

Background: Cadherin interactions with catenins are crucial for intercellular adhesion.
Results: αE-catenin and vinculin cooperate to promote the time-dependent reinforcement of cadherin-mediated adhesions.
Conclusion: αE-catenin and vinculin form a mechanoresponsive link between cadherin and the underlying actin cytoskeleton.
Significance: The force-dependent modulation of α-catenin and vinculin recruitment contributes to the development of cadherin adhesion strength.
Keywords: Adhesion, Cadherins, Catenin, Cytoskeleton, Mechanotransduction
Abstract
Maintaining cell cohesiveness within tissues requires that intercellular adhesions develop sufficient strength to support traction forces applied by myosin motors and by neighboring cells. Cadherins are transmembrane receptors that mediate intercellular adhesion. The cadherin cytoplasmic domain recruits several partners, including catenins and vinculin, at sites of cell-cell adhesion. Our study used force measurements to address the role of αE-catenin and vinculin in the regulation of the strength of E-cadherin-based adhesion. αE-catenin-deficient cells display only weak aggregation and fail to strengthen intercellular adhesion over time, a process rescued by the expression of αE-catenin or chimeric E-cadherin·αE-catenins, including a chimera lacking the αE-catenin dimerization domain. Interestingly, an αE-catenin mutant lacking the modulation and actin-binding domains restores cadherin-dependent cell-cell contacts but cannot strengthen intercellular adhesion. The expression of αE-catenin mutated in its vinculin-binding site is defective in its ability to rescue cadherin-based adhesion strength in cells lacking αE-catenin. Vinculin depletion or the overexpression of the αE-catenin modulation domain strongly decreases E-cadherin-mediated adhesion strength. This supports the notion that both molecules are required for intercellular contact maturation. Furthermore, stretching of cell doublets increases vinculin recruitment and α18 anti-αE-catenin conformational epitope immunostaining at cell-cell contacts. Taken together, our results indicate that αE-catenin and vinculin cooperatively support intercellular adhesion strengthening, probably via a mechanoresponsive link between the E-cadherin·β-catenin complexes and the underlying actin cytoskeleton.
Introduction
Cellular adhesion is one of the crucial processes governing cell behavior and morphogenesis (1, 2). Cadherins are transmembrane receptors that mediate homophilic intercellular adhesion via their extracellular domain and recruit protein partners at their cytoplasmic domain. They bridge the intracellular actin cytoskeleton with the surface of neighboring cells, thereby contributing to mechanical coupling between cells within a monolayer. This allows cells to sense and react to external forces and regulate adhesion strength (3–6).
Recent work has outlined that cadherin adhesions transmit forces (7) and are themselves regulated in a force-dependent manner (5). Indeed, the recruitment of cadherin-associated complexes, as well as of actin filaments in adhesion plaques, is dependent both on the external forces applied to the contact and on the activity of myosin II. Cadherin-based adhesion strength has been quantified in terms of separation force measurements by the dual pipette assay, which shows that adhesion strength enhances with time (6, 8) and requires both a cadherin-actin cytoskeleton connection and actomyosin contractility. Several catenins, including p120-catenin, β-catenin, and α-catenin, are recruited at sites of cadherin-based adhesion, where they are required for cadherin anchoring to the cytoskeleton and for the development of adherens junctions (AJs)5 (for a review, see Ref. 9).
Cells expressing chimeric cadherin, where the cytoplasmic domain has been replaced by the full-length αE-catenin, exhibit significant intercellular adhesion strength (6, 10); this presents an argument for the critical role of αE-catenin in the connection of cadherin to F-actin during the strengthening of intercellular adhesions.
αE-catenin is an actin-binding protein that can interact with numerous partners (11). It carries a homodimerization site within the vinculin homology 1 (VH1) domain that overlaps with the binding site for β-catenin, ajuba, and spectrin. The VH2 domain contains a binding site for α-actinin, formin, afadin, and vinculin. In particular, vinculin binds to the first half of the VH2 domain (VH2a) of αE-catenin within amino acids (aa) 327–402 (12–14), allowing the formation of α2-Vinc2 heterotetramers in vitro through a second αE-catenin dimerization motif located in this region (13, 14). The second half of the VH2 domain (VH2b) carries an adhesion modulation domain located between aa 510 and 633 (15, 16), whereas the VH3 domain carries overlapping binding sites for F-actin, ZO-1, and eplin. Eplin (17–19) and formin (20) have been suggested to promote indirect links between cadherin and actin.
An initially proposed model for cadherin-mediated adhesion involved a direct physical link between cadherins and the actin cytoskeleton through a tripartite complex comprising the cytoplasmic tail of cadherin, β-catenin, and α-catenin, which is recruited by β-catenin to bind to actin filaments. More recently, however, this role of α-catenin in the direct binding of cadherin·catenin complexes to actin has been called into question, with reconstituted in vitro systems outlining the inability of α-catenin to bind β-catenin and actin simultaneously (21). Instead, it has been suggested that an increased concentration of monomeric α-catenin at adhesion sites promotes the formation of soluble α-catenin dimers acting as bundling agents for F-actin, inhibitors of the ARP2/3 complex (22). This results in modulation of the actin cytoskeletal structure and dynamics in the vicinity of the cadherin tail. However, this cannot explain the observed anchoring of cadherin·catenin complexes to actin filaments (23–25).
More recent studies implicate vinculin in the anchoring of cadherin·catenin complexes to actin (26, 27). Vinculin is a component of integrin-associated complexes (28) and plays a role in mechanotransduction at focal adhesions (29). Vinculin is also found at AJs (30) and acts as a binding partner for α-catenin in mature cadherin-based AJs (31, 32). The vinculin head binds to the VH2a domain of αE-catenin, probably in response to conformational changes in both vinculin and αE-catenin; this, in turn, leads to the formation of a putative heterotetramer where both vinculin and αE-catenin C-terminal domains can bind F-actin with high affinity (13, 14). Structural and biochemical analysis of the VH2 domain suggests a possible conformational change in the αE-catenin VH2 domain from a closed to an open conformation, where the VH2b domain unfurls from the VH2a domain, thereby unmasking the vinculin binding domain (33, 34). This conformational change of αE-catenin, controlled by a force-dependent mechanism, may be responsible for myosin II-dependent recruitment of vinculin in mature AJs (4, 12, 26, 27). These data suggest that αE-catenin and vinculin may be part of the tension transducer that allows the strengthening of AJs, although the involvement of this process in the regulation of cadherin adhesion strength has never been demonstrated.
To address the functional role of αE-catenin and vinculin in the regulation of cadherin-based adhesion strength, we used cell lines expressing E-cadherin at their surface and manipulated the levels of α-catenin or vinculin or expressed α-catenin mutants in these cells. We measured and compared the force required to separate a cell doublet (separation force (SF)) using the dual pipette assay. Our data indicate that αE-catenin and vinculin cooperate to promote the time-dependent reinforcement of cadherin-mediated adhesions by forming a mechanoresponsive link between cadherin and the underlying actin cytoskeleton.
EXPERIMENTAL PROCEDURES
Cell Lines and Transfectants
Ecad and EcadGFP cells are S180 cells stably transfected to express chicken E-cadherin and mouse E-cadherin·GFP, respectively (6, 35). These S180 transfectants express a functional cadherin·catenin complex. DLD-1-R2/7 variants are invasive non-epithelioid carcinoma cell lines expressing E-cadherin but not αE-catenin or αN-catenin (36). These cells were transiently transfected, as described previously (6, 8).
Cell Cultures and Knockdown Experiments
S180 transfectants were maintained in DMEM with 10% FCS (Lonza, Verviers, Belgium). DLD-1-R2/7 cells were maintained in the same culture medium, with an additional 10% NCS (Sigma). Confluent cultures were routinely passaged by treatment with 0.05% trypsin + 0.02% EDTA (Invitrogen). For SF measurements, cell dissociation was performed in TC buffer (0.01% trypsin + 10 mm calcium), as described previously (6). Cells were transferred in working medium (CO2-independent medium; Invitrogen) and used immediately. For aggregation assays, cell monolayers were dissociated using cell dissociation buffer (enzyme-free; Invitrogen), as described previously (37). Knockdown experiments were performed using siRNA transfection with HiPerfect reagent (Qiagen, Courtaboeuf, France), according to the manufacturer's guidelines. Cadherin expression was analyzed by flow cytometry with a specific antibody directed against the extracellular domain.
