acids, and yellow highlights represent identical residues. The highlighted red box denotes the identified functional microdomain residues 16–45. The labeled amino acids are the subject of six different alanine-scanning mutants. B, Western blot analysis of cell lysates treated with either control dsRNA or IntS12 dsRNA#2 that were then cotransfected with U4-GFP reporter and IntS12* plasmids containing mutations as described in A using anti-GFP and anti-IntS12 antibodies. An ∼60-kDa cross-reacting band with anti-IntS1 polyclonal antibodies is used as the loading control (Con.). Resc., rescue plasmid; VA, vector alone.
The IntS12 Microdomain Is Required for Incorporation of IntS12 into the Integrator Complex
Given the discrete and conserved nature of the IntS12 microdomain, we hypothesize that it may function as a protein-protein interaction motif required for association with other member(s) of the Integrator complex. To test this possibility, we cloned full-length RNAi-resistant IntS12* cDNAs encoding either full-length wild-type (FL), Mt3, or Mt5 proteins in-frame with an N-terminal FLAG tag to facilitate immunoprecipitation studies. Analogous constructs comprising a deletion of the first 45 amino acids of IntS12* (ΔN) or the first 45 amino acids at the N terminus (N45) were also generated. We observed that the FLAG-tagged proteins were well expressed and effectively restored 3′ end processing of both the U7-GFP and U4-GFP reporters in an identical fashion to the myc-tagged proteins described above (Fig. 5A). We further determined the effect of these FLAG-tagged proteins on endogenous snRNA processing in stable cells depleted of IntS12. We generated stable cell lines for these proteins, treated them with IntS12 dsRNA#2 and measured endogenous snRNA misprocessing using the quantitative real-time PCR assay described in Fig. 1C. We observed that in the control stable line expressing the FLAG only, depletion of IntS12 led to a 4- to 5-fold increase in the levels of misprocessed endogenous U2 or U5 snRNA. Consistent with the reporter observations, stable expression the FLAG-IntS12* (FL) in IntS12-depleted cells fully reverted the misprocessed endogenous U2 and U5 snRNAs to similar basal levels (Fig. 5B). In contrast, depletion of IntS12 in stable lines expressing FLAG-tagged Mt3, Mt5, or ΔN led to a 3- to 5-fold increase in misprocessed levels of endogenous U2 or U5 snRNA (Fig. 5B). These data demonstrate that the IntS12 microdomain is necessary to mediate the 3′ end formation of both reporter and endogenous snRNAs, thereby ruling against any reporter-derived bias.
FIGURE 5.

Loss-of-function mutations in the IntS12 microdomain disrupt IntS12 interaction with endogenous integrator subunits. A, Western blot analysis of cell lysates from S2 cells treated with either control dsRNA or IntS12 dsRNA#2 followed by cotransfection of FLAG-tagged rescue plasmids and either the U4-GFP or U7-GFP reporter. Resc., rescue plasmid; VA, vector alone. B, quantitative real-time PCR measuring levels of misprocessed endogenous U2 or U5 snRNA in stable cell lines treated with dsRNA targeting IntS12. Control (Con.) represents S2 cells expressing FLAG only, and cell lines stably expressing FLAG-tagged IntS12 proteins are labeled on the x axis. All results are plotted as fold increase relative to LacZ dsRNA treatment and normalized to RpS17 mRNA levels. C, Western blot analysis of immunoprecipitations (IP) using anti-FLAG-agarose from nuclear extracts prepared from cell lines stably expressing FLAG-tagged IntS12* proteins. Inp, input. D, Western blot analysis of cell lysates from S2 cells treated with either control dsRNA or IntS12 dsRNA#2 followed by cotransfection with U4-GFP and FLAG-mCherry plasmids with or without the N-terminal 45 amino acids of IntS12. E, Western blot analysis of immunoprecipitations using anti-FLAG-agarose from nuclear extracts purified from cell lines stably expressing FLAG-tagged IntS12* proteins and FLAG-mCherry proteins with and without the IntS12 microdomain (N45). The upper panels show probes for endogenous IntS1/9, and the bottom panel shows probes with anti-FLAG antibody to confirm pull-down. In A and D, an ∼60-kDa cross-reacting band with anti-IntS1 polyclonal antibodies is used as the loading control.
