

Background: Nascent VLDL exits the ER in a specialized large vesicle, the VTV.
Results: CideB interacts with COPII proteins, and CideB ablation abrogates VTV biogenesis.
Conclusion: CideB forms an intricate COPII coat and regulates the formation of the VTV.
Significance: New physiological role of CideB provides new insight into the mechanism that controls intracellular VLDL trafficking and secretion.
Keywords: Apolipoproteins, Endoplasmic Reticulum (ER), Lipoprotein, Transport, Vesicles, VLDL Transport Vesicle, CideB, Coat Complex II, Very Low Density Lipoprotein
Abstract
Nascent very low density lipoprotein (VLDL) exits the endoplasmic reticulum (ER) in a specialized ER-derived vesicle, the VLDL transport vesicle (VTV). Similar to protein transport vesicles (PTVs), VTVs require coat complex II (COPII) proteins for their biogenesis from the ER membranes. Because the size of the VTV is large, we hypothesized that protein(s) in addition to COPII components might be required for VTV biogenesis. Our proteomic analysis, supported by Western blotting data, shows that a 26-kDa protein, CideB, is present in the VTV but not in other ER-derived vesicles such as PTV and pre-chylomicron transport vesicle. Western blotting and immunoelectron microscopy analyses suggest that CideB is concentrated in the VTV. Our co-immunoprecipitation data revealed that CideB specifically interacts with VLDL structural protein, apolipoprotein B100 (apoB100), but not with albumin, a PTV cargo protein. Confocal microscopic data indicate that CideB co-localizes with apoB100 in the ER. Additionally, CideB interacts with COPII components, Sar1 and Sec24. To investigate the role of CideB in VTV biogenesis, we performed an in vitro ER budding assay. We show that the blocking of CideB inhibits VTV budding, indicating a direct requirement of CideB in VTV formation. To confirm our findings, we knocked down CideB in primary hepatocytes and isolated ER and cytosol to examine whether they support VTV budding. Our data suggest that CideB knockdown significantly reduces VTV biogenesis. These findings suggest that CideB forms an intricate COPII coat and regulates the VTV biogenesis.
Introduction
Intracellular transport of newly synthesized lipoproteins from the ER2 to the Golgi is of utmost importance because abnormalities associated with this transport step lead to the pathogenesis of various metabolic diseases (1, 2). The liver and the small intestine are two organs that primarily produce lipoproteins: VLDL and chylomicrons, respectively. In liver, the biogenesis of VLDLs occurs in the ER, and this process is facilitated by microsomal triglyceride transfer protein (3–9). Once synthesized in the ER lumen, nascent VLDLs are exported to the Golgi, where several essential modifications occur to VLDL particles (10–14). Their structural protein, apolipoprotein B100 (apoB100), gets further glycosylated and phosphorylated (12–14). Moreover, it has been proposed that additional triglycerides are added to the nascent VLDL in the Golgi lumen (11, 15–17). Based on a number of biochemical and histological data, it has been suggested that Golgi is the site of VLDL maturation; however, this supposition is still a subject of debate (11–20). Regardless of their maturation site, the transport of nascent VLDL particles from the ER to the Golgi is imperative and determines the rate of VLDL secretion from the liver (10).
Transport of nascent VLDL from the ER is mediated by a specialized vesicle, the VTV, which buds off the hepatic ER membranes (21). Although these vesicles have been shown to be morphologically and biochemically different from classical ER-derived PTVs, their biogenesis requires COPII proteins (22–28). COPII proteins consist of five cytosolic proteins Sar1, Sec23-Sec24, and Sec13-Sec31 (29, 30). These five proteins have been shown to be sufficient to select cargo proteins and facilitate vesicle formation from the ER membrane. Sar1, a COPII component that initiates vesicle biogenesis, has two mammalian homologs: Sar1a and Sar1b (31). Several studies have shown that Sar1b is involved in lipoprotein trafficking and secretion because mutations in Sar1b lead to chylomicron retention disease (32–36). The definitive role of Sar1 in VLDL exit from the hepatic ER has been evident from our data and those of others (15, 21). Using H89, an inhibitor of Sar1 recruitment to the ER membrane, we found a significant reduction in VTV formation (21). Overexpression of dominant negative Sar1 (Sar1T39N) in rat hepatoma cells significantly blocks ER-to-Golgi transport of apoB100, a core component of VLDL (15).
That both nascent VLDL and secretory proteins exit the same ER in two different kinds of vesicles that require the same initiator for their genesis, Sar1, and other COPII components, has been shown in many studies (15, 21). These observations, however, raise the possibility of potential involvement of an additional factor(s) that is required for either cargo selection or vesicle biogenesis. It has been demonstrated that several cargoes require additional cytosolic proteins for their exit from the ER (37–46). For example, the formation of pre-chylomicron transport vesicle (PCTV) from the intestinal ER requires four cytosolic proteins in addition to five COPII proteins (43). These nine proteins form a prebudding complex that leads to the formation of the PCTVs, which contain pre-chylomicrons and are able to fuse with and deliver their cargo to the Golgi lumen (43, 44). It is possible that the generation of PCTV requires extra proteins because of its very large size and that these additional proteins facilitate the formation of a larger cage. However, the requirement of protein(s) other than COPII is not limited to larger cargoes or their carrier vesicles; smaller cargoes such as amyloid precursor protein need additional cytosolic factor(s) for their exit from the ER (47). These findings suggest that proteins other than COPII are required either for sorting and packaging of specific cargoes into vesicles or for the genesis of specialized vesicles that transport specific cargoes to the Golgi.
