

Background: Modulation of host miRNAs coincides with increased pathogenicity in various infectious diseases.
Results: miR-99b is up-regulated in M. tuberculosis-infected dendritic cells, which inhibits production of proinflammatory cytokines.
Conclusion: Our findings unfold a novel immune evasion strategy of M. tuberculosis by modulating miRNAs.
Significance: Our study opens up the possibility to design vaccines and immunotherapies for tuberculosis by targeting specific miRNAs.
Keywords: Innate immunity, MicroRNA, Mycobacterium tuberculosis, T cell, Tumor necrosis factor (TNF), DCs
Abstract
Mycobacterium tuberculosis resides and replicates within host phagocytes by modulating host microbicidal responses. In addition, it suppresses the production of host protective cytokines to prevent activation of and antigen presentation by M. tuberculosis-infected cells, causing dysregulation of host protective adaptive immune responses. Many cytokines are regulated by microRNAs (miRNAs), a newly discovered class of small noncoding RNAs, which have been implicated in modulating host immune responses in many bacterial and viral diseases. Here, we show that miRNA-99b (miR-99b), an orphan miRNA, plays a key role in the pathogenesis of M. tuberculosis infection. We found that miR-99b expression was highly up-regulated in M. tuberculosis strain H37Rv-infected dendritic cells (DCs) and macrophages. Blockade of miR-99b expression by antagomirs resulted in significantly reduced bacterial growth in DCs. Interestingly, knockdown of miR-99b in DCs significantly up-regulated proinflammatory cytokines such as IL-6, IL-12, and IL-1β. Furthermore, mRNA and membrane-bound protein data indicated that inhibition of miR-99b augments TNF-α and TNFRSF-4 production. Thus, miR-99b targets TNF-α and TNFRSF-4 receptor genes. Treatment of anti-miR-99b-transfected DCs with anti-TNF-α antibody resulted in increased bacterial burden. Thus, our findings unveil a novel host evasion mechanism adopted by M. tuberculosis via miR-99b, which may open up new avenues for designing miRNA-based vaccines and therapies.
Introduction
Mycobacterium tuberculosis, the causative agent of tuberculosis (TB),3 remains one of the biggest global health problems, with an estimated one-third of the global population latently infected and more than 2 million deaths every year (1). M. tuberculosis successfully combats microbicidal mechanisms of phagocytes to promote infection and replication and even outmaneuvers these mechanisms to favor its persistence in infected cells (2). Phagocytes, the primary innate immune cells responding to M. tuberculosis, engulf microorganisms into phagosomes, which later fuse with lysosomes (3). The acidic environment of the phagolysosome degrades the harbored organisms, and this process also makes antigens available for priming of T cell responses (4, 5). M. tuberculosis resides and replicates within phagocytes of susceptible hosts by avoiding phagocytic destruction (6, 7). To survive within innate immune cells, M. tuberculosis has evolved mechanisms to inhibit phagolysosome fusion and to neutralize the acidic environment of the phagolysosomal compartment (8). In addition to its capacity to avoid phagolysosomal degradation, M. tuberculosis successfully inhibits production of proinflammatory cytokines, such as tumor necrosis factor-α (TNF-α) and interleukin-1 (IL-1), as a means to prevent autocrine activation of infected cells and to ensure persistence within the cells (9–12).
Immune responses to various infectious microorganisms and cancers are regulated at the post-transcriptional level by various microRNAs (miRNAs) (13). It is now well documented that production of several cytokines is regulated by miRNAs, and these regulatory RNAs have therefore emerged as key players in the immune system (14). miRNAs are an abundant class of highly conserved small (18–25 nucleotides long) noncoding RNAs that suppress gene expression by binding to the 3′-untranslated region (UTR) of target mRNAs (15). A direct role of miRNAs in innate immunity was revealed by the identification of miR146a as a negative feedback regulator in Toll-like receptor signaling by targeting IL-1R-associated kinase 1 (IRAK1) and TNF receptor-associated factor 6 (TRAF6). miR-146a inhibits the expression of IRAK1 and TRAF6 and impairs nuclear factor-κB (NF-κB) activity, which in turn suppresses the expression of IL-6, IL-8, IL-1β, and TNF-α (13, 15–18). A recent study further suggested that lipomannan from virulent M. tuberculosis but not from avirulent Mycobacterium smegmatis is a potent inhibitor of TNF-α biosynthesis in human macrophages, by regulating miR-125b and miR-155 expression via the inositol phosphatase SHIP-1 (19). Taken together, these data suggest a correlation between miRNA expression and cytokine regulation during M. tuberculosis infection.