To test interactions of other integrator subunits with IntS12, we generated nuclear extracts from these same stable cells lines and immunoprecipitated IntS12-associated proteins using anti-FLAG-agarose beads (Fig. 5C). We observed robust and significant levels of endogenous IntS1 and IntS9 associating with FLAG-tagged full-length (FL) IntS12 relative to control pull-downs (Fig. 5C, lane 4 versus lane 2). IntS12 protein expressed from Mt3- or Mt5-containing constructs poorly associated with endogenous IntS1 or IntS9, and this association was completely absent in cells expressing the ΔN IntS12 microdomain truncation (Fig. 5C, lanes 6, 8, and 10 versus lane 4). These data demonstrate that mutations introduced into the IntS12 N-terminal microdomain compromise integrator activity in snRNA 3′ end processing by reducing the ability of IntS12 to interact with other integrator subunits.
As the data presented in Figs. 3E and 5A demonstrate, transient expression of only the first 45 amino acids of IntS12 is sufficient to rescue depletion of the endogenous protein. We next asked whether the IntS12 microdomain alone (N45) is sufficient to mediate interaction with endogenous integrator subunits. To circumvent the issue of low expression of the N45 peptide in stable cell lines, we generated stable cell lines expressing either FLAG-mCherry (Ch.) or FLAG-N45mCherry (N45Ch.), where the first 45 amino acids of IntS12 were fused to the N terminus of mCherry. To confirm that fusion to mCherry did not generate any unintended effects, we transiently cotransfected the U4-GFP reporter with Ch., N45Ch., or IntS12* (FL) into cells pretreated with IntS12 dsRNA#2. Western blot analysis determined that both Ch. and N45Ch. were expressed at comparable levels. However, only N45Ch. was capable of rescuing the snRNA processing defects associated with depletion of endogenous IntS12 (Fig. 5D, lane 2 versus lane 3). FLAG-tagged proteins from cell lines stably expressing these constructs were then immunoprecipitated utilizing anti-FLAG-agarose and probed for their ability to pull down endogenous IntS1 and IntS9. We found robust levels of both these subunits interacting with the N45Ch. relative to the Ch. control (Fig. 5E, lane 2 versus lane 4). We also noted that the N45Ch. was not as efficient as FLAG-tagged full-length IntS12* in interacting with either the IntS1 or IntS9 subunit, suggesting that residues beyond the first 45 amino acids are also likely contribute to the interaction between IntS12 and other members of the complex. This observation was consistent with the inability of N45Ch. to fully restore endogenous snRNA processing in stable cells expressing FLAG-N45mCherry depleted with endogenous IntS12 (Fig. 5B). Nevertheless, the N-terminal 45 amino acid IntS12 microdomain is capable of binding to IntS1 and IntS9 at levels significantly above background and able to function in snRNA 3′ end formation nearly as efficiently as the full-length protein.
