

Background: Tropomodulin is a tropomyosin-dependent actin-capping protein.
Results: Mutations in tropomodulin-1 that reduce its affinity for tropomyosin (R11K, D12N, Q144K) reduced inhibition of actin pointed-end polymerization in vitro and decreased assembly of tropomodulin-1 in skeletal myocytes.
Conclusion: The tropomyosin-binding ability of tropomodulin-1 directly influences its actin filament regulatory activity.
Significance: Creating a tool for studying the roles of different tropomodulin isoforms in living cells.
Keywords: Actin, Circular Dichroism (CD), Fluorescence, Site-directed Mutagenesis, Tropomyosin, Actin-capping, Intrinsically Disordered Regions, Protein-Protein Interactions, Tropomodulin
Abstract
Tropomodulin (Tmod) is an actin-capping protein that binds to the two tropomyosins (TM) at the pointed end of the actin filament to prevent further actin polymerization and depolymerization. Therefore, understanding the role of Tmod is very important when studying actin filament dependent processes such as muscle contraction and intracellular transport. The capping ability of Tmod is highly influenced by TM and is 1000-fold greater in the presence of TM. There are four Tmod isoforms (Tmod1–4), three of which, Tmod1, Tmod3, and Tmod4, are expressed in skeletal muscles. The affinity of Tmod1 to skeletal striated TM (stTM) is higher than that of Tmod3 and Tmod4 to stTM. In this study, we tested mutations in the TM-binding sites of Tmod1, using circular dichroism (CD) and prediction analysis (PONDR). The mutations R11K, D12N, and Q144K were chosen because they decreased the affinity of Tmod1 to stTM, making it similar to that of affinity of Tmod3 and Tmod4 to stTM. Significant reduction of inhibition of actin pointed-end polymerization in the presence of stTM was shown for Tmod1 (R11K/D12N/Q144K) as compared with WT Tmod1. When GFP-Tmod1 and mutants were expressed in primary chicken skeletal myocytes, decreased assembly of Tmod1 mutants was revealed. This indicates a direct correlation between TM-binding and the actin-capping abilities of Tmod. Our data confirmed the hypothesis that assembly of Tmod at the pointed-end of the actin filament depends on its TM-binding affinity.
Introduction
Actin filaments are dynamic structures of the cytoskeleton responsible for cellular processes such as cell motility, division, and intracellular transport (for review, see Refs. 1 and 2). Actin filaments are a major component in muscle cells. For example, actin forms thin filaments in sarcomeres, the contractile structure in striated muscle cells. In addition, actin filaments are part of many diverse structures and bind to many different proteins. Actin filaments are formed by the polymerization of globular actin (G-actin)4 monomers. Polymerization is ATP-dependent and occurs differentially on either fast- (barbed) or slow-growing (pointed) ends of the actin filament. Polymerization is tightly controlled by a wide variety of proteins. In striated muscles, the length of the thin filaments depends on actin polymerization/depolymerization at the pointed ends that are capped by the tropomyosin-tropomodulin (TM-Tmod) complex (3–6).
The TM protein family contains a wide variety of isoforms encoded, in vertebrates, by four genes (α, β, γ, and δ) that undergo alternative splicing (for review, see Ref. 7). TM is a coiled-coil protein consisting of two α-helical coils. TM binds head to tail along the actin filament on both sides. One strand of TM spans six or seven actin monomers depending on the TM isoform. TM plays a key role in regulating actin/myosin binding and the stiffness of the actin filament. Striated muscle TM (stTM) is the long α-TM isoform most commonly found in muscle sarcomeres. The N terminus of TM faces the pointed end of actin filaments and interacts with Tmod to influence capping (for review, see Ref. 8).
There are four Tmod isoforms (Tmod1–4) specific to different tissues (9). Skeletal muscles contain Tmod1, Tmod3, and Tmod4, which are 60% identical and 70% similar in amino acid sequence. Tmod1 and Tmod4 cap α-actin thin filament pointed ends. Tmod3 localizes at the pointed ends of γ-actin filaments associated with the sarcoplasmic reticulum compartment (10). However, the sequence similarity enables Tmod isoforms to replace each other at the pointed ends of the different actin filaments. For example, it was shown that knocking out Tmod1 in embryonic mice skeletal muscle resulted in replacement of Tmod1 with Tmod3 and Tmod4 at the pointed ends of α-actin filaments (11). Furthermore, both Tmod3 and Tmod4 were detected at the sarcomeric pointed ends in adult skeletal muscles after Tmod1 knock-out.
The Tmod molecule can be divided into two functionally and structurally distinct halves, an intrinsically disordered N-terminal domain and a highly conserved compact C-terminal domain (12). The N-terminal domain contains three binding sites: TM-binding sites 1 and 2 (residues 1–38) and (residues 109–144), respectively, and the TM-dependent actin-capping site (residues 48–92).
TM binding to Tmod is isoform-specific; for example, Tmod1 binds to α-TM more strongly than all other Tmod isoforms (13). We hypothesized that changes in TM-binding ability should affect the actin-capping properties of Tmod. In this study, we designed and tested mutations that potentially could impair the TM-binding ability of Tmod1. Three mutations, R11K and D12N in site 1 and Q144K in site 2 of Tmod1 were found to decrease the TM-binding ability of Tmod1, making it similar to that of Tmod3 and Tmod4. Supporting our hypothesis, these mutations decreased both the actin filament capping abilities of Tmod1 in pyrene-actin polymerization assays and the localization of Tmod1 at the pointed ends in chicken skeletal myocytes.
EXPERIMENTAL PROCEDURES
Intrinsic Disorder Prediction
Predictions of intrinsic disorder in Tmod proteins and their mutations were performed using a PONDR® VLXT predictor, access to which was provided by Molecular Kinetics, Inc. PONDR® (Predictor of Natural Disordered Regions) is a set of neural network predictors of disordered regions; the predictors are based on local amino acid composition, flexibility, hydropathy, coordination number, and other factors. These predictors classify each residue within a sequence as either ordered or disordered. PONDR® VLXT integrates three feed forward neural networks: the VL1 (variously characterized long version 1) predictor (14); the XN (x-ray crystallography characterized N terminus) predictor (15), and the XC (x-ray crystallography characterized C terminus) predictor (15). Output for the VL1 predictor starts and ends 11 amino acids from the termini. The XT predictors output provides predictions up to 14 amino acids from their respective ends. A simple average is taken for the overlapping predictions; and a sliding window of nine amino acids is used to smooth the prediction values along the length of the sequence. Unsmoothed prediction values from the XT predictors are used for the first and last four sequence positions.