Antibodies and Reagents
The ECCD-2 mAb directed against E-cadherin and 7F9 mAb directed against vinculin were from Takara Biomedicals (Shiga, Japan) and Millipore (Billerica, MA), respectively. The mAb, which recognizes the chicken E-cadherin and was developed by W. Gallin, was obtained from the Hybridoma Bank (Iowa City, IA). The rabbit polyclonal antibody directed against the extracellular domain of chicken E-cadherin has been described elsewhere (38). The mAbs directed against β-catenin and p120ctn as well as secondary antibodies conjugated to HRP were obtained from BD Biosciences. The mAbs directed against α-catenin and α-tubulin (clone DM 1A) were obtained from Sigma-Aldrich. The mAb directed against αE-catenin was purchased from Abcam (Cambridge, UK). The mAb (α18) antibody against αE-catenin was a generous gift of Dr. A. Nagafuchi (39). Secondary antibodies coupled to phycoerythrin and the anti-human FC antibodies were from Interchim (Asnières, France). Fluorescently (Alexa-488, -555, and -647) coupled secondary antibodies and phalloidin were purchased from Invitrogen. The human Ecad-Fc chimera was from R&D Systems. Polylysine solution was from Sigma. The pEcadGFP and pEαMC were a generous gift of M. Ozawa (10). The ON-TARGETplusTM SMARTpool siRNAs for ctnna1 and vinculin (Vcl) and the siGLO RISC-Free and non-targeting control (Ctrl) siRNA were purchased from Dharmacon RNA Technologies (Perbio Sciences, Brebière, France).
Chimeric Cadherin Constructs
The construct encoding the wild type (WT) αE-catenin fused to GFP at the C terminus (αEcatGFP(c)) and carrying an HA tag epitope at the N terminus was kindly provided by A. Bershadsky (Weismann Institute, Rehovot, Israel). The plasmid encoding the WT E-cadherin fused to GFP at the C terminus (EcadGFP) and pEαMC were kindly provided by M. Ozawa (10, 40). The pEαMC encodes an E-cadherin·αMCE-catenin chimeric protein lacking the C-terminal catenin-binding domain fused in frame with the aa 301–906 sequence of αE-catenin (EαMC). The construct encoding the chimeric E-cadherin·αE-catenin·GFP (EcadαEcatGFP) was obtained by in-frame cloning the αEcatGFP sequence downstream of the E-cadherin transmembrane domain (EcadαEcatGFP). Briefly, a PCR product for the αEcatGFP sequence carrying an RsrII site at the 3′- and 5′-extremities was amplified from pαEcatGFP with 5′-gacgcggaccgttattacttgtacagctcgtccat-3′ and 5′-cttctcggtccgatggcttctagctatccttatga-3′ primers. The PCR product was digested by RsrII, purified using the Nucleospin Extract II kit (Macherey Nagel, Düren, Germany), and ligated into the corresponding site of pEcadΔcyto (6). The integrity of the construct was verified by restriction enzyme digestion and analysis of the DNA sequence. A schematic representation of the chimeric cadherins used is shown in Fig. 1A.
FIGURE 1.

Schematic representation of chimeric E-cadherins, αE-catenin, and its mutants. A, schematic representation of chimeric cadherins: E-cadherin fused to GFP (EcadGFP), E-cadherin fused after its transmembrane domain with WT αE-catenin (EcadαEcatGFP), or E-cadherin fused after its p120ctn binding site with an αE-catenin lacking the VH1 domain (EαMC). B, schematic representation of the WT αE-catenin fused to GFP at either the N or C terminus (αEcatGFP and αEcatGFP(c), respectively) and the respective GFP mutants, with GFP located at the N terminus.
αE-catenin Mutant Constructs
GFP-tagged αE-catenin mutant constructs aa1–510, aa1–670, and aa491–660 were derived from a full-length mouse αE-catenin fused to GFP at its N terminus (αEcatGFP) in the pEGFC2 vector (41). The aa1–670 construct was obtained by excising a BamHI (position site 2300 in the αE-catenin cDNA sequence)-BamHI (multiple cloning site) fragment. The construct aa1–510 was obtained by subcloning the fragment XmaI (multiple cloning site)-XmaI (position site 2049 in the αE-catenin cDNA sequence) in a new pEGFPC2 vector. The aa491–660 construct was obtained by cloning into pEGFPC2 between EcoRI and BamHI sites a PCR fragment amplified from the mouse GFP-αE-catenin chimeric cDNA as a template, using the following primers: 5′-tatagaattcaagcaagtccgtgttctcacagatgctgttgatgac-3′ and 5′-tataggatccttgtctggacactggtcctgcttctgacatcaaagt-3′. All constructs were validated by DNA sequencing. The plasmid encoding the EαcatmutVincGFP mutant was derived from the plasmid encoding the full-length WT mouse αE-catenin fused to GFP at the N terminus in the pEGFC2 vector (41), in which a unique KpnI site was introduced in the αE-catenin coding sequence at position 799 (between the aa 273 codon and the aa 274 codon) and a unique ApaI site at position 2200 (between the aa 734 codon and the aa 735 codon). Then the central KpnI-ApaI fragment of the WT αE-catenin sequence was replaced by digestion and religation by the corresponding mutated DNA segment obtained by direct gene synthesis (Proteogenix SAS, Oberhausbergen, France). In the mutated DNA, the basic and acidic aa were replaced by acidic and basic aa, respectively, as follows: R326D, D328R, R332D, E336K, N338D, R341D, Q345E, D346R, E350K, N354D, and K358E. The final construction was checked by restriction mapping and by complete sequencing. The schematic representation of the chimeric αE-catenin and mutants are shown in Fig. 1B.
Preparation of Adhesion Substrate and Cell Spreading Assays
Plasma-treated glass coverslips were coated overnight with anti-human Fc antibody at 9 μg/cm2, rinsed, and incubated with human Ecad-Fc chimera at 4 μg/cm2, as described previously (42).
Immunofluorescence Microscopy
Samples were fixed in 4% paraformaldehyde in PBS + Ca2+/Mg2+ (PBS+) or in cold methanol. For detection of vinculin and α18 epitope, cells were fixed in the presence of Triton (1% paraformaldehyde + 0.5% Triton X-100 in PBS+) for 3 min, rinsed, and then fixed for 10 min in 1% paraformaldehyde. Immunodetection was performed as described previously (6). Cells were viewed by an epifluorescent (DM6000, Leica Microsystems, Wetzlar, Germany) or inverted confocal microscope (Eclipse Ti, Nikon, Tokyo, Japan). Cadherin expression was analyzed on living cells or fixed cells by flow cytometry, as described previously (8). Total internal reflection fluorescence microscopy analysis was performed on an Eclipse TE2000 inverted microscope (Nikon).
Cell Sorting, Preparation of Cell Extracts, Co-immunoprecipitation, and Immunoblotting
The characterization of αE-catenin mutants was performed by GST pull-down, co-immunoprecipitation, and Western blot analysis using lysates from transfected DLD-1-R2/7 cells with anti-β-catenin antibody, as described previously (12). DLD-1-R2/7 cells were transiently transfected to express GFP-tagged αE-catenin and its mutants, GFP-tagged E-cadherin or chimeric cadherins (EcadαEcatGFP or EαMC (where cells were co-transfected with plasmid encoding GFP in a 1:10 ratio to that of plasmid encoding EαMC)) and dissociated with cell dissociation buffer. GFP+ cells were sorted using FacsVantage Diva (BD Biosciences), and cells were extracted with Laemmli 1× buffer (Bio-Rad), boiled for 5 min, and analyzed on SDS-PAGE. After the transfer, membranes were incubated with Ponceau Red. Western blot analysis was performed (8) using SuperSignal West Pico Chemoluminescent for detection (Thermo Scientific). The extraction of cell monolayers was performed as described previously (8). Quantitative analysis was done on two independent experiments using the ImageJ software (National Institutes of Health, Bethesda, MD). The α-tubulin content was used to normalize for the protein content. We calculated the total levels of β-catenin, p120, or vinculin in cells expressing mutant catenins or chimeric cadherins as compared with cells transfected with αEcatGFP or EcadGFP, respectively. (In cells, the levels of proteins were considered as 100%.)
Doublet Stretching
Ecad cell doublets were deposited on silicon substrates (Poly-Di-Methyl-Siloxane (PDMS) and Gel-Pak PF-60-X4, thickness 150 μm, Teltek (Sonora, CA)) micropatterned with polylysine-coated parallel lines (10-μm width) and incubated at 37 °C for 20 min. The substrates were stretched using a manual uniaxial stretcher combined with micropatterning, as described previously (43). This method fixes the orientation of the force field exerted perpendicular to the cell-cell contact. We increased the original length of the substrate by 20% for 2 min. After 2 min of stretching, doublets were immediately fixed, and the silicon substrates were removed from the stretcher. Four independent experiments were performed. Samples were immunostained with vinculin, E-cadherin, and α18 epitope antibodies, mounted with Immumount medium (Thermo Shandon, Pittsburgh, PA), and analyzed by confocal microscopy. The ratio of intensities (expressed in arbitrary units) between vinculin and Ecad or α18 and Ecad was calculated using ImageJ software at the cell-cell contact area from compilation of the three-dimensional stack of confocal images. We compared 11 non-stretched and 16 stretched doublets.
