

Background: Cu,Zn-superoxide dismutase (SOD1) possesses a highly conserved intramolecular disulfide bond.
Results: Structural destabilization of SOD1 scrambles the intramolecular disulfide to form cross-linked oligomers with an intermolecular disulfide bond.
Conclusion: Disulfide scrambling is a key to understand the folding/misfolding process of SOD1.
Significance: A disulfide-scrambling model provides a molecular pathomechanism describing the formation of disulfide-linked SOD1 oligomers in amyotrophic lateral sclerosis.
Keywords: Amyotropic Lateral Sclerosis (Lou Gehrig's Disease), Disulfide, Protein Aggregation, Protein Misfolding, Superoxide Dismutase (SOD)
Abstract
Dominant mutations in Cu,Zn-superoxide dismutase (SOD1) are a cause of a familial form of amyotrophic lateral sclerosis. Wild-type SOD1 forms a highly conserved intra-molecular disulfide bond, whereas pathological SOD1 proteins are cross-linked via intermolecular disulfide bonds and form insoluble oligomers. A thiol-disulfide status in SOD1 will thus play a regulatory role in determining its folding/misfolding pathways; however, it remains unknown how pathogenic mutations in SOD1 affect the thiol-disulfide status to facilitate the protein misfolding. Here, we show that the structural destabilization of SOD1 scrambles a disulfide bond among four Cys residues in an SOD1 molecule. The disulfide scrambling produces SOD1 monomers with distinct electrophoretic mobility and also reproduces the formation of disulfide-linked oligomers. We have also found that the familial form of amyotrophic lateral sclerosis-causing mutations facilitate the disulfide scrambling in SOD1. Based upon our results, therefore, scrambling of the conserved disulfide bond will be a key event to cause the pathological changes in disease-associated mutant SOD1 proteins.
Introduction
In many proteins, correct formation of a disulfide bond is required for folding into the native structures, and aberrant reduction/formation of disulfide bonds often results in protein misfolding (1). A thiol-disulfide status in proteins can thus function as an important branch point for determining the folding/misfolding pathways. Cu,Zn-superoxide dismutase (SOD1)2 is one of such proteins in which the folding/misfolding is regulated by the thiol-disulfide status (2–4). The enzymatic activity of SOD1 is to catalyze the removal of superoxide anion radical at the bound copper ion (5), and the formation of an intramolecular disulfide bond is required for the stabilization of matured SOD1 (6). Reduction of the disulfide bond, in contrast, significantly decreases the conformational stability (7) and leads to misfolding and aggregation of SOD1 (8), which are thought to play a central role in a familial form of amyotrophic lateral sclerosis (fALS).
More than a hundred mutations in the SOD1 gene have been reported as a cause of fALS, where aggregation of mutant SOD1 proteins in spinal motor neurons is a pathological hallmark (9). fALS mutations have been proposed to accumulate relatively unstable SOD1 species in the cell by retarding either one of the maturation processes of SOD1; i.e. the binding of copper and zinc ions and the formation of a disulfide bond (7, 8). In particular, fALS mutations render SOD1 proteins in vitro susceptible to disulfide reduction with a disulfide-reducing agent, tris(2-carboxyethyl)phosphine (10); furthermore, mutant SOD1 has been reported to be incompetent for disulfide formation in the transgenic mice expressing human SOD1 with a G85R mutation (11). The disulfide-reduced state of demetallated (apo) SOD1 easily forms amyloid-like fibrillar aggregates in vitro, which would describe the formation of thioflavin S-positive inclusions in fALS model mice (8). Given highly reducing environment of the cytosol (12), reduction of the intramolecular disulfide bond in mutant SOD1 thus appears to be a key event for triggering the SOD1 aggregation inside the cell.
An SOD1 aggregation model triggered by the disulfide reduction has, however, been recently challenged by several experimental results (11, 13, 14). As mentioned, almost all of SOD1G85R remain disulfide-reduced in the model mice; however, the disulfide-reduced protein is a minority (∼10%) in the mice expressing other fALS-causing mutant SOD1 such as SOD1D90A and SOD1G93A (11, 14). Comparable amounts of the disulfide-reduced species have been also detected even in the transgenic mice expressing wild-type (WT) SOD1, but no aggregation of WT SOD1 occurs (11, 14). In addition, neuropathological abnormalities were not altered in G37R, G85R, and G93A mutant SOD1 mice by genetic deletion of a protein, CCS, which is responsible for the disulfide formation in SOD1 in vivo (15). Notably, however, significant amounts of SOD1 that appears to be in the disulfide-reduced state have been reported to constitute the insoluble aggregates observed in fALS model mice (11, 14), where non-native intermolecular disulfide bonds have been further found to cross-link the SOD1 oligomers (14, 16, 17). Abnormal redox chemistry of Cys residues in SOD1, therefore, appears to operate in misfolding and aggregation of mutant SOD1, but it remains unknown how the thiol-disulfide status of SOD1 is changing in the course of the disease.
Here, we have shown for the first time that abnormal scrambling of a disulfide bond is involved in a molecular mechanism for misfolding of mutant SOD1 proteins. Structural destabilization of the apo-SOD1 with a canonical intramolecular disulfide bond (Cys57–Cys146) triggers the nucleophilic attack of Cys6/111 to the disulfide bond between Cys57 and Cys146. Such disulfide isomerization first occurs within an SOD1 molecule and then proceeds between the molecules to form insoluble disulfide-linked oligomers. These changes in the disulfide linkage in SOD1 were found to be facilitated by fALS-causing mutations. We therefore propose that the disulfide reduction is not always required for SOD1 misfolding; rather, the disulfide scrambling by intra- and intermolecular isomerization also constitutes a relevant pathway for the aggregation of mutant SOD1 proteins.
EXPERIMENTAL PROCEDURES
Expression and Purification of Apo-SOD1S-S Proteins
A human SOD1 cDNA was cloned in a vector, pET15b (Novagen), using NdeI and SalI sites (8); thus, a tag containing six consecutive His (a His6 tag) was fused at the N terminus of SOD1. Mutations were introduced by QuikChange mutagenesis (Stratagene), and all constructs used in this study were confirmed by DNA sequencing. Escherichia coli, Rosetta (Novagen), was transformed with the plasmid, and the expression of SOD1 proteins was induced with 0.1 mm isopropyl 1-thio-β-d-galactopyranoside at 37 °C for 4 h. At the induction of protein expression, 3 mm CuSO4 and 30 μm ZnSO4 were also added. For fALS-causing mutant SOD1s, induction of the protein expression was performed at 20 °C for 19 h.
Cells were lysed with a cycle of freeze and thaw and resuspended in PBS containing 2% Triton X-100, 5 mm MgCl2 and 67.5 mg/liter DNase I. Using a soluble supernatant by centrifugation at 14,000 rpm for 10 min, the ammonium sulfate precipitation was performed; the pellets resulting from the addition of 35–80% ammonium sulfate was collected and redissolved in a wash buffer containing 50 mm Tris, 500 mm NaCl, 10 mm imidazole, pH 8.0. The proteins were further purified with Ni2+-affinity chromatography using the Proteus IMAC Midi kit (Pro-Chem, Inc.), and the bound SOD1 proteins were washed with the wash buffer and then eluted from a spin column with an elution buffer (50 mm Tris, 500 mm NaCl, 250 mm imidazole, pH 8.0).