Protein Sequences
Sequences of Tmod1–4 from selected vertebrates were downloaded from UniProt, Ensembl, or NCBI. Below is a list of analyzed Tmod1, Tmod2, Tmod3, and Tmod4 isoforms together with their corresponding accession numbers: Gallus gallus (NP_990358.1; XP_413805.1; NP_001005813.1; NP_990105.1), Homo sapiens (NP_001159588.1; AAF31668.1; NP_055362.1; NP_037485.2), Mus musculus (NP_068683.1; AAF31669.1; NP_058659.1; NP_057921.1), Rattus norvegicus (NP_037176.2; NP_113801.1; NP_001011997.1; NP_001099919.1), Bos taurus (NP_001073108.1; XP_610595.3; NP_001069455.1; NP_001068683.1), Canis familiaris (XP_532002.2; XP_535484.2; XP_862973.1; XP_861607.1).
Peptides
Tmod fragments, WT and mutated, corresponding to the first and second TM-binding sites, and N-acetylated TM peptide (αTM1aZip) were synthesized by the Tufts University Core Facility (Boston, MA). The chimeric TM peptide designed for structural and functional studies (16) contained 14 N-terminal residues of long α-TM, encoded by exon 1a. To stabilize the coiled-coil structure, the TM peptide contained an additional 18 C-terminal residues of the GCN4 leucine zipper domain. The quality of the synthetic peptides was confirmed using mass spectroscopy; molecular weights of the peptides were the same as the predicted ones.
Plasmid Construction
The plasmid for chicken Tmod1, pET(His)Tmod1 (17), was used as the template to obtain pET(His)Tmod1(Q144K). pET(His)Tmod1(Q144K) was then used as the template plasmid to construct pET(His)Tmod1(D12N/Q144K), which was further used as a template plasmid to construct pET(His)Tmod1(R11K/D12N/Q144K). Site-directed mutagenesis was performed using a QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). The plasmids encoding Tmod1 were amplified by PCR using Pfu Turbo DNA polymerase (Agilent Technologies) and two complementary sets of oligonucleotides. The original plasmid was digested using DpnI (New England Biolabs), and the mixture was used to transform Escherichia coli (max efficiency DH5α). To change Gln-144 to lysine, Asp-12 to asparagine, and Arg-11 to lysine, the following oligonucleotides were used, respectively: 5′-CAC ACC TTG ATG AGT AAC AAG CAG TAC TAC GAG GCA CTG-3′ (gTmod1 Q144K); 5′-CTG GAA AAA TAC CGG AAC CTG-GAT GAA GAC AAG ATC-3′ (gTmod1 D12K); and 5′-GAA CTG GAA AAA TAC AAGAAC CTG GAT GAA GAC AAG ATC CTC-3′ (gTmod1 R11K/D12N). Mutated triplets are underlined.
For transfection experiments, the plasmid for the mouse Tmod1 (accession no. NM_21883) was subcloned into pEGFP-C1 (18). To insert Q144K, D12N/Q144K, and R11K/D12N/Q144K mutations in GFP-Tmod1, the following oligonucleotides were used, respectively: 5′-CAC ACT CAT GAG CAA CAA GCA GTA CTA CCA AGC C-3′ (mTmod1 Q144K); 5′-GAG AGC TGG AGA AAT ATC GAA ACC TGG ATG AGG ATG-3′ (mTmod1 D12N); and 5′-CAG ACG AGA GCT GGA GAA ATA TAA AAA CCT GGA TGA GGA TG-3′ (mTmod1 R11K).
Synthesis of all designed oligonucleotides was done at Integrated DNA Technologies, Inc. After plasmid purification, the presence of the mutations was confirmed by DNA sequencing (GENEWIZ, Inc.).
CD Measurements
Far-UV CD spectra of Tmod1 (WT and mutants) was measured using an Aviv Model 400 spectropolarimeter in 1-mm cuvettes. Changes in helical content during temperature denaturation were monitored at 222 nm. Measurements were done using 10 μm Tmod1 and 10 μm αTM1azip in 100 mm NaCl and 10 mm sodium phosphate buffer pH 7.0 as described in Ref. 19. Binding constants were calculated as described previously (20).
Protein Expression and Purification
Tmod1 (WT and mutants) were overexpressed in E. coli BL21(DE3) pLysE using the method described (21) and purified according to Ref. 17.
Chicken pectoral skeletal muscle G-actin was purified from acetone powder as described in Refs. 22 and 23 and used fresh in fluorescence experiments. Pyrene iodoacetamide-labeled G-actin was prepared (24), labeling ratios were calculated according to Ref. 25, and protein was stored in liquid nitrogen. Before experiments, pyrene actin was defrosted at 37 °C and centrifuged to remove aggregates.
N-Acetylated stTM was purified from chicken pectoral muscles by modified protocol (26). 10 g of alcohol ether powder were extracted with 200 ml of buffer A (25 mm Tris-HCl, pH 7.5, containing 1 m KCl, 1 mm DTT, 1 mm EDTA, 1 mm pefabloc, 1 mm NaN3) overnight at 4 °C. The mixture was filtered through a Whatman filter under vacuum. Liquid was collected, and residue was re-extracted for 1 h at room temperature with 100 ml of the same buffer. Combined supernatants were acidified to pH 4.6–4.65 with 7.5% acetic acid (on ice), dropwise, with stirring. The precipitate was allowed to settle for 1 h and then collected by centrifugation at 20,000 rpm (Sorvall SA-300 Rotor), 4 °C, for 10 min. The pellet was resuspended in buffer A, and pH was adjusted up to 7.0 with 1 m Tris. The solution was kept on ice for 40 min and centrifuged at 20,000 rpm (Sorvall SA-300 Rotor), 4 °C, for 20 min. The supernatant was further fractionated by precipitation with (NH4)2SO4. At 40% saturation, the mixture was incubated for 30 min at 4 °C, and precipitate was removed by centrifugation at 20,000 RPM (Sorvall SA-300 Rotor), 4 °C, for 20 min. The (NH4)2SO4 concentration was further increased to 70% saturation, and the mixture was stirred overnight at 4 °C. The precipitate contained stTM and was centrifuged (20,000 rpm, Sorvall SA-300 Rotor, 4 °C, 20 min), resuspended in buffer B (20 mm Tris-HCl, pH 7.5; 0.5 mm DTT) and dialyzed against buffer B overnight at 4 °C. The dialyzed stTM solution was loaded onto a DE52 preswollen microgranular DEAE cellulose (Whatman), pre-equilibrated with buffer B. The column was first washed with buffer B, and then stTM was eluted by linear 0–1 m NaCl gradient. Fractions containing stTM were combined, dialyzed against 50 mm NH4HCO3, and lyophilized. The protein concentration was determined by measuring difference spectra in 6 m guanidine HCl between pH 12.5 and 7.0 (27), using extinction coefficients of 2357 per tyrosine and 830 per tryptophan (28).