Measurement of Separation Forces between Cells
We used the dual pipette assay as described previously (6). Briefly, cells were brought into contact with two micropipettes at 37 °C, each held by a micromanipulator connected to a combined hydraulic-pneumatic system and a pressure sensor. The pipettes were then moved apart to detach the adherent cells. If a doublet was pulled intact from the left pipette, it was moved back to the orifice of that pipette, and the degree of aspiration was then increased. The cycle was repeated until the nth cycle, at which the cells separated. Aspiration values recorded for the last two cycles (Pn − 1 and Pn) were used to calculate the SF for each doublet using the equation, SF = π(d/2)2(Pn − 1 + Pn)/2, where d is the internal diameter of the left pipette. The results for 30–40 measurements were used to obtain the mean SF for a specific contact time in at least three independent experiments. SF values indicated in the figures are the mean SF, and the error values are the S.E. For transiently transfected cells expressing fluorescent proteins, we selected cells presenting a similar intensity of the fluorescent protein signal. The level of cadherin expression was monitored in parallel by flow cytometry on a cell suspension sample.
RESULTS
The Strengthening of E-cadherin-based Adhesion Is Rescued by the Expression of αE-catenin Constructs in αE-catenin-defective Cells
We used the DLD-1-R2/7 cells and the dual pipette assay to quantitatively evaluate the contribution of αE-catenin to adhesion strength. This assay, originally developed by Prof. S. Chien and co-workers (44), has been modified to determine the force required to separate adherent cells (SF), and several studies have been published demonstrating the validity of these SF measurements to quantify adhesion strength (45) and analyze the molecular mechanisms involved in cadherin adhesion strengthening (6, 8, 46–48).
DLD-1-R2/7 cells expressed significant levels of E-cadherin at their surface (Fig. 2A) and expressed β-catenin, vinculin, and p120ctn (32, 36) (data not shown). The absence of αE-catenin in these cells did not impede the initiation of cadherin-driven intercellular adhesion, as revealed by the formation of cell aggregates in suspension (Fig. 2B). However, these aggregates were easily dissociated in single cells by pipetting (Fig. 2C), indicating that cadherin adhesions lack strength. αE-catenin-depleted cell doublets exhibited a detectable but low SF at 4 min of contact (Fig. 2D, left) that was severely reduced compared with the classical E-cadherin-based adhesion strength (6). The expression of WT αE-catenin fused to GFP at the C terminus (αEcatGFP(c)), but not of GFP alone, increased the SF significantly at 4 min of contact (Fig. 2D, left). After 30 min of contact, DLD-1-R2/7 cells exhibited a small increase in the SF (Fig. 2D, right). In contrast, when they expressed αEcatGFP(c), DLD-1-R2/7 cells displayed a 2-fold increase in the SF at 30 min of contact (Fig. 2D, right).
FIGURE 2.

DLD-1-R2/7 aggregation properties and determination of the separation force in αE-catenin-depleted cells and transfectants. A, flow cytometry analysis of cell surface E-cadherin expression levels in isolated DLD-1-R2/7 cells before commencement of the aggregation assay. B, morphology of the DLD-1-R2/7 aggregates after 120 min in the suspension shaker assay. C, disruption of the DLD-1-R2/7 aggregates in single cells by a gentle pipetting indicates that these aggregates exhibit a low cohesiveness between cells. D, mean SF measured at 4 min (left) and 30 min (right) of contact for DLD-1-R2/7 cells and for DLD-1-R2/7 transfectants expressing GFP alone, αEcatGFP(c) (C-terminal tagged GFP), EcadGFP cadherin, and chimeric cadherins (EcadαEcatGFP or EαMC) (See Fig. 1). High E-cadherin-based adhesion strength was rescued by the expression of full-length αE-catenin and chimeric E-cadherin·αE-catenin but not by the expression of GFP alone or E-cadherin·GFP. E, Western blot analysis of lysates of flow cytometry-sorted GFP+DLD-1-R2/7 cells transfected to express EcadGFP or EcadαEcatGFP or to co-express EαMC and GFP (see “Experimental Procedures”). The α-tubulin content was used to normalize for the protein content. Black and gray asterisks in the left panel correspond to the statistical analysis of the SF value compared with those of untransfected DLD-1-R2/7 or transfected to express EcadGFP, respectively. ***, p < 0.001; **, p < 0.01; *, p < 0.05 (two-tailed). Scale bar (B), 30 μm. Error bars, S.E.
We expressed various E-cadherin·αE-catenin chimeric proteins (Fig. 1A) to compare the effect of soluble αE-catenin and membrane-targeted αE-catenin. E-cadherin·αE-catenin·GFP (EcadαEcatGFP) targeted at sites of cadherin adhesions and E-cadherin·αMCE-catenin (EαMC), which lacks the dimerization domain, promoted a strong SF at 4 min of contact (Fig. 2D, left); this is similar to the SF that was observed with αE-catenin·GFP. The expression of EcadαEcatGFP induced a significant increase in the SF with time (Fig. 2D, right).
As these chimeric cadherins increased the level of E-cadherin at the cell surface, we analyzed the consequence of E-cadherin·GFP (EcadGFP) overexpression and showed that it only produced a small increase in the SF of DLD-1-R2/7 cells (Fig. 2D) at 4 min of contact. This indicates that the strong increase in SF induced by the chimeric cadherins is mostly due to the presence of the αE-catenin sequence in the cytoplasmic domain of these cadherins.
Next, we checked if the expression of exogenous E-cadherin and chimeric cadherins in DLD-1-R2/7 cells affected the endogenous levels of β-catenin and p120ctn (p120) (Fig. 2E). Transfected DLD-1-R2/7 cells were sorted based on their GFP positivity (see “Experimental Procedures”), extracted, and analyzed by Western blotting. We found that DLD-1-R2/7 cells expressing EcadαEcatGFP or EαMC (GFP+; see “Experimental Procedures”) exhibited 112 ± 43 and 117 ± 44% of β-catenin, 130 ± 10 and 95 ± 25% of p120, and 75 ± 6 and 79 ± 4% of vinculin contents, respectively, compared with cells expressing EcadGFP (set as 100% levels; Fig. 2E). In another set of experiments, we used DLD-1-R2/7 cells expressing GFP alone as a control. We found that DLD-1-R2/7 cells expressing EcadGFP exhibited a 1.4-fold increase in β-catenin and p120 contents as compared with control cells. This indicates that the expression of endogenous β-catenin and p120 is increased in DLD-1-R2/7 cells expressing exogenous cadherins and is increased slightly more in DLD-1-R2/7 cells expressing chimeric E-cadherin·αE-catenins than in those expressing WT E-cadherin (Fig. 2E).
Together these results indicate that the high E-cadherin-based adhesion strength of αE-catenin-depleted cells is recovered by the expression of WT αE-catenin and membrane-targeted E-cadherin·αE-catenin proteins.
Effects of αE-catenin Deletion Mutants on Cadherin Adhesion Phenotype
To further analyze the mechanisms of αE-catenin-dependent rescue of cadherin-based adhesion, we prepared various αE-catenin·GFP-deleted constructs (see “Experimental Procedures”). We first assessed the capability of these deletion mutants to interact with β-catenin, which recruits αE-catenin at cadherin-based adhesions. DLD-1R2/7 cells were transfected with the plasmids encoding GFP alone or the N terminus of the GFP-tagged WT αE-catenin (αEcatGFP) and the aa491–660 αEcatGFP, aa1–510 αEcatGFP, or aa1–670 αEcatGFP mutants. Cell lysates were incubated with anti-β-catenin, and immunoprecipitates were analyzed by Western blotting with an anti-GFP (Fig. 3A). The GFP and the αE-catenin modulation domain (aa491–660 αEcatGFP mutant) did not co-precipitate with β-catenin, in contrast to other αE-catenin mutants, which carry the β-catenin binding site located in their VH1 domain.
FIGURE 3.

Characterization of αE-catenin mutants and their effect on αE-catenin-depleted cell-cell junction formation, cell spreading, and the formation of cadherin adhesion on Ecad-Fc-coated surfaces. A, characterization of αE-catenin mutants for their capacity to bind to β-catenin. Cell lysates of transfected DLD-1-R2/7 cells expressing GFP alone, the αE-catenin mutants (aa491–660 αEcatGFP, aa1–510 αEcatGFP, and aa1–670 αEcatGFP), or αEcatGFP were immunoprecipitated (IP) with an anti-β-catenin antibody and analyzed by Western blot with an anti-GFP antibody (right). The arrows indicate the position of the band corresponding to GFP-tagged αE-catenin and the mutants that co-precipitate with β-catenin. Note that GFP alone and the αE-catenin mutant corresponding to the modulation domain (aa491–660 αEcatGFP) do not co-precipitate with β-catenin. The corresponding cell lysate inputs are presented in the left panel. B, localization of GFP (top panels) and β-catenin (bottom panels) in DLD-1-R2/7 cells transiently transfected with plasmids encoding GFP alone, αEcatGFP, and aa1–510 or aa1–670 αEcatGFP mutant. GFP-transfected and non-transfected cells did not form cell-cell contacts. However, cells transfected with a αEcatGFP construct readily formed cell contacts and recruited β-catenin at their boundaries. Cells transfected with the aa1–510 αEcatGFP mutant were indistinguishable from αEcatGFP-transfected cells. Surprisingly, the slightly longer mutant (aa1–670) restored neither cell contact formation nor the recruitment of β-catenin. C, localization of GFP (top panels) and β-catenin (bottom panels) in transiently transfected cells to express αEcatGFP, aa1–510 αEcatGFP, or aa1–670 αEcatGFP mutant and GFP after spreading for 16 h on Ecad-Fc-coated surfaces. Scale bar, 10 μm.