The IntS12 Microdomain Is Likely to Bind IntS1 to Promote Efficient snRNA 3′ End Processing
To explore the potential mechanism for IntS12 microdomain function in snRNA 3′ end formation, we utilized a directed, pairwise yeast two-hybrid assay to determine the potential integrator binding partner for IntS12, because Saccharomyces cerevisiae does not encode any orthologous integrator proteins. We expressed full-length Drosophila IntS12 fused to the Gal4 transcriptional activation domain (AD-IntS12) and all other members of the fly integrator complex as individual fusions to the Gal4 DNA binding domain (BD-IntS1 through BD-IntS12). When individual BD fusions were coexpressed in cells along with AD-IntS12, only those expressing IntS1 and IntS10 supported growth on nutritional selection plates lacking histidine (Fig. 6A). Using this system, we were unable to conclusively determine the existence of an IntS10/12 association, as expression of BD-IntS10 alone supported growth on media lacking histidine. To determine whether the yeast two-hybrid interaction between IntS1 and IntS12 was sensitive to mutations within the N-terminal microdomain of IntS12, we expressed AD- fusions of full-length, ΔN, Mt3-, or Mt5-containing full-length IntS12 in the presence of BD-Ints1. We observed a total absence of growth on selective media lacking histidine for all IntS12 protein fusions (AD-ΔN, -Mt3, and -Mt5) that contained inactivating mutations within the N terminus identified previously as unable to complement the effects of endogenous IntS12 knockdown (Fig. 6B). Importantly, Western blotting confirmed that loss of growth was indicative of loss of interaction rather than loss of expression (Fig. 6C). Lastly, when fused to the Gal4 activation domain, the N-terminal IntS12 microdomain (AD-N45) was found to partially support yeast growth on selective media, suggesting a weak or transient interaction between the IntS12 microdomain and IntS1 (Fig. 6B). Collectively, these results demonstrate that the IntS12 microdomain is necessary and partially sufficient to interact with IntS1 in the absence of other integrators, which is likely essential for the activity of the integrator complex in snRNA 3′ end processing.
FIGURE 6.

The IntS12 microdomain is necessary and partially sufficient to mediate interaction with IntS1 in the absence of other integrator subunits. A, S. cerevisiae (AH109) was cotransformed with plasmids encoding hybrid proteins containing IntS12 fused to the Gal4 DNA activation domain (AD-IntS12) and each of the other integrator subunits fused to the Gal4 DNA binding domain (BD-IntSs). A dilution series of overnight cultures was spotted on either SC/-Leu/-Trp plates or the same media without histidine to test interaction. All BD constructs were tested for autoactivation. B, similar A, except that AH109 yeast was transformed with full-length IntS12 containing mutations 3 or 5, a deletion of the N-terminal 45 amino acids (ΔN), or the first 45 amino acids of IntS12 (N45). C, Western blot analysis confirming the expression of HA-tagged IntS12 constructs in yeast strain AH109. Con., control.
To further determine the importance of the interaction identified above, we performed a codepletion assay to test the requirement of IntS12 microdomain for IntS1 expression. As codepletion is commonly observed between interacting proteins and has been found to occur between several members of the histone pre-mRNA processing complex, including Symplekin and CPSF73 (24), we first asked whether depletion of endogenous IntS12 had a similar effect on other integrator subunits, especially IntS1. S2 cells were treated in duplicate with non-overlapping dsRNAs for each of the integrator genes for which we had available antibodies (IntS1, IntS9, IntS11, and IntS12) or with a control dsRNA (LacZ). Depletion of integrator subunits was equally effective with either dsRNAs relative to control dsRNA-treated cells (Fig. 7A). In addition, both dsRNAs targeting IntS12 were as effective in depleting IntS1 protein levels as were dsRNAs directly targeting IntS1, and, reciprocally, dsRNAs targeting IntS1 were capable of codepleting IntS12. In contrast, IntS9 and IntS11 levels were unaffected by depletion of IntS1 or IntS12, and targeting either IntS9 or IntS11 did not impact the levels of IntS1 or IntS12. These results demonstrate that the expressions of IntS1 and IntS12 are interdependent. We then repeated the codepletion experiment by depletion of the endogenous IntS12 proteins in cell lines stably expressing RNAi-resistant full-length IntS12* (FL); Mt3- or Mt5-containing full-length IntS12; or FLAG-N45mCherry. We observed that expression of the resistant full-length IntS12* blocked the codepletion of IntS1 in response to treatment of cells with IntS12 dsRNA#2, whereas neither Mt3- or Mt5-containing constructs were effective in maintaining IntS1 stability (Fig. 7B, lane 2 versus lanes 6 and 8). Consistent with the microdomain binding studies (Figs. 5 and 6), stable expression of N45Ch. was sufficient to stabilize IntS1 in cells where endogenous IntS12 had been depleted. Collectively, these data demonstrate that the IntS12 N-terminal microdomain is essential to maintain the stability of IntS1 in cells through a potential direct interaction and that this interaction is important for integrator complex function.