We have performed a detailed investigation of the role of miRNAs in M. tuberculosis infection and pathogenesis. Microarray analysis suggested that eight different miRNAs were up-regulated in M. tuberculosis-infected DCs. However, only miR-99b was highly up-regulated by M. tuberculosis-infected DCs but not by lipopolysaccharide (LPS)-stimulated DCs, and this was dependent on MyD88 signaling. Knockdown of miR-99b in DCs significantly reduced the bacterial burden. Moreover, blocking miR-99b by synthetic antagomirs enhanced the production of IL-6, IL-12, and IL-1β. We further showed that miR-99b targets various mRNA transcripts, including TNFRSF-4 and TNF-α, to regulate expression of various cytokines and transcription factors involved in T cell lineage differentiation pathways and in the clearance of M. tuberculosis bacteria. Blockade of TNF-α in anti-miR-99b-transfected DCs significantly increased the burden of M. tuberculosis infection, providing evidence for a critical role of TNF-α as a target for miR-99b. Therefore, our findings provide evidence that M. tuberculosis inhibits the activation of infected cells by up-regulating miR-99b expression, which represents a novel strategy adopted by M. tuberculosis to evade host protective immune responses that allows these organisms to survive within phagocytes.
EXPERIMENTAL PROCEDURES
Mice
C57BL/6 mice (6–8 weeks of age) were initially purchased from The Jackson Laboratory. MyD88 knock-out mice were a kind gift from Prof. Ruslan Medzhitov, Yale University, School of Medicine. All animals were subsequently bred and maintained in our specific pathogen-free animal facility at the International Centre for Genetic Engineering and Biotechnology (ICGEB), New Delhi, India.
DC Preparation
DCs were prepared as described earlier (20) and in the supplemental Methods.
H37Rv Bacterial Infections
DCs were prepared from WT or MyD88-deficient C57BL/6 mice as described above. Infection experiments were performed in 24-well plates (Nunc). DCs were infected in complete RPMI medium without antibiotics at a final multiplicity of infection of 10 H37Rv M. tuberculosis bacteria (multiplicity of infection of 1:10; DCs:H37Rv). DCs were incubated for another 24 h at 37 °C in a 5% CO2 humidified incubator. For control experiments, DCs were also cultured in the presence or absence of LPS at 1 μg/ml. Culture supernatants were collected at 24 h from uninfected, LPS-treated, H37Rv-infected and LPS+H37Rv-treated DCs and kept at −80 °C until used for cytokine analysis.
miRCURY LNATM-based miRNA Microarrays
Total RNA was isolated by miRNeasy isolation kit (Qiagen) according to the manufacturer's instructions, and microRNA profiling analysis was performed by EXIQON, Vedbaek, Denmark.
q-PCR Analysis of miRNA Expression
Total RNA, including miRNAs, was isolated by miRNeasy isolation kit (Qiagen), and cDNA was synthesized separately for miRNAs and mRNA for real-time quantitative PCR (q-RT-PCR). q-RT-PCR analysis was performed using Bio-Rad CFX96 real-time thermal cycler (Bio-Rad) and miRCURY LNA universal reverse transcriptase microRNA PCR SYBR Green master mix (EXIQON) for miRNA (miR-146a miR-125a-5p and miR-99b) amplification and iQTM Bio-Rad SYBR Green master mix (Bio-Rad) for TNF-α and TNFRSF-4 expression as recommended by the manufacturers. Detailed protocols are provided in the supplemental Methods.