Because the size of the VTV (100–120 nm) is larger than the size of a normal COPII vesicle (∼55–70 nm), it is likely that generation of the VTV requires supplementary protein(s). We recently carried out a detailed proteomic analysis of the VTV to find out proteins present exclusively in VTV (48). Of several important proteins, which are not present in other ER-derived vesicles such as PCTV and PTV, one protein was identified as CideB (cell death-inducing DFF45-like effector b) (48). Interestingly, CideB has been shown to be involved in secretion of VLDL (49, 50). CideB belongs to the CIDE family, which includes CideA, CideB, and CideC, also called as Fsp27 in mice, and these proteins are reported to play important roles in lipoprotein metabolism (51). CideB is expressed in liver and kidney and is associated with maturation and secretion of VLDL and apoptosis (51, 52). It has been reported that CideB−/− mice have reduced secretion of VLDL particles, which are also small in size (49). However, the presence of CideB or its functional role in VTV biogenesis has not been investigated so far.
In the current study, we sought to identify the role of CideB in VTV budding. We found that CideB interacts with apoB100 in the ER, whereas it does not interact with albumin, which is excluded from the VTV. Our data suggest that CideB interacts with Sar1 at the ER level. The interaction of CideB with VLDL cargo protein, apoB100, and COPII component, Sar1, indicates its role in VTV biogenesis and thus VLDL exit from the ER. We further examined the effect of blocking the function of CideB or CideB knockdown on VTV generation from the ER. We report that CideB forms a specialized complex with COPII proteins that leads to the biogenesis of the VTV.
EXPERIMENTAL PROCEDURES
Reagents
[3H]Oleic acid (45.5 Ci/mm) was obtained from PerkinElmer Life Sciences. Reagents used for immunoblotting were purchased from Bio-Rad. ECL (enhanced chemiluminescence) reagents were purchased from GE Healthcare. Protease inhibitor mixture tablets were obtained from Roche Applied Science. Albumin was purchased from Sigma. Other biochemical reagents used were of analytical grade and were purchased from local companies. Sprague-Dawley rats, 150–200 g, were obtained from Harlan Laboratories (Indianapolis, IN). All procedures involving animals were conducted according to the guidelines of the University of Central Florida Institutional Animal Care and Use Committee (IACUC) and strictly following the IACUC approved protocol.
Antibodies
Goat polyclonal antibodies to CideB, calnexin, Ykt6, and GOS28; rabbit polyclonal antibodies to syntaxin 5, Sec13, Sec23, Sec31, and Sec24; and goat anti-rabbit IgG Texas Red and bovine anti-goat IgG-fluorescein isothiocyanate-conjugated antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Rabbit polyclonal anti-Sar1 antibodies were generated commercially and have been described previously (21). Rabbit polyclonal antibodies against rat VAMP7 (vesicle-associated membrane protein 7; amino acids 105–123) were described earlier (53). Mouse monoclonal antibodies to rBet1 and membrin were procured from StressGen (Vancouver, Canada). Rabbit anti-rat albumin antibody was purchased from Bethyl Laboratories, Inc. (Montgomery, TX), and rabbit polyclonal anti-apoB antibodies were a gift from Dr. Larry Swift (Department of Pathology, Vanderbilt University, Nashville, TN). Rabbit anti-goat IgG and goat anti-rabbit IgG conjugated with agarose beads were purchased from Sigma.
Primary Hepatocyte Culture
Primary hepatocytes were isolated from adult male Sprague-Dawley rats (Harlan Laboratories, Indianapolis, IN). Hepatocytes were isolated using collagen perfusion as described previously (54, 55). Briefly, liver was perfused with Krebs buffer (121 mm NaCl, 25 mm NaHCO3, 4.8 mm KCl, 2 mm MgSO4, and 1.2 mm KH2PO4; pH 7.2). This was followed by perfusion with type II collagenase (Worthington Biochemical Corp.). Cell suspension was obtained in RPMI 1640 (Life Technologies), supplemented with 1 μm dexamethasone (Sigma), 100 nm insulin (Sigma), 5% FBS (Life Technologies), 1% penicillin streptomycin (Sigma). Cells were seeded on a 60-mm collagen-coated dish (BD Biosciences) with the density of 3 × 106 cells/plate. After 4 h, unattached cells were removed, and fresh medium was added followed by overnight incubation.
Preparation of Radiolabeled Hepatic ER and cis- and trans-Golgi
ER labeled with [3H]triacylglycerol (TAG) was prepared from rat liver using the same method as we have described previously (21). Briefly, primary hepatocytes in buffer B (136 mm NaCl, 11.6 mm KH2PO4, 8 mm Na2HPO4, 7.5 mm KCl, 0.5 mm dithiothreitol; pH 7.2) were incubated with BSA-bound [3H]oleate (100 μCi) for 35 min at 37 °C and washed twice with 2% BSA in PBS to wash the excess of [3H]oleate. Cells were then homogenized in 0.25 m sucrose in 10 mm Hepes, 50 mm EDTA, and protease inhibitor (Roche Diagnostics GmbH, Mannheim, Germany) in a Parr bomb at 1,100 psi for 40 min followed by isolation of ER and cis- and trans-Golgi in a sucrose step gradient (21, 42, 56).