FIGURE 7.
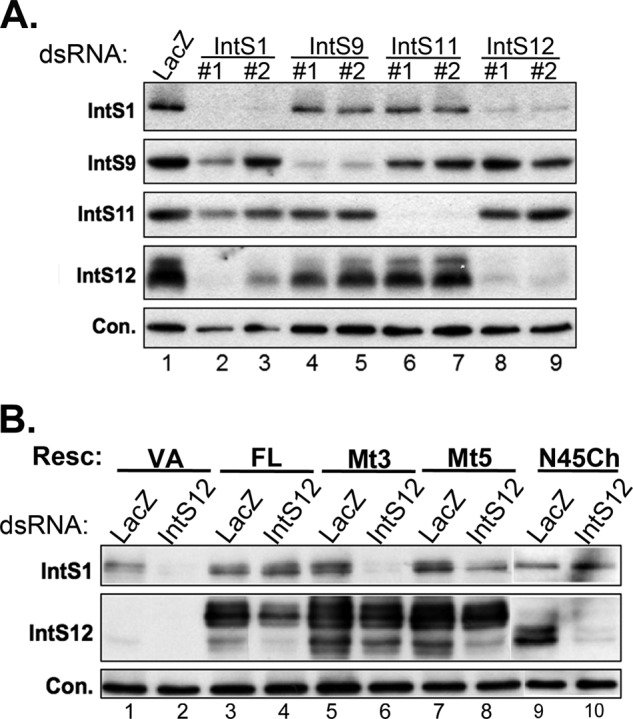
Expression of Drosophila IntS1 and IntS12 is interdependent, and stability of IntS1 requires an intact IntS12 microdomain. A, Western blot analysis of endogenous integrator subunit expression from S2 cells treated with various dsRNA targeting IntSs. An ∼60-kDa cross-reacting band with anti-IntS1 polyclonal antibodies is used as the loading control (Con.). B, Western blot analysis of endogenous IntS1 expression in S2 cells stably expressing FLAG-tagged IntS12* proteins containing mutations within the IntS12 microdomain. An ∼130-kDa cross-reacting band with anti-IntS1 polyclonal antibodies is used as the loading control. Resc., rescue plasmid; VA, vector alone.
DISCUSSION
Here we present a detailed functional analysis of the Drosophila IntS12 protein through characterizing its role in snRNA 3′ end formation. Unexpectedly, our investigations determined that the highly conserved PHD finger is not needed for IntS12 to promote nascent reporter snRNA processing (Fig. 3). Rather, we identified a small, highly conserved N-terminal 45 amino acid microdomain that is both necessary and sufficient for the restoration of integrator function in IntS12-depleted cells (Figs. 3–5). Residues within the IntS12 microdomain are also required for interaction with other endogenous integrator subunits and, when mutated, disrupt their stable interaction (Figs. 5–7). These data establish a critical role for IntS12 in regulating the activity of the integrator complex, mediated through binding to and stabilization of IntS1, the largest integrator subunit. Additionally, our approach of using dual snRNA GFP reporters in combination with functional rescue through expression of RNAi-resistant cDNAs represents a powerful tool for going forward to elucidate the function of the other integrator subunits.