Eluted protein samples were further precipitated with 20% trichloroacetic acid (TCA) to remove bound metal ions, and the resultant pellets were washed with acetone and resolubilized with a pH 7.4 buffer containing 100 mm sodium phosphate, 100 mm NaCl, and 5 mm EDTA (NNE buffer). The addition of EDTA to the protein solution inhibits binding of metal ions present as trace contaminants in buffers and reagents. To cleave the His6 tag from SOD1, a solution (150 μl) containing a His6-tagged SOD1 purified above (100 μm) was incubated with 2 units of thrombin from human plasma (Sigma, SIGT1063) at 37 °C for 2 h. The samples were then purified with a gel-filtration column (KW803, Shodex®, 8.0 and 300 mm of its inner diameter and length, respectively) equilibrated with an NNE buffer (flow rate, 1.0 ml/min); thereby, the cleaved His6 tag was removed. Eluted fractions containing a tag-free SOD1 were collected, concentrated, and used as apo-SOD1S-S in this study. SDS-PAGE as well as MALDI-TOF mass spectrometry has confirmed complete removal of a His6 tag and also the presence of the disulfide bond in our apo-SOD1S-S samples.
For preparation of fALS-mutant (G37R and G93R) as well as WT SOD1s for the experiments shown in Fig. 6, the proteins purified with Ni2+-affinity chromatography were first precipitated with TCA, washed with acetone, and then redissolved in an NNE buffer containing 6 m guanidine hydrochloride (GdnHCl). The proteins were refolded by 100-fold dilution in an NNE buffer with stirring at 4 °C for 16 h. The refolded samples were concentrated and further purified with gel filtration column chromatography (KW803 mentioned above) equilibrated with an NNE buffer (flow rate, 1.0 ml/min).
FIGURE 6.

fALS-causing mutations facilitate the formation of disulfide-linked oligomers through scrambling of a disulfide bond. A, aggregation kinetics of 20 μm apo-SOD1S-S proteins in an NNE buffer containing 0.3 m GdnHCl were followed by monitoring the absorbance change at 350 nm. Compared with the WT protein (open circles), apo-SOD1S-S with G37R (filled circles) and G93R (filled squares) exhibited larger increase in the solution turbidity. B, samples after agitation at 37 °C for 2,880 min were fractionated into soluble supernatant (s) and insoluble pellets (p) by ultracentrifugation and analyzed by non-reducing SDS-PAGE using a 12.5% polyacrylamide gel. G93R mutation has been known to increase the electrophoretic mobility of SOD1, which was indicated by arrows shown on the right of the figure. C, analysis of the thiol-disulfide state in the aggregates of apo-SOD1S-S with G37R mutation. Free thiol groups in the aggregates were first modified with 10 mm IA in the presence of 6 m GdnHCl, which was then analyzed by MALDI-TOF mass spectrometry with (+IA/+DTT) or without (+IA) addition of 50 mm DTT. A control experiment was also performed, in which the aggregates were first reduced with 5 mm DTT and then modified with 20 mm IA (+DTT/+IA). Mass peaks corresponding to unmodified SOD1, SOD1+IA, SOD1+2IA, SOD1+3IA, and SOD1+4IA were indicated on top of the figure. D, the disulfide scrambling between two molecules of apo-SOD1S-S theoretically produces a disulfide-linked SOD1 dimer, which consists of SOD1 with three free Cys residues and SOD1 with only one free Cys residue. This schematic representation thus describes appearance of the mass peaks corresponding to SOD1+IA and SOD1+3IA shown in C.
Protein concentration was spectroscopically determined from the absorbance at 280 nm using 5,500 cm−1 m−1 as an extinction coefficient. Throughout this study, the spectroscopic measurements of absorbance at 280 nm were performed in the presence of 6 m GdnHCl, where SOD1 proteins are unfolded (18).
Aggregation of SOD1 Proteins
150 μl of 20 μm apo-SOD1S-S in an NNE buffer with and without either 5 mm dithiothreitol (DTT) or 1 m GdnHCl was set per well in a 96-well plate and shaken at 1,200 rpm, 37 °C using a plate shaker (MBR-022UP, TAITEC). To increase the shaking efficiency, a POM ball (3/32 inch, SANPLATEC) per well was further included. Solution turbidity was measured in a plate reader (Epoch, BioTek) by measuring the absorbance at 350 nm. After the turbidity change reached a plateau (∼24 h), a sample solution was collected and ultracentrifuged at 110,000 × g for 15 min to obtain soluble supernatant and insoluble pellets. Proteins in soluble supernatant were further collected as pellets by precipitation with TCA, and these soluble and insoluble proteins were analyzed by either reducing or non-reducing SDS-PAGE (see below).
To examine effects of fALS-causing mutations on the kinetics of SOD1 aggregation (see Fig. 6), 150 μl of 20 μm mutant SOD1s purified above were incubated with 1 unit of thrombin (Sigma, SIGT1063) at 37 °C for 24 h without any agitation. Then, the aggregation reaction was started by shaking the samples at 1,200 rpm, 37 °C with a POM ball (3/32 inch) in a plate shaker.
SDS-PAGE Analysis
In reducing SDS-PAGE, the protein samples were mixed with an SDS-PAGE sample buffer containing 100 mm DTT and boiled at 100 °C for 5 min before loaded on an SDS-polyacrylamide gel. In non-reducing SDS-PAGE, protein samples were further incubated with 100 mm iodoacetamide (IA) in an NNE buffer containing 2% SDS at 37 °C for 1 h. Then, an SDS-PAGE sample buffer without any reductants was added, and after boiling at 100 °C for 5 min, the samples were loaded on an SDS-polyacrylamide gel. In both reducing and non-reducing SDS-PAGE, the protein bands were stained with Coomassie Brilliant Blue.
Solubility Assays of SOD1 Aggregates
To examine temperature dependence of the aggregate solubility (19), SOD1 aggregates were collected as pellets by ultracentrifugation at 110,000 × g for 15 min and resuspended/sonicated in an NNE buffer. 70 μl of 50 μm (monomer-based) SOD1 aggregates was set in a 0.2-ml PCR tube, mixed with 7 μl of 10% sarkosyl in water and then incubated at 37, 60, 80, or 100 °C for 1 h by using a PCR thermal cycler (PTC-225, MJ Research). After incubation, the sample solution was again fractionated into soluble supernatant and insoluble pellets by ultracentrifugation at 110,000 × g for 15 min. Insoluble pellets were resuspended in an NNE buffer, and both fractions were analyzed by reducing SDS-PAGE (see below). Intensity of SOD1 bands was analyzed by NIH ImageJ software. When band intensities of soluble and insoluble fractions were denoted as Is and Ii, indicating the fraction solubilized, F (%), is calculated as Is/(Is + Ii) × 100, respectively. F is then plotted against incubation temperature, T, and estimation of T½ was performed by fitting the data with a following sigmoidal function using Igor Pro (version 4.0, Wavemetrics),
 |
where a is a coefficient. Experiments were repeated three times to estimate errors.