Knockdown of miRNAs Using Antagomirs
For transfection of anti-miR-99b (antagomirs) and scramble control (EXIQON), DCs were transfected at day 4 of culture in antibiotic-free RPMI 1640 using LipofectamineTM 2000 (Invitrogen) reagents. Briefly, at day 4 of culture, DCs were washed once with antibiotic-free RPMI 1640 medium and cultured in 500 μl of antibiotic-free RPMI 1640 medium for 4 h in a humidified CO2 incubator at 37 °C. Four hours later, 100 nm of antagomirs or scramble control were added into 50 μl of Opti-MEM medium (Invitrogen) according to the manufacturer's instructions. The next day, medium was changed, and DCs were subjected to H37Rv bacterial infections. These DCs were called knockdown DCs. After 24 h of bacterial infection, DCs were harvested for RNA preparation and quantitative real-time PCR for miR-99b, TNFRSF-4, and TNF-α.
Determination of H37Rv Colony-forming Units in DC Cultures
DCs (0.5 × 106) were grown in 24-well plates (Nunc), transfected with anti-miR99b or scramble control antagomirs, and infected with H37Rv bacteria as described above. In some experiments 10 μg/ml anti-TNF-α antibody (Santa Cruz Biotechnology) was added to the DC cultures. After 48 h, 250 μl of lysis buffer (0.05% SDS in 7H9 medium) was added and mixed well, and the cells were left in contact with lysis buffer. After 5 min of incubation, 10- and 100-fold dilutions were made in a 96-well plate, and 10 μl of each dilution was plated on square plates (Bio-Rad) using trailing methods.
Detection of Cytokines
DC cytokines in the culture supernatant were assayed by a Luminex microbead-based multiplexed (Qiagen Luminex LiquiChip) assay using commercially available kits according to the manufacturer's protocol (Bio-Plex, Bio-Rad).
Statistical Analysis
Paired t tests and analysis of variance and Tukey's post hoc test were performed to compare the statistical significance between various groups. p values were considered significant if p ≤ 0.05, and S.D. is shown in all figures unless stated otherwise. In some cases, GraphPad Prism®5.0 software was used for statistical analyses.
RESULTS AND DISCUSSION
It has been well established that M. tuberculosis infects macrophages and DCs. DCs are the most potent antigen-presenting cells of the immune system and thus determine the outcome of adaptive immune responses during infection. Therefore, we infected DCs with M. tuberculosis and determined the expression of miRNAs by miRCURY LNATM miRNA microarray profiling. We generated DCs from bone marrow precursor cells following culture with IL-4 and GM-CSF as described elsewhere (20). At day 5, DCs were infected with virulent H37Rv bacteria. As a control, parallel DC cultures were activated with LPS. The miRCURYTM LNA miRNA microarray data revealed that eight miRNAs (miR-710, miR-881*, miR-882, miR-877, miR-146a, miR-125a-5p, miR-99b, and miR-222) were up-regulated in the M. tuberculosis-infected DCs. Among these, miRNAs miR-146a and miR-125a-5p were LPS- and H37Rv-induced, and the other miRNAs, miR-710, miR-881*, miR-882, miR-877, and miR-99b, appeared to be preferentially induced by H37Rv as compared with LPS (Fig. 1B). Interestingly, only miR-99b was highly up-regulated in DCs by M. tuberculosis infection but not by LPS, and this was also dependent on MyD88 expression, as demonstrated with DCs from MyD88-deficient mice (Fig. 1A). Previously, it has been shown that MyD88-deficient animals are hypersusceptible to M. tuberculosis infection (21, 22). Our findings therefore raised the possibility that miR-99b regulates cytokine expression and determines susceptibility or resistance to M. tuberculosis infection. Therefore, we mainly focused on the role of miR-99b in the pathogenesis of M. tuberculosis infection. Some of the miRNAs, such as miR-762, miR-290-5p, and miR-665, were down-regulated in M. tuberculosis-infected DCs. To corroborate our miRNA microarray profiling data, we performed q-PCR for detecting the expression levels of these miRNAs in infected DCs. Consistent with the microarray profiling results, both miR-146a and miR-125a-5p were up-regulated in the presence of H37Rv bacteria (miR-146a, p = 0.02; miR-125a-5p, p = 0.03) or LPS (miR-146a, p = 0.03; miR-125a-5p, p = 0.04) (Fig. 1C). Furthermore, when both LPS and H37Rv were present, we observed a cumulative induction of these miRNAs (miR-146a, p = 0.02; miR-125a-5p, p = 0.01), which confirmed the miRNA profiling data (Fig. 1B). These data therefore suggested that M. tuberculosis H37Rv bacteria modulate the expression of miRNAs to promote survival inside host cells. Furthermore, q-PCR data confirmed that miR-99b was the least sensitive to LPS induction, as predicted by miRNA microarray analysis, and this is consistent with results from a recent study performed with macrophages (23). However, miR-99b was significantly up-regulated in DCs in the presence of H37Rv bacteria (p = 0.01) (Fig. 1B), indicating that induction of miR-99b in DCs was specific to M. tuberculosis infection.