Potential Functions of the IntS12 Microdomain
Our results demonstrate a strong correlation between the IntS12 microdomain binding to IntS1 and its ability to promote snRNA 3′ end formation. This argues that this small interaction motif is critical for integrator complex function. Moreover, the results shown in Fig. 5, B and D, reveal that the microdomain, when fused to mCherry, is nearly as effective as full-length IntS12 in restoring the processing of the endogenous and reporter snRNAs, thereby excluding a simple localization sequence function. However, as the FLAG-N45mCherry (N45Ch.) protein was not as effective at pulling down endogenous integrator subunits as the full-length protein, other residues of IntS12 may play an additional role in achieving maximal stability of the IntS1/IntS12 complex (Fig. 5E).
Position Specific Iterated predication (PSIPRED) secondary structure prediction posits that the IntS12 microdomain forms a helix-coil-helix structure (25–27) similar to the hepatocyte nuclear factor 1-α dimerization domain and the dimerization domain required for the heterodimeric association of the SinR-SinI antirepressor complex (28, 29). In both of these cases, small helix-coil-helix domains act as crucial protein-protein interaction motifs that elicit dimerization. On the basis of these examples, we propose that the microdomain of IntS12 binds to an as-yet-unidentified complementary domain within IntS1. This IntS1/IntS12 interaction is not only essential for IntS1 function but is also required for the stability of both IntS1 and IntS12 (Fig. 7). In this respect, the IntS12 microdomain is behaving similarly to the RNase E microdomain that interacts with the glycolytic enzyme enolase (30–32). RNase E is the essential component of the Escherichia coli RNA degradosome and contains a N-terminal catalytic domain as well as a long C-terminal scaffolding domain (reviewed in Ref. 33). The scaffold domain binds to the RhlB helicase, the phosphorolytic exoribonuclease polynucleotide phosphorylase, and enolase. The RNase E microdomain resides within the scaffolding section and consists of a ∼28 amino acid conserved region that binds to a dimeric interface between two enolase proteins to promote the decay of specific mRNAs in E. coli (30, 31, 34, 35). Given the large size of IntS1, it may simultaneously interact with multiple other members of the integrator complex in an analogous way to the scaffolding domain of RNase E. Our data support a model where binding of IntS12 to IntS1 alters IntS1 confirmation, allowing further interaction(s) with other proteins and activation of the complex.
Why Has Evolution Selected the Central IntS12 PHD Finger?
Aside from the conserved β-CASP/β-lactamase domains of IntS9/11, the central PHD finger region of IntS12 is one of the few domains found among the integrator subunits whose function can be readily inferred from sequence conservation. Members of the PHD finger family of proteins are involved in a diverse range of biological functions, yet typically bind to specific histone H3 posttranslational modifications (reviewed in Refs. 18, 19). The prediction that IntS12 forms a canonical PHD finger topology is supported by NMR structure analysis of the mouse IntS12 PHD finger (PDB code 1WEV)3. We have also experimentally confirmed that recombinant Drosophila and human IntS12 PHD finger proteins bind histone H3 in vitro4. Although nascent transcription from an snRNA promoter is essential for integrator function, a central role for chromatin in 3′ end formation has not been established (3, 5, 7, 36). Although the presence of a stable nucleosome located between the Distal Sequence Element (DSE) and Proximal Sequence Element (PSE) in human cells has been reported by several groups (37–40), the preponderance of evidence argues for a general deficiency of histones within snRNA genes, with the snRNA promoter establishing a perpetual “open” transcription state (38, 41). In Drosophila, the lack of a DSE (42) and our own observations that the Proximal Sequence Element A (PSEA) alone is sufficient to impart integrator sensitivity to our reporters are inconsistent with the prediction of a stable nucleosome at fly snRNA promoters4. These data, together with evidence described here that the PHD domain is dispensable for snRNA 3′ end processing, argues against the IntS12 PHD finger coupling the integrator complex to the snRNA promoter via histone binding.