Preparation of Hepatic Cytosol
Hepatic cytosol was prepared by following the same method as described previously (21). After washing with Krebs buffer, cells were washed in cytosol buffer (25 mm Hepes, 125 mm KCl, 2.5 mm MgCl2, 0.5 mm DTT, and protease inhibitors; pH 7.2) and homogenized using a Parr bomb at 1,100 psi for 40 min. This was followed by centrifugation at 40,000 rpm for 95 min (Beckman rotor 70.1 Ti). Supernatant was dialyzed overnight against ice-cold fresh cytosol buffer and concentrated using a Centricon filter (Amicon, Beverly, MA) and ultrafiltration membrane (Millipore, Billerica, MA) with a cut-off of 10 kDa to a final concentration of protein to 17 mg/ml.
In Vitro VTV Formation
The in vitro VTV formation was carried out as established previously in our laboratory (21, 48, 56). In brief, ER having [3H]TAG (500 μg) was incubated at 37 °C for 30 min with hepatic cytosol (1 mg of protein), an ATP-regenerating system, 5 mm Mg2+, 5 mm Ca2+, 5 mm DTT, 1 mm GTP, 1 mm E600. Reaction mixture volume was adjusted to 500 μl by the addition of transport buffer (30 mm Hepes, 250 mm sucrose, 2.5 mm MgOAc, 30 mm KCl; pH 7.2). Next, the reaction mixture was placed on a sucrose continuous gradient made from 0.2 and 2.1 m sucrose, respectively, and centrifuged using a Beckman rotor SW41 at 25,900 rpm for 2 h at 4 °C, resulting in resolution of VTV in lighter fractions. Fractions (500 μl) having VTV were separated from sucrose continuous gradient.
Measurement of Radioactivity
Radioactivity associated with [3H]TAG was measured in terms of dpm by using a Tri-Carb 2910TR liquid scintillation analyzer (PerkinElmer Life Sciences) (21, 42).
Co-immunoprecipitation
ER membranes (250 μg) were solubilized in ice-cold PBS containing 2% (v/v) Triton X-100 (Fisher Scientific) at 4 °C for 15 min. Next, rabbit anti-apoB100 antibodies were added and incubated for 4 h at 4 °C. Similarly, parallel experiments were performed with goat anti-CideB and rabbit anti-albumin. After 4 h, either anti-goat or anti-rabbit IgGs bound to agarose beads were added and incubated overnight at 4 °C. Beads bound to immunocomplexes were washed 12 times with ice-cold PBS (21, 43).
Preparation of Cell Extract
Rat hepatocytes were lysed using radioimmunoprecipitation assay buffer (Thermo Scientific) supplemented with protease inhibitor. Lysed cell extract was centrifuged at 13,000 × g for 15 min. Supernatant obtained was used to determine protein concentration.
SDS-PAGE and Immunoblot Analysis
Concentration of protein in ER and whole cell lysate was determined by the Bradford method (21). Protein samples were separated by SDS-PAGE followed by transblotting onto a nitrocellulose membrane (Bio-Rad). Detection of protein was done by ECL Western blot detection reagent (GE Healthcare) and autoradiography film (MIDSCI, St. Louis, MO).
Effect of Antibody Treatment on VTV Budding
ER containing [3H]TAG (450 μg of protein) was incubated with same amount of indicated antibody (figure legends) or preimmune IgG for 1 h at 4 °C as described previously (43). The ER was washed with cold 0.1 m sucrose in Hepes buffer to remove unbound antibody. The ER pellet was resuspended in transport buffer (30 mm Hepes, 250 mm sucrose, 2.5 mm MgOAc, 30 mm KCl; pH 7.2) and used in an in vitro VTV budding assay.
Transfection with siRNA
Rat primary hepatocytes were transfected with CideB siRNA (Silencer select predesigned SiRNA, Life Technologies). The sequence of siRNA was 5′CAUGAGCUGCGAUUUUCAATT3′. Transfection was carried out by Lipofectamine by following the method according to manufacturer's protocol (Life Technologies).
Immunocytochemistry
Primary rat hepatocytes were plated on 22-mm round coverslips coated with collagen type I (BD Biosciences). Cells were washed three times with PBS, fixed with 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA) for 10 min, and permeabilized with 0.2% Triton X-100 (Fisher Scientific) for 10 min at room temperature. After washing three times with PBS, cells were blocked with 5% BSA solution in PBS for 30 min at room temperature. Cells were then double-labeled with goat anti-CideB (1:100) and either rabbit anti-apoB100 (1:100) or rabbit anti-calnexin antibodies (1:100) for 1 h at room temperature and washed three times with PBS. Next, cells were incubated in dark with species-specific secondary antibody, bovine anti-goat IgG-FITC (1:500), and goat anti-rabbit IgG-Texas Red (1:500) for 45 min at room temperature. After washing with PBS, cells were mounted on a glass slide using mounting medium (Electron Microscopy Sciences, Hatfield, PA) and visualized using the Zeiss spinning disk confocal microscope and velocity image analyzer software.