Characterization of the Thiol-disulfide Status in SOD1 by Mass Spectrometry
SOD1 aggregates (20 μm, monomer-based) collected as pellets by ultracentrifugation were redissolved in an NNE buffer containing 6 m GdnHCl and 10 mm IA and incubated at 37 °C for 1 h. If necessary, the intermolecular disulfide bonds were reduced by adding 50 mm DTT before mass analysis. As a control, any disulfide bonds in SOD1 aggregates were first reduced by incubation in an NNE buffer containing 6 m GdnHCl and 5 mm DTT for 1 h at 37 °C. Twenty mm of IA was then added to the samples, which were further incubated at 37 °C for 1 h. The samples (50 μl, 45 μg) were mixed with 0.1% trifluoroacetic acid (TFA) and purified with ZipTip C18 (Millipore). Sinapinic acid was chosen as a matrix, and MALDI-TOF mass spectra were acquired using UltraflexTM (Bruker) in linear mode. Myoglobin (m/z, 16951.5 (from equine heart)) and cytochrome c (m/z, 12,361 (from horse heart)) were used as internal mass standards.
Disulfide-mapping Experiments by Mass Spectrometry
In the course of apo-SOD1S-S aggregation, SOD1 remained soluble until 2 h after agitation but became insoluble in 24 h (see Fig. 3A). Soluble SOD1 proteins at 0 and 2 h were thus collected by precipitation with TCA, whereas insoluble SOD1 proteins at 24 h were obtained as pellets by ultracentrifugation. Both soluble and insoluble samples (45 μg) were then redissolved in an NNE buffer containing 4 m urea and 100 mm IA and incubated at 37 °C for 1 h to alkylate free Cys residues. The samples were further treated with 0.2 μg of trypsin (from porcine pancreas, Wako) and again incubated at 37 °C for 1 h. After addition of 0.1% TFA and 6 m GdnHCl, the trypsinized samples were purified with ZipTip C18 and spotted on a mass plate with sinapinic acid as a matrix. MALDI-TOF mass spectra were acquired using UltraflexTM in linear mode, and human ACTH(18–39) (WAKO, m/z of 2,466.6) and bovine pancreas insulin (nacalai tesque, m/z of 5,734.5) were used as external mass standards.
FIGURE 3.

Electrophoretic mobility of SOD1 was retarded upon aggregation without reduction of a disulfide bond. A, twenty μm apo-SOD1S-S was shaken at 1,200 rpm, 37 °C in an NNE buffer containing 1 m GdnHCl (Gdn), and at indicated time, the samples were fractionated into soluble supernatant (s) and insoluble pellets (p) by ultracentrifugation. Soluble proteins in supernatant were further precipitated by TCA. Free Cys residues in both soluble and insoluble proteins were alkylated by incubation in an NNE buffer containing 2% SDS and 100 mm IA at 37 °C for 1 h. SDS-PAGE analysis of the alkylated samples was then performed in non-reducing conditions using a 12.5% polyacrylamide gel. B, insoluble aggregates of apo-SOD1S-S, which formed by agitation for 24 h in the presence of 1 m GdnHCl (Fig. 1A), were collected by ultracentrifugation, reacted with 10 mm IA in the presence of 6 m GdnHCl. After incubation at 37 °C for 1 h, analysis by MALDI-TOF mass spectrometry was performed (+IA). As a control (+DTT/+IA), the aggregates were first treated with 5 mm DTT in the presence of 6 m GdnHCl to reduce disulfide bonds, alkylated by adding 20 mm IA, and then analyzed by a MALDI-TOF mass spectrometry.
RESULTS
Distinct Aggregation Pathways of Apo-SOD1 with and without a Conserved Disulfide Bond
An enzymatically active form of SOD1 is equipped with copper and zinc ions and an intramolecular disulfide bond (6). Without all of these post-translational processes, such unmodified SOD1 polypeptides have been shown to form fibrillar aggregates (8). Indeed, under near-physiological conditions, apo-SOD1 with the conserved disulfide bond (apo-SOD1S-S) was highly resistant to aggregation, and further reduction of the disulfide bond was required to form the insoluble aggregates (Fig. 1, A and B). In contrast, when a chaotropic agent, GdnHCl, was included in the sample solution (20), we found that apo-SOD1S-S formed insoluble aggregates with increased turbidity (Fig. 1, A (filled circles) and B) but did not in the presence of Zn2+ ion (Fig. 1A, open triangles). Reduction of a disulfide bond hence appears not to be a prerequisite for SOD1 aggregation; however, it remains obscure whether the apo-SOD1S-S aggregates possess distinct properties from those of the disulfide-reduced apo-SOD1 (apo-SOD1SH) aggregates.
FIGURE 1.

Apo-SOD1 forms aggregates with distinct properties that are dependent upon the thiol-disulfide status at Cys57 and Cys146. A, aggregation kinetics of SOD1 proteins monitored by turbidity changes at 350 nm. 20 μm apo-SOD1S-S in an NNE buffer was agitated in the absence (open circles) and presence of either 5 mm DTT (filled triangles) or 1 m GdnHCl (Gdn; filled circles). Effects of Zn2+ ions on the aggregation were also examined by agitating 20 μm apo-SOD1S-S in a buffer at pH 7.4 containing 100 mm sodium phosphate, 100 mm NaCl, and 20 μm ZnSO4 (open triangles). B, samples agitated for 1,440 min were fractionated into soluble supernatant (s) and insoluble pellets (p) by ultracentrifugation at 110,000 × g and loaded onto a 12.5% SDS-polyacrylamide gel after boiling in the presence of DTT. C, aggregates of apo-SOD1S-S prepared in the presence of either 5 mm DTT (upper panel) or 1 m GdnHCl (lower panel) were incubated with 1% sarkosyl at the indicated temperature for 1 h. Soluble supernatant (s) and insoluble pellets (p) were obtained by ultracentrifugation and analyzed by a 12.5% SDS-polyacrylamide gel in reducing conditions. D, fractions of solubilized SOD1 were further estimated from its relative band intensities between supernatant and pellet and plotted against incubation temperature: aggregates of apo-SOD1S-S in the presence of either 5 mm DTT (open circles) or 1 m GdnHCl (filled circles). T½ was obtained by fitting these data with a sigmoidal function (dashed curves, see “Experimental Procedures”).