We next analyzed the potential of the αE-catenin mutants to rescue the phenotype of a normal cadherin intercellular adhesion in DLD-1-R2/7 cells. As expected, GFP-transfected cells did not form cell-cell contacts in a monolayer (Fig. 3B, left panel), and both β-catenin and E-cadherin remained uniformly distributed at the plasma membrane. In contrast, cells transfected with a WT αE-catenin construct fused to GFP at its N terminus formed extensive cell contacts and recruited β-catenin (Fig. 3B, right panels) and E-cadherin (data not shown) to their boundaries. The expression of a αE-catenin (aa1–510) mutant, lacking both the actin binding and modulation domains, also restored normal cell-cell contacts that were indistinguishable from those formed in cells transfected with WT αE-catenin (Fig. 3B, middle left panels). Interestingly, the slightly longer mutant (aa1–670), lacking only the actin-binding domain but containing the modulation domain, restored neither DLD-1-R2/7 intercellular contacts nor recruitment of β-catenin (Fig. 3B, middle right panels). To characterize the rescue of cadherin-based adhesion by these two mutants, we evaluated their potency to induce αE-catenin-depleted cell spreading on Ecad-Fc-coated surfaces and form cadherin-mediated adhesions (49) (Fig. 3C and Table 1). As expected, DLD-1-R2/7 cells did not undergo spreading on Ecad-Fc, whereas, when expressing WT αE-catenin, they showed extensive spreading and formed classical radial cadherin adhesions (Fig. 3C and Table 1) similar to those described previously (50). However, whereas the αE-catenin (aa1–510) mutant restored spreading and promoted the formation of cadherin adhesions on Ecad-Fc, the αE-catenin (aa1–670) mutant did not restore any of these phenotypes. Very similar results were observed on Ncad-Fc-coated surfaces when αE-catenin-depleted cells were co-transfected with these mutants and the N-cadherin·RFP construct (data not shown). These results indicate that a lack of the actin-binding domain does not interfere with the ability of the αE-catenin mutant to contribute to the formation of cadherin-mediated intercellular contacts and cadherin adhesion formation, whereas the presence of the VH2b modulation domain in the mutant impedes this process.
TABLE 1.
Quantification of the effect of the αE-catenin mutants on cadherin adhesions formed on Ecad-Fc-coated surfaces
| Transfection | DLD-1-R2/7 cells spread with cadherin adhesions | Spread DLD-1-R2/7 cells | Non -spread DLD-1-R2/7 cells | Total observed cells |
|---|---|---|---|---|
| αEcatGFP | 21 | 55 | 12 | 88 |
| aa1–510 αEcatGFP | 46 | 34 | 13 | 93 |
| aa1–670 αEcatGFP | 1 | 3 | 78 | 82 |
| GFP | 88 | 88 |
Effects of αE-catenin Deletion Mutants on Vinculin Recruitment at Cell-Cell Contacts and Adhesion Strength
Vinculin is a partner of αE-catenin and is recruited during cadherin-based adhesion. It binds to the VH2a domain; this process is negatively regulated when αE-catenin is in its closed conformation by interaction with the modulation domain (33, 34).
We searched for the presence of vinculin and α18 antibody immunoreactivity at cell-cell contacts restored by the expression of WT αE-catenin and mutant forms in DLD-1-R2/7 cells. α18 antibody has been described as recognizing an epitope close to the vinculin binding site that is putatively unmasked upon the conformational change of αE-catenin when VH2a and VH2b cease to interact (12). Cells transfected with WT αE-catenin·GFP displayed vinculin and weak α18 staining at cell-cell contacts (Fig. 4A, top panels). Interestingly, when transfected to express the αE-catenin (aa1–510) mutant, these cells exhibited an increased vinculin and α18 staining at cell-cell contacts (Fig. 4A, middle panels). We quantified the fluorescence intensity ratio of GFP, vinculin, and α18 epitope stainings at cell-cell contacts of DLD-1-R2/7 expressing WT αE-catenin·GFP and αE-catenin (aa1–510) mutant (Fig. 4B). We observed much higher vinculin/αEcatGFP and α18/αEcatGFP staining intensity ratios in cells expressing the αE-catenin (aa1–510) mutant as compared with those expressing WT αE-catenin. These results suggest that the αE-catenin (aa1–510) mutant can induce the recruitment of vinculin at cadherin-mediated cell-cell contacts due to its unmasked central binding site, a process contributing to the rescue of DLD-1 R2/7 intercellular contact and cadherin adhesions formation. Together, these experiments show that αE-catenin is necessary and sufficient in these cells for the recruitment of vinculin at cell-cell contacts. This recruitment does not require the presence of the C-terminal actin-binding domain of αE-catenin but is impaired by the adhesion modulation domain.
FIGURE 4.

Detection of αE-catenin·GFP variants, α18-specific epitope, and vinculin in transfected DLD-1-R2/7 cells and their effect on E-cadherin-mediated cell adhesion strength. A, localization of GFP (left panels), vinculin (middle left panels), and α18 epitope (middle right panels) in cells transiently transfected to express αEcatGFP (top panels), aa1–510 αEcatGFP mutant (middle panels), or αEcatMutVincGFP mutant (bottom panels). Merged images are shown in the right panels. Scale bar, 10 μm. Arrows point to some of the intercellular adhesions between transfected cells. B, mean ratio between vinculin and catenin (GFP) (vinc./αcat, blue bars) intensity and α18 and catenin (GFP) intensity (α18/αcat, pink bars) were measured on confocal images at the cell contact area of DLD-1-R2/7 cells transiently transfected to express either αEcatGFP or the mutants aa1–510 αEcatGFP or αEcatMutVincGFP. The number of cell-cell contacts analyzed was 12, 14, and 11, respectively. Error bars, S.E. ***, p < 0.001 (two-tailed) by comparing with the values of cells expressing αEcatGFP. C, mean SF measured at 4 min of contact for αE-catenin-depleted cells transiently transfected to express the αEcatGFP (black bar), αEcatmutVincGFP mutant (blue bar), or aa1–510 αEcatGFP mutant (red bar). Error bars, S.E.M. ***, p < 0.001 (two-tailed). D, Western blot analysis of lysates of flow cytometry-sorted GFP+DLD-1-R2/7 cells transfected to express αEcatGFP, αEcatmutVincGFP, and aa1–510 αEcatGFP (see “Experimental Procedures”).
The αE-catenin (aa1–510) mutant could not rescue a strong SF at 4 min of contact in αE-catenin-depleted cells (Fig. 4C); the measured values are not statistically different from the SF obtained for cells expressing GFP alone (Fig. 2D) but they are 3-fold lower than the SF of cells expressing αEcatGFP or αEcatGFP(c) (Fig. 2D). A similar observation was made after 10 min of contact with an SF of 5.9 ± 0.2 and 32.2 ± 8.6 nanonewtons measured for cells expressing αE-catenin (aa1–510) mutant and WT αE-catenin, respectively; this result indicates that the aa1–510 mutant cannot promote a strong intercellular adhesion or restore normal cadherin adhesion strengthening over time, although it permits vinculin recruitment at cell-cell contacts. These results thus indicate a specific requirement for the αE-catenin C-terminal domain for the development of normal E-cadherin adhesion strength. Overall, our data indicate that strengthening of cadherin adhesions require both the availability of the central vinculin-binding domain and the presence of the C-terminal domain of αE-catenin.