FIGURE 1.
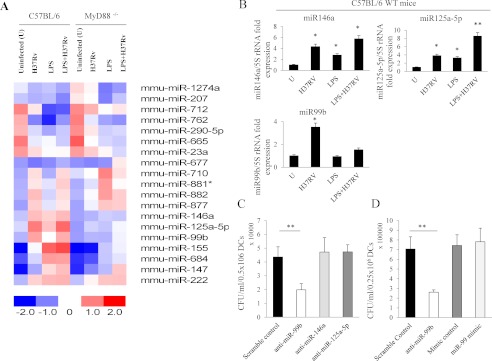
Differential expression of miRNAs in M. tuberculosis-infected dendritic cells. A, heat map diagram showing the result of two-way hierarchical clustering of genes and samples. Each row represents an miRNA, and each column represents a sample. The clustering is performed on log2 (Hy3/Hy5) ratios, which passed the filtering criteria on variation across samples within each group: Δ log median ratio ≥ ± 1.0 equal to -fold change >2.0. Only 19 out of 599 miRNAs passed the filtering criteria on variation across samples. The color scale shown at the bottom illustrates the relative expression level of an miRNA across all samples; red color represents an expression level above the mean, and blue color represents expression lower than the mean. Eight different miRNAs were up-regulated after M. tuberculosis infection as compared with uninfected (U) control, and two miRNAs were down-regulated as compared with control in C57BL/6 mice. Most of the miRNAs that were up-regulated in C57BL/6 mice were induced at lower levels in MyD88−/− mice. B, real-time expression of miR-146a, miR-125a-5p, and miR-99b in WT dendritic cells after M. tuberculosis (H37Rv) infection, LPS treatment, or treatment with LPS+H37Rv. Student's t test was performed to measure the statistical significance between uninfected cells, infected cells, LPS-treated cells, or cells treated with both LPS and H37Rv. *, p ≤ 0.05, **, p ≤ 0.01. All miRNAs were detected by real-time RT-PCR, and 5 S rRNA was used as an internal control to normalize the relative amount of miRNA. The 2−ΔΔCt method was used to calculate the -fold expression. Data shown here are mean ± S.D. for one representative experiment of at least 3–4 independent experiments. C, DCs were subjected to antagomirs for blocking miR-99b, miR-146a, or miR-125a-5p expression, and scrambled antagomirs were used as a control. After 48 h of H37Rv bacterial infection, cells were lysed, and 10- and 100-fold dilutions were made and then plated on square plates using trailing methods. Student's t test was performed; **, p = 0.01. Data shown here are representative of 2 independent experiments. D, DCs were subjected to miR mimics for overexpression of miR-99b and mimic control. In parallel, DCs were subjected to antagomirs for blocking miR-99b and a scramble control. After 48 h of H37Rv bacterial infection, cells were lysed, and 10- and 100-fold dilutions were made and then plated on square plates using trailing methods. Student's t test was performed; **, p = 0.01. Data shown here are representative of 2 independent experiments.