A possible difference in biochemical properties of protein aggregates will be clarified by testing a temperature dependence of the aggregate solubility (19). As shown in Fig. 1C, increasing temperature is found to facilitate the solubilization of SOD1 aggregates, but it is notable that aggregates of apo-SOD1S-S are more resistant to solubilization than those of apo-SOD1SH. This can be quantitatively confirmed by estimating the temperature (T½) that is required for solubilization of half-amounts of aggregates (Fig. 1D and also see “Experimental Procedures”), and the aggregates of apo-SOD1S-S exhibit higher T½ (86.7 °C) than that of apo-SOD1SH (73.9 °C). These results have thus suggested that the thiol-disulfide status in SOD1 is an important determinant for the pathways to form aggregates with distinct properties.
Intermolecular Cross-linking via Disulfide Bonds Is a Prominent Feature of the SOD1S-S Aggregates
One of the relevant features in pathological SOD1 aggregates is the non-native intermolecular disulfides that cross-link SOD1 proteins (16, 17). Importantly, non-reducing SDS-PAGE analysis has shown that the disulfide-linked SOD1 oligomers were clearly confirmed in our WT apo-SOD1S-S aggregates (Fig. 2A, −DTT) but absent in the apo-SOD1SH aggregates (data not shown). These oligomeric structures disappeared upon treatment with a reductant, DTT, before electrophoresis (Fig. 2A, +DTT). In this experiment, free thiols in the samples were protected by a thiol-specific modifier, IA; therefore, it is unlikely that the disulfide cross-links occur during the electrophoresis. Alternatively, apo-SOD1SH aggregates were prepared in the presence of DTT, which would inhibit the disulfide cross-links. The apo-SOD1SH aggregates were therefore isolated by ultracentrifugation and again agitated in the absence of any reductants under aerobic conditions; however, formation of the disulfide-linked oligomers was not observed (data not shown).
FIGURE 2.

Cys residues at positions 6 and 111 are involved in the formation of disulfide-linked oligomers upon aggregation of apo-SOD1S-S. A, non-reducing SDS-PAGE analysis of the aggregates of apo-SOD1S-S with Cys mutations. Aggregates (10 μg) were collected by ultracentrifugation, redissolved in an NNE buffer with 2% SDS and 100 mm IA to alkylate any free Cys residues, and then analyzed by a 5–20% gradient SDS-polyacrylamide gel (left). Addition of DTT before electrophoresis has collapsed the high-molecular weight oligomer bands into a single monomer band (right), confirming that cross-links occur through intermolecular disulfide bonds. Even after treatment with DTT, a faint band remained at the position indicated as SOD1S-S, suggesting that the reduction of a disulfide bond is occasionally incomplete. B, a schematic representation of the Cys position in SOD1. A conserved disulfide bond forms between Cys57 and Cys146 in native SOD1. C–E, aggregation kinetics of apo-SOD1S-S with C6S (C), C111S (D), and C6S/C111S (E) mutations were monitored by changes of solution turbidity at 350 nm. 20 μm of mutant apo-SOD1S-S in an NNE buffer was agitated at 1,200 rpm, 37 °C in the absence (open circles) and presence of either 5 mm DTT (filled triangles) or 1 m GdnHCl (Gdn; filled circles). Similar to the WT protein, these Cys mutant apo-SOD1S-S proteins were resistant to the insoluble aggregation but became aggregated upon either reduction of the disulfide with DTT or destabilization of the structure with GdnHCl.
SOD1 possesses four Cys residues of total; a highly conserved disulfide forms between Cys57 and Cys146, whereas Cys6 and Cys111 usually remain in the free thiol/thiolate state (Fig. 2B) (21). We thus initially expected that Cys6 and Cys111 are directly involved in the disulfide cross-links of apo-SOD1S-S with the Cys57–Cys146 disulfide intact. Indeed, apo-SOD1S-S with C6S/C111S double mutations formed insoluble aggregates in the presence of 1 m GdnHCl (Fig. 2E) but with no disulfide-linked oligomers (Fig. 2A). Furthermore, C6S mutation in apo-SOD1S-S leaves only a single free Cys residue at position 111, resulting in the favorable formation of disulfide-linked dimers upon insoluble aggregation (Fig. 2, A and C). It is, however, notable that apo-SOD1S-S with C111S mutation reproduces the disulfide-linked oligomerization even with only a single free Cys residue (Cys6) (Fig. 2, A and D). If the disulfide bond between Cys57 and Cys146 remains intact during the aggregation of apo-SOD1S-S (C111S), this is quite enigmatic because the formation of disulfide-linked structures larger than dimers will require at least two free Cys residues. Accordingly, we supposed that disulfide cross-links between SOD1 molecules may not be caused simply by oxidation between free Cys residues.
An Aggregation Process Experiences an Abnormal SOD1 Monomer That Exhibits Distinct Electrophoretic Mobility
As evidenced by the SDS-PAGE analysis (Fig. 2A, −DTT), our apo-SOD1S-S aggregates were dissolved in an SDS-containing buffer with boiling and found to be composed of monomers and disulfide-linked oligomers. Reduction of the conserved disulfide bond has been known to retard the electrophoretic mobility of SOD1 (6), and interestingly, the monomers in apo-SOD1S-S aggregates exhibited similar electrophoretic mobility to that of the disulfide-reduced SOD1 (WT in Fig. 2A, −DTT). As shown in Fig. 3A, furthermore, such alternative SOD1 monomers with retarded mobility (SOD1S-S*) were observed before the solution turbidity started to increase (also see Fig. 1A), and those soluble monomers appeared to selectively form insoluble pellets. Given the retardation of electrophoretic mobility, therefore, we have next tested whether the disulfide bond in apo-SOD1S-S is somehow reduced even in the absence of any reductants.
For that purpose, we have attempted to detect a fully reduced form of SOD1 in the aggregates by mass spectrometry. The apo-SOD1S-S aggregates formed after agitation for 24 h (Fig. 3A) were first completely resolubilized in a buffer containing 6 m GdnHCl with 10 mm IA, by which free thiols in the aggregated SOD1 were alkylated. Because there are four Cys residues of total in SOD1, a fully reduced SOD1, if any, in the aggregates will increase its mass by 58.0 × 4 Da due to the addition of four acetamido groups. As shown in Fig. 3B (+IA), however, we could not find a mass peak corresponding to SOD1 modified with four acetamido groups (SOD1 + 4IA); instead, SOD1 was modified with two molecules of IA (SOD1 + 2IA), consistent with not reduction but retention of a disulfide bond. As a control, we have confirmed the mass peak of SOD1 + 4IA, when the aggregates were first reduced with DTT in the presence of 6 m GdnHCl and then alkylated with IA (Fig. 3B, +DTT/+IA). SOD1S-S* was, therefore, found to retain disulfide bond(s), even though it exhibited similar electrophoretic mobility with the disulfide-reduced SOD1.