αE-catenin Mutant Carrying Mutations in the Vinculin Binding Site Is Defective in Rescuing Cadherin Adhesion Strength
To further support the hypothesis that the αE-catenin·vinculin interaction is required for the production of high adhesion strength, we prepared a αE-catenin mutant with mutations into the vinculin-binding site (αEcatmutVincGFP; Fig. 1B) by exchanging positively charged aa residues with negatively charged ones and vice versa at 12 positions between aa 326 and 348. DLD-1-R2/7 cells transfected to express this mutant displayed a positive staining for vinculin at cell-cell contacts, indicating that this αE-catenin mutant was still able to interact with vinculin. Staining with α18 antibody was detected at intercellular adhesions (Fig. 4A, bottom panels). We quantified the fluorescence intensity ratio of GFP, vinculin, and α18 epitope staining at the cell-cell contacts of DLD-1-R2/7 cells expressing WT αE-catenin, αE-catenin (aa1–510), or αEcatmutVincGFP mutants (Fig. 4B). We observed similar vinculin/αEcatGFP staining intensity ratios in cells expressing WT αE-catenin or αEcatmutVinc mutant but a much higher ratio in cells expressing the αE-catenin (aa1–510) mutant (Fig. 4B). The α18/αEcatGFP intensity ratios were similar for cells expressing αEcatmutVinc or αE-catenin (aa1–510) mutant but much lower in cells expressing WT αE-catenin (Fig. 4B). This suggests that the mutations introduced in the αEcatmutVincGFP mutant may alter the closed conformation by modifying the interaction between the modulation domain (VH2b) and the vinculin-binding domain (VH2a) rather than affecting the vinculin-αE-catenin interaction.
The expression of the αEcatmutVincGFP mutant in DLD-1-R2/7 cells did not rescue strong cell adhesion strength at 4 min of contact; the values are not statistically different from the SF obtained for cells expressing GFP alone (p > 0.05, two-tailed). However, the difference is significant when compared with the SF produced for cells expressing αEcatGFP (Fig. 4C).
We next checked if the expression of αEcatGFP, αEcatmutVincGFP, or aa1–510 αEcat in DLD-1-R2/7 cells affected the endogenous levels of β-catenin, vinculin, and p120 (Fig. 4D), calculating the levels of these three proteins in lysates of DLD-1-R2/7 cells expressing the respective mutants compared with cells expressing αEcatGFP (set as 100%; see “Experimental Procedures”). We found that DLD-1-R2/7 cells expressing αEcatmutVincGFP mutant exhibited that same content (100%) of β-catenin and 110% of p120 as compared with cells expressing αEcatGFP. Similarly, DLD-1-R2/7 cells expressing aa1–510 αEcatGFP mutant exhibited 110% of β-catenin and p120 contents. Vinculin levels were increased by 2.0- and 2.1-fold in DLD-1-R2/7 cells expressing αEcatmutVincGFP or aa1–510 αEcatGFP mutant, respectively, as compared with αEcatGFP-expressing cells. Taken together, our results suggest that the defect in adhesion strength measured for cells expressing the αEcatmutVincGFP mutant arises from a perturbation of the αE-catenin interaction with vinculin at cell-cell contacts.
The Depletion of αE-catenin and Vinculin in Cells Exhibiting Classical E-cadherin-mediated Adhesion Reduces Adhesion Strength
To ensure that these results were not cell line-specific, we analyzed αE-catenin and vinculin depletion in Ecad cells, an S180-derived cell line expressing β-catenin, α-catenin, p120ctn, and E-cadherin. These cells have previously been shown to exhibit a classical cadherin-based adhesion strengthening over time (6).
When treated with ctnna1 (αE-catenin) siRNA, Ecad cells displayed a strong decrease in the level of αE-catenin (Fig. 5A) but expressed significant levels of E-cadherin at their surface (Fig. 5B). Western blot analyses with antibodies directed against E-cadherin, β-catenin, α-catenin, and p120ctn were used to quantify the levels of these proteins in ctnna1 siRNA-treated Ecad cells. Protein levels in Ctrl siRNA-treated cells were set at 100%. ctnna1 siRNA-treated Ecad cells expressed very low levels of αE-catenin (3%) but similar levels of E-cadherin and β-catenin (87 and 97%, respectively), as observed previously (51). The levels of p120ctn were slightly reduced to about 69% of the control levels. Treatment with ctnaa2 (αN-catenin) siRNA did not affect α-catenin levels or E-cadherin levels (data not shown), indicating that αE-catenin is the major form of α-catenin expressed in Ecad cells. The depletion of αE-catenin induced a 4-fold reduction in the mean SF at 4 min compared with Ctrl siRNA-treated Ecad cell doublets (p < 0.001, two-tailed).
FIGURE 5.

Effect of αE-catenin or vinculin depletion on E-cadherin-mediated cell adhesion strength. A, Western blot analysis on lysates of Ecad cells treated for 72 h with 12 nm ctnna1 (αE-catenin) siRNA or non-targeting Ctrl smart pool siRNA. B, flow cytometry analysis of surface levels of E-cadherin in ctnna1 or Ctrl siRNA-treated cells. C, Western blot analysis of Ecad cell lysates after the cells were treated for 72 h with 16 nm of Vcl siRNA or Ctrl siRNA. D, flow cytometry analysis of surface levels of E-cadherin in Vcl (dark gray) or Ctrl (light gray) siRNA-treated cells. E, mean SF measured at 4 min of contact for Vcl siRNA-treated (dark gray bar) or Ctrl siRNA-treated Ecad cells (light gray bar). α-tub, α-tubulin. Error bars, S.E.
The treatment of Ecad cells with Vcl siRNA produced a 66% decrease in the total vinculin levels as compared with Ctrl siRNA-treated cells (Fig. 5C). E-cadherin total levels and its expression at the cell surface were unchanged (103% of the Ctrl protein level; Fig. 5, C and D). Vcl siRNA-treated cells exhibited 80, 79, and 93% expression levels of β-catenin, p120ctn, and αE-catenin, respectively, as compared with Ctrl siRNA-treated cells. Vcl siRNA-treated Ecad cells exhibited a 3-fold decrease in SF at 4 min compared with Ctrl siRNA-treated cells (Fig. 5E); this argues for the involvement of vinculin in the development of cadherin-based adhesion strength. Taken together, these results show that both αE-catenin and vinculin are required for reinforcement of cadherin-mediated adhesions in cells exhibiting classical cadherin-based adhesion.
To further determine if the recently proposed conformation-dependent interaction between the two molecules was involved in this process, we expressed in cells the aa491–660 αEcatGFP mutant (Fig. 1B), which has been proposed to mask the vinculin-binding site required for myosin II-dependent vinculin recruitment at AJs (12, 26). When we expressed aa491–660 αEcatGFP in Ecad cells, we observed a reduced vinculin staining at cell-cell contacts (Fig. 6A), but focal adhesions were still visible. Co-immunoprecipitation experiments revealed that this mutant was able to interact with WT αE-catenin (data not shown). It was found mostly located in the cytoplasm. The plot profile of fluorescence intensity for vinculin staining along a line perpendicular to the cell-cell contact (Fig. 6B) revealed more a higher intensity staining intensity in the cytoplasm and a reduced intensity at the site of cell-cell contact when cells expressed aa491–660 αEcatGFP mutant as compared with GFP-expressing cells. The ratio of intensity quantified at the contact site was significantly reduced by 28% in cells (n = 16) expressing the modulation domain (n = 16) compared with GFP-expressing cells (n = 15) (p value < 0.001). This reduction in vinculin recruitment at cell-cell contacts coincided with a 36% decrease in the SF at 4 min measured for aa491–660 αEcatGFP-expressing Ecad cells, as compared with Ctrl-transfected cells, with 36.1 ± 6.2 and 56.8 ± 9.2 nanonewtons measured (p < 0.05, two-tailed), respectively. This suggests that the modulation domain of αE-catenin interferes with normal cell-cell adhesion strength, probably by perturbing the binding of vinculin to endogenous αE-catenin. Western blot analysis revealed that Ecad cells expressing the αE-catenin modulation domain exhibited 90 ± 10% of the vinculin content (Fig. 6C) and exhibited 130 ± 20% of β-catenin content and 135% of p120 content as compared with cells expressing GFP alone.
FIGURE 6.

Detection of vinculin in Ecad cells expressing the modulation domain of αE-catenin. A, GFP signals and immunolocalization of vinculin in Ecad cells transfected to express GFP alone or aa491–660 αEcatGFP mutant corresponding to the modulation domain. Arrows, cell-cell contacts between transfected Ecad cells (GFP+) and their neighbors. B, a representative plot profile showing the fluorescence intensity (expressed in arbitrary units (AU)) of vinculin along a line perpendicular to the cell-cell contact (corresponding to the cell-cell contact marked by an asterisk in the bottom panels in A) for Ecad cells expressing GFP (black line) or aa491–660 αEcatGFP mutant (gray line). C, Western blot analysis of lysates of Ecad cells transfected to express GFP or aa491–660 αEcatGFP mutant with anti-vinculin and α-tubulin antibodies. Error bars, S.E.
Taken together, our results using the DLD-1-R2/7 and Ecad cells suggest that the defect in adhesion strength measured for cells expressing the modulation domain of αE-catenin or the αEcatmutVincGFP mutant arises both from a perturbation of the αE-catenin interaction with vinculin at cell-cell contacts and from perturbations to the cadherin-actin coupling through an altered αE-catenin conformation.