To address the question whether enhanced miRNA expression supports the growth of bacteria, we performed loss-of-function miRNA experiments. For this purpose, we employed synthetic antagomirs to investigate the effect of miRNA knockdown on the production of various cytokines produced by DCs. Transfection efficiency of miR-99b antagomirs was >95% in all experiments (supplemental Fig. 1A). To examine the knockdown (KD) efficiency, we performed miRNA-q-PCR and found statistically significant inhibition of all three miRNAs (miR-99b, miR-146a, and miR-125a-5p) in different treatments (supplemental Fig. 1B). Once we confirmed the KD efficiency for different miRNAs, we investigated the bacterial burden in antagomir-transfected DCs infected with H37Rv and found that inhibition of miR-99b enhanced the clearance of bacteria from infected cells as compared with scramble controls, whereas KD of other miRNAs (miR-146a and miR-125-5p) had no effect on bacterial growth. At 48 h after infection, there was a statistically significant reduction in the growth of H37Rv bacteria (p = 0.01) in DCs treated with anti-miR-99b antagomirs as compared with the scramble control (Fig. 1C). Furthermore, when we overexpressed miR-99b by miRNA mimics, we failed to observe a statistically significant enhancement in bacterial growth as compared with mimic controls (Fig. 1D). We speculate that bacteria themselves induce miR-99b levels in the infected DCs and that this miRNA might therefore already have reached a saturated level. Thus, overexpression of miRNA does not enhance bacterial growth. Overall, our findings suggest that KD of miR-99b is essential for reduced survival of bacteria.
In the preceding section, we showed that blockade of miR99b expression resulted in restriction of bacterial growth. Therefore, we hypothesized that miRNA knockdown would enhance cytokine secretion by DCs. Previous studies have suggested that various M. tuberculosis strains (virulent and avirulent) differentially induce production of distinct cytokines such as IL-6, IL-12, IL-1β, and TNF-α (20, 24, 25). In agreement with previous findings, we found comparable production of IL-6 and IL-12 in DCs infected with the H37Rv strain of M. tuberculosis (Fig. 2A) (20). Surprisingly, KD of miR-99b in uninfected DCs also significantly up-regulated IL-6 (p = 0.04), IL-12p40 (p = 0.001), IL-12p70 (p = 0.03), and IL-1β (p = 0.05) production (Fig. 2A). Thus, these data suggested that even at basal levels, inhibition of miR-99b can cause an enhancement in cytokine production. Further, we have found that KD of miR-99b in H37Rv-infected DCs significantly up-regulates IL-6 (p = 0.01), IL-12p40 (p = 0.01), IL-12p70 (p = 0.04), and IL-1β (p = 0.04) production (Fig. 2A). These data suggested that miR-99b plays a critical role in regulating cytokine production in DCs.
FIGURE 2.

Blockade of miRNA expression by antagomirs up-regulates multiple cytokines. A, dendritic cells were treated with miR99b antagomirs after M. tuberculosis (H37Rv) infection. Culture supernatants were analyzed for cytokines by multiplex Luminex method. Student's t test was performed; *, p ≤ 0.05, **, p ≤ 0.01. Data shown here are mean ± S.D. for a representative experiment of at least 4 independent experiments. U, uninfected. B, dendritic cells were treated with miR-99b antagomirs and then infected with M. tuberculosis (H37Rv). Gene expression of candidate targets of miR-99b was assessed by q-RT-PCR after 24 h of H37Rv infection. TNFRSF-4 and TNF-α mRNA transcript is shown. Student's t test was performed to compare mRNA transcript expression levels between the scramble control and miR-99b knockdown samples; *, p ≤ 0.05, **, p ≤ 0.01. DCs were treated with miR-99b antagomirs, and scramble antagomirs were used as a control. C, Western blotting for TNF-α was performed from DCs infected with M. tuberculosis and treated with the scramble control or miR-99b KD samples. Protein blot data suggested that miR-99b KD samples have higher TNF-α expression as compared with the scramble control. A similar trend was also observed for TNFRSF-4 (48 kDa) protein expression. Protein was quantified using ImageJ software (TNF-α/β-actin or TNFRSF-4/β-actin). Data shown here are representative of 2 independent experiments. D, as shown in the figure, when the 3′-UTR of TNF and miR-99 were present in the cells, a 17% reduction in luciferase activity was observed as compared with control miR as well as control pGL-3 vector co-transfected with miR-99 (*, p = 0.03). There was no change in luciferase activity induced by miR-21 co-transfected with pGL-3 or pGL-3-TNF-3′-UTR vectors. Data shown here summarize 3 independent experiments. E, after 48 h of H37Rv bacterial infection, bacterial growth was measured. Scramble-treated DCs act as a control, showing higher bacterial burden as compared with DCs treated with miR-99b LNA-anti-miR probes (*, p = 0.04). miR-99b knockdown samples were supplemented with anti-TNF-α antibody to block TNF-α, which showed increased H37Rv (Rv) bacterial growth as compared with the miR-99b knockdown samples as well as scramble control samples (*, p = 0.02). Data shown here are representative of 2 independent experiments.