Immunoelectron Microscopy
In an attempt to examine the localization of CideB on VTVs by immunoelectron microscopy, we adopted the negative staining approach and followed the same methodology for immunogold labeling of the VTVs as described previously (46, 48, 56). In brief, VTVs adsorbed on Formvar/carbon-coated nickel grids were incubated with 10% (w/v) BSA containing either anti-rabbit preimmune IgG (1:100) or rabbit polyclonal anti-CideB antibodies (1:100) for 4 h. Samples were washed with PBS and incubated with anti-rabbit IgG (1:50) conjugated with 15-nm colloidal gold. After washing with PBS, samples were subsequently fixed in 1% (w/v) glutaraldehyde in PBS for 10 min, stained with 0.5% aqueous uranyl acetate for 1 min, and examined at 12,000× magnification.
Statistical Analysis
Data were compared using a one-way analysis of variance (ANOVA) using GraphPad software (GraphPad Prism 5 Software for Mac OS X version).
RESULTS
CideB Is Present in the Hepatic ER, Golgi, Cytosol, and VTV
Our first goal was to determine the distribution of CideB in various subcellular organelles in primary hepatocytes. To achieve this, we separated the ER, Golgi, and cytosol from primary hepatocytes. Because the purity of subcellular fractions isolated from primary hepatocytes is considered crucial for the success of our in vitro assays, we carried out Western blotting utilizing organelle-specific protein markers for ER and Golgi to establish their purity as reported previously (21, 56). Our results show that ER did not contain recognizable GOS28, a Golgi marker protein, whereas Golgi was free from calnexin, an ER marker protein (Fig. 1A). Both ER and Golgi membranes were found to be devoid of Rab11, an endosomal/lysosomal marker protein (data not shown). After confirming that our subcellular organelles are of adequate purity, we performed a pre-established in vitro VTV budding assay to prepare VTVs, which were purified and characterized as we reported earlier (21). To make sure that our vesicular fraction contains bona fide VTVs, we probed for VTV markers, Sar1 and apoB100. As shown in Fig. 1A, both proteins were concentrated in VTV fractions, suggesting that we have adequate VTV fractions to perform further analysis. To examine that CideB is present in the VTV, ER, Golgi, and cytosol, we performed Western blotting using specific CideB antibodies. The data presented in Fig. 1A show that hepatic ER, Golgi, and cytosol contain CideB, which is consistent with previous studies (21). However, CideB was found to be the least in cytosolic fraction (Fig. 1A). Interestingly, we observed that CideB is concentrated in VTV fractions as compared with their parent membrane, hepatic ER (Fig. 1A).
FIGURE 1.
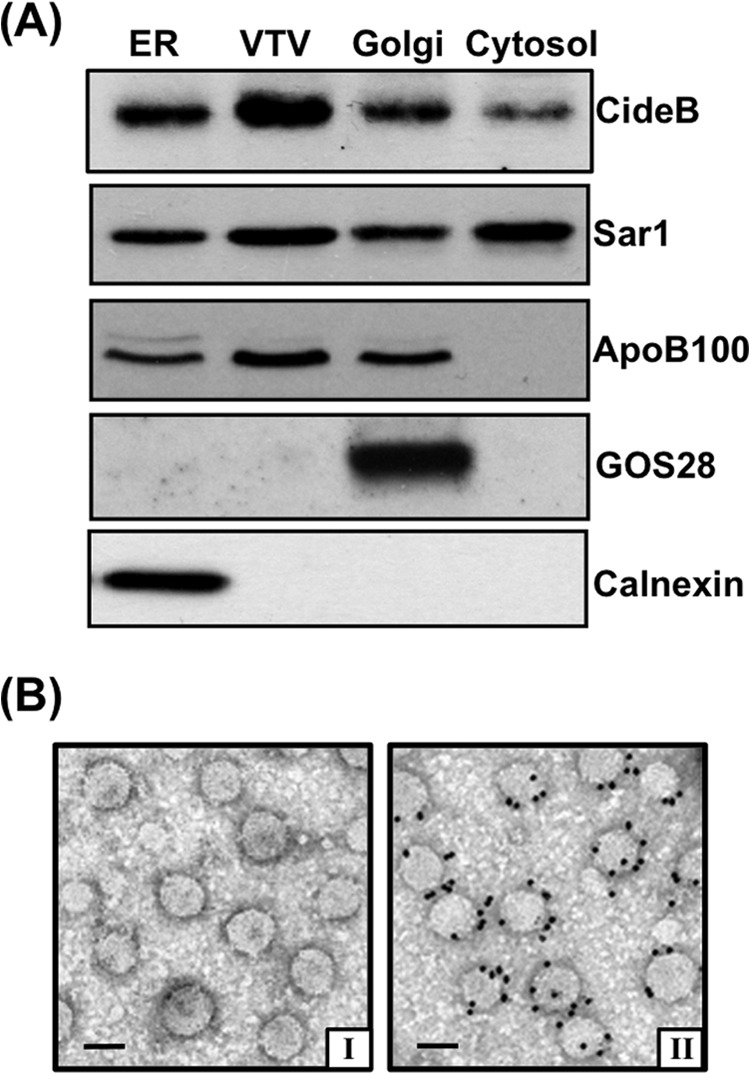
CideB concentrates in VTV. A, protein samples of purified fractions of ER, VTV, Golgi, and cytosol (each sample contains 35 μg of protein) were separated by 12% SDS-PAGE (except for apoB100; a 4–20% gel was utilized), transblotted onto a nitrocellulose membrane, and probed with specific antibodies against the indicated proteins. ECL reagents were used to detect proteins. The data are representative of three independent experiments. B, visualization of CideB localization on VTVs by immunoelectron microscopy using the negative staining. Freshly prepared VTVs were adsorbed on Formvar/carbon-coated nickel grids, incubated with either anti-rabbit preimmune IgG (panel I) or rabbit polyclonal anti-CideB (panel II) antibodies, and probed with anti-rabbit IgG conjugated with 15-nm gold particles. Bar = 100 nm.