Fluorescence Measurements
The rates of actin polymerization were determined using the change in pyrene actin fluorescence using a Photon Technology International fluorometer (Lawrenceville, NJ) (excitation, 366 nm; emission, 387 nm, with a 2-nm slit). To measure polymerization of actin at the pointed end, short filaments capped at the barbed ends with gelsolin were prepared by polymerization of 3 μm G-actin in the presence of 15 nm gelsolin. Polymerization was monitored by the increase in fluorescence when the filaments were diluted 5-fold with G-actin (10% pyrenyl actin) in F-buffer (100 mm KCl, 2 mm MgCl2, 1 mm EGTA, 0.5 mm DTT, 0.2 mm ATP, 0.2 mm CaCl2, 1 mm NaN3, 10 mm imidazole, pH 7.0), containing a saturated concentration of stTM (1 μm) and Tmod1 (3, 5, 10, 15, and 20 nm). The final concentrations of F- and G-actin, after dilution, were 0.6 μm and 1.1 μm, respectively. Exponential curves were fit to the polymerization data using SigmaPlot, and initial rates (R) were calculated as the first derivatives at time zero according to Ref. 29. Recombinant human gelsolin was a generous gift from Dr. John H. Hartwig (Brigham and Women's Hospital & Hematology Division, Boston, MA).
To measure actin nucleation, 1 μm G-actin (10% pyrenyl actin) was mixed with 20, 50, and 100 nm Tmod1 (WT and mutants), and the reaction was initiated by addition of polymerization buffer (as described above). Spontaneous actin nucleation in the absence of Tmod was measured as control for each set of Tmod concentrations.
Cell Culture and Transfection Procedure
Skeletal muscle cultures were prepared from day 11–12 chick embryonic pectoralis muscle and maintained as described (30, 31). Isolated cells were plated in 35-mm tissue culture dishes containing 12-mm round glass coverslips coated with Matrigel (Fisher Scientific, Pittsburgh, PA) for staining (∼1 × 106 cells/dish). Transfection was performed using Effectene (Qiagen, Valencia, CA) as described (18).
Immunofluorescence Microscopy
Four days after transfection, myotubes were incubated in relaxing buffer (150 mm KCl, 5 mm MgCl2, 10 mm MOPS (pH 7.4), 1 mm EGTA, and 4 mm ATP) for 20 min and fixed with 4% paraformaldehyde in relaxing buffer for 20 min. Coverslips were washed and stored in PBS at 4 °C until staining. Transfected chick myotubes were stained as described previously (32). Briefly, the fixed cells were permeabilized in 0.2% Triton X-100/PBS for 20 min. The cells were incubated first in blocking solution (2% BSA plus 1% normal donkey serum in PBS) and then for 1 h with primary monoclonal anti-sarcomeric α-actinin antibodies (1:10,000) (EA-53, Sigma-Aldrich). The cells were then incubated with Alexa Fluor 350-conjugated goat anti-mouse IgG (1:350) (A21049, Invitrogen) in PBS for 1 h. All coverslips were mounted on slides using Aqua Poly/Mount (Polysciences, Warren, PA). More than 200 cells were observed for each treatment/condition on a Zeiss Axiovert microscope using a 100× (numerical aperture, 1.3) objective. The cells analyzed were chosen based on 1) evidence of relaxation (distance between Z-lines via α-actinin staining) and 2) the presence of moderate GFP expression levels. GFP-positive skeletal myocytes were scored based upon the thin filament pointed end localization of GFP-Tmod1 and its mutants: “consistent” refers to clear and well defined striated staining (>90% of sarcomeres contain GFP striations); “inconsistent” refers to faint and partial striated staining (<90% of sarcomeres contain GFP striations) with high levels of diffuse cytoplasmic staining; and “diffuse” refers to no detectable striated staining. The data are presented as the percentage of the total number of GFP-positive skeletal myocytes in each group (mean ± S.D.). Data are from three independent cultures. Representative images were captured using a Deltavision RT system (Applied Precision) with an inverted microscope (IX70; Olympus), a 100× (NA 1.3) objective, and a charge-coupled device camera (CoolSNAP HQ; Photometrics) using SoftWoRx software (version 3.5.1, Applied Precision). The images were then deconvolved using SoftWoRx and processed using Photoshop CS (Adobe).
RESULTS
Using Alignment to Exclude Residues Not Important for Affinity Differences
The aim was to alter Tmod binding to TM by mutating a minimal number of residues in Tmod. We chose residues in Tmod1 for which the mutation may decrease stTM binding. To determine the residues that may be responsible for difference in TM-binding affinity, sequences of all isoforms (Tmod1–4) from different orthologs (mammals and avians) were analyzed. In Fig. 1, the alignments of fragments corresponding to TM-binding sites 1 and 2 that were used in our experiments are presented.
FIGURE 1.

Alignment of TM-binding site 1 (A) and site 2 (B) for Tmod1, Tmod3, and Tmod4 from Gallus gallus and Tmod2 from Mus musculus. Dark gray, identical; light gray, may have identical residues (in other orthologs); underlined, variable within orthologs for the same isoform. Asterisks show position of residues that should not be responsible for isoform differences; arrows show residues that were tested.
Identical residues are marked by dark gray. Light gray shows positions of residues that are identical in orthologs different from those shown in this picture. As a result of the alignment, we excluded 19 of 38 residues in site 1, and 19 of 36 residues in site 2 as positions that should not be responsible for differences in TM-binding affinity (indicated by asterisks). Residues that were variable within orthologs for the same isoform were underlined.