To determine which key signaling molecules are affected by miR-99b during M. tuberculosis infection in DCs, we explored possible targets related to Toll-like receptor signaling, cytokine signaling, and autophagolysosomal pathways. For this purpose, we have employed various computational algorithms to identify potential targets (supplemental Fig. 2). It has been established that based on the predicted seed-match sequences (7–8-nucleotide homology matches), each miRNA has several potential false positive target sites. We found that TNF-α and TNFRSF-4 have potential matches for miR-99b (supplemental Fig. 3A). Therefore, we next performed q-PCR and Western blot analyses for TNF-α and TNFRSF-4 to test the targets of miR-99b.
Consistent with previous findings suggesting that miRNAs are post-transcriptional regulators of mRNAs (13, 14), we found that miR-99b has a TNF receptor superfamily member-4 (TNFRSF-4, OX40, or CD134) as a target site, which was significantly up-regulated (p = 0.03) in the presence of bacteria after miR-99b KD (Fig. 2B). Inhibition/overexpression of miR-99b affects TNFRSF-4 protein expression as well (Fig. 2C and supplemental Fig. 3, B and D). Furthermore, mRNA transcripts for TNF-α, a key molecule in eliminating the bacterial burden from the infected cells, were also significantly up-regulated (p = 0.01) after M. tuberculosis infection (Fig. 2B). TNF-α was further validated as a target for miR-99b at the protein level by immunoblotting of M. tuberculosis-infected DCs with scramble control or miR-99b antagomirs, which suggested that KD of miR-99b enhanced total TNF-α protein production (Fig. 2C and supplemental Fig. 3, B and C). To further confirm that TNF-α is targeted by miR-99b, we performed luciferase assays to measure 3′-UTR luciferase protein activity. To perform the luciferase assay, we have cloned the 3′-UTR of TNF-α into pGL-3 vector downstream of the luciferase gene. 293T cells were transfected with miR-99b construct, control vectors and pGL-3-3′-UTR-TNF vector as described under “Experimental Procedures.” Luciferase activity was 17% reduced when the 3′-UTRs of TNF-α and miR-99b were both present as compared with control miRNAs (miR-C) or nontargeted miR-21 in the presence of TNF-α. This experiment revealed that TNF-α is indeed a bona fide target of miR-99b (Fig. 2D). Furthermore, we overexpressed miR-99b in CD4+ T cells, differentiated these cells into T helper (Th1) cells, and measured TNF-α and IFN-γ production. As anticipated, overexpression of miR-99b reduced expression of TNF-α protein but did not alter IFN-γ production (supplemental Fig. 4). Thus, TNF-α expression is regulated upon KD and overexpression of miR-99b. Hence, our data provide substantial evidence that TNF-α is one of the vital genes targeted by miR-99b. Nevertheless, despite observing increased levels of mRNA transcripts and enhanced levels of membrane-bound and intracellular TNF-α protein, we were unable to detect significant changes in secreted TNF-α protein upon infection of DCs with M. tuberculosis (supplemental Fig. 3E), for reasons that remain unclear. Thus, although there is unequivocal evidence that miR-99b regulates TNF-α, there are likely additional factors that influence TNF-α secretion or regulatory feedback mechanisms. This might also partly explain why treatment with mimic miR-99b does not lead to a significant increase in CFU in H37Rv-infected DCs (Fig. 1D). Nonetheless, it has previously been shown that TNF-α-deficient mice are hypersusceptible to M. tuberculosis infection (12). Therefore, KD of certain miRNAs might permit the host to resist M. tuberculosis infection. Thus, our results indicate that various miRNAs control different signaling pathways and that modulation of their expression by M. tuberculosis permits these organisms to evade host immunity and survive inside DCs and macrophages.