To illustrate the localization of CideB to the VTVs morphologically, we carried out immunogold labeling of CideB on the VTVs and probed the results by electron microscopy using the negative staining technique. As shown in Fig. 1B (panel II), CideB is localized on the surface of the VTVs as established by immunogold labeling. By contrast, control experiments using preimmune IgG displayed no immunogold labeling (Fig. 1B, panel I). Together, these biochemical and morphological data strongly suggest that CideB is not only present on the surface of the VTV but is concentrated in the VTVs.
CideB Is Not Present in ER-derived PTV and PCTV
Our next aim was to find out whether CideB is present in other ER-derived vesicles such as PTVs, which contain nascent secretory proteins, and PCTVs, which transport nascent lipoproteins of intestinal origin, the pre-chylomicrons (41, 42). We isolated and purified these vesicles as discussed previously (53). Our data reveal that CideB is not present in either PTV or PCTV (Fig. 2); however, we observed a strong band of CideB in the VTV fractions (Fig. 2), indicating that CideB is present in VTVs only but not in other ER-derived vesicles. When we probed for PTV marker protein, albumin (a hepatic secretory protein), we found a strong band of albumin in the PTV fraction. In addition, we immunoblotted for VAMP7, which is an endosomal or post-Golgi protein but uniquely present in ER-derived PCTVs (46). As expected, VAMP7 is enriched in the PCTV fraction (Fig. 2); however, both VTV and PTV do not have identifiable VAMP7 (Fig. 2), which is consistent with previous studies (21). Taken together, these data suggest that CideB is specifically present in the VTVs.
FIGURE 2.
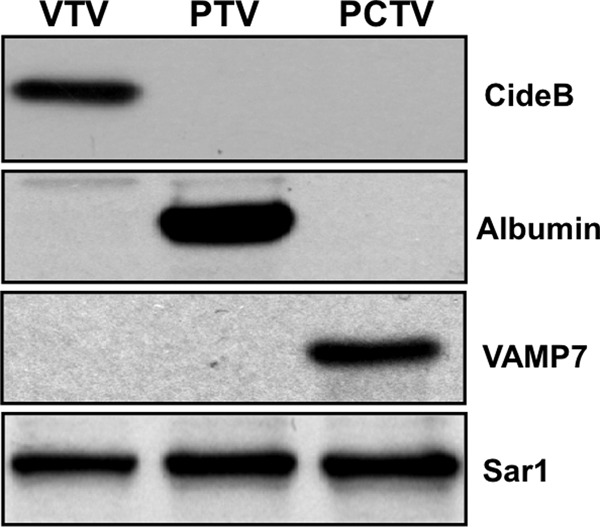
CideB is present in VTV but not in PTV or PCTV. Protein samples of purified VTV, PTV, and PCTV (each sample containing 40 μg of protein) were separated by 12% SDS-PAGE, transblotted onto a nitrocellulose membrane, and probed with specific antibodies against the indicated proteins. Proteins were detected using ECL reagents. The data are representative of four independent experiments.
CideB Interacts with apoB100 but Not with Albumin
Because both PTV and VTV are budding off the same ER at the same time and CideB is not present in the PTV but is enriched in VTV, we questioned whether it interacts with albumin (PTV cargo protein) and apoB100 (a VTV cargo protein) at the ER level. To address this issue, we carried out co-immunoprecipitation experiments utilizing ER membranes solubilized in 2% Triton X-100 and specific CideB antibodies. Our results demonstrate that CideB co-immunoprecipitates apoB100 but not albumin; however, albumin is present in the ER (Fig. 3A). These findings suggest that CideB specifically interacts with VTV cargo protein but not with PTV cargo, indicating its potential role in VLDL transport from the ER to the Golgi.
FIGURE 3.