Using Circular Dichroism Spectroscopy to Evaluate Protein-Protein Interactions
To test the effect of changes in the Tmod1 sequence on αTM1azip binding, CD spectroscopy was used as a very sensitive technique to analyze protein-protein interactions (e.g. Ref. 33). TM and Tmod1 fragments were synthesized. The chimeric peptide (αTM1azip) contained 14 N-terminal residues of long α-TM and an additional 18 C-terminal residues of the GCN4 leucine zipper domain, which helped to stabilize the TM coiled-coil structure (34). Tmod fragments, Tmod1s1 and Tmod1s2, corresponded to TM-binding site 1 and site 2 of Tmod1 with designed changes, respectively.
The far-UV CD spectra were measured to monitor formation of complexes of TM and Tmod fragments. We successfully used this approach in our previous studies (13). The CD spectra of all Tmod1 peptides (WT and mutated) were typical for the mostly unfolded polypeptide; the CD spectrum of αTM1azip was typical for the α-helical polypeptides. Spectra of mixed Tmod and TM fragments differed from the sums of their individual curves. The increase in helicity indicated complex formation and likely resulted from folding of the Tmod fragment upon binding to the TM peptide.
Stability of the complexes of TM and Tmod fragments was analyzed using changes in CD during temperature denaturation at 222 nm (Fig. 2 and see Fig. 4). Full-length Tmod cannot be used in this experiment because it aggregates irreversibly upon heat denaturation (35). The increase in the melting temperature of the mixtures (ΔTm) was calculated by subtracting the melting temperature of the sum of curves for individual peptides from the melting temperature of the mixture curves. The dissociation constants (Kd) were calculated from unfolding curves of the complexes compared with the curves for Tmod and TM peptides alone.
FIGURE 2.
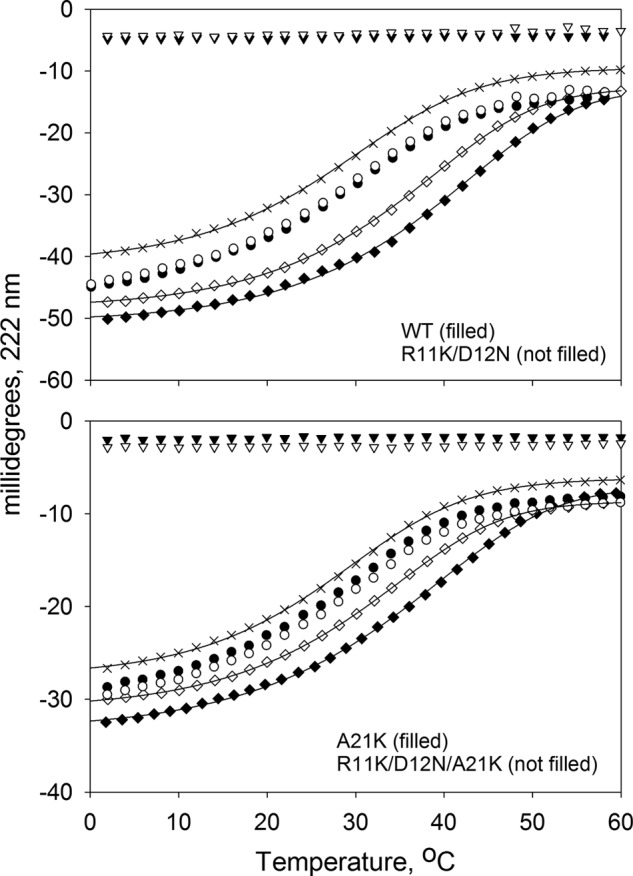
R11K/D12N mutations in the TM-binding site 1 of Tmod1 decrease its binding to αTM1azip. Binding of Tmod1s1 fragments, WT and mutants, to αTM1azip was measured using CD spectroscopy. The graph shows the unfolding curves for individual peptides (×, αTM1azip; inverted triangle, Tmod fragments), the unfolding curves of the mixture (diamonds), and sums of the individual unfolding curves of αTM1azip with Tmod peptides (circles).
FIGURE 4.
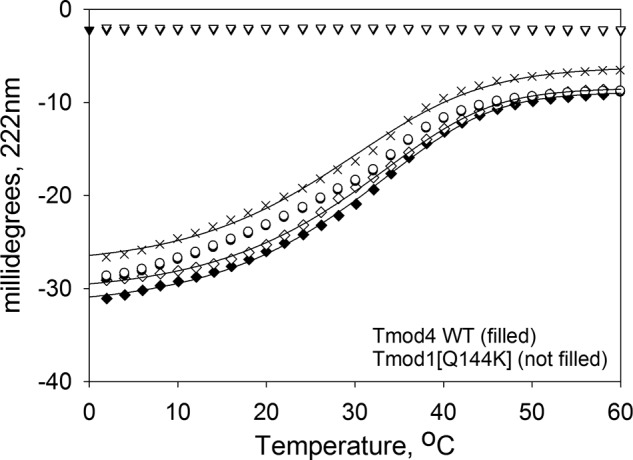
Q144K mutation in the TM-binding site 2 of Tmod1 decreases its binding to αTM1azip. Binding of Tmod4s2 WT and Tmod1s2(Q144K) fragments to αTM1azip measured using CD spectroscopy. The graph shows the unfolding curves for individual peptides (×, αTM1azip; triangle, Tmod fragments), the unfolding curves of the mixture (diamonds), and sums of the individual unfolding curves of αTM1azip with Tmod peptides (circles).
Mutations Chosen for Site 1
It was shown that Tmod binding to TM is isoform dependent, and dissociation constants (Kd) for all possible complexes between Tmod isoforms and TM isoforms were determined (13). For example, the affinity of αTM1azip for Tmod2s1 was the lowest, whereas the affinity of αTM1azip was the highest for Tmod1s1 among other isoforms (Kd = 1.1 ± 0.4 μm, 4.7 ± 0.4 μm, 1.9 ± 0.3 μm, and 2.4 ± 0.6 μm for Tmod1s1, Tmod2s1, Tmod3s1, and Tmod4s1, respectively). In a previous study, we made changes in the Tmod1s1 sequence to increase its binding affinities for fragments of γ- and δ-TMs. The Tmod1s1 fragment with R11K, D12N, and A21K substitutions was tested with γTM1bzip and δTM1bzip and had no effect.5 However, this fragment had a drastic effect on binding to αTM1azip (fragment of α-TM), increasing the Kd to 2.3 ± 0.1 μm (Fig. 2).