To determine whether TNF-α plays a critical role in controlling bacterial growth following miR-99b KD, we neutralized TNF-α production in the cultures with anti-TNF-α antibody. In Fig. 2E, scrambled control sample infected with H37Rv was employed as a positive control for the experiment testing the effect of anti-miR-99b on bacterial burden of H37Rv in DCs, as well as for scrambled control sample treated with anti-TNF-α. Our data suggested that treatment of scrambled control sample with anti-TNF-α antibody modestly enhanced bacterial growth as compared with DCs treated with scrambled control only. Knockdown of miR-99b in DCs significantly reduced bacterial growth because of an increase in the production of various proinflammatory cytokines (e.g. IL-1β, TNF-α, etc.). To determine whether TNF-α has a specific role in bacterial survival, we added an anti-TNF-α antibody to H37Rv-infected DCs with miR-99b knockdown (Fig. 2E). Neutralization of TNF-α resulted in an increase in bacterial burden in these cultures. Hence, TNF-α is critical for reduced bacterial burden as it was produced in the absence of miR-99b. Thus, altogether these data confirmed that miR-99b affects bacterial growth in M. tuberculosis-infected DCs by modulating TNF-α production, which activates DCs to clear the engulfed bacteria.
In summary, our data reveal a novel role of miR-99b in modulating host immunity after M. tuberculosis infection by controlling TNF-α production. Furthermore, miR-99b could potentially serve as a therapeutic and diagnostic tool in TB infection. However, to design vaccines and DC-based therapies for TB, further in vivo studies will be needed to confirm the potential roles of miR-99b and other miRNAs in active TB infections in murine models and human patients.
Acknowledgments
We thank the staff of the BSL-3 containment facility (DBT-supported Tuberculosis Aerosol Challenge Facility (DBT-TACF) at the ICGEB, New Delhi, India) and the animal facility.
This research was supported by a grant from the Wellcome Trust/Department of Biotechnology (DBT) alliance.

This article contains supplemental Methods and Figs. 1–4.
- TB
- tuberculosis
- miRNA
- microRNA
- miR-99b
- miRNA-99b
- DC
- dendritic cell
- KD
- knockdown
- q-PCR
- quantitative PCR
- q-RT-PCR
- real-time quantitative PCR.
REFERENCES
- 1. Barnes P. F., Cave M. D. (2003) Molecular epidemiology of tuberculosis. N. Engl. J. Med. 349, 1149–1156 [DOI] [PubMed] [Google Scholar]
- 2. Krutzik S. R., Modlin R. L. (2004) The role of Toll-like receptors in combating mycobacteria. Semin. Immunol. 16, 35–41 [DOI] [PubMed] [Google Scholar]
- 3. Wilson M., Seymour R., Henderson B. (1998) Bacterial perturbation of cytokine networks. Infect. Immun. 66, 2401–2409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Behar S. M., Martin C. J., Nunes-Alves C., Divangahi M., Remold H. G. (2011) Lipids, apoptosis, and cross-presentation: links in the chain of host defense against Mycobacterium tuberculosis. Microbes Infect. 13, 749–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kaufmann S. H. (2005) Recent findings in immunology give tuberculosis vaccines a new boost. Trends Immunol. 26, 660–667 [DOI] [PubMed] [Google Scholar]
- 6. Flannagan R. S., Cosío G., Grinstein S. (2009) Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat. Rev. Microbiol. 7, 355–366 [DOI] [PubMed] [Google Scholar]
- 7. Meena L. S., Rajni (2010) Survival mechanisms of pathogenic Mycobacterium tuberculosis H37Rv. FEBS J. 277, 2416–2427 [DOI] [PubMed] [Google Scholar]
- 8. Rohde K., Yates R. M., Purdy G. E., Russell D. G. (2007) Mycobacterium tuberculosis and the environment within the phagosome. Immunol. Rev. 219, 37–54 [DOI] [PubMed] [Google Scholar]
- 9. Flynn J. L., Goldstein M. M., Chan J., Triebold K. J., Pfeffer K., Lowenstein C. J., Schreiber R., Mak T. W., Bloom B. R. (1995) Tumor necrosis factor-α is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity 2, 561–572 [DOI] [PubMed] [Google Scholar]