CideB interacts with apoB100 but not with albumin. A, ER (250 μg of protein) were solubilized in 2% (v/v) Triton X-100 and incubated with anti-goat CideB antibody (10 μg) for 4 h at 4 °C. Anti-goat IgGs bound to agarose beads were added and incubated overnight at 4 °C. Immunocomplexes bound to agarose beads were isolated and washed 10 times with ice-cold PBS to remove unbound proteins. Protein sample was separated by SDS-PAGE (8–16% gel) and probed with anti-CideB, anti-apoB100, and anti-albumin antibodies. IP, immunoprecipitation. B, CideB co-localizes with apoB100 in hepatic ER. Primary hepatocytes were double-labeled with either CideB (FITC, green) and apoB100 (Texas Red, red) (upper and middle panels) or CideB (FITC, green) and calnexin, an ER marker (Texas Red, red) (lower panel). In the middle panel, we used higher magnification and less saturation of both channels. Arrowheads in the middle panel show punctate vesicular staining in the VTVs. The nucleus is stained with DAPI (blue). Merged figures show co-localization of CideB with apoB100 and calnexin.
To substantiate our observations pertaining to CideB-apoB100 interaction at the ER level, we sought to provide additional morphological evidence using confocal microscopy. We used a double-labeled immunofluorescence technique. As can be seen in Fig. 3B, CideB co-localizes with apoB100. The middle panel of Fig. 3B clearly shows the co-localization of apoB100 with CideB in the characteristic reticular structure of the ER. Interestingly, we observed punctate co-staining most likely in VTVs because this is characteristic staining of the ER-derived vesicles (Fig. 3B, middle panel, arrowheads). To show that this interaction occurs at the ER level, we used an ER marker, calnexin. The data presented in the lower panels of Fig. 3B indicate that CideB co-localizes with calnexin. Together, these data confirm that the interaction between CideB and apoB100 is specific and occurs at the level of the ER.
CideB Interacts with COPII Components
Because the VTV originates from hepatic ER membrane in a COPII-dependent fashion and CideB is specifically present in the VTV, it is likely that CideB may interact with VTV coat proteins i.e. COPII proteins. Another reason for this assumption is that VTVs are larger in size than the typical COPII-coated PTVs, suggesting that the assembly of VTV coat may require additional protein(s) to form a larger/modified COPII cage. To find out whether CideB interacts with COPII proteins, we performed co-immunoprecipitation experiments. Because all five COPII proteins (i.e. Sar1, Sec23, Sec24, Sec13, and Sec31) are cytosolic, we used hepatic cytosol anti-CideB antibodies in our co-immunoprecipitation assays.
To have high confidence in our results, we reprobed the same membrane with different antibodies against each of the COPII proteins. The immunoblotting data presented in Fig. 4 suggest that CideB strongly interacts with Sar1 and Sec24; however, its binding with Sec23 was minimal. Also, we did not observe a strong signal for CideB interaction with Sec13 and Sec31 (Fig. 4). These data suggest that CideB interacts with select COPII components (Sar1 and Sec24). Interaction between CideB and Sec24 indicates that these two proteins form a heterodimer, which in turn facilitates the assembly of an intricate COPII coat necessary for VTV formation.
FIGURE 4.
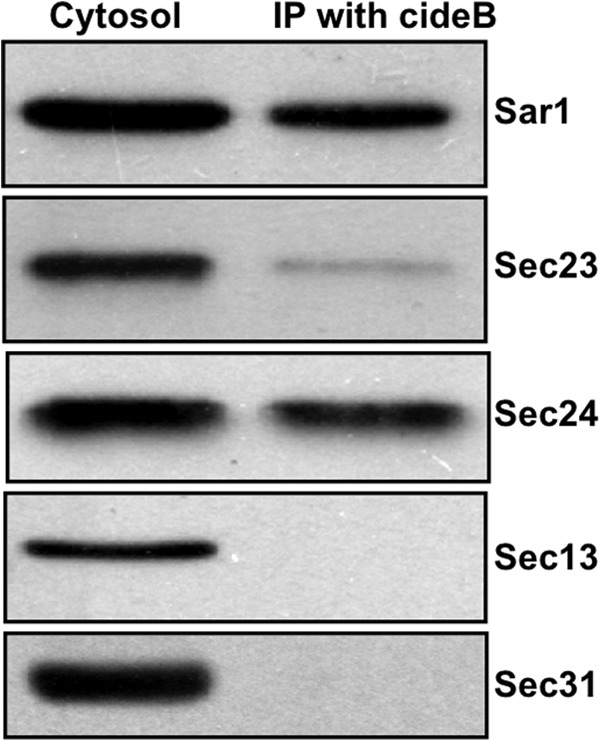
CideB interacts with COPII components. Rat hepatic cytosol (150 μg of protein) was incubated with anti-goat CideB antibody bound to agarose beads at 4 °C overnight. Immunocomplexes bound to agarose beads were isolated and washed thoroughly. The proteins were separated by SDS-PAGE (5–15%) and probed with antibodies against the indicated proteins. A single membrane was used, which was sequentially probed with the indicated antibodies after washing. Protein detection was carried out using ECL reagents. The results are representative of four experiments. IP, immunoprecipitation.