Although we cannot exclude the possibility that the IntS12 PHD finger is involved in recruiting the integrator complex to chromatin at non-snRNA genes, there is presently no indication that the integrator complex functions elsewhere in the genome. Whether the endogenous target of the PHD finger is indeed histone H3 in vivo has also yet to be determined. There are numerous examples of PHD fingers that associate with non-histone substrates (19), including the MLL1 methyltransferase PHD3 finger bound to the RNA recognition motif of nuclear cyclophilin Cyp33 (43). The IntS12 PHD finger may behave similarly and interact with a non-histone partner, perhaps another integrator subunit. Nevertheless, on the basis of data presented here, this interaction is not critical for IntS12 function in snRNA 3′ end formation.
Acknowledgments
We thank Phil Carpenter and members of the Wagner laboratory for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant 5R00GM080447 (to E. J. W.). This work was also supported by the T. C. Hsu faculty development award and by the Queensland Cancer Fund (to E. J. W.), and by the National Health and Medical Research Council (NHMRC) through the Australian Drosophila Biomedical Research Support Facility.

This article contains supplemental Figs. S1 and S2.
F. He, Y. Muto, M. Inoue, T. Kigawa, M. Shirouzu, T. Terada, and S. Yokoyama, unpublished data.
J. Chen and E. J. Wagner, unpublished data.
- RNAPII
- RNA polymerase II
- IntS1
- integrator subunit 1
- PHD
- plant homeodomain
- FL
- full-length
- snRNA
- small nuclear RNA
- CASP
- CPSF/Artemis, SNM1, PSO2
- HEAT
- Huntingtin, Elongation factor 3, protein phosphatase 2A, TOR1
- dsRNA
- double stranded RNA
- MMLV
- Moloney Murine Leukemia Virus.
REFERENCES
- 1. Egloff S., O'Reilly D., Murphy S. (2008) Expression of human snRNA genes from beginning to end. Biochem. Soc. Trans. 36, 590–594 [DOI] [PubMed] [Google Scholar]
- 2. Matera A. G., Terns R. M., Terns M. P. (2007) Non-coding RNAs. Lessons from the small nuclear and small nucleolar RNAs. Nat. Rev. Mol. Cell Biol. 8, 209–220 [DOI] [PubMed] [Google Scholar]
- 3. de Vegvar H. E., Lund E., Dahlberg J. E. (1986) 3′ end formation of U1 snRNA precursors is coupled to transcription from snRNA promoters. Cell 47, 259–266 [DOI] [PubMed] [Google Scholar]
- 4. Hernandez N. (1985) Formation of the 3′ end of U1 snRNA is directed by a conserved sequence located downstream of the coding region. EMBO J. 4, 1827–1837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hernandez N., Weiner A. M. (1986) Formation of the 3′ end of U1 snRNA requires compatible snRNA promoter elements. Cell 47, 249–258 [DOI] [PubMed] [Google Scholar]
- 6. Ach R. A., Weiner A. M. (1987) The highly conserved U small nuclear RNA 3′-end formation signal is quite tolerant to mutation. Mol. Cell Biol. 7, 2070–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ezzeddine N., Chen J., Waltenspiel B., Burch B., Albrecht T., Zhuo M., Warren W. D., Marzluff W. F., Wagner E. J. (2011) A subset of Drosophila integrator proteins is essential for efficient U7 snRNA and spliceosomal snRNA 3′-end formation. Mol. Cell Biol. 31, 328–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Egloff S., Szczepaniak S. A., Dienstbier M., Taylor A., Knight S., Murphy S. (2010) The integrator complex recognizes a new double mark on the RNA polymerase II carboxyl-terminal domain. J. Biol. Chem. 285, 20564–20569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Egloff S., O'Reilly D., Chapman R. D., Taylor A., Tanzhaus K., Pitts L., Eick D., Murphy S. (2007) Serine-7 of the RNA polymerase II CTD is specifically required for snRNA gene expression. Science 318, 1777–1779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Baillat D., Hakimi M. A., Näär A. M., Shilatifard A., Cooch N., Shiekhattar R. (2005) Integrator, a multiprotein mediator of small nuclear RNA processing, associates with the C-terminal repeat of RNA polymerase II. Cell 123, 265–276 [DOI] [PubMed] [Google Scholar]