Notably, unlike the WT protein, apo-SOD1S-S(C6S/C111S) did not exhibit retardation of the electrophoretic mobility of monomeric SOD1 during aggregation (C6S/C111S in Fig. 2A, −DTT), consistent with the retention of a canonical intramolecular disulfide bond. Instead, the presence of either one of free Cys residues, i.e. Cys6 or Cys111, in apo-SOD1S-S appears to be sufficient for the formation of monomeric species with retarded electrophoretic mobility (C6S and C111S in Fig. 2A, −DTT). When treated with DTT before electrophoresis, the monomeric species in all of these Cys mutant SOD1S-S aggregates exhibited the same mobility with that of disulfide-reduced SOD1 (Fig. 2A, +DTT). These results have thus led us to suspect that, within an SOD1 molecule, thiolate anions at Cys6 and Cys111 attack the sulfur atoms forming the disulfide bond between Cys57 and Cys146. In other words, such disulfide isomerization will break the conserved disulfide bond (Cys57–Cys146) and start to scramble the disulfide among all four Cys residues involving Cys6 and Cys111.
A Disulfide Bond Is Scrambled during the Aggregation of Apo-SOD1S-S
To test whether the disulfide scrambling really occurs during the aggregate formation, we have further attempted to map the disulfide(s) in SOD1 proteins. In our disulfide-mapping experiments, SOD1 samples are first dissolved in 4 m urea with IA and then digested with trypsin (see “Experimental Procedures”). The tryptic fragments are further analyzed by MALDI-TOF mass spectrometry, and we have monitored the thiol-disulfide status at Cys57 and Cys146 in SOD1 by inspecting two peaks observed at m/z 3,521.9 (peak 1) and 4,406.6 (peak 5), in particular (Fig. 4). Peak 5 indicates a tryptic fragment (Gly37–Arg69 + Leu144–Gln153) connected by a disulfide between Cys57 and Cys146, whereas reduction of the disulfide bond produces a tryptic fragment (Gly37–Arg69) represented by peak 1.
FIGURE 4.

Mapping of a disulfide bond in SOD1 reveals intra- and intermolecular scrambling of a disulfide bond upon aggregation. A, MALDI-TOF mass analysis was performed on tryptic fragments of the apo-SOD1S-S samples that were agitated in an NNE buffer containing 1 m GdnHCl for 0, 2, and 24 h. Numbers (1–29) indicated on top of the figure correspond to the tryptic peptides shown in B and supplemental Table S1. Mass peaks that could not be assigned to any tryptic fragments from SOD1 proteins were indicated with asterisks. B, based upon the observed mass peaks shown in A, tryptic peptides were identified by MS-Bridge (University of California, San Francisco, CA) and summarized together with their observed and theoretical m/z values. A difference between observed and theoretical m/z values was indicated as Δm/z. A disulfide bond in each tryptic peptide was also indicated. A list of the assigned tryptic fragments with relatively small intensity in A can be found in supplemental Table S1.
As shown in Fig. 4A (0 h), tryptic digestion of soluble apo-SOD1S-S before aggregation gave rise to peak 5 but not peak 1, consistent with the presence of the disulfide bond between Cys57 and Cys146. We have then performed the disulfide-mapping experiments on monomeric species with alternative electrophoretic mobility (SOD1S-S*, Fig. 3A), which is converted from apo-SOD1S-S within 2 h of agitation. Surprisingly, treatment of SOD1S-S* with trypsin produced peak 1 (Fig. 4A, 2 h), even though no reductants were added during the experiments. Concomitantly, several DTT-sensitive mass peaks were newly emerged in tryptic fragments of SOD1S-S* at m/z 4,440.7, 4,608.0, and 5,578.9 (peaks 6, 11, and 22 in Fig. 4), which correspond to the peptides harboring a non-native disulfide bond between Cys6–Cys146, Cys111–Cys146, and Cys6–Cys57, respectively. Given that smaller amounts of disulfide-linked oligomers are produced after 2 h of agitation, therefore, these results provide evidence to show that intramolecular isomerization of a disulfide bond occurs and leads to form non-native disulfide bonds.
In disulfide-linked SOD1 oligomers, which were obtained by agitation of apo-SOD1S-S for 24 h (Figs. 1A and 2A), a tryptic fragment containing the Cys57–Cys146 disulfide bond (peak 5 in Fig. 4) became a minor species; instead, mass peaks indicating the non-native disulfide bonds were observed with increased intensity (peaks 6, 11, 22, 23, 27, 29 in Fig. 4). These changes in the pattern of tryptic digestions were not observed during the aggregation of apo-SOD1S-S with C6S/C111S mutations (Fig. 5), which did not associate with the scrambling of a disulfide bond (Fig. 2A). Based upon these results, therefore, we have confirmed that structural destabilization of apo-SOD1S-S allows free Cys residues at position 6 and 111 to attack the disulfide bond between Cys57 and Cys146 and scrambles a disulfide bond among four Cys residues in SOD1. Such disulfide scrambling will then lead to the formation of abnormal monomers and disulfide-linked oligomers of SOD1.
FIGURE 5.

Disulfide-mapping experiments support no changes in the disulfide linkage connectivity in apo-SOD1S-S with C6S/C111S mutations during aggregation. A, MALDI-TOF mass analysis was performed on tryptic fragments of the apo-SOD1S-S (C6S/C111S) samples that were agitated in an NNE buffer containing 1 m GdnHCl for 0, 2, and 24 h. Numbers (1–12) indicated on top of the figure correspond to the tryptic peptides shown in B. Mass peaks that could not be assigned to any tryptic fragments from SOD1 proteins were indicated with asterisks. B, based upon the observed mass peaks shown in A, tryptic peptides were identified by MS-Bridge and summarized together with their observed and theoretical m/z values. A difference between observed and theoretical m/z values was indicated as Δm/z. A disulfide bond in each tryptic peptide was also indicated.
FALS-causing Mutations Facilitate the Scrambling of a Disulfide Bond in SOD1
We have next attempted to show effects of fALS-causing mutations on the disulfide scrambling in SOD1. As shown in Fig. 1A, formation of disulfide-linked oligomers from WT apo-SOD1S-S was dependent upon the concentration of added GdnHCl (1 m) in solution; indeed, upon addition of 0.3 m GdnHCl, WT apo-SOD1S-S still remained soluble with small changes in solution turbidity even after agitation at 37 °C for 2,880 min (Fig. 6, A and B). Furthermore, in these conditions, electrophoretic mobility of the agitated sample suggested no scrambling of a disulfide bond in soluble WT apo-SOD1S-S (Fig. 6B). When fALS-causing mutations, G37R and G93R, were introduced in apo-SOD1S-S, however, agitation of those mutant SOD1s increased the solution turbidity in the presence of 0.3 m GdnHCl (Fig. 6A). Non-reducing SDS-PAGE analysis on those mutant SOD1s has also shown the formation of insoluble oligomers cross-linked via disulfide bonds (Fig. 6B). Furthermore, monomeric mutant SOD1 consisting of the insoluble pellets exhibited the electrophoretic mobility very similar to that of the disulfide-reduced form, even though no reducing reagents were added to the solution. As mentioned above (Fig. 3), however, these results do not indicate auto-reduction of the disulfide bond in SOD1; rather, the disulfide bond was considered to scramble, leading to the formation of SOD1S-S*.