Blockade of CideB Reduces VTV Formation
That CideB is specifically present in ER-derived VTVs and its interaction with VTV cargo and VTV coat proteins raise the possibility of its active involvement in the process of VTV biogenesis, and thus, ER exit of nascent VLDL particles. To determine whether CideB has some role in VTV budding from hepatic ER, we first decided to examine the effect of blocking CideB using specific antibodies on VTV budding. To accomplish this, we performed an in vitro VTV budding assay that we have standardized previously (21). To block CideB on the ER, [3H]TAG-containing hepatic ER membranes were preincubated at 4 °C with an equal amount of either control IgG or anti-CideB antibodies. After antibody treatment, excess antibodies were removed by washing. Because cytosol also contains a small amount of CideB, we treated cytosol with anti-CideB antibodies or the preimmune IgGs (control) bound to agarose beads prior to the budding assay, and the excess antibodies were removed. Anti-CideB antibody-treated ER membranes and cytosol were used in the VTV budding assay (see “Experimental Procedures”). As presented in Fig. 5A, the treatment of hepatic ER with anti-CideB antibodies significantly inhibited VTV generation. However, IgG treatment did not have any effect on VTV budding activity (Fig. 5A). These results show that CideB blockade results in reduced VTV formation, suggesting an active role of CideB in VTV biogenesis.
FIGURE 5.
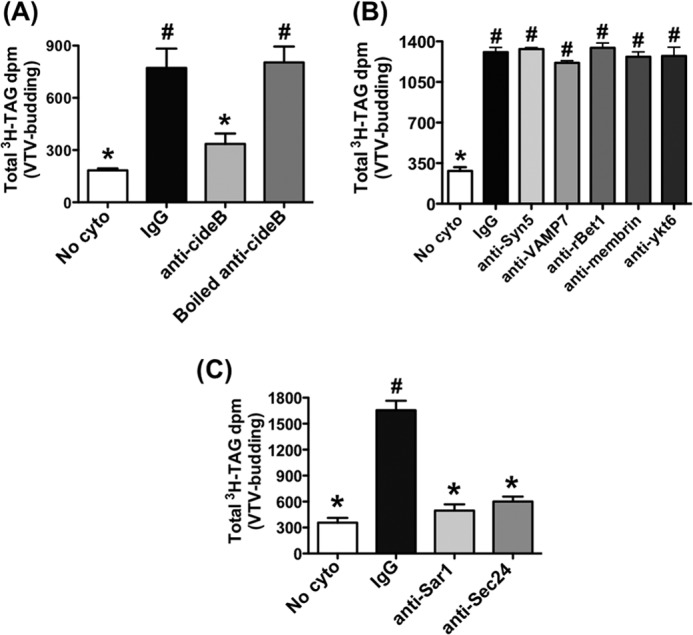
The effects of CideB blockade on VTV formation. VTV budding assays were performed in which hepatic ER containing [3H]TAG was incubated with or without rat hepatic cytosol in the presence of an ATP-regenerating system. After incubation, the reaction mixture was resolved on a continuous sucrose density gradient, and fractions (0.5 ml) were collected. The [3H]TAG dpm values in all fractions (500 μl) were measured. A, prior to budding assay, the ER containing [3H]TAG was incubated at 4 °C for 2 h with preimmune IgG, anti-CideB, or boiled anti-CideB antibodies, and antibodies in each case were removed by washing. Cytosol was pretreated at 4 °C for 2 h with preimmune IgG, anti-CideB, or boiled anti-CideB antibodies bound to agarose beads, and the antibodies were removed by centrifugation. Results are mean ± S.D. (n = 4). Bars labeled with different symbols show p < 0.01 using one-way ANOVA. No cyto, absence of cytosol. B, the ER containing [3H]TAG and the hepatic cytosol similar to panel A were treated prior to performing budding assays, with the indicated antibodies. Data are mean ± S.D. (n = 4). Bars labeled with different symbols are p < 0.005 using one-way ANOVA. C, ER containing [3H]TAG and hepatic cytosol were pretreated with the indicated antibodies and used in VTV budding assays as described above. For details, see “Experimental Procedures.” Values are mean ± S.D. (n = 4). Bars labeled with different symbols have p < 0.001 using one-way ANOVA.
It is, however, possible that antibody treatment results in nonspecific inhibition of the VTV budding process due to a number of factors. To rule out the possibility of nonspecific inhibition, we treated the ER and cytosol with a variety of antibodies against proteins that are known to be present on hepatic ER and involved in ER-to-Golgi transport of proteins and lipoproteins. We used antibodies against syntaxin5, rbet1, ykt6, and membrin, which have been well characterized to be present on hepatic ER (21). We followed exactly the same protocol of antibody treatment as we used for CideB antibody treatment. Our data reveal that the treatment with each of these antibodies did not have any effect on VTV formation (Fig. 5B), suggesting that the blocking effect of CideB on VTV generation was specific. Moreover, we treated hepatic ER with VAMP7, which is required for the generation of PCTVs from small intestinal ER (46). As shown in Fig. 5B, there was no decrease in VTV budding as compared with control when VAMP7 antibodies were used. To rule out the possibility of steric hindrance caused by CideB antibody binding on the ER surface, we boiled these antibodies and used them in our budding assay. As expected, boiled CideB antibodies did not inhibit VTV formation (Fig. 5A). Because both Sar1 and Sec24 interact with CideB and apoB100, it is possible that this interaction triggers the process of VTV formation. To test this speculation, we decided to immunodeplete hepatic cytosol with specific antibodies against Sar1 and Sec24 as we have done successfully previously (57). Also, we washed our ER membranes with urea to remove peripheral Sar1 and Sec24 as we have done in the past (21). As can be seen in Fig. 5C, the Sar1- and Sec24-depleted system did not support the formation of the VTV, which is consistent with published studies (56, 57). Taken together, these data strongly suggest that CideB plays an important role in the biogenesis of the VTV.