We wanted to find out what residue is responsible for decreasing the efficiency of the αTM1azip binding. Of three substitutions, the choice that looked obvious was the residue at position 21 in the Tmod1 sequence. There is a big difference in physicochemical properties between the hydrophobic Ala-21 residue in Tmod1 and charged residues Lys-21 (in Tmod2 and Tmod3) and Glu-21 (in Tmod4) (Fig. 1A). We tested the Tmod1s1 fragment with an Ala to Lys substitution. Surprisingly, no changes were observed in TM-binding ability; the increase in the melting temperature (ΔTm) resulting from the complex formation of WT Tmod1 and Tmod1(A21K) with αTM1azip was the same, 9.1 ± 2.4 °C and 8.9 ± 1.5 °C, respectively (Fig. 2). Therefore, our next choice was both Asp-12 and Arg-11 of the Tmod1 sequence. Negatively charged Asp is the same residue for Tmod3 and Tmod4, but Tmod2 has an uncharged Asn in the corresponding position. Arg-11 is identical for both Tmod1 and Tmod4, but there is a Lys in the corresponding position in Tmod2 and Tmod3.
The Tmod1s1 fragment with R11K and D12N replacements was tested for complex formation (Fig. 2). The difference in melting curves clearly shows that binding of the double mutant Tmod1(R11K/D12N) to αTM1azip is weaker than that of the WT Tmod1. These substitutions decreased the ability of Tmod1s1 to bind αTM1azip almost 2-fold. To minimize the number of residues to mutate, we used the R11K/D12N replacement for actin-capping and cell experiments.
Mutations Chosen for Site 2
The TM-binding dissociation constant of Tmod2s2 is 4.4 ± 0.5 μm; it is the highest among other isoforms (1.3 ± 0.3 μm, 2.1 ± 0.4 μm, and 3.6 ± 1.2 μm for Tmod1s2, Tmod3s2, and Tmod4s2, respectively) (13).
For the second TM-binding site (s2), we took into consideration only residues in the C-terminal half of the Tmod1s2 peptide. According to our antiparallel model of TM-Tmod interaction in site 2, only the C-terminal half of Tmod1s2 should interact with the TM sequence in the chimeric peptide (36). The clear candidate for change was the residue in position 144 that is different in all isoforms (Gln for Tmod1, Pro for Tmod2, Asp for Tmod3, and Lys for Tmod4). However, because this residue was the last one in the fragment, we were not sure whether changing it would produce effective modulation of the Tmod-TM interactions. Therefore, we decided to use the disorder prediction method (PONDR® VLXT) to evaluate the potential role of this mutation on the predicted disorder propensity of this peptide. We were looking for differences between Tmod1 and Tmod4 because among muscle Tmod isoforms that bind stTM, Tmod4 has the lowest affinity. Therefore two other residues (position 128 and 138) were chosen for testing. Residues in position 128 (Lys) and 138 (His) of the Tmod1 sequence are identical for the corresponding residues in Tmod2 and Tmod3, whereas the Tmod4 sequence contains Met and Tyr, respectively, at the corresponding positions. The Tmod1s2 sequences with L128M, H138Y, and Q144K were analyzed by a disoder prediction method PONDR® VLXT that is known to be sensitive to functional disordered regions (Fig. 3) (37–39). In fact, the PONDR® VLXT predictions might identify potential binding sites as short regions of predicted order bounded by regions of predicted disorder. Based on these observations, it has been proposed that such “signatures” in the PONDR® prediction plots, where short regions of predicted order are bounded by extended regions of predicted disorder, might correspond to potential binding sites, called molecular recognition features, which undergo disorder-to-order transitions upon complex formation (37–39). We have already utilized this approach to compare different Tmod isoforms from nine species (13). That earlier analysis revealed that the N-terminal region of Tmods possess a large variation in the number and appearance of “dips” and that for a given Tmod, the disorder distribution was mostly conserved even for proteins from distant species. Importantly, such conservation was obvious not only for the ordered C-terminal domains but also for the significant portions of the disordered N-terminal domains of various Tmods (13).
FIGURE 3.

Predicted disorder distribution profiles of sequences corresponding to the site 2 of the wild type Tmod1, Tmod4, and three mutants Tmod1(L128M), Tmod1(H138Y), and Tmod1(Q144K).
Fig. 3 represents the results of the PONDR® VLXT analysis of the region corresponding to site 2 of WT Tmod1, WT Tmod4, and three mutants Tmod1(L128M), Tmod1(H138Y), and Tmod1(Q144K). This figure clearly shows that the L128M mutation has a minor effect on the disorder profile, whereas the Tmod1(H138Y) disorder profile was different from profiles from both WT Tmod1 and WT Tmod4. Finally, the Q144K mutation in Tmod1 resulted in a dramatic change in the disorder profile, making it similar to that of the WT Tmod4. Based on these observations, we hypothesized that both H138Y and Q144K mutations in Tmod1 might have significant effects on the binding affinity of this protein to αTM1azip, with the Q144K mutation expected to have αTM1azip binding parameters similar to those of the WT Tmod4.
This hypothesis was tested using the CD spectroscopy-based approach described above. CD experiments were performed using Tmod1(H138Y) (data not shown) and Tmod1(Q144K) (Fig. 4). This analysis revealed that as a result of a single change of His-138 to Tyr, the affinity of Tmod1 to αTM1azip decreased 2-fold. The Kd was increased from 1.3 ± 0.3 μm (for the WT Tmod1) to 2.8 μm, making it close to the affinity of Tmod3s2 (2.1 ± 0.4 μm). Another single change, Q144K, caused a 3-fold increase in the Kd value, from 1.3 ± 0.3 μm (for the WT Tmod1) to 3.5 ± 0.2 μm and made it close to the affinity of Tmod4s2 to αTM1azip (3.6 ± 1.2 μm). Therefore, our CD results were in a good agreement with the results of disorder predictions. Based on the results of this analysis, we chose R11K, D12N, and Q144K mutations to introduce into full-length Tmod1 for further studies.
Correlation between TM-binding and Actin-capping Abilities of Tmod1
To investigate the correlation between reduced TM-binding affinity and actin capping, mutations D12N/Q144K and R11K/D12N/Q144K were introduced into full-length Tmod1 and the two mutants were tested in the pyrene actin polymerization assay.