Vinculin and α18 Epitope Staining Are Increased at Cell-Cell Contacts in Stretched Doublets
It has been postulated that internal mechanical strain induces αE-catenin conformational switching and vinculin recruitment, a mechanism required for force-dependent AJ maturation (4, 12). We analyzed whether the α18 epitope and vinculin immunostaining at cell-cell contacts were enhanced upon external force application. To do this, we deposited cell doublets expressing E-cadherin (Ecad cell doublets) on a deformable polylysine-coated silicon for 20 min and subjected the doublets to a 20% unidirectional stretching for 2 min (Fig. 7, A and B). Non-stretched Ecad cell doublets exhibited vinculin recruitment but a faint staining for α18 epitope at cell-cell contacts (Fig. 7C). We calculated the ratios of staining intensity for vinculin/Ecad and α18/Ecad at cell-cell contacts for non-stretched doublets (n = 11) (Fig. 7D). The quantification performed on stretched doublets (n = 16) revealed a significant increase in both of these ratios. Thus, upon stretching, more α18 epitope-specific αE-catenin is found at cell-cell contacts, correlating with an increased recruitment of vinculin at these sites. This finding is in agreement with the hypothesis of a force-dependent change in αE-catenin conformation that promotes vinculin recruitment and the subsequent strengthening of the mechanical link between cadherins and the actin cytoskeleton.
FIGURE 7.

Effect of stretching of cell doublets on vinculin and α18 epitope recruitment at Ecad cell-cell contacts. A and B, schematic representation of the stretching experiment. Shown are a top view (A) and side view (B) of the experimental procedure. Cell doublets (green ovals with blue nucleus) were deposited on silicon substrates (blue) micropatterned with polylysine-coated parallel lines (pink lines) and incubated at 37 °C for 20 min. The left and right panels represent the non-stretched and stretched conditions, respectively. The orientation of the force field exerted perpendicular to the cell-cell contact is represented by the red dashed arrow and induced an increase in the original substrate length by 20% (dashed blue line in the right panel indicates the position of the original relaxed border of the substrate). C, z projection of confocal images showing E-cadherin (green), vinculin (red), and α-catenin α18 conformational epitope (cyan) localization on Ecad cell doublets deposited on polylysine-coated silicon substrates and exposed (bottom panel) or not (top panel) to 2 min of stretching before fixation. D, mean ratios between α18 and E-cadherin intensity or vinculin and E-cadherin intensity were measured at the cell contact area of non-stretched (white bars; n = 11) and stretched (black bars; n = 16) doublets. Error bars, S.D. for non-stretched versus stretched doublets. ***, p value < 0.0001 (two-tailed). Scale bar and double arrow in C represent 10 μm and the orientation of the stretch, respectively.
DISCUSSION
Connections between cadherins and the actin cytoskeleton are crucial for the development and maintenance of adherens junctions. Numerous studies have analyzed the molecular basis of these connections, and recent work has uncovered that, contrary to previous beliefs, cadherins do not interact with actin filaments merely via the tripartite cadherin tail·β-catenin·α-catenin complex. Binding to F-actin requires additional partners, such as vinculin (4, 12) and eplin (18, 19). These proteins are recruited at mature AJs and have also been implicated in the maturation process, although their role in the development of cell-cell contact strength has not been demonstrated.
In this study, we used the dual pipette assay and cell stretching to analyze the roles of α-catenin and vinculin in the development of intercellular adhesion force in two different cell lines (Table 2). The DLD-1R2/7 cells do not express α-catenin but possess the other components of the cadherin adhesion complex. By comparison, the Ecad cells exhibit a typical E-cadherin-based adhesion and have been well studied using the dual pipette assay (6, 8). If we take into consideration (a) the dual function of vinculin at cell-cell and cell-matrix adhesion sites (30); (b) the role of vinculin in mechanotransduction at focal adhesion sites; and (c) the additional role of αE-catenin in cell migration independently of cadherins (52), it is important to probe the requirement for vinculin and αE-catenin in cadherin-based adhesion strengthening in a context where cells are not engaged in focal adhesions. Moreover, focal adhesion contributes to increased cellular tension (53) and stimulates cadherin-based adhesions (47).
TABLE 2.
Summary of the effects observed on Ecad and DLD-1-R2/7 cells by the modulation of αE-catenin, E-cadherin, or vinculin content or by the expression of αE-catenin mutants and E-cadherin/αE-catenin chimeras on the adhesion strength (SF) and vinculin and a18 epitope staining at cell-cell contacts
*, compared with cells expressing GFP alone; #, compared with non-stretched Ecad cells; n.d., not determined.
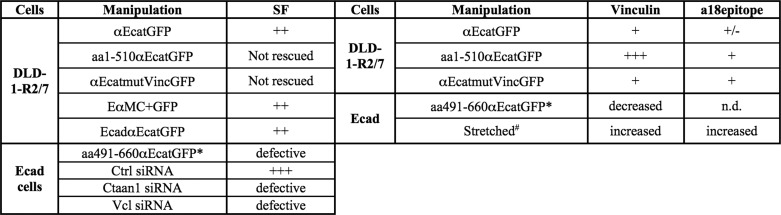
αE-catenin-depleted cells form aggregates in suspension and cell-cell contacts in doublets. However, using SF measurements, we show here that the absence of α-catenin dramatically affects the initial and time-dependent strength of cadherin-based adhesions. αE-catenin-depleted cell doublets exhibit an SF of a few nanonewtons, similar to those forces measured for cells expressing truncated cadherins (lacking the cytoplasmic domain) or in cells with impaired actin polymerization (6). The SF of the Ecad cell doublets was also substantially reduced following α-catenin depletion, indicating that α-catenin is required to produce strong adhesion strength. Interestingly, the rescue obtained with αE-catenin re-expression in αE-catenin-depleted cells is similar to that obtained with chimeric cadherin·α-catenin proteins, where the cytoplasmic domain of E-cadherin was replaced with either the full-length αE-catenin (αEcatGFP) or the VH2-VH3 domain of αE-catenin (EαMC) without the dimerization site. These results indicate that the observed rescue is not due to the influence of the soluble form of α-catenin on actin dynamics (22), at least in the 30 min following cell-cell contact initiation, but rather to a direct effect of α-catenin on cadherin adhesion complexes.
These chimeric proteins, as well as the full-length α-catenin, can recruit partners, such as formin (54) or eplin (17), to nucleate or anchor actin at the contact site. The aa1–510 αEcatGFP mutant carrying the formin binding site was unable to rescue the adhesion strength defect in 4 and 10 min in DLD-1R2/7 cell doublets, suggesting that formin does not play a major role in the early development of cadherin-based adhesion in these cells. Taguchi et al. (18) showed that eplin was absent from immature peripheral AJs in the parental DLD1 cell line, but it was recruited and required for the formation of mature AJs. Chervin-Pétinot et al. (19) recently reported the role of eplin in bridging the VE-cadherin·α-catenin complex to the actin cytoskeleton in a manner that is independent of actomyosin contractility and its role in promoting vinculin recruitment at mature AJs in endothelial cells. Accordingly, our observations indicate that the C-terminal domain of αE-catenin bearing the eplin binding site is required for the rescue of adhesion strength either by re-expression of soluble αE-catenin or the chimeric αE-catenin·cadherin in αE-catenin-depleted cell doublets. However, the same C-terminal domain of the protein has been also reported to possess an F-actin binding domain (10). Our results do not allow a conclusion as to whether it is the binding to eplin or actin or both that determines cadherin adhesion strengthening.
The aa1–510 αEcatGFP mutant possesses binding sites for β-catenin, spectrin, ajuba, and α-actinin and can be recruited during cadherin-based adhesion and can promote cell-cell contact in DLD-1R2/7 cells. These results indicate that, although these partners play a role in α-catenin recruitment at cell junctions and contact initiation, they are not sufficient for adhesion strengthening. It has been proposed that α-catenin exists as an open or a closed conformation (55). In its closed conformation, the central binding site for vinculin is masked by its interaction with the modulation domain. In the αE-catenin (aa1–510) mutant, this modulation domain is absent, thereby removing any restriction based on a conformational modulation on the vinculin-binding site to restore cadherin adhesion. Indeed, we observed strong staining for vinculin and the α18 epitope at intercellular contacts of DLD-1-R2/7 cells expressing this mutant. However, this was not enough to rescue high adhesion strength. By comparison, the slightly longer α-catenin (aa1–670) mutant was unable to restore cadherin adhesions, suggesting some hindrance in the central vinculin-binding site that renders the protein defective to initiate adhesion. We produced an αEcatmutVincGFP mutant carrying several mutations in the VH2a domain that did not affect the recruitment of vinculin, suggesting that this mutant may be rather impaired for VH2a-VH2b interaction. Interestingly, this mutant was unable to rescue the adhesion strength of αE-catenin-depleted cells, suggesting that the αE-catenin conformational change may be necessary for adhesion strengthening independent of vinculin binding.