Silencing of CideB Abrogates VTV Biogenesis
In an attempt to provide more concrete evidence that CideB is crucial for the genesis of the VTV, we sought to knock down CideB in primary rat hepatocytes. We decided to use siRNA with Lipofectamine because this approach has been used successfully in primary rat hepatocytes (58). After transfection, the level of CideB knockdown was ascertained in whole cell lysate, ER, and cytosol from hepatocytes. As shown in Fig. 6A, 50 nm concentration of CideB siRNA significantly decreased CideB protein levels as compared with control siRNA in rat primary hepatocytes, whereas there was no effect on the expression of β-actin (Fig. 6A). Next, we isolated the ER and the cytosol from these hepatocytes and examined the presence of CideB using Western blotting. Fig. 6B shows that the ER, isolated from CideB siRNA-treated hepatocytes, contains significantly low level of CideB, whereas there is no effect on calnexin, an ER resident protein. Similarly, a reduced level of CideB protein was found in cytosol prepared from CideB knockdown hepatocytes, whereas there was no effect on β-actin level in the same cytosol (Fig. 6C). Treatment of control siRNA had no effect on the expression of CideB in whole cell lysate, ER, or cytosol of hepatocytes (Fig. 6, A–C).
FIGURE 6.
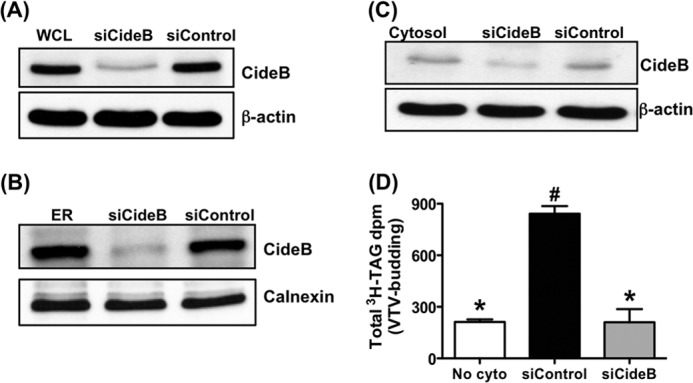
Knockdown of CideB in primary hepatocytes abrogates VTV formation. Primary hepatocytes were transfected with either CideB siRNA (5′CAUGAGCUGCGAUUUUCAATT3′) or control siRNA (Silencer select siRNA, Ambion). ER containing [3H]TAG and cytosol were prepared and purified from both sets of hepatocytes as described under “Experimental Procedures.” A, protein samples of whole cell lysate (WCL). siCideB, CideB siRNA; siControl, control siRNA. B, hepatic ER membranes. C, cytosol prepared from untreated primary hepatocytes or treated with either CideB siRNA (siCideB) or control siRNA (siControl) was probed with specific anti-CideB, anti-β-actin, or anti-calnexin antibodies. D, VTV budding assays were performed using ER containing [3H]TAG and cytosol, which were isolated from primary hepatocytes treated with either CideB siRNA (siCideB) or control siRNA (siControl). As a negative control, ER containing [3H]TAG isolated from untreated hepatocytes was incubated in the absence of cytosol (No cyto). Results are mean ± S.D. (n = 4). Bars labeled with different symbols depict p < 0.001 using one-way ANOVA.
To find out the effect of knocking down CideB on VTV formation process, we carried out an in vitro VTV budding assay. As we expected, VTV budding activity was significantly decreased when CideB was knocked down as shown in Fig. 6D. In contrast, there was no effect on VTV formation when control siRNA was used (Fig. 6D). These data strongly suggest a functional role for CideB in VTV budding.
DISCUSSION
Increased secretion of very low density lipoproteins from the liver is associated with hyperlipidemia, which is a major risk factor for the development of various cardiovascular diseases. The assembly of VLDL occurs in the ER lumen, and this step is mediated by microsomal triglyceride transfer protein, which possesses binding sites for both apoB100 and triglycerides (4, 5). After their biogenesis in the ER, newly synthesized VLDL particles are exported to the Golgi, and this step determines the rate of VLDL secretion from the liver. The transport of nascent VLDL particles from the ER to the Golgi is mediated by specialized vesicles, the VTVs (10, 21). Our previous studies have shown that the VTVs are different from the classical ER-derived COPII-coated PTVs that carry nascent proteins to the Golgi (21). Despite their distinct cargoes, size, and protein compositions, both VTVs and PTVs utilize the COPII system for their biogenesis (21). This indicates that the formation of the VTVs requires other protein(s) in addition to the COPII machinery. Taking these observations into account, we reasoned that the additional protein(s) that interacts specifically with VLDL core protein (apoB100) forms an intricate COPII complex