To measure polymerization of actin at the pointed end only, seeds (short filaments capped at the barbed ends with gelsolin) were prepared by polymerization of G-actin in the presence of gelsolin. Pyrene actin assays were run upon the addition of 1 μm G-actin in the presence of 1 μm stTM and Tmod1 at various concentrations (3, 5, 10, 15, and 20 nm). Polymerization was monitored by the increase in fluorescence of pyrene actin (Fig. 5, A–E). Exponential polymerization curves were fit to the polymerization data and derivatives of curves at time 0 were taken to calculate initial rates (Fig. 5F). Decreased actin-capping ability was observed for both mutants as compared with WT Tmod1. A 50% inhibition effect for WT Tmod1 was observed at 5 nm, whereas for Tmod1(D12N/Q144K) and Tmod1(R11K/D12N/Q144K), it was observed at 10 and 15 nm, respectively (Fig. 5F). These data support our hypothesis that the actin-capping ability of Tmod isoforms is influenced by their TM-binding affinity.
FIGURE 5.

A–E, time courses of pointed end elongation of gelsolin-capped actin filaments in the absence and presence of stTM (1 μm) and Tmod1 (3, 5, 10, 15, and 20 nm) detected as increase of fluorescence. F, dependence of inhibition of actin polymerization on Tmod1 concentration. Initial rates were calculated as the first derivatives at time zero after fitting. The inhibition of polymerization was calculated as Rexp/Rcontrol, where Rcontrol = 2 (in the absence of TM and Tmod). Black circle, control, no stTM, or no Tmod; red triangle, with stTM and Tmod1 WT; green square, with stTM and Tmod1(D12N/Q144K); yellow diamond, with stTM and Tmod1(R11K/D12N/Q144K).
Interestingly, in our pyrene actin polymerization assay the shape of the polymerization curve for 20 nm Tmod1(R11K/D12N/Q144K) differs from the other curves. After a few minutes of inhibition, the fluorescence started to increase rapidly so the curve looked like there was a lag phase. This type of curve can be obtained when two simultaneous processes are happening: capping and nucleation. During nucleation, Tmod may bind two actin monomers that cause formation of new barbed ends and therefore faster polymerization happens at these ends. Indeed, the nucleation effect of the mutant Tmod1 was higher than that of wild type (data not shown).
Effect of Mutations in the TM-binding Sites on Assembly of Tmod1 in Skeletal Myocytes
For expression in chicken skeletal muscle cells, the same three mutations were introduced into GFP-Tmod1. Three mutants GFP-Tmod1(Q144K), GFP-Tmod1(D12N/Q144K), and GFP-Tmod1(R11K/D12N/Q144K) were used for transfection. It was shown that single, double, and triple mutations in Tmod1 decreased its assembly at the pointed ends, correlating with our in vitro data (Fig. 6). Consistent assembly of Tmod1 with the single mutation Q144K in the second TM-binding site was ∼10% lower compared with wild-type Tmod1, whereas consistent assembly of Tmod1(D12N/Q144K) and Tmod1(R11K/D12N/Q144K) was ∼30 and ∼35% lower, respectively.
FIGURE 6.

Effect of the TM-binding mutations on the assembly of Tmod1 in primary chicken skeletal myocytes. A, graph shows the percentage of myocytes demonstrating consistent, inconsistent, or diffuse thin filament pointed end localization (mean ± S.D.); categories are as described under “Experimental Procedures.” B, representative images of primary chicken skeletal myocytes expressing GFP-Tmod1 (WT or triple mutant) or GFP alone that were stained for sarcomeric α-actinin to label the Z-discs. GFP-Tmod1 was consistently localized to the thin filament pointed ends (90.7 ± 0.58%), whereas GFP-Tmod1(R11K/D12N/Q144K) did not assemble as well with 34.0 ± 3.61% localized to the thin filament pointed ends (i.e. inconsistent). Arrows mark the pointed end assembly. Scale bar, 10 μm.
DISCUSSION
Skeletal muscle cells contain three of four Tmod isoforms: Tmod1, Tmod3, and Tmod4. It was shown earlier that Tmod1 binds to the long muscle α-TM, stTM, tighter than any other Tmod isoform (11, 13). The aim of this study was to find correlations between TM-binding ability, actin-capping activity, and pointed end localization of Tmod.
We introduced point mutations to both TM-binding sites of Tmod1 to decrease its ability to bind stTM and to study the consequences of this decrease. We expected that changing A21K in Tmod1 should decrease stTM affinity in site 1 because this residue differs from corresponding residues in other isoforms. However, the A21K replacement had no effect on stTM binding. The double substitution R11K/D12N turned out to decrease TM binding. Interestingly, although A21K had no effect by itself, adding this change to R11K/D12N decreased stTM-binding affinity even more. We suggest that not one but many residues are involved in the attenuation of stTM binding in Tmod isoforms. Multiple residues might work in a cooperative manner to produce an affinity specific for each isoform.
According to the previously published possible binding models of Tmod1 to TM, the Tmod molecule loops around the N-terminal domain of TM and bundles it at site 1 (36). The formation of this bundle incorporates many residues and provides a binding environment for TM at the same time. Therefore, in this site, the isoform-dependent difference in binding may need cooperation of several residues in the Tmod1 sequence.
The model for site 2 suggests that the Tmod molecule lies along the N terminus of the second TM molecule in an antiparallel fashion (36). Because a change in the nature of an amino acid seems to be non-compensable due to the interaction of Tmod with two strands of TM in this site, a single mutation may reduce TM-binding affinity significantly. We showed that introduction of an extra positive charge by the Q144K mutation decreased the affinity of Tmod1 greatly. Gln-144 belongs to a region in site 2 where ionic interactions dominate. Introducing positively charged Lys instead of Gln made a significant impact on TM-binding ability of Tmod1.
Introducing mutations into full-length Tmod, which decreases its TM-binding affinity, resulted in significantly decreased inhibition of actin polymerization by the mutants when compared with wild type. This result supports the hypothesis that the actin-capping ability of Tmod isoforms is a function of their affinity for TM. Unexpectedly, mutations in TM-binding sites also caused increased nucleating activity. Our mutations in the first TM-binding site of Tmod1 made it act similar to Tmod2 and Tmod3, which are known to bind and nucleate actin monomers (40, 41). Fisher et al. (41) suggested that residues 31–40 of Tmod3, which are within the TM-binding site 1 (42), could be responsible for G-actin binding. It is possible that mutations in positions 11 and 12 led to a conformational change that enhanced the ability of Tmod1 to bind and nucleate actin monomers. However, the exact structure of the TM-Tmod complex is not known yet and it must be investigated furthermore to verify the interrelation between the TM- and actin-binding sites of Tmod.