Indeed, mass spectrometric analysis on the aggregated apo-SOD1S-S(G37R, G93R) was performed after modification of free thiol groups with IA, but a mass peak corresponding to SOD1+4IA was not confirmed (Fig. 6C, +IA). Instead, the observed mass peak (m/z of 16,417) was consistent with modification of SOD1 with just two molecules of IA (calculated m/z of 16,432), suggesting not reduction but retention of a disulfide bond (Fig. 6C, +IA). As a control, we confirmed that the IA modification after reducing the aggregated samples with DTT produced a mass peak observed at an m/z of 16,541, corresponding to SOD1 + 4IA (calculated m/z of 16,548) (Fig. 6C, +DTT/+IA). Furthermore, when mass analysis was performed after reduction of the IA-modified samples with excess amounts of DTT, mass peaks corresponding to SOD1+IA and SOD1+3IA were observed in addition to SOD1+2IA (Fig. 6C, +IA/+DTT). As schematically represented in Fig. 6D, this is consistent with the fact that isomerization from an intra- to an intermolecular disulfide bond between two molecules of apo-SOD1S-S results in the formation of SOD1s with three and one free Cys residues. These results thus support our idea that fALS-causing mutations facilitate abnormal scrambling of a disulfide bond and thereby lead to the formation of insoluble oligomers cross-linked via disulfide bonds.
DISCUSSION
Abnormal accumulation of SOD1-immunoreactive inclusions in spinal motor neurons is known as a major pathological change in fALS cases with SOD1 mutations (9). Various mechanisms have been proposed to describe the formation of insoluble SOD1 aggregates in vitro and in vivo, which have, however, yet to reach a consensus (22). One of the major controversial issues is whether any abnormalities in the thiol-disulfide status of SOD1 are involved in a pathomechanism of SOD1 aggregation. In this study, we have for the first time revealed that misfolding of SOD1 proteins scrambles a conserved intramolecular disulfide bond. As discussed below, this new disulfide-scrambling mechanism for SOD1 aggregation can describe several important aspects of molecular pathologies in SOD1-related fALS cases.
SOD1 Aggregation Model Triggered by Disulfide Reduction
So far, many researchers have agreed with the decreased stability of an SOD1 structure upon reduction of the disulfide bond (7, 23, 24). Consistently, under near-physiological conditions in vitro, disulfide-reduced apo-SOD1 has been characterized as the most facile state for fibrillar aggregation (8). It has hence been speculated that fALS-causing mutations decrease stability of the disulfide bond in SOD1 and then facilitate its fibrillar aggregation. Indeed, the disulfide bond between Cys57 and Cys146 cannot form in some of the pathogenic mutant SOD1s, where the thiol group is absent at position 146 due to missense (C146R) or truncation (e.g. L126Z) mutations. Reducing environment of the cytosol (12), where SOD1 usually localizes, also appears to fit with the aggregation model triggered by reduction of a disulfide bond (the disulfide-reduction model, route I in Fig. 7).
FIGURE 7.
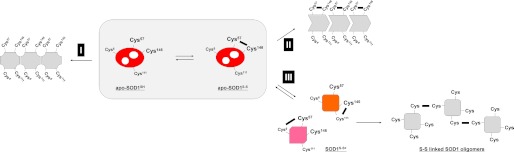
An overview of our SOD1 aggregation model regulated by the thiol-disulfide status. Apo-SOD1 has been shown to exhibit the highest propensities for fibrillar aggregation upon the reduction of a disulfide bond (route I). Even without disulfide reduction, however, apo-SOD1S-S was found to become susceptible to insoluble aggregation in the presence of fALS-causing mutations (routes II and III). In particular, we have found here that the scrambling of a disulfide bond is triggered by the structural destabilization of apo-SOD1S-S (route III). Intermolecular scrambling of a disulfide bond further describes the formation of insoluble disulfide-linked SOD1 oligomers, which is one of the important pathological changes in fALS cases.
Alternative Models for SOD1 Aggregation Require No Reduction of a Disulfide Bond
However, the disulfide reduction model alone does not completely describe the aggregate formation of fALS mutant SOD1 proteins under pathological conditions. In transgenic mice expressing G37R and G93A mutant SOD1, for example, the intramolecular disulfide bond was found to be introduced into ∼90% of soluble mutant SOD1 molecules, which was comparable with that of WT SOD1 overexpressed in mice (11, 14). In cultured cells, furthermore, quadruple mutations of all four Cys residues in fALS-mutant SOD1 can preclude the disulfide formation but actually suppress the intracellular formation of insoluble SOD1 (25). As reproduced by apo-SOD1S-S with C6S/C111S mutations (Fig. 2A), SOD1 can form insoluble aggregates while retaining the intramolecular disulfide between Cys57 and Cys146 (the disulfide-retention model, route II in Fig. 7). Reduction of the disulfide bond is, therefore, not an absolute requirement for the aggregate formation of mutant SOD1, which has initially led us to suspect that the disulfide retention model represents another aggregation pathway of SOD1 with pathogenic mutations.
Despite this, the disulfide retention model is still insufficient for the complete description of the mutant SOD1 aggregation. Previous studies have shown that insoluble SOD1 aggregates purified from G37R and G93A SOD1 mice are electrophoretically migrated at almost the same position with that of the disulfide-reduced SOD1 (14). This implies reduction of the conserved disulfide bond in the SOD1 aggregates, which contradicts the disulfide retention model for SOD1 aggregation. Such apparent contradiction can, however, be resolved by taking into account the disulfide-scrambling model of SOD1 aggregation that we have proposed here (route III in Fig. 7). In this model, scrambling of a disulfide bond forms an alternative SOD1 monomer with a non-native disulfide bond; importantly, it was found that the disulfide-scrambled SOD1 exhibited similar electrophoretic mobility to that of the disulfide-reduced SOD1 (Fig. 3A). Indeed, by using a highly resolving polyacrylamide gel, Karch et al. (14) have noticed the presence of alternative SOD1 monomers with a distinct electrophoretic mobility in several mutant SOD1 mice. In the previously reported aggregates of fALS model mice, therefore, the “apparent” disulfide-reduced SOD1 might actually represent the disulfide-scrambled protein.
Free Cys Residues Play Key Roles in the Disulfide Scrambling That Describes Pathological Changes of Mutant SOD1
In our disulfide-scrambling model, structural destabilization of SOD1 first allows free Cys residues (Cys6 and Cys111) to attack the conserved disulfide bond between Cys57 and Cys146. Intermolecular scrambling of a disulfide bond is then considered to produce disulfide-linked SOD1 oligomers. Indeed, when both Cys6 and Cys111 in SOD1 were replaced with Ser, a disulfide bond remained intact, and the disulfide-linked oligomerization was not observed (Fig. 2A). Besides, important roles of free Cys residues in the disulfide-linked oligomerization can be supported by previous studies using cultured cells. For example, Niwa et al. (25) have systematically examined effects of all four Cys residues on the aggregation propensities of mutant SOD1 in cultured Neuro2a cells and found that Cys6 is indispensable to the formation of SOD1 oligomers with extensive disulfide cross-links. This is completely consistent with our in vitro studies showing that, compared with C6S, C111S mutation better reproduces the extensive formation of disulfide-linked oligomers upon aggregation of apo-SOD1S-S (Fig. 2A). Although both Cys residues at 6 and 111 are considered to participate in the disulfide scrambling, Cys6 hence appears to play more critical roles in the disulfide-linked oligomerization of SOD1.