- 11. Malovannaya A., Lanz R. B., Jung S. Y., Bulynko Y., Le N. T., Chan D. W., Ding C., Shi Y., Yucer N., Krenciute G., Kim B. J., Li C., Chen R., Li W., Wang Y., O'Malley B. W., Qin J. (2011) Analysis of the human endogenous coregulator complexome. Cell 145, 787–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Malovannaya A., Li Y., Bulynko Y., Jung S. Y., Wang Y., Lanz R. B., O'Malley B. W., Qin J. (2010) Streamlined analysis schema for high-throughput identification of endogenous protein complexes. Proc. Natl. Acad. Sci. U.S.A. 107, 2431–2436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen J., Ezzeddine N., Albrecht T. R., Waltenspiel B., Warren W. D., Marzluff W. F., Wagner E. J. (2012) An RNAi screen identifies additional members of the Drosophila integrator complex and a requirement for cyclin C/Cdk8 in snRNA 3′ end formation. RNA 18, 2148–2156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Albrecht T. R., Wagner E. J. (2012) snRNA 3′ end formation requires heterodimeric association of integrator subunits. Mol. Cell Biol. 32, 1112–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dominski Z., Yang X. C., Purdy M., Wagner E. J., Marzluff W. F. (2005) A CPSF-73 homologue is required for cell cycle progression but not cell growth and interacts with a protein having features of CPSF-100. Mol. Cell Biol. 25, 1489–1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dominski Z. (2007) Nucleases of the metallo-β-lactamase family and their role in DNA and RNA metabolism. Crit. Rev. Biochem. Mol. Biol. 42, 67–93 [DOI] [PubMed] [Google Scholar]
- 17. Chen J., Wagner E. J. (2010) snRNA 3′ end formation. The dawn of the Integrator complex. Biochem. Soc. Trans. 38, 1082–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bienz M. (2006) The PHD finger, a nuclear protein-interaction domain. Trends Biochem. Sci. 31, 35–40 [DOI] [PubMed] [Google Scholar]
- 19. Musselman C. A., Kutateladze T. G. (2011) Handpicking epigenetic marks with PHD fingers. Nucleic Acids Res. 39, 9061–9071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schulz J. G., David G., Hassan B. A. (2009) A novel method for tissue-specific RNAi rescue in Drosophila. Nucleic Acids Res. 37, e93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dominski Z., Sumerel J., Hanson R. J., Marzluff W. F. (1995) The polyribosomal protein bound to the 3′ end of histone mRNA can function in histone pre-mRNA processing. RNA 1, 915–923 [PMC free article] [PubMed] [Google Scholar]
- 22. Dominski Z., Yang X. C., Purdy M., Marzluff W. F. (2003) Cloning and characterization of the Drosophila U7 small nuclear RNA. Proc. Natl. Acad. Sci. U.S.A. 100, 9422–9427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hernandez G., Jr., Valafar F., Stumph W. E. (2007) Insect small nuclear RNA gene promoters evolve rapidly yet retain conserved features involved in determining promoter activity and RNA polymerase specificity. Nucleic Acids Res. 35, 21–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sullivan K. D., Steiniger M., Marzluff W. F. (2009) A core complex of CPSF73, CPSF100, and Symplekin may form two different cleavage factors for processing of poly(A) and histone mRNAs. Mol. Cell 34, 322–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Buchan D. W., Ward S. M., Lobley A. E., Nugent T. C., Bryson K., Jones D. T. (2010) Protein annotation and modelling servers at University College London. Nucleic Acids Res. 38, W563–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McGuffin L. J., Bryson K., Jones D. T. (2000) The PSIPRED protein structure prediction server. Bioinformatics 16, 404–405 [DOI] [PubMed] [Google Scholar]