The assembly experiments using chicken skeletal myocytes showed decreased assembly of Tmod1 mutants at the pointed ends of thin filaments. Tmod1 containing only the mutation Q144K in site 2 had a slight decrease in assembly compared with WT Tmod1. When the affinity of Tmod1 for TM was weakened in both TM-binding sites, its assembly became more perturbed. Interestingly, adding the third mutation R11K did not cause a dramatic difference in localization compared with Tmod1(D12N/Q144K), although it significantly decreased the actin-capping ability and TM-binding affinity of Tmod1 in vitro. We conclude that TM-binding is important for localization of Tmod1 in living myocytes.
When residues Leu-27 and Ile-131, which were shown to be crucial for TM binding were mutated, the mutation I131D in site 2 caused an ∼90% decrease in assembly with more than half of Tmod1 observed to be diffusely localized, whereas mutation L27E caused an ∼20% decrease in assembly (18). Introduction of both mutations caused a dramatic loss of consistent assembly, with most staining appearing diffuse (e.g. soluble). Here, the mutation in site 2 had only a slight effect, suggesting that we did not disrupt residues essential for TM binding but only residues important for isoform affinity.
We conclude that the TM-binding property of Tmod regulates its interaction with actin for capping, and tight binding of Tmod to TM results in proper capping function. The binding affinity of Tmod for TM is also an important factor for the localization of Tmod in cells. Weakening the interaction between Tmod and TM instigates improper targeting of Tmod to the pointed ends of thin filaments. Our ability to alter the affinity of Tmod for TM, making one isoform similar to others, creates a tool for further studies of the roles of different isoforms in living cells.
Acknowledgment
We thank the Rutgers Aresty Research Center for Undergraduates that allowed R. Azevedo to work in the Kostyukova laboratory.
This work was supported by National Institutes of Health Grants GM081688 (to A. S. K.) and HL083146 and HL108625 (to C. C. G.).
A. S. Kostyukova, unpublished data.
- G-actin
- globular actin
- Tmod
- tropomodulin
- TM
- tropomyosin
- CD
- circular dichroism
- stTM
- striated TM.
REFERENCES
- 1. Pollard T. D., Borisy G. G. (2003) Cellular motility driven by assembly and disassembly of actin filaments. Cell 112, 453–465 [DOI] [PubMed] [Google Scholar]
- 2. Cooper J. A., Schafer D. A. (2000) Control of actin assembly and disassembly at filament ends. Curr. Opin. Cell Biol. 12, 97–103 [DOI] [PubMed] [Google Scholar]
- 3. Gregorio C. C., Weber A., Bondad M., Pennise C. R., Fowler V. M. (1995) Requirement of pointed-end capping by tropomodulin to maintain actin filament length in embryonic chick cardiac myocytes. Nature 377, 83–86 [DOI] [PubMed] [Google Scholar]
- 4. Sussman M. A., Baqué S., Uhm C. S., Daniels M. P., Price R. L., Simpson D., Terracio L., Kedes L. (1998) Altered expression of tropomodulin in cardiomyocytes disrupts the sarcomeric structure of myofibrils. Circ. Res. 82, 94–105 [DOI] [PubMed] [Google Scholar]
- 5. Mudry R. E., Perry C. N., Richards M., Fowler V. M., Gregorio C. C. (2003) The interaction of tropomodulin with tropomyosin stabilizes thin filaments in cardiac myocytes. J. Cell Biol. 162, 1057–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Littlefield R., Almenar-Queralt A., Fowler V. M. (2001) Actin dynamics at pointed ends regulates thin filament length in striated muscle. Nat. Cell Biol. 3, 544–551 [DOI] [PubMed] [Google Scholar]
- 7. Gunning P. W., Schevzov G., Kee A. J., Hardeman E. C. (2005) Tropomyosin isoforms: divining rods for actin cytoskeleton function. Trends Cell Biol. 15, 333–341 [DOI] [PubMed] [Google Scholar]
- 8. Sung L. A., Lin J. J. (1994) Erythrocyte tropomodulin binds to the N-terminus of hTM5, a tropomyosin isoform encoded by the γ-tropomyosin gene. Biochem. Biophys. Res. Commun. 201, 627–634 [DOI] [PubMed] [Google Scholar]
- 9. Yamashiro S., Gokhin D. S., Kimura S., Nowak R. B., Fowler V. M. (2012) Tropomodulins: pointed-end capping proteins that regulate actin filament architecture in diverse cell types. Cytoskeleton 69, 337–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gokhin D. S., Fowler V. M. (2011) The sarcoplasmic reticulum: Actin and tropomodulin hit the links. Bioarchitecture 1, 175–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gokhin D. S., Lewis R. A., McKeown C. R., Nowak R. B., Kim N. E., Littlefield R. S., Lieber R. L., Fowler V. M. (2010) Tropomodulin isoforms regulate thin filament pointed-end capping and skeletal muscle physiology. J. Cell Biol. 189, 95–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kostyukova A. S. (2008) Tropomodulins and tropomodulin/tropomyosin interactions. Cell. Mol. Life Sci. 65, 563–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Uversky V. N., Shah S. P., Gritsyna Y., Hitchcock-DeGregori S. E., Kostyukova A. S. (2011) Systematic analysis of tropomodulin/tropomyosin interactions uncovers fine-tuned binding specificity of intrinsically disordered proteins. J. Mol. Recognit. 24, 647–655 [DOI] [PubMed] [Google Scholar]
- 14. Romero P., Obradovic Z., Li X., Garner E. C., Brown C. J., Dunker A. K. (2001) Sequence complexity of disordered protein. Proteins 42, 38–48 [DOI] [PubMed] [Google Scholar]
- 15. Li X., Romero P., Rani M., Dunker A. K., Obradovic Z. (1999) Predicting Protein Disorder for N-, C-, and Internal Regions. Genome Inform. Ser Workshop Genome Inform. 10, 30–40 [PubMed] [Google Scholar]
- 16. Greenfield N. J., Huang Y. J., Palm T., Swapna G. V., Monleon D., Montelione G. T., Hitchcock-DeGregori S. E. (2001) Solution NMR structure and folding dynamics of the N terminus of a rat non-muscle α-tropomyosin in an engineered chimeric protein. J. Mol. Biol. 312, 833–847 [DOI] [PubMed] [Google Scholar]