An ability of SOD1 to scramble a disulfide bond will be dependent upon the solvent exposure of free Cys residues. In both holo and apo states of SOD1, Cys111 has been known to be highly solvent-exposed; indeed, Cys111 is susceptible to oxidative modifications (26) and also to disulfide formation with exogenous thiol compounds such as glutathione (27). Besides, recent H/D exchange experiments have revealed that Cys6 is buried in the holo state of SOD1 but dramatically increases its solvent accessibility upon the dissociation of bound metal ions (28). Indeed, in this study, addition of Zn2+ ions was found to suppress the aggregation of SOD1S-S in the presence of 1 m GdnHCl (Fig. 1A), although no direct evidence showing the presence of apo-SOD1 in affected tissues of fALS patients or animal models have not been available. Nonetheless, given that affinity of SOD1 for copper and zinc ions in vitro is significantly decreased by most of the fALS-causing mutations (29), such pathogenic mutations in SOD1 will increase the structural flexibility at Cys6, which are considered to start scrambling of the disulfide bond and form insoluble cross-linked oligomers.
As discussed above, Cys6 seems to play triggering roles in the disulfide scrambling but has been also known as a site for pathogenic mutations (i.e. C6F and C6G). In our disulfide-scrambling model, mutations at Cys6 are supposed to suppress extensive cross-linking of SOD1s via disulfide bonds (Fig. 2A); however, transient expression of C6F SOD1 in NSC-34 cells has been shown to reproduce the formation of insoluble disulfide-linked oligomers (30). Notably, C6F mutation significantly destabilizes apo-SOD1 and impairs the formation of a disulfide bond (31, 32); therefore, C6F mutant SOD1 can be speculated to proceed through the disulfide-reduction model (route I in Fig. 7) to form insoluble aggregates, which may then be cross-linked via disulfide bonds. To further test our disulfide-scrambling model, future characterization will be very promising on the thiol-disulfide status of fALS-causing mutant SOD1 with a C6S background in rodent models.
In summary, depending upon the thiol-disulfide status in SOD1, we have identified at least three distinct routes for SOD1 aggregation (Fig. 7). It remains to be further investigated whether these three pathways are mutually exclusive or co-exist to form pathological aggregates. Our preliminary experiments using lumber spinal cord of a transgenic mouse expressing human SOD1 with G93A mutation have suggested the presence of both disulfide-reduced and disulfide-bonded SOD1s in insoluble inclusions (data not shown), supporting multiple pathways of SOD1 aggregation in vivo (Fig. 7). In such in vivo conditions, it will be somewhat difficult to test our disulfide-scrambling model because a change in a pair of Cys residues forming a disulfide bond (i.e. scrambling of a disulfide bond) cannot be easily followed in a time-dependent manner. Among multiple aggregation pathways, nonetheless, a disulfide-scrambling route is found to be triggered by structural destabilization of apo-SOD1S-S with either addition of a chaotropic reagent or introduction of a fALS-causing mutation, which reproduces the formation of insoluble SOD1 oligomers cross-linked via disulfide bonds (route III in Fig. 7). Further in vivo studies will be also required to test whether those disulfide-linked SOD1 oligomers exert toxicity in spinal motor neurons. Free Cys residues, if any, in the other disulfide-containing proteins can essentially attack a disulfide bond, through which the resultant non-native disulfide bond may stabilize/lock a misfolded state of protein molecules. Our scrambling model may, therefore, not be limited to the SOD1 aggregation but could be a general mechanism to describe aggregation of the disulfide-containing proteins.
This work was supported by Grants-in-aid 24111542 (to Y. F.) and 23111006 (to K. Y.) for Scientific Research on Innovative Areas and 24657093 (to Y. F.) for Challenging Exploratory Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan, grants-in-aid from the Research Committee of CNS Degenerative Diseases from the Ministry of Health, Labor, and Welfare of Japan and from CREST, Japan Science and Technology Agency (to K. Y.), a grant from Japan ALS association (to Y. F.), and a grant from the Mochida Memorial Foundation for Medical and Pharmaceutical Research (to Y. F.).

This article contains supplemental Table S1.
- SOD1
- Cu,Zn-superoxide dismutase
- fALS
- a familial form of amyotrophic lateral sclerosis
- apo-SOD1S-S
- demetallated SOD1 with a conserved disulfide bond
- apo-SOD1SH
- demetallated and disulfide-reduced SOD1
- GdnHCl
- guanidine hydrochloride
- IA
- iodoacetamide.
REFERENCES
- 1. Sevier C. S., Kaiser C. A. (2002) Formation and transfer of disulphide bonds in living cells. Nat. Rev. Mol. Cell Biol. 3, 836–847 [DOI] [PubMed] [Google Scholar]
- 2. Chattopadhyay M., Valentine J. S. (2009) Aggregation of copper-zinc superoxide dismutase in familial and sporadic ALS. Antioxid. Redox Signal 11, 1603–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Furukawa Y., O'Halloran T. V. (2006) Posttranslational modifications in Cu,Zn-superoxide dismutase and mutations associated with amyotrophic lateral sclerosis. Antioxid. Redox Signal 8, 847–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Seetharaman S. V., Prudencio M., Karch C., Holloway S. P., Borchelt D. R., Hart P. J. (2009) Immature copper-zinc superoxide dismutase and familial amyotrophic lateral sclerosis. Exp. Biol. Med. 234, 1140–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McCord J. M., Fridovich I. (1969) Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 244, 6049–6055 [PubMed] [Google Scholar]
- 6. Furukawa Y., Torres A. S., O'Halloran T. V. (2004) Oxygen-induced maturation of SOD1: a key role for disulfide formation by the copper chaperone CCS. EMBO J. 23, 2872–2881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Furukawa Y., O'Halloran T. V. (2005) Amyotrophic lateral sclerosis mutations have the greatest destabilizing effect on the apo- and reduced form of SOD1, leading to unfolding and oxidative aggregation. J. Biol. Chem. 280, 17266–17274 [DOI] [PubMed] [Google Scholar]
- 8. Furukawa Y., Kaneko K., Yamanaka K., O'Halloran T. V., Nukina N. (2008) Complete loss of post-translational modifications triggers fibrillar aggregation of SOD1 in the familial form of amyotrophic lateral sclerosis. J. Biol. Chem. 283, 24167–24176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bruijn L. I., Miller T. M., Cleveland D. W. (2004) Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu. Rev. Neurosci. 27, 723–749 [DOI] [PubMed] [Google Scholar]