- 27. Jones D. T. (1999) Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292, 195–202 [DOI] [PubMed] [Google Scholar]
- 28. Colledge V. L., Fogg M. J., Levdikov V. M., Leech A., Dodson E. J., Wilkinson A. J. (2011) Structure and organisation of SinR, the master regulator of biofilm formation in Bacillus subtilis. J. Mol. Biol. 411, 597–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Johnen G., Kaufman S. (1997) Studies on the enzymatic and transcriptional activity of the dimerization cofactor for hepatocyte nuclear factor 1. Proc. Natl. Acad. Sci. U.S.A. 94, 13469–13474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chandran V., Luisi B. F. (2006) Recognition of enolase in the Escherichia coli RNA degradosome. J. Mol. Biol. 358, 8–15 [DOI] [PubMed] [Google Scholar]
- 31. Nurmohamed S., McKay A. R., Robinson C. V., Luisi B. F. (2010) Molecular recognition between Escherichia coli enolase and ribonuclease E. Acta Crystallogr. D Biol. Crystallogr. 66, 1036–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Py B., Higgins C. F., Krisch H. M., Carpousis A. J. (1996) A DEAD-box RNA helicase in the Escherichia coli RNA degradosome. Nature 381, 169–172 [DOI] [PubMed] [Google Scholar]
- 33. Carpousis A. J. (2007) The RNA degradosome of Escherichia coli. An mRNA-degrading machine assembled on RNase E. Annu. Rev. Microbiol. 61, 71–87 [DOI] [PubMed] [Google Scholar]
- 34. Bernstein J. A., Khodursky A. B., Lin P. H., Lin-Chao S., Cohen S. N. (2002) Global analysis of mRNA decay and abundance in Escherichia coli at single-gene resolution using two-color fluorescent DNA microarrays. Proc. Natl. Acad. Sci. U.S.A. 99, 9697–9702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bernstein J. A., Lin P. H., Cohen S. N., Lin-Chao S. (2004) Global analysis of Escherichia coli RNA degradosome function using DNA microarrays. Proc. Natl. Acad. Sci. U.S.A. 101, 2758–2763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cazalla D., Xie M., Steitz J. A. (2011) A primate herpesvirus uses the integrator complex to generate viral microRNAs. Mol. Cell 43, 982–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Boyd D. C., Greger I. H., Murphy S. (2000) In vivo footprinting studies suggest a role for chromatin in transcription of the human 7SK gene. Gene 247, 33–44 [DOI] [PubMed] [Google Scholar]
- 38. Pavelitz T., Bailey A. D., Elco C. P., Weiner A. M. (2008) Human U2 snRNA genes exhibit a persistently open transcriptional state and promoter disassembly at metaphase. Mol. Cell Biol. 28, 3573–3588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stünkel W., Kober I., Seifart K. H. (1997) A nucleosome positioned in the distal promoter region activates transcription of the human U6 gene. Mol. Cell Biol. 17, 4397–4405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhao X., Pendergrast P. S., Hernandez N. (2001) A positioned nucleosome on the human U6 promoter allows recruitment of SNAPc by the Oct-1 POU domain. Mol. Cell 7, 539–549 [DOI] [PubMed] [Google Scholar]
- 41. Egloff S., Al-Rawaf H., O'Reilly D., Murphy S. (2009) Chromatin structure is implicated in “late” elongation checkpoints on the U2 snRNA and β-actin genes. Mol. Cell Biol. 29, 4002–4013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hernandez N. (2001) Small nuclear RNA genes. A model system to study fundamental mechanisms of transcription. J. Biol. Chem. 276, 26733–26736 [DOI] [PubMed] [Google Scholar]
- 43. Fair K., Anderson M., Bulanova E., Mi H., Tropschug M., Diaz M. O. (2001) Protein interactions of the MLL PHD fingers modulate MLL target gene regulation in human cells. Mol. Cell Biol. 21, 3589–3597 [DOI] [PMC free article] [PubMed] [Google Scholar]