- 10. Fremond C. M., Togbe D., Doz E., Rose S., Vasseur V., Maillet I., Jacobs M., Ryffel B., Quesniaux V. F. (2007) IL-1 receptor-mediated signal is an essential component of MyD88-dependent innate response to Mycobacterium tuberculosis infection. J. Immunol. 179, 1178–1189 [DOI] [PubMed] [Google Scholar]
- 11. Juffermans N. P., Florquin S., Camoglio L., Verbon A., Kolk A. H., Speelman P., van Deventer S. J., van Der Poll T. (2000) Interleukin-1 signaling is essential for host defense during murine pulmonary tuberculosis. J. Infect. Dis. 182, 902–908 [DOI] [PubMed] [Google Scholar]
- 12. Roach D. R., Bean A. G., Demangel C., France M. P., Briscoe H., Britton W. J. (2002) TNF regulates chemokine induction essential for cell recruitment, granuloma formation, and clearance of mycobacterial infection. J. Immunol. 168, 4620–4627 [DOI] [PubMed] [Google Scholar]
- 13. Lu L. F., Liston A. (2009) MicroRNA in the immune system, microRNA as an immune system. Immunology 127, 291–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Asirvatham A. J., Magner W. J., Tomasi T. B. (2009) miRNA regulation of cytokine genes. Cytokine 45, 58–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Taganov K. D., Boldin M. P., Chang K. J., Baltimore D. (2006) NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. U.S.A. 103, 12481–12486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Larner-Svensson H. M., Williams A. E., Tsitsiou E., Perry M. M., Jiang X., Chung K. F., Lindsay M. A. (2010) Pharmacological studies of the mechanism and function of interleukin-1β-induced miRNA-146a expression in primary human airway smooth muscle. Respir. Res. 11, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nahid M. A., Pauley K. M., Satoh M., Chan E. K. (2009) miR-146a is critical for endotoxin-induced tolerance: implication in innate immunity. J. Biol. Chem. 284, 34590–34599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Starczynowski D. T., Kuchenbauer F., Argiropoulos B., Sung S., Morin R., Muranyi A., Hirst M., Hogge D., Marra M., Wells R. A., Buckstein R., Lam W., Humphries R. K., Karsan A. (2010) Identification of miR-145 and miR-146a as mediators of the 5q− syndrome phenotype. Nat. Med. 16, 49–58 [DOI] [PubMed] [Google Scholar]
- 19. Rajaram M. V., Ni B., Morris J. D., Brooks M. N., Carlson T. K., Bakthavachalu B., Schoenberg D. R., Torrelles J. B., Schlesinger L. S. (2011) Mycobacterium tuberculosis lipomannan blocks TNF biosynthesis by regulating macrophage MAPK-activated protein kinase 2 (MK2) and microRNA miR-125b. Proc. Natl. Acad. Sci. U.S.A. 108, 17408–17413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chatterjee S., Dwivedi V. P., Singh Y., Siddiqui I., Sharma P., Van Kaer L., Chattopadhyay D., Das G. (2011) Early secreted antigen ESAT-6 of Mycobacterium tuberculosis promotes protective T helper 17 cell responses in a Toll-like receptor-2-dependent manner. PLoS Pathog. 7, e1002378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fremond C. M., Yeremeev V., Nicolle D. M., Jacobs M., Quesniaux V. F., Ryffel B. (2004) Fatal Mycobacterium tuberculosis infection despite adaptive immune response in the absence of MyD88. J. Clin. Invest. 114, 1790–1799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sugawara I., Yamada H., Mizuno S., Takeda K., Akira S. (2003) Mycobacterial infection in MyD88-deficient mice. Microbiol. Immunol. 47, 841–847 [DOI] [PubMed] [Google Scholar]
- 23. Monk C. E., Hutvagner G., Arthur J. S. (2010) Regulation of miRNA transcription in macrophages in response to Candida albicans. PLoS One 5, e13669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cooper A. M., Khader S. A. (2008) The role of cytokines in the initiation, expansion, and control of cellular immunity to tuberculosis. Immunol. Rev. 226, 191–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cooper A. M., Mayer-Barber K. D., Sher A. (2011) Role of innate cytokines in mycobacterial infection. Mucosal Immunol. 4, 252–260 [DOI] [PMC free article] [PubMed] [Google Scholar]