

Background: New antibacterial compounds are urgently needed; DNA gyrase is a well-validated target.
Results: Diospyrin and other naphthoquinones inhibit DNA gyrase by binding to a novel site in the B subunit.
Conclusion: Naphthoquinones are inhibitors of gyrase with a novel mechanism of action.
Significance: Naphthoquinones have potential as antibacterial compounds against TB.
Keywords: Antibiotics, Antibiotic Action, DNA Gyrase, DNA Topoisomerase, Mycobacterium tuberculosis, Tuberculosis, Diospyrin, Naphthoquinones
Abstract
Tuberculosis and other bacterial diseases represent a significant threat to human health. The DNA topoisomerases are excellent targets for chemotherapy, and DNA gyrase in particular is a well-validated target for antibacterial agents. Naphthoquinones (e.g. diospyrin and 7-methyljuglone) have been shown to have therapeutic potential, particularly against Mycobacterium tuberculosis. We have found that these compounds are inhibitors of the supercoiling reaction catalyzed by M. tuberculosis gyrase and other gyrases. Our evidence strongly suggests that the compounds bind to the N-terminal domain of GyrB, which contains the ATPase active site, but are not competitive inhibitors of the ATPase reaction. We propose that naphthoquinones bind to GyrB at a novel site close to the ATPase site. This novel mode of action could be exploited to develop new antibacterial agents.
Introduction
Infectious diseases caused by bacterial pathogens are a serious and growing problem. For example, tuberculosis (TB)4 is the most deadly infectious disease in the world with ∼2 billion people infected and >1 million deaths every year (1). Current treatments involve therapy over a long period (∼6 months), and there are serious problems with drug-resistant strains (multidrug-resistant TB and extensively drug resistant-TB). One strategy for developing new antibacterials is to seek new targets. However, despite more than a decade of research driven by genomics and proteomics, and enormous expenditure, very few promising new targets have emerged. It appears that Nature, through millions of years of evolutionary warfare, has already identified the “good bacterial targets,” which include the ribosome, cell wall synthesis enzymes, and DNA gyrase (2).
DNA gyrase is a DNA topoisomerase that is present in bacteria and plants, but not animals, and has been widely exploited as a target for antimicrobial chemotherapy (3, 4). DNA topoisomerases are enzymes that catalyze changes in the topology of DNA and are essential to all cells (5). They are classified into two types, depending upon whether their reactions involve transient single- (type I) or double- (type II) strand breaks in DNA. All topoisomerases can relax supercoiled DNA but gyrase, a type II enzyme, can also introduce negative supercoils in a reaction coupled to ATP hydrolysis (4, 5). Gyrase consists of two subunits, GyrA and GyrB, which form an A2B2 complex in the active enzyme in which a segment of DNA is wrapped around the protein. The GyrA subunit interacts with DNA and contains the active-site tyrosine responsible for DNA cleavage and the formation of a protein-DNA covalent bond during the reaction cycle. GyrB also interacts with DNA and contains the ATPase active site. Gyrase shares a number of features with other type II topoisomerases but is distinct in its ability to wrap DNA and harness the free energy of ATP hydrolysis to introduce negative supercoils into DNA. The uniqueness of gyrase has made it a successful target for antibacterial agents. Fluoroquinolones (e.g. ciprofloxacin, moxifloxacin) target gyrase and are highly successful clinical agents that have been used against tuberculosis. However, despite their efficacy, fluoroquinolone-resistant TB is a serious problem (6).
Most of our current information about DNA gyrase concerns the Escherichia coli enzyme, but it is clear that enzymes from other bacteria have important differences that need to be investigated to exploit their potential as drug targets for specific diseases. DNA gyrase from Mycobacterium tuberculosis has a number of distinct features that warrant investigation in their own right and that may be exploitable for the targeting of this enzyme (7, 8). For example, M. tuberculosis gyrase has been found to be a potent decatenase, in contrast to most other gyrases (8–10), reflecting the fact that M. tuberculosis lacks topo IV, which is the predominant decatenating enzyme in most bacteria (11). Recent advances in M. tuberculosis gyrase have included the structures of the N- and C-terminal domains of GyrA and the C-terminal domain of GyrB (12–14), the identification of DNA-binding residues in the C-terminal domain of GyrA (15), and the development of monoclonal antibodies that specifically target the enzyme as potential therapeutic agents (16). Recently a potential Ca2+-binding site has been identified in M. tuberculosis GyrA, which may have a regulatory role (17).
Extracts from plants used in traditional medicine provide a source for novel compounds that may have antibacterial properties. Lall and Meyer have analyzed the antibacterial properties of the South African tree Euclea natalensis. This plant, also known as the “toothbrush tree” (native South Africans use its twigs as toothbrushes), has been used extensively to treat a variety of medical complaints (e.g. bronchitis, pleurisy, venereal disease) and in oral health (18). Lall and Meyer showed that crude extracts from E. natalensis were active against drug-sensitive and drug-resistance strains of M. tuberculosis (19). They later reported that the active component in these extracts was the naphthoquinone diospyrin (Fig. 1) (20). Naphthoquinones are widely distributed in nature, and their presence in many plants is the basis for some folk medicines (21). They have been implicated in the treatment of a variety of diseases including urinary tract infections, trypanosome diseases, and tuberculosis (21, 22). Diospyrin, a bisnaphthoquinone (Fig. 1), has previously been found to be an inhibitor of DNA topoisomerase (topo) I from Leishmania donovani and can stabilize the topo I-DNA cleavage complex (23); isodiospyrin was found to be an inhibitor of human topo I, but did not stabilize the cleavage complex (24); 7-methlyjuglone was shown to be a subversive substrate for M. tuberculosis mycothiol disulfide reductase (25). In addition, there are a number of reports of quinolones interacting with eukaryotic type II topoisomerases (e.g. human topo IIα). For example, several naphthoquinones, including juglone, have been shown to inhibit topo II and stabilize the cleavage complex (26); these compounds react with thiol groups on the protein.