The depletion of vinculin (56) or αE-catenin (57) alters AJs in MDCK cells. In endothelial cells, vinculin does not play a crucial role during the formation and maintenance of intercellular adhesion but contributes to junction remodeling (58). Here, we show that the depletion of αE-catenin or vinculin in Ecad cells strongly decreases the SF measured at 4 min of contact, indicating that both α-catenin and vinculin are required for the development of cadherin-based adhesion strength. In addition, the expression of an αE-catenin fragment carrying the modulation domain alone reduces the SF by 2-fold, indicating that it can interfere with endogenous α-catenin mechanosensing activity. One hypothesis is that it associates with the full-length endogenous α-catenin, masking the vinculin-binding domain and thus preventing the recruitment of vinculin at adhesion sites. Taken together, our data show that the availability of the VH2 domain of α-catenin and its interaction with vinculin are necessary for the development of cadherin adhesion strength. This process possibly defines the basis of AJ mechanosensing. To support this premise, we observed vinculin and α-catenin α18 epitope staining at cell-cell contacts of DLD-1R2/7 cells transfected to express WT αE-catenin and at the intercellular contacts of cell doublets. Furthermore, we showed that the stretching of cell doublets induced a significant increase in this staining at cell-cell contacts. Overall, our results provide the first direct evidence to support the contribution of a force-dependent modulation of α-catenin conformation in the development of cadherin adhesion strength (Fig. 8). Our results thus further unravel the cooperation between vinculin and α-catenin and highlight the requirement for the α-catenin C-terminal domain in the process of cadherin-based cell adhesion strengthening.
FIGURE 8.

Model for the role of α-catenin central and C-terminal domain and the cooperation with vinculin in the process of cadherin-based cell adhesion development, strengthening, and coupling to the actin cytoskeleton.
Acknowledgments
We acknowledge A. Nagafuchi for the generous gift of the mAb α18, M. Lambert for advice and help in the construction of the αE-catenin expression vectors, and C. Carlier for technical help. We acknowledge Rebecca Jackson for critical reading of the manuscript. We acknowledge Z. Maciorowski and S. Grondin from the Flow cytometry facility, L. Sengmanivong from the Nikon Imaging Centre-Institut Curie, and V. Fraisier and F. Waharte from the PICT-IBiSA facility for help with cell sorting, imaging, and computerized video microscopy, respectively. Part of the wide field microscopy was also performed at the Institut du Fer à Moulin Cell Imaging Facility. Hybridomas obtained from the Developmental Studies Hybridoma Bank were developed under the auspices of the NICHD, National Institutes of Health, and maintained by the University of Iowa (Iowa City, IA).
This work was supported, in whole or in part, by the National Institutes of Health, NCI/Institut National du Cancer Joint Research Project Fellowship Program. This work was also supported by grants from the Agence National de la Recherche (ANR) (Physique et Chimie du Vivant I (PCVI) program, Grant PCV07-186757; BLANC program, Grant 1515 02), Programme Incitatif et Coopératif (PIC) of Institut Curie, and Fondation Pierre Gilles de Gennes, as well as by grants from the Association pour la Recherche sur le Cancer (ARC), Association Française contre les Myopathies, and Institute of Molecular and Cell Biology A*STAR core funding.
- AJ
- adherens junction
- VH1–VH3
- vinculin homology domain 1–3, respectively
- SF
- separation force
- aa
- amino acid(s)
- Ctrl
- control
- Vcl
- vinculin.
REFERENCES
- 1. Papusheva E., Heisenberg C. P. (2010) Spatial organization of adhesion. Force-dependent regulation and function in tissue morphogenesis. EMBO J. 29, 2753–2768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Takeichi M. (1991) Cadherin cell adhesion receptors as a morphogenetic regulator. Science 251, 1451–1455 [DOI] [PubMed] [Google Scholar]
- 3. Wang N., Butler J. P., Ingber D. E. (1993) Mechanotransduction across the cell surface and through the cytoskeleton. Science 260, 1124–1127 [DOI] [PubMed] [Google Scholar]
- 4. le Duc Q., Shi Q., Blonk I., Sonnenberg A., Wang N., Leckband D., de Rooij J. (2010) Vinculin potentiates E-cadherin mechanosensing and is recruited to actin-anchored sites within adherens junctions in a myosin II-dependent manner. J. Cell Biol. 189, 1107–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ladoux B., Anon E., Lambert M., Rabodzey A., Hersen P., Buguin A., Silberzan P., Mège R.-M. (2010) Strength dependence of cadherin-mediated adhesions. Biophys. J. 98, 534–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chu Y. S., Thomas W. A., Eder O., Pincet F., Perez E., Thiery J. P., Dufour S. (2004) Force measurements in E-cadherin-mediated cell doublets reveal rapid adhesion strengthened by actin cytoskeleton remodeling through Rac and Cdc42. J. Cell Biol. 167, 1183–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ganz A., Lambert M., Saez A., Silberzan P., Buguin A., Mège R. M., Ladoux B. (2006) Traction forces exerted through N-cadherin contacts. Biol. Cell 98, 721–730 [DOI] [PubMed] [Google Scholar]
- 8. Chu Y. S., Eder O., Thomas W. A., Simcha I., Pincet F., Ben-Ze'ev A., Perez E., Thiery J. P., Dufour S. (2006) Prototypical type I E-cadherin and type II cadherin-7 mediate very distinct adhesiveness through their extracellular domains. J. Biol. Chem. 281, 2901–2910 [DOI] [PubMed] [Google Scholar]
- 9. Mège R.-M., Gavard J., Lambert M. (2006) Regulation of cell-cell junctions by the cytoskeleton. Curr. Opin. Cell Biol. 18, 541–548 [DOI] [PubMed] [Google Scholar]
- 10. Ozawa M. (1998) Identification of the region of α-catenin that plays an essential role in cadherin-mediated cell adhesion. J. Biol. Chem. 273, 29524–29529 [DOI] [PubMed] [Google Scholar]
- 11. Perez-Moreno M., Fuchs E. (2006) Catenins. Keeping cells from getting their signals crossed. Dev. Cell 11, 601–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yonemura S., Wada Y., Watanabe T., Nagafuchi A., Shibata M. (2010) α-Catenin as a tension transducer that induces adherens junction development. Nat. Cell Biol. 12, 533–542 [DOI] [PubMed] [Google Scholar]
- 13. Rangarajan E. S., Izard T. (2012) The cytoskeletal protein α-catenin unfurls upon binding to vinculin. J. Biol. Chem. 287, 18492–18499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Choi H. J., Pokutta S., Cadwell G. W., Bobkov A. A., Bankston L. A., Liddington R. C., Weis W. I. (2012) Proc. Natl. Acad. Sci. 109, 8576–8581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yonemura S. (2011) Cadherin-actin interactions at adherens junctions. Curr. Opin. Cell Biol. 23, 515–522 [DOI] [PubMed] [Google Scholar]
- 16. Imamura Y., Itoh M., Maeno Y., Tsukita S., Nagafuchi A. (1999) Functional domains of α-catenin required for the strong state of cadherin-based cell adhesion. J. Cell Biol. 144, 1311–1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Abe K., Takeichi M. (2008) EPLIN mediates linkage of the cadherin catenin complex to F-actin and stabilizes the circumferential actin belt. Proc. Natl. Acad. Sci. U.S.A. 105, 13–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Taguchi K., Ishiuchi T., Takeichi M. (2011) Mechanosensitive EPLIN-dependent remodeling of adherens junctions regulates epithelial reshaping. J. Cell Biol. 194, 643–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chervin-Pétinot A., Courçon M., Almagro S., Nicolas A., Grichine A., Grunwald D., Prandini M. H., Huber P., Gulino-Debrac D. (2012) Epithelial protein lost in neoplasm (EPLIN) interacts with α-catenin and actin filaments in endothelial cells and stabilizes vascular capillary network in vitro. J. Biol. Chem. 287, 7556–7572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kobielak A., Fuchs E. (2004) α-Catenin. At the junction of intercellular adhesion and actin dynamics. Nat. Rev. Mol. Cell Biol. 5, 614–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yamada S., Pokutta S., Drees F., Weis W. I., Nelson W. J. (2005) Deconstructing the cadherin-catenin-actin complex. Cell 123, 889–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Drees F., Pokutta S., Yamada S., Nelson W. J., Weis W. I. (2005) α-Catenin is a molecular switch that binds E-cadherin-β-catenin and regulates actin-filament assembly. Cell 123, 903–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lambert M., Choquet D., Mège R. M. (2002) Dynamics of ligand-induced, Rac1-dependent anchoring of cadherins to the actin cytoskeleton. J. Cell Biol. 157, 469–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ozawa M., Ringwald M., Kemler R. (1990) Uvomorulin-catenin complex formation is regulated by a specific domain in the cytoplasmic region of the cell adhesion molecule. Proc. Natl. Acad. Sci. U.S.A. 87, 4246–4250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sako Y., Nagafuchi A., Tsukita S., Takeichi M., Kusumi A. (1998) Cytoplasmic regulation of the movement of E-cadherin on the free cell surface as studied by optical tweezers and single particle tracking. Corralling and tethering by the membrane skeleton. J. Cell Biol. 140, 1227–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Miyake Y., Inoue N., Nishimura K., Kinoshita N., Hosoya H., Yonemura S. (2006) Actomyosin tension is required for correct recruitment of adherens junction components and zonula occludens formation. Exp. Cell Res. 312, 1637–1650 [DOI] [PubMed] [Google Scholar]