- 10. Tiwari A., Hayward L. J. (2003) Familial amyotrophic lateral sclerosis mutants of copper/zinc superoxide dismutase are susceptible to disulfide reduction. J. Biol. Chem. 278, 5984–5992 [DOI] [PubMed] [Google Scholar]
- 11. Jonsson P. A., Graffmo K. S., Andersen P. M., Brännström T., Lindberg M., Oliveberg M., Marklund S. L. (2006) Disulphide-reduced superoxide dismutase-1 in CNS of transgenic amyotrophic lateral sclerosis models. Brain 129, 451–464 [DOI] [PubMed] [Google Scholar]
- 12. Hwang C., Sinskey A. J., Lodish H. F. (1992) Oxidized redox state of glutathione in the endoplasmic reticulum. Science 257, 1496–1502 [DOI] [PubMed] [Google Scholar]
- 13. Karch C. M., Borchelt D. R. (2008) A limited role for disulfide cross-linking in the aggregation of mutant SOD1 linked to familial amyotrophic lateral sclerosis. J. Biol. Chem. 283, 13528–13537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Karch C. M., Prudencio M., Winkler D. D., Hart P. J., Borchelt D. R. (2009) Role of mutant SOD1 disulfide oxidation and aggregation in the pathogenesis of familial ALS. Proc. Natl. Acad. Sci. U.S.A. 106, 7774–7779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Subramaniam J. R., Lyons W. E., Liu J., Bartnikas T. B., Rothstein J., Price D. L., Cleveland D. W., Gitlin J. D., Wong P. C. (2002) Mutant SOD1 causes motor neuron disease independent of copper chaperone-mediated copper loading. Nat. Neurosci. 5, 301–307 [DOI] [PubMed] [Google Scholar]
- 16. Deng H. X., Shi Y., Furukawa Y., Zhai H., Fu R., Liu E., Gorrie G. H., Khan M. S., Hung W. Y., Bigio E. H., Lukas T., Dal Canto M. C., O'Halloran T. V., Siddique T. (2006) Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc. Natl. Acad. Sci. U.S.A. 103, 7142–7147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Furukawa Y., Fu R., Deng H. X., Siddique T., O'Halloran T. V. (2006) Disulfide cross-linked protein represents a significant fraction of ALS-associated Cu,Zn-superoxide dismutase aggregates in spinal cords of model mice. Proc. Natl. Acad. Sci. U.S.A. 103, 7148–7153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rumfeldt J. A., Stathopulos P. B., Chakrabarrty A., Lepock J. R., Meiering E. M. (2006) Mechanism and thermodynamics of guanidinium chloride-induced denaturation of ALS-associated mutant Cu,Zn superoxide dismutases. J. Mol. Biol. 355, 106–123 [DOI] [PubMed] [Google Scholar]
- 19. Furukawa Y., Kaneko K., Yamanaka K., Nukina N. (2010) Mutation-dependent polymorphism of Cu,Zn-superoxide dismutase aggregates in the familial form of amyotrophic lateral sclerosis. J. Biol. Chem. 285, 22221–22231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chattopadhyay M., Durazo A., Sohn S. H., Strong C. D., Gralla E. B., Whitelegge J. P., Valentine J. S. (2008) Initiation and elongation in fibrillation of ALS-linked superoxide dismutase. Proc. Natl. Acad. Sci. U.S.A. 105, 18663–18668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bordo D., Djinović K., Bolognesi M. (1994) Conserved patterns in the Cu,Zn superoxide dismutase family. J. Mol. Biol. 238, 366–386 [DOI] [PubMed] [Google Scholar]
- 22. Furukawa Y. (2012) Protein aggregates in pathological inclusions of amyotrophic lateral sclerosis in Amyotrophic lateral sclerosis (Maurer M. H., ed.), pp. 335–356, InTech, Rijeka, Croatia [Google Scholar]
- 23. Rodriguez J. A., Shaw B. F., Durazo A., Sohn S. H., Doucette P. A., Nersissian A. M., Faull K. F., Eggers D. K., Tiwari A., Hayward L. J., Valentine J. S. (2005) Destabilization of apoprotein is insufficient to explain Cu,Zn-superoxide dismutase-linked ALS pathogenesis. Proc. Natl. Acad. Sci. U.S.A. 102, 10516–10521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vassall K. A., Stubbs H. R., Primmer H. A., Tong M. S., Sullivan S. M., Sobering R., Srinivasan S., Briere L. A., Dunn S. D., Colón W., Meiering E. M. (2011) Decreased stability and increased formation of soluble aggregates by immature superoxide dismutase do not account for disease severity in ALS. Proc. Natl. Acad. Sci. U.S.A. 108, 2210–2215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Niwa J., Yamada S., Ishigaki S., Sone J., Takahashi M., Katsuno M., Tanaka F., Doyu M., Sobue G. (2007) Disulfide bond mediates aggregation, toxicity, and ubiquitylation of familial amyotrophic lateral sclerosis-linked mutant SOD1. J. Biol. Chem. 282, 28087–28095 [DOI] [PubMed] [Google Scholar]
- 26. Fujiwara N., Nakano M., Kato S., Yoshihara D., Ookawara T., Eguchi H., Taniguchi N., Suzuki K. (2007) Oxidative modification to cysteine sulfonic acid of Cys111 in human copper-zinc superoxide dismutase. J. Biol. Chem. 282, 35933–35944 [DOI] [PubMed] [Google Scholar]
- 27. Wilcox K. C., Zhou L., Jordon J. K., Huang Y., Yu Y., Redler R. L., Chen X., Caplow M., Dokholyan N. V. (2009) Modifications of superoxide dismutase (SOD1) in human erythrocytes: a possible role in amyotrophic lateral sclerosis. J. Biol. Chem. 284, 13940–13947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Banci L., Bertini I., Boca M., Calderone V., Cantini F., Girotto S., Vieru M. (2009) Structural and dynamic aspects related to oligomerization of apo SOD1 and its mutants. Proc. Natl. Acad. Sci. U.S.A. 106, 6980–6985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hayward L. J., Rodriguez J. A., Kim J. W., Tiwari A., Goto J. J., Cabelli D. E., Valentine J. S., Brown R. H., Jr. (2002) Decreased metallation and activity in subsets of mutant superoxide dismutases associated with familial amyotrophic lateral sclerosis. J. Biol. Chem. 277, 15923–15931 [DOI] [PubMed] [Google Scholar]
- 30. Cozzolino M., Amori I., Pesaresi M. G., Ferri A., Nencini M., Carrì M. T. (2008) Cysteine 111 affects aggregation and cytotoxicity of mutant Cu,Zn-superoxide dismutase associated with familial amyotrophic lateral sclerosis. J. Biol. Chem. 283, 866–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bruns C. K., Kopito R. R. (2007) Impaired post-translational folding of familial ALS-linked Cu,Zn superoxide dismutase mutants. EMBO J. 26, 855–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lindberg M. J., Tibell L., Oliveberg M. (2002) Common denominator of Cu/Zn superoxide dismutase mutants associated with amyotrophic lateral sclerosis: decreased stability of the apo state. Proc. Natl. Acad. Sci. U.S.A. 99, 16607–16612 [DOI] [PMC free article] [PubMed] [Google Scholar]