- 17. Kostyukova A., Maeda K., Yamauchi E., Krieger I., Maéda Y. (2000) Domain structure of tropomodulin: distinct properties of the N-terminal and C-terminal halves. Eur. J. Biochem. 267, 6470–6475 [DOI] [PubMed] [Google Scholar]
- 18. Tsukada T., Kotlyanskaya L., Huynh R., Desai B., Novak S. M., Kajava A. V., Gregorio C. C., Kostyukova A. S. (2011) Identification of residues within tropomodulin-1 responsible for its localization at the pointed ends of the actin filaments in cardiac myocytes. J. Biol. Chem. 286, 2194–2204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kostyukova A. S., Choy A., Rapp B. A. (2006) Tropomodulin binds two tropomyosins: a novel model for actin filament capping. Biochemistry 45, 12068–12075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Greenfield N. J. (2004) Circular dichroism analysis for protein-protein interactions. Methods Mol. Biol. 261, 55–78 [DOI] [PubMed] [Google Scholar]
- 21. Studier F. W. (2005) Protein production by auto-induction in high density shaking cultures. Prot. Exp. Purif. 41, 207–234 [DOI] [PubMed] [Google Scholar]
- 22. Spudich J. A., Watt S. (1971) The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. J. Biol. Chem. 246, 4866–4871 [PubMed] [Google Scholar]
- 23. MacLean-Fletcher S., Pollard T. D. (1980) Identification of a factor in conventional muscle actin preparations which inhibits actin filament self-association. Biochem. Biophys. Res. Commun. 96, 18–27 [DOI] [PubMed] [Google Scholar]
- 24. Kouyama T., Mihashi K. (1981) Fluorimetry study of N-(1-pyrenyl)iodoacetamide-labelled F-actin. Local structural change of actin protomer both on polymerization and on binding of heavy meromyosin. Eur. J. Biochem. 114, 33–38 [PubMed] [Google Scholar]
- 25. Cooper J. A., Walker S. B., Pollard T. D. (1983) Pyrene actin: documentation of the validity of a sensitive assay for actin polymerization. J. Muscle Res. Cell Motil. 4, 253–262 [DOI] [PubMed] [Google Scholar]
- 26. Gerhard M. D., DiGirolamo P. M., Hitchcock-DeGregori S. E. (1985) Isolation and characterization of a tropomyosin binding protein from human blood platelets. J. Biol. Chem. 260, 3221–3227 [PubMed] [Google Scholar]
- 27. Edelhoch H. (1967) Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry 6, 1948–1954 [DOI] [PubMed] [Google Scholar]
- 28. Fasman G. D. (1989) Protein conformational prediction. Trends Biochem. Sci. 14, 295–299 [DOI] [PubMed] [Google Scholar]
- 29. Kostyukova A. S., Hitchcock-DeGregori S. E. (2004) Effect of the structure of the N terminus of tropomyosin on tropomodulin function. J. Biol. Chem. 279, 5066–5071 [DOI] [PubMed] [Google Scholar]
- 30. Nawrotzki R., Fischman D. A., Mikawa T. (1995) Antisense suppression of skeletal muscle myosin light chain-1 biosynthesis impairs myofibrillogenesis in cultured myotubes. J. Muscle Res. Cell Motil. 16, 45–56 [DOI] [PubMed] [Google Scholar]
- 31. Gilbert R., Kelly M. G., Mikawa T., Fischman D. A. (1996) The carboxyl terminus of myosin binding protein C (MyBP-C, C-protein) specifies incorporation into the A-band of striated muscle. J. Cell Sci. 109, 101–111 [DOI] [PubMed] [Google Scholar]
- 32. Pappas C. T., Bhattacharya N., Cooper J. A., Gregorio C. C. (2008) Nebulin interacts with CapZ and regulates thin filament architecture within the Z-disc. Mol. Biol. Cell 19, 1837–1847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Greenfield N. J. (2006) Using circular dichroism collected as a function of temperature to determine the thermodynamics of protein unfolding and binding interactions. Nat. Protoc. 1, 2527–2535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Greenfield N. J., Montelione G. T., Farid R. S., Hitchcock-DeGregori S. E. (1998) The structure of the N-terminus of striated muscle α-tropomyosin in a chimeric peptide: nuclear magnetic resonance structure and circular dichroism studies. Biochemistry 37, 7834–7843 [DOI] [PubMed] [Google Scholar]
- 35. Kostyukova A. S., Tiktopulo E. I., Maéda Y. (2001) Folding properties of functional domains of tropomodulin. Biophys. J. 81, 345–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kostyukova A. S., Hitchcock-Degregori S. E., Greenfield N. J. (2007) Molecular basis of tropomyosin binding to tropomodulin, an actin-capping protein. J. Mol. Biol. 372, 608–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Garner E., Romero P., Dunker A. K., Brown C., Obradovic Z. (1999) Predicting Binding Regions within Disordered Proteins. Genome Inform. Ser Workshop Genome Inform. 10, 41–50 [PubMed] [Google Scholar]
- 38. Oldfield C. J., Cheng Y., Cortese M. S., Romero P., Uversky V. N., Dunker A. K. (2005) Coupled folding and binding with α-helix-forming molecular recognition elements. Biochemistry 44, 12454–12470 [DOI] [PubMed] [Google Scholar]
- 39. Cheng Y., LeGall T., Oldfield C. J., Dunker A. K., Uversky V. N. (2006) Abundance of intrinsic disorder in protein associated with cardiovascular disease. Biochemistry 45, 10448–10460 [DOI] [PubMed] [Google Scholar]
- 40. Yamashiro S., Speicher K. D., Speicher D. W., Fowler V. M. (2010) Mammalian tropomodulins nucleate actin polymerization via their actin monomer binding and filament pointed end-capping activities. J. Biol. Chem. 285, 33265–33280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fischer R. S., Yarmola E. G., Weber K. L., Speicher K. D., Speicher D. W., Bubb M. R., Fowler V. M. (2006) Tropomodulin 3 binds to actin monomers. J. Biol. Chem. 281, 36454–36465 [DOI] [PubMed] [Google Scholar]
- 42. Greenfield N. J., Kostyukova A. S., Hitchcock-DeGregori S. E. (2005) Structure and tropomyosin binding properties of the N-terminal capping domain of tropomodulin 1. Biophys. J. 88, 372–383 [DOI] [PMC free article] [PubMed] [Google Scholar]