- 27. Maddugoda M. P., Crampton M. S., Shewan A. M., Yap A. S. (2007) Myosin VI and vinculin cooperate during the morphogenesis of cadherin cell cell contacts in mammalian epithelial cells. J. Cell Biol. 178, 529–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Geiger B., Tokuyasu K. T., Dutton A. H., Singer S. J. (1980) Vinculin, an intracellular protein localized at specialized sites where microfilament bundles terminate at cell membranes. Proc. Natl. Acad. Sci. U.S.A. 77, 4127–4131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Geiger B., Bershadsky A. (2001) Assembly and mechanosensory function of focal contacts. Curr. Opin. Cell Biol. 13, 584–592 [DOI] [PubMed] [Google Scholar]
- 30. Geiger B., Volk T., Volberg T. (1985) Molecular heterogeneity of adherens junctions. J. Cell Biol. 101, 1523–1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Weiss E. E., Kroemker M., Rüdiger A. H., Jockusch B. M., Rüdiger M. (1998) Vinculin is part of the cadherin-catenin junctional complex. Complex formation between α-catenin and vinculin. J. Cell Biol. 141, 755–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Watabe-Uchida M., Uchida N., Imamura Y., Nagafuchi A., Fujimoto K., Uemura T., Vermeulen S., van Roy F., Adamson E. D., Takeichi M. (1998) α-Catenin-vinculin interaction functions to organize the apical junctional complex in epithelial cells. J. Cell Biol. 142, 847–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yang J., Dokurno P., Tonks N. K., Barford D. (2001) Crystal structure of the M-fragment of α-catenin. Implications for modulation of cell adhesion. EMBO J. 20, 3645–3656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pokutta S., Drees F., Takai Y., Nelson W. J., Weis W. I. (2002) Biochemical and structural definition of the l-afadin- and actin-binding sites of α-catenin. J. Biol. Chem. 277, 18868–18874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mege R. M., Matsuzaki F., Gallin W. J., Goldberg J. I., Cunningham B. A., Edelman G. M. (1988) Construction of epithelioid sheets by transfection of mouse sarcoma cells with cDNAs for chicken cell adhesion molecules. Proc. Natl. Acad. Sci. U.S.A. 85, 7274–7278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vermeulen S. J., Bruyneel E. A., Bracke M. E., De Bruyne G. K., Vennekens K. M., Vleminckx K. L., Berx G. J., van Roy F. M., Mareel M. M. (1995) Transition from the noninvasive to the invasive phenotype and loss of α-catenin in human colon cancer cells. Cancer Res. 55, 4722–4728 [PubMed] [Google Scholar]
- 37. Dufour S., Beauvais-Jouneau A., Delouvée A., Thiery J. P. (1999) Differential function of N-cadherin and cadherin-7 in the control of embryonic cell motility. J. Cell Biol. 146, 501–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Thiery J. P., Delouvée A., Gallin W. J., Cunningham B. A., Edelman G. M. (1984) Ontogenetic expression of cell adhesion molecules. L-CAM is found in epithelia derived from the three primary germ layers. Dev. Biol. 102, 61–78 [DOI] [PubMed] [Google Scholar]
- 39. Nagafuchi A., Tsukita S. (1994) The loss of expression of α-catenin, the 102 kD cadherin associated protein, in central nervous tissue during development. Dev. Growth Differ. 36, 59–71 [DOI] [PubMed] [Google Scholar]
- 40. Ozawa M. (2003) p120-independent modulation of E-cadherin adhesion activity by the membrane-proximal region of the cytoplasmic domain. J. Biol. Chem. 278, 46014–46020 [DOI] [PubMed] [Google Scholar]
- 41. Fukata M., Nakagawa M., Itoh N., Kawajiri A., Yamaga M., Kuroda S., Kaibuchi K. (2001) Involvement of IQGAP1, an effector of Rac1 and Cdc42 GTPases, in cell-cell dissociation during cell scattering. Mol. Cell. Biol. 21, 2165–2183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gavard J., Lambert M., Grosheva I., Marthiens V., Irinopoulou T., Riou J. F., Bershadsky A., Mège R. M. (2004) Lamellipodium extension and cadherin adhesion. Two cell responses to cadherin activation relying on distinct signalling pathways. J. Cell Sci. 117, 257–270 [DOI] [PubMed] [Google Scholar]
- 43. Azioune A., Carpi N., Fink J., Chehimi M. M., Cuvelier D., Piel M. (2011) Robust method for high-throughput surface patterning of deformable substrates. Langmuir 27, 7349–7352 [DOI] [PubMed] [Google Scholar]
- 44. Sung K.-L., Sung L. A., Crimmins M., Burakoff S. J., Chien S. (1986) Determination of junction avidity of cytolytic T cell and target cell. Science 234, 1405–1408 [DOI] [PubMed] [Google Scholar]
- 45. Chu Y. S., Dufour S., Thiery J. P., Perez E., Pincet F. (2005) Johnson-Kendall-Roberts theory applied to living cells. Phys. Rev. Lett. 94, 028102 [DOI] [PubMed] [Google Scholar]
- 46. Martinez-Rico C., Pincet F., Perez E., Thiery J. P., Shimizu K., Takai Y., Dufour S. (2005) Separation force measurements reveal different types of modulation of E-cadherin-based adhesion by nectin-1 and -3. J. Biol. Chem. 280, 4753–4760 [DOI] [PubMed] [Google Scholar]
- 47. Martinez-Rico C., Pincet F., Thiery J. P., Dufour S. (2010) Integrins stimulate E-cadherin-mediated intercellular adhesion by regulating Src-kinase activation and actomyosin contractility. J. Cell Sci. 123, 712–722 [DOI] [PubMed] [Google Scholar]
- 48. Stockinger P., Maître J. L., Heisenberg C. P. (2011) Defective neuroepithelial cell cohesion affects tangential branchiomotor neuron migration in the zebrafish neural tube. Development 138, 4673–4683 [DOI] [PubMed] [Google Scholar]
- 49. Gavard J., Marthiens V., Monnet C., Lambert M., Mège R. M. (2004) N-cadherin activation substitutes for the cell contact control in cell cycle arrest and myogenic differentiation. Involvement of p120 and β-catenin. J. Biol. Chem. 279, 36795–36802 [DOI] [PubMed] [Google Scholar]
- 50. Lambert M., Thoumine O., Brevier J., Choquet D., Riveline D., Mège R. M. (2007) Nucleation and growth of cadherin adhesions. Exp. Cell Res. 313, 4025–4040 [DOI] [PubMed] [Google Scholar]
- 51. Kümper S., Ridley A. J. (2010) p120ctn and P-cadherin but not E-cadherin regulate cell motility and invasion of DU145 prostate cancer cells. PloS One 5, e11801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Vasioukhin V., Bauer C., Degenstein L., Wise B., Fuchs E. (2001) Hyperproliferation and defects in epithelial polarity upon conditional ablation of α-catenin in skin. Cell 104, 605–617 [DOI] [PubMed] [Google Scholar]
- 53. Parsons J. T., Horwitz A. R., Schwartz M. A. (2010) Cell adhesion. Integrating cytoskeletal dynamics and cellular tension. Nat. Rev. Mol. Cell Biol. 11, 633–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kobielak A., Pasolli H. A., Fuchs E. (2004) Mammalian formin-1 participates in adherens junctions and polymerization of linear actin cables. Nat. Cell Biol. 6, 21–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yonemura S. (2011) A mechanism of mechanotransduction at the cell-cell interface. Emergence of α-catenin as the center of a force-balancing mechanism for morphogenesis in multicellular organisms. BioEssays 33, 732–736 [DOI] [PubMed] [Google Scholar]
- 56. Sumida G. M., Tomita T. M., Shih W., Yamada S. (2011) Myosin II activity dependent and independent vinculin recruitment to the sites of E-cadherin-mediated cell-cell adhesion. BMC Cell Biol. 12, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Capaldo C. T., Macara I. G. (2007) Depletion of E-cadherin disrupts establishment but not maintenance of cell junctions in Madin-Darby canine kidney epithelial cells. Mol. Biol. Cell 18, 189–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Huveneers S., Oldenburg J., Spanjaard E., van der Krogt G., Grigoriev I., Akhmanova A., Rehmann H., de Rooij J. (2012) Vinculin associates with endothelial VE-cadherin junctions to control force-dependent remodeling. J. Cell Biol. 196, 641–652 [DOI] [PMC free article] [PubMed] [Google Scholar]