FIGURE 1.

Structures of naphthoquinones.
The lack of a clear target definition and the observation of the efficacy of diospyrin against drug-sensitive and drug-resistance strains of M. tuberculosis (19), prompted us to test this and other naphthoquinones against M. tuberculosis DNA gyrase. We have found that these compounds can inhibit gyrase and that they target the enzyme by a novel mechanism, raising the possibility of developing these compounds as potential anti-TB agents.
EXPERIMENTAL PROCEDURES
Enzymes and DNA
M. tuberculosis and E. coli gyrases and the N-terminal domain of E. coli GyrB (GyrB43) were prepared as described previously (17, 27, 28). Staphylococcus aureus WCUH29 gyrA, gyrB, parC, and parE genes in pET vectors were gifts from Hiroshi Hiasa (University of Minnesota). They were re-cloned into plasmid pET11 (Novagen) and the proteins expressed in E. coli Rosetta 2 (DE3) pLysS (Novagen). Both proteins were purified using an Äkta system (GE Healthcare) using Q-Sepharose, heparin-Sepharose, and phenyl-Sepharose columns to >95% purity.
Enzyme Assays
M. tuberculosis gyrase supercoiling, relaxation, and decatenation assays were carried out as described previously (17); cleavage assays were carried out as per relaxation assays (±ATP) except that following incubation at 37 °C, SDS and proteinase K were added (to 0.2% and 0.1 mg/ml, respectively) and the incubation was continued at 37 °C for 30 min before loading onto an agarose gel for analysis. E. coli gyrase supercoiling assays were performed as described (29). S. aureus gyrase and topo IV assays were carried out as described (30), apart from the buffer exchange step prior to electrophoresis, which was omitted; samples were loaded onto agarose gels and left for 30 min prior to electrophoresis to allow diffusion of salt. ATPase assays were performed using a linked assay as described (27), except that assays were carried out in microtiter plates, and A340 values were measured continuously using an absorbance plate reader. Limited proteolysis experiments were performed as described previously (17).
Mass Spectrometry and SPR
Nanoflow electrospray ionization mass spectrometry (nano-ESI MS) and surface plasmon resonance (SPR) experiments were performed as described previously (31, 32), with the exception that SPR data collection used a Biacore T-100 instrument (GE Healthcare), and regeneration was carried out with 4.5 mm sodium hydroxide. Protein and drug stock solutions were filtered through 0.1-μm spin filters (Merck Millipore) prior to injection over the chip surface to prevent damage to the microfluidics caused by insoluble aggregates. Data were analyzed was using the BIAEvaluation software and displayed using Sigma Plot (Systat Software Inc.).
RESULTS
Inhibition of DNA Gyrase by Naphthoquinones
To determine whether diospyrin and 7-methyljugolone might target M. tuberculosis DNA gyrase, we assessed their effect on the gyrase supercoiling reaction (Fig. 2). Both compounds inhibit this reaction with IC50 values of ∼15 μm (diospyrin) and ∼30 μm (7-methyljugolone); other naphthoquinones were also tested in this reaction (Table 1; the IC50 values of novobiocin and ciprofloxacin against M. tuberculosis gyrase are given for comparison). We tested diospyrin against M. tuberculosis gyrase in relaxation reactions and found that this reaction is also inhibited by this compound with an IC50 similar to that for supercoiling (supplemental Fig. S1). We also tested diospyrin and 7-methyljuglone for their ability to inhibit DNA supercoiling by E. coli and S. aureus gyrases. We found that both of these enzymes were inhibited by these compounds with approximate IC50 values as follows: E. coli, 4 μm (diospyrin) and 30 μm (7-methyljuglone); S. aureus, 8 μm (diospyrin) and 60 μm (7-methyljuglone) (Table 2 and supplemental Fig. S2). As our assays contained 4 mm DTT (i.e. reducing conditions), and it has been reported that quinone inhibitors of topo II are less active under reducing conditions (33), we attempted to carry out experiments without reducing agents. However, we found that M. tuberculosis gyrase was inactive in the absence of reducing agents and even with 50 μm DTT showed no activity (data not shown). M. tuberculosis gyrase supercoiling assays carried out with diospyrin and 7-methyljugone in the presence of 2 mm β-mercaptoethanol gave results similar to those carried out with 4 mm DTT. In contrast we have found that S. aureus gyrase is active in the presence of a low concentration of DTT (50 μm), and we found inhibition by diospyrin and 7-methyljuglone similar to that we had found at 4 mm DTT (data not shown). Taken together, it seems that there is no significant effect of reducing agents on the inhibitory effects of diospyrin and 7-methyljuglone for gyrase.
FIGURE 2.
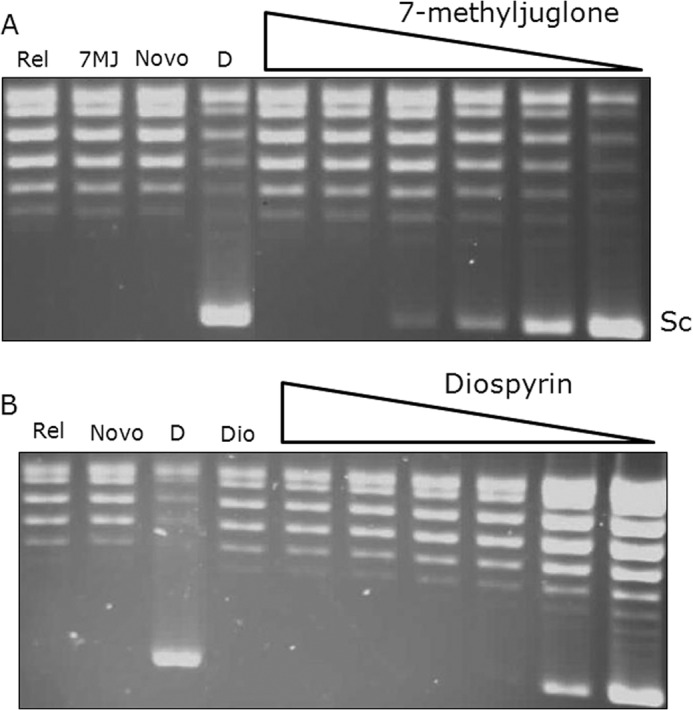
Inhibition of M. tuberculosis gyrase supercoiling by 7-methyljuglone and diospyrin. A, inhibition of supercoiling in the presence of 7-methyljuglone (1, 10, 30, 50, 100, 200 μm); Rel is relaxed DNA; 7MJ is 7-methyljuglone (no enzyme); Novo is gyrase in the presence of 5 μm novobiocin; D is gyrase in the presence of 3% dimethyl sulfoxide; Sc indicates the position of the supercoiled DNA band. All 7-methyljuglone samples also contained 3% dimethyl sulfoxide. B, inhibition of supercoiling in the presence of diospyrin (concentrations as above).
TABLE 1.
IC50 values for the inhibition of supercoiling by M. tuberculosis gyrase by naphthoquinones
| Compound | IC50 |
|---|---|
| μm | |
| Diospyrin | 15 |
| 7-Methyljuglone | 30 |
| Neodiospyrin | 50 |
| Isodiospyrin | 100 |
| Menadione | >200 |
| Shinanolone | >200 |
| Ciprofloxacin | 10 |
| Novobiocin | 1 |
TABLE 2.
Inhibition of DNA gyrases from S. aureus, E. coli, and M. tuberculosis by diospyrin and 7-methyljuglone
| Compound | IC50 for gyrase supercoiling inhibition |
||
|---|---|---|---|
| S. aureus | E. coli | M. tuberculosis | |
| μm | μm | μm | |
| Diospyrin | 8 | 4 | 15 |
| 7-Methyljuglone | 60 | 30 | 30 |
DNA topoisomerase IV (topo IV) is also the target of a number of antibacterial agents (34, 35), however M. tuberculosis lacks this enzyme (36); M. tuberculosis gyrase has evolved to carry out activities catalyzed by gyrase and topo IV in other bacteria. We tested naphthoquinones against S. aureus topo IV and found that in general naphthoquinones favored gyrase over topo IV, for example the IC50 for diospyrin is 4–8 times higher for S. aureus topo IV than for gyrase (supplemental Table S1).
The success of fluoroquinolones, such as ciprofloxacin, as antibacterial agents is due to the fact that they stabilize the cleavage complex between gyrase and DNA (37–39), in which the GyrA protein is covalently attached to the 5′-end of the cleaved DNA. We tested diospyrin and 7-methyljuglone in cleavage-complex assays under conditions (±ATP) where ciprofloxacin stabilizes the cleavage complex between M. tuberculosis gyrase and DNA. We found no evidence of stabilization of the gyrase-DNA cleavage complex by these agents (data not shown); this is in contrast with results obtained with naphthoquinones (e.g. juglone) and topo II (26).
The fact that diospyrin inhibits ATP-independent relaxation by gyrase suggests that naphthoquinones are unlikely to be ATPase inhibitors. We tested diospyrin in gyrase supercoiling reactions at a range of ATP concentrations (Fig. 3). (This approach has been used previously to show that simocyclinones are not inhibitors of the ATPase reaction of gyrase (32).) We found that whereas inhibition by novobiocin (a well-established inhibitor of the gyrase ATPase reaction) could be abrogated by ATP, inhibition by diospyrin and simocyclinone could not, supporting the contention that diospyrin is not a competitive inhibitor of the gyrase ATPase reaction. We directly tested the effect of diospyrin on the ATPase reaction of M. tuberculosis GyrB, but we found that the intrinsic ATPase activity of this protein to be very low, making this experiment problematic (data not shown). However, we found that the ATPase activity of S. aureus GyrB could be measured more readily and showed that, whereas this reaction is completely inhibited by novobiocin, diospyrin could only inhibit the reaction by ∼50% (Fig. 4); similar results were obtained with S. aureus GyrB and 7-methyljuglone (data not shown). For novobiocin, the dependence of rate on inhibitor concentration was sigmoidal, so the data were fitted to a scheme involving the binding of two ligands to the enzyme to achieve inhibition; in the case of diospyrin the data showed a hyperbolic dependence on inhibitor concentration consistent with the binding of one ligand molecule/enzyme to achieve inhibition (Fig. 4). These data suggest that diospyrin is not a competitive inhibitor of the ATPase reaction of GyrB, but that its site of action is likely to be within the GyrB N-terminal domain and can allosterically affect the ATPase reaction (see below).
FIGURE 3.
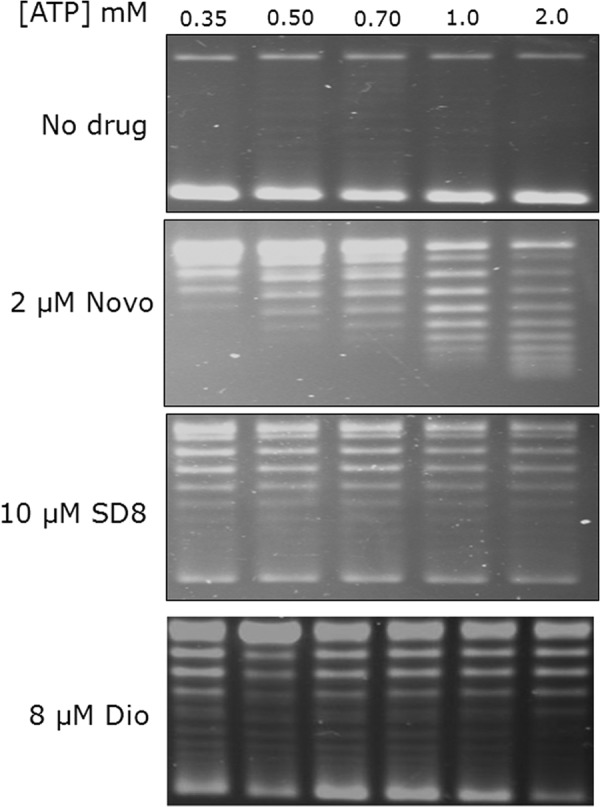
Effect of ATP on the inhibition of M. tuberculosis gyrase by novobiocin, simocyclinone, and diospyrin. The indicated concentrations of ATP were added to M. tuberculosis gyrase supercoiling reactions containing the indicated amounts of novobiocin (Novo), simocyclinone D8 (SD8), and diospyrin (Dio).
FIGURE 4.
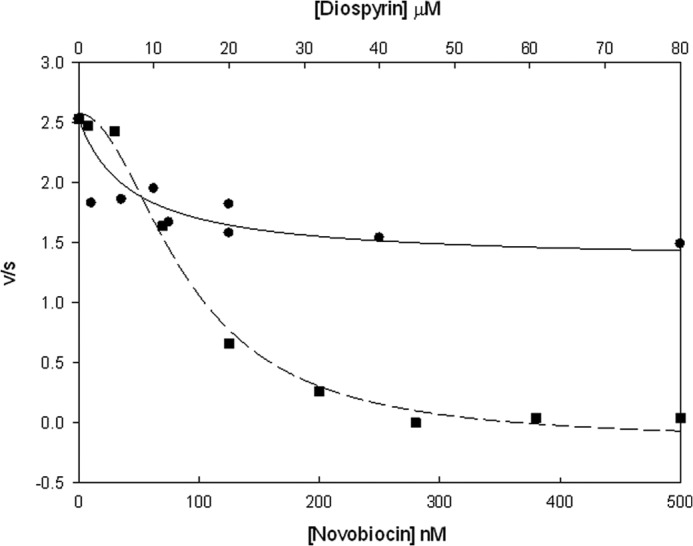
Inhibition of S. aureus GyrB ATPase by novobiocin and diospyrin. The rates of ATP hydrolysis (s−1) by GyrB (0.6 μm) in the presence of novobiocin (squares) and diospyrin (circles) were determined. Data were fitted using SigmaPlot 12.3 to schemes involving the binding of two ligands to inhibit ATPase activity (novobiocin) or a single ligand (diospyrin).
The Binding Site of Diospyrin Is in GyrB
The biochemical data presented above suggest that diospyrin can inhibit the topoisomerase reaction of gyrase and that it may act by binding to the GyrB subunit. We used three further approaches to explore this. In previous work we have used limited tryptic digestion to probe ligand-binding sites in gyrase (17, 40–42). Limited digestion of M. tuberculosis GyrA with trypsin revealed little or no difference with and without diospyrin (supplemental Fig. S3A), whereas differences were observed in the case of GyrB (supplemental Fig. S3B). In the case of E. coli GyrB43 (the N-terminal domain of GyrB), significant differences in the digestion pattern were also observed with and without diospyrin (supplemental Fig. S3C). These data support the idea that diospyrin may bind to the N-terminal domain of GyrB, which houses the ATPase active site (27, 44).
Nano-ESI MS has been used successfully to analyze the noncovalent interaction between proteins and ligands (45). Recently, we have used this method to characterize the interaction between the N-terminal domain of E. coli GyrA and the antibiotic simocyclinone D8 (29, 31). We have now analyzed the interaction between diospyrin and gyrase by this method (Fig. 5; note that all nano-ESI MS experiments were performed in the absence of DTT). We found no evidence for interaction between diospyrin and M. tuberculosis GyrA (data not shown), but spectra of samples with M. tuberculosis GyrB and diospyrin showed additional peaks consistent with a complex between the two (Fig. 5A). In each spectrum two sets of charged species are apparent (indicated by red and yellow circles), which correspond to the GyrB monomer: observed molecular mass = 79,876 ± 7 Da (calculated = 79,263) and a lower molecular mass contaminant, which is likely to be a truncated form of GyrB. In the spectra in the presence of diospyrin, additional species are present (red stars), which correspond to the GyrB monomer with bound ligand: observed molecular masses = 80,381 ± 18 Da and 80,320 ± 6 Da (the molecular mass of diospyrin is 374 Da). E. coli GyrB43 shows three different charged species the largest of which corresponds to the GyrB43 monomer (observed molecular mass = 43,272 ± 2 Da (calculated = 43,024); the other two minor species are likely to correspond to protein fragments or contaminants. In the presence of diospyrin a further species (MW 43,313 ± 10 Da) appears corresponding to the GyrB43 monomer with bound diospyrin (Fig. 5B). No evidence for interaction between diospyrin and the C-terminal domain of E. coli GyrB (GyrB47) was found (supplemental Fig. S4). Taken together, it seems very likely that the binding site for diospyrin lies within the N-terminal domain of GyrB.
FIGURE 5.

Mass spectrometry analysis of the binding of diospyrin to GyrB. A: top, M. tuberculosis GyrB (red and yellow circles indicate two different charged species); middle, M. tuberculosis GyrB with 8-fold excess diospyrin (red stars indicate species containing diospyrin); bottom, M. tuberculosis GyrB with 16-fold excess diospyrin. B: top, E. coli GyrB43 (N-terminal domain; red, yellow, and blue circles indicate three different charged species); bottom, E. coli GyrB43 with 16-fold excess diospyrin (red stars indicate species containing diospyrin).
To further evaluate the GyrB-diospyrin interaction, SPR was employed. Control experiments showed that novobiocin bound to E. coli GyrB43 and M. tuberculosis GyrB with equilibrium binding constant (KD) values of 10−8 to 10−7 m (supplemental Fig. S5), consistent with earlier SPR studies on the interaction of this drug with E. coli GyrB (46). In experiments with diospyrin, we observed a specific interaction with E. coli GyrB43 (Fig. 6), with an upper limit for the KD determined to be 10−4 m. Further refinement of this value was impractical due to the tendency of diospyrin to aggregate on the E. coli GyrB chip surface at high concentrations (>50 μm; supplemental Fig. S5).
FIGURE 6.

SPR analysis of the binding of diospyrin to E. coli GyrB43. Sensograms showing the interaction of diospyrin at selected concentrations (5 (black), 15 (red), 25 (blue), and 45 (green) μm) with E. coli GyrB43.
From these data it seems very likely that the binding site for diospyrin lies in the N-terminal domain of GyrB, but does not overlap the ATP-binding site. This suggests a mode of interaction that is distinct from that of other known gyrase inhibitors.
DISCUSSION
There is a significant and immediate threat from infections due to pathogenic bacteria that is exacerbated by the prevalence of antibiotic-resistant bacteria and diminution in the drug-discovery efforts of large pharmaceutical companies (47, 48). This means that there is an urgent need for new compounds with antibacterial potential. Plants are a rich and largely untapped source of antibacterials (49–51), although plant extracts have been used medicinally for centuries. The toothbrush tree (E. natalensis), for example, has been traditionally utilized for its medicinal properties by native South Africans: the roots are chewed for oral hygiene and for the treatment of bronchitis, pleurisy, and chronic asthma (18). Extracts from this plant have been found to show antibacterial activities, particularly against Gram-positive organisms (18), including M. tuberculosis (19). Diospyrin isolated from E. natalensis was shown to have activity against drug-sensitive and drug-resistant M. tuberculosis strains (20). Other naphthoquinones were also tested against M. tuberculosis strains and showed the following MIC values (in order of potency): 7-methyljuglone (0.5 μg/ml; 2.6 μm), diospyrin (8 μg/ml; 21.4 μm), isodiospyrin (10 μg/ml; 26.7 μm), and neodiospyrin (10 μg/ml; 26.7 μm). However, the in vivo target for these compounds is not clear, with 7-methyljuglone having been identified as a subversive substrate for mycothiol disulfide reductase (25), diospyrin found to be an inhibitor of topo I from L. donovani (23), and isodiospyrin found to be an inhibitor of human topo I (24). In an attempt to address this issue, we have tested diospyrin, 7-methyljgulone, and other naphthoquinones against gyrases from M. tuberculosis, S. aureus, and E. coli.
We have found that diospyrin and other naphthoquinones are inhibitors of the DNA supercoiling reaction of DNA gyrase with IC50 values that are comparable with the reported MICs. For example, we found the IC50 value for diospyrin against M. tuberculosis gyrase to be ∼15 μm, which compares with an MIC of ∼20 μm (8 μg/ml). Interestingly diospyrin also inhibits the DNA relaxation reaction of gyrase despite our evidence suggesting binding to the ATPase domain of GyrB, which is not required for relaxation activity. Our ATPase experiments suggest that diospyrin is an allosteric inhibitor of this reaction, and we propose that it binds to the N-terminal domain of GyrB close to but not overlapping with the ATPase active site. Its mode of action may involve stabilization of a conformation of the N-terminal domain of GyrB that prevents strand passage and thus inhibits both supercoiling and relaxation by gyrase. This mode of action is similar to that proposed by Bender et al. (52) for the action of the quinone metabolites of polychlorinated biphenyls on human topo IIα. In the case of the action of naphthoquinones on gyrase, the mechanism would need to involve binding of the inhibitor to the GyrB-N-terminal domain in a manner that would prevent strand passage in either direction and not just prevent clamp closure.
The work in this paper provides evidence that gyrase is a target for naphthoquinones. However, we have not proven that it is the primary in vivo target in the antibacterial action of these compounds. One way to show this would be to isolate target-based naphthoquinone-resistant mutations and to establish a correlation between potency and target mutations. Our efforts to isolate such mutations in E. coli and Mycobacterium smegmatis have so far been unsuccessful. Most gyrase inhibitors found so far either stabilize the DNA cleavage complex by binding to a pocket comprising residues from GyrA, GyrB, and DNA (such as quinolones), or bind to the ATPase site (such as aminocoumarins) (3). Diospyrin does not appear to emulate either of these modes of action but would appear to bind to a novel binding pocket in the N-terminal domain of GyrB, i.e. it has a completely new mode of action.
Given that naphthoquinones and other quinone compounds have been found to inhibit eukaryotic topo II, it is interesting to compare their modes of action with that of diospyrin on gyrase. Several naphthoquinones, including juglone, have been shown to inhibit topo II and stabilize the cleavage complex (26); these compounds react with thiol groups on the protein. Lindsey et al. (33) found that 1,4-benzoquinone is a strong cleavage-complex stimulator and that the presence of reducing agents led to a reduction in potency, suggesting that that the oxidized form of the compound was the active species. Further, benzoquinone was found to form adducts with Cys residues within the ATPase domain of topo II (Cys-392 and Cys-405 of human topo IIα); mutation of these residues to Ala led to 50% reduction in the potency of the compound (53). More recently, Jacob et al. showed that etoposide quinone, a metabolite of etoposide (itself a cleavage-complex inhibitor of topo II (43)), is effective at stabilizing the cleavage complex under nonreducing conditions. Taken together, this work suggests that compounds of this class are cleavage-complex-stabilizing inhibitors of eukaryotic topo II that may act via the formation of adducts with Cys residues in the N terminus (ATPase) domain, although it is unclear as to why mutation of the two key Cys residues in human topo IIα only leads to a ∼50% drop in potency.
In comparison, our experiments do not support a similar mode of action. Our results suggest that the naphthoquinone-binding site is within the N-terminal domain of GyrB, but as one of the enzymes examined (S. aureus gyrase) lacks Cys residues in the ATPase domain, it is unlikely that covalent adducts with Cys residues form part of the mode of binding. Moreover, in a control experiment, using mass spectrometry, we found no evidence of adduct formation with M. tuberculosis gyrase, and, in particular, the Cys residue in the N-terminal domain is unmodified. In addition, we have also found no evidence of diospyrin and other naphthoquinones stabilizing the gyrase cleavage complex.
A structure of the diospyrin-GyrB complex would clearly be of great value, but, so far, crystallization trials with diospyrin and GyrB and fragments thereof have not yet yielded crystals suitable for diffraction studies. Identification of the naphthoquinone-binding pocket in GyrB would potentiate the development of these compounds as potential anti-TB agents and the design of other molecules that exploit this mode of action.
Acknowledgments
We thank Chris Hamilton (University of East Anglia) and Nick Burton (Inspiralis Ltd.) for providing materials and useful advice, and Anisha Das for contributions to these experiments.
This work was supported by an Early Stage Research Training Scholarship (EU, FP7), by Grant BB/J004561/1 from the Biotechnology and Biological Sciences Research Council (UK) and the John Innes Foundation, and by the National Research Foundation of South Africa.

This article contains supplemental Figs. S1–S5, Table S1, and additional references.
- TB
- tuberculosis
- MIC
- minimum inhibitory concentration
- nano-ESI
- nanoflow electrospray ionization
- SPR
- surface plasmon resonance
- topo
- topoisomerase.
REFERENCES
- 1. Young D. B., Perkins M. D., Duncan K., Barry C. E., 3rd (2008) Confronting the scientific obstacles to global control of tuberculosis. J. Clin. Invest. 118, 1255–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Walsh C. (2000) Molecular mechanisms that confer antibacterial drug resistance. Nature 406, 775–781 [DOI] [PubMed] [Google Scholar]
- 3. Collin F., Karkare S., Maxwell A. (2011) Exploiting bacterial DNA gyrase as a drug target: current state and perspectives. Appl. Microbiol. Biotechnol. 92, 479–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nöllmann M., Crisona N. J., Arimondo P. B. (2007) Thirty years of Escherichia coli DNA gyrase: from in vivo function to single-molecule mechanism. Biochimie 89, 490–499 [DOI] [PubMed] [Google Scholar]
- 5. Bates A. D., Maxwell A. (2005) DNA Topology, Oxford University Press, Oxford [Google Scholar]
- 6. Ginsberg A. M., Spigelman M. (2007) Challenges in tuberculosis drug research and development. Nat. Med. 13, 290–294 [DOI] [PubMed] [Google Scholar]
- 7. Mdluli K., Ma Z. (2007) Mycobacterium tuberculosis DNA gyrase as a target for drug discovery. Infect. Disord. Drug Targets 7, 159–168 [DOI] [PubMed] [Google Scholar]
- 8. Aubry A., Fisher L. M., Jarlier V., Cambau E. (2006) First functional characterization of a singly expressed bacterial type II topoisomerase: the enzyme from Mycobacterium tuberculosis. Biochem. Biophys. Res. Commun. 348, 158–165 [DOI] [PubMed] [Google Scholar]
- 9. Manjunatha U. H., Dalal M., Chatterji M., Radha D. R., Visweswariah S. S., Nagaraja V. (2002) Functional characterisation of mycobacterial DNA gyrase: an efficient decatenase. Nucleic Acids Res. 30, 2144–2153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Manjunatha U. H., Madhusudan K., Unniraman S., Sikder D., Chatterjee M., Bhaduri T., Radha D. R., Nagaraja V. (2006) DNA topoisomerases from Mycobacterium tuberculosis and Mycobacterium smegmatis. J. Ind. Inst. Sci. 86, 751–761 [Google Scholar]
- 11. Zechiedrich E. L., Cozzarelli N. R. (1995) Roles of topoisomerase IV and DNA gyrase in DNA unlinking during replication in Escherichia coli. Genes Dev. 9, 2859–2869 [DOI] [PubMed] [Google Scholar]
- 12. Tretter E. M., Schoeffler A. J., Weisfield S. R., Berger J. M. (2010) Crystal structure of the DNA gyrase GyrA N-terminal domain from Mycobacterium tuberculosis. Proteins 78, 492–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fu G., Wu J., Liu W., Zhu D., Hu Y., Deng J., Zhang X. E., Bi L., Wang D. C. (2009) Crystal structure of DNA gyrase B′ domain sheds lights on the mechanism for T-segment navigation. Nucleic Acids Res. 37, 5908–5916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tretter E. M., Berger J. M. (2012) Mechanisms for defining supercoiling set point of DNA gyrase orthologs. II. The shape of the GyrA subunit C-terminal domain (CTD) is not a sole determinant for controlling supercoiling efficiency. J. Biol. Chem. 287, 18645–18654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huang Y. Y., Deng J. Y., Gu J., Zhang Z. P., Maxwell A., Bi L. J., Chen Y. Y., Zhou Y. F., Yu Z. N., Zhang X. E. (2006) The key DNA-binding residues in the C-terminal domain of Mycobacterium tuberculosis DNA gyrase A subunit (GyrA). Nucleic Acids Res. 34, 5650–5659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Manjunatha U. H., Maxwell A., Nagaraja V. (2005) A monoclonal antibody that inhibits mycobacterial DNA gyrase by a novel mechanism. Nucleic Acids Res. 33, 3085–3094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Karkare S., Yousafzai F., Mitchenall L. A., Maxwell A. (2012) The role of Ca2+ in the activity of Mycobacterium tuberculosis DNA gyrase. Nucleic Acids Res. 40, 9774–9787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lall N., Meyer J. J. (2000) Antibacterial activity of water and acetone extracts of the roots of Euclea natalensis. J. Ethnopharmacol. 72, 313–316 [DOI] [PubMed] [Google Scholar]
- 19. Lall N., Meyer J. J. (1999) In vitro inhibition of drug-resistant and drug-sensitive strains of Mycobacterium tuberculosis by ethnobotanically selected South African plants. J. Ethnopharmacol. 66, 347–354 [DOI] [PubMed] [Google Scholar]
- 20. Lall N., Meyer J. J. (2001) Inhibition of drug-sensitive and drug-resistant strains of Mycobacterium tuberculosis by diospyrin, isolated from Euclea natalensis. J. Ethnopharmacol. 78, 213–216 [DOI] [PubMed] [Google Scholar]
- 21. Pinto A. V., de Castro S. L. (2009) The trypanocidal activity of naphthoquinones: a review. Molecules 14, 4570–4590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. van der Kooy F., Meyer J. J., Lall N. (2006) Antimycobacterial activity and possible mode of action of newly isolated neodiospyrin and other naphthoquinones from Euclea natalensis. S. Afr. J. Botany 72, 349–352 [Google Scholar]
- 23. Ray S., Hazra B., Mittra B., Das A., Majumder H. K. (1998) Diospyrin, a bisnaphthoquinone: a novel inhibitor of type I DNA topoisomerase of Leishmania donovani. Mol. Pharmacol. 54, 994–999 [DOI] [PubMed] [Google Scholar]
- 24. Ting C. Y., Hsu C. T., Hsu H. T., Su J. S., Chen T. Y., Tarn W. Y., Kuo Y. H., Whang-Peng J., Liu L. F., Hwang J. (2003) Isodiospyrin as a novel human DNA topoisomerase I inhibitor. Biochem. Pharmacol. 66, 1981–1991 [DOI] [PubMed] [Google Scholar]
- 25. Mahapatra A., Mativandlela S. P., Binneman B., Fourie P. B., Hamilton C. J., Meyer J. J., van der Kooy F., Houghton P., Lall N. (2007) Activity of 7-methyljuglone derivatives against Mycobacterium tuberculosis and as subversive substrates for mycothiol disulfide reductase. Bioorg. Med. Chem. 15, 7638–7646 [DOI] [PubMed] [Google Scholar]
- 26. Wang H., Mao Y., Chen A. Y., Zhou N., LaVoie E. J., Liu L. F. (2001) Stimulation of topoisomerase II-mediated DNA damage via a mechanism involving protein thiolation. Biochemistry 40, 3316–3323 [DOI] [PubMed] [Google Scholar]
- 27. Ali J. A., Jackson A. P., Howells A. J., Maxwell A. (1993) The 43-kDa N-terminal fragment of the gyrase B protein hydrolyses ATP and binds coumarin drugs. Biochemistry 32, 2717–2724 [DOI] [PubMed] [Google Scholar]
- 28. Maxwell A., Howells A. J. (1999) in DNA Topoisomerase Protocols. I. DNA Topology and Enzymes (Bjornsti M.-A., Osheroff N., eds) pp. 135–144, Humana Press, Totowa, NJ [Google Scholar]
- 29. Edwards M. J., Flatman R. H., Mitchenall L. A., Stevenson C. E., Le T. B., Fiedler H. P., McKay A. R., Clarke T. A., Buttner M. J., Lawson D. M., Maxwell A. (2009) A crystal structure of the bifunctional antibiotic, simocyclinone D8, bound to DNA gyrase. Science 326, 1415–1418 [DOI] [PubMed] [Google Scholar]
- 30. Alt S., Mitchenall L. A., Maxwell A., Heide L. (2011) Inhibition of DNA gyrase and DNA topoisomerase IV of Staphylococcus aureus and Escherichia coli by aminocoumarin antibiotics. J. Antimicrob. Chemother. 66, 2061–2069 [DOI] [PubMed] [Google Scholar]
- 31. Edwards M. J., Williams M. A., Maxwell A., McKay A. R. (2011) Mass spectrometry reveals that the antibiotic simocyclinone D8 binds to DNA gyrase in a “bent-over” conformation: evidence of positive cooperativity in binding. Biochemistry 50, 3432–3440 [DOI] [PubMed] [Google Scholar]
- 32. Flatman R. H., Howells A. J., Heide L., Fiedler H.-P., Maxwell A. (2005) Simocyclinone D8: an inhibitor of DNA gyrase with a novel mode of action. Antimicrob. Agents Chemother. 49, 1093–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lindsey R. H., Jr., Bromberg K. D., Felix C. A., Osheroff N. (2004) 1,4-Benzoquinone is a topoisomerase II poison. Biochemistry 43, 7563–7574 [DOI] [PubMed] [Google Scholar]
- 34. Pan X.-S., Ambler J., Mehtar S., Fisher L. M. (1996) Involvement of topoisomerase IV and DNA gyrase as ciprofloxacin targets in Streptococcus pneumoniae. Antimicrob. Agents Chemother. 40, 2321–2326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Khodursky A. B., Zechiedrich E. L., Cozzarelli N. R. (1995) Topoisomerase IV is a target of quinolones in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 92, 11801–11805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cole S. T., Brosch R., Parkhill J., Garnier T., Churcher C., Harris D., Gordon S. V., Eiglmeier K., Gas S., Barry C. E., 3rd, Tekaia F., Badcock K., Basham D., Brown D., Chillingworth T., Connor R., Davies R., Devlin K., Feltwell T., Gentles S., Hamlin N., Holroyd S., Hornsby T., Jagels K., Krogh A., McLean J., Moule S., Murphy L., Oliver K., Osborne J., Quail M. A., Rajandream M. A., Rogers J., Rutter S., Seeger K., Skelton J., Squares R., Squares S., Sulston J. E., Taylor K., Whitehead S., Barrell B. G. (1998) Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544 [DOI] [PubMed] [Google Scholar]
- 37. Gellert M., Mizuuchi K., O'Dea M. H., Itoh T., Tomizawa J. (1977) Nalidixic acid resistance: a second genetic character involved in DNA gyrase activity. Proc. Natl. Acad. Sci. U.S.A. 74, 4772–4776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sugino A., Peebles C. L., Kreuzer K. N., Cozzarelli N. R. (1977) Mechanism of action of nalidixic acid: purification of Escherichia coli nalA gene product and its relationship to DNA gyrase and a novel nicking-closing enzyme. Proc. Natl. Acad. Sci. U.S.A. 74, 4767–4771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Willmott C. J., Maxwell A. (1993) A single point mutation in the DNA gyrase A protein greatly reduces the binding of fluoroquinolones to the gyrase-DNA complex. Antimicrob. Agents Chemother. 37, 126–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gormley N. A., Orphanides G., Meyer A., Cullis P. M., Maxwell A. (1996) The interaction of coumarin antibiotics with fragments of the DNA gyrase B protein. Biochemistry 35, 5083–5092 [DOI] [PubMed] [Google Scholar]
- 41. Kampranis S. C., Maxwell A. (1998) Conformational changes in DNA gyrase revealed by limited proteolysis. J. Biol. Chem. 273, 22606–22614 [DOI] [PubMed] [Google Scholar]
- 42. Kampranis S. C., Howells A. J., Maxwell A. (1999) The interaction of DNA gyrase with the bacterial toxin CcdB: evidence for the existence of two gyrase-CcdB complexes. J. Mol. Biol. 293, 733–744 [DOI] [PubMed] [Google Scholar]
- 43. Jacob D. A., Mercer S. L., Osheroff N., Deweese J. E. (2011) Etoposide quinone is a redox-dependent topoisomerase II poison. Biochemistry 50, 5660–5667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wigley D. B., Davies G. J., Dodson E. J., Maxwell A., Dodson G. (1991) Crystal structure of an N-terminal fragment of the DNA gyrase B protein. Nature 351, 624–629 [DOI] [PubMed] [Google Scholar]
- 45. Loo J. A. (2000) Electrospray ionization mass spectrometry: a technology for studying noncovalent macromolecular complexes. Int. J. Mass Spectrom. 200, 175–186 [Google Scholar]
- 46. Kampranis S. C., Gormley N. A., Tranter R., Orphanides G., Maxwell A. (1999) Probing the binding of coumarins and cyclothialidines to DNA gyrase. Biochemistry 38, 1967–1976 [DOI] [PubMed] [Google Scholar]
- 47. Wright G. D. (2011) Molecular mechanisms of antibiotic resistance. Chem. Comm. 47, 4055–4061 [DOI] [PubMed] [Google Scholar]
- 48. Livermore D. M. (2009) Has the era of untreatable infections arrived? J. Antimicrob. Chemother. 64, i29–36 [DOI] [PubMed] [Google Scholar]
- 49. Lewis K., Ausubel F. M. (2006) Prospects for plant-derived antibacterials. Nat. Biotechnol. 24, 1504–1507 [DOI] [PubMed] [Google Scholar]
- 50. Sibanda T., Okoh A. I. (2007) The challenges of overcoming antibiotic resistance: plant extracts as potential sources of antimicrobial and resistance modifying agents. Afr. J. Biotechnol. 6, 2886–2896 [Google Scholar]
- 51. Cowan M. M. (1999) Plant products as antimicrobial agents. Clin. Microbiol. Rev. 12, 564–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bender R. P., Lehmler H. J., Robertson L. W., Ludewig G., Osheroff N. (2006) Polychlorinated biphenyl quinone metabolites poison human topoisomerase IIα: altering enzyme function by blocking the N-terminal protein gate. Biochemistry 45, 10140–10152 [DOI] [PubMed] [Google Scholar]
- 53. Bender R. P., Ham A. J., Osheroff N. (2007) Quinone-induced enhancement of DNA cleavage by human topoisomerase IIα: adduction of cysteine residues 392 and 405. Biochemistry 46, 2856–2864 [DOI] [PMC free article] [PubMed] [Google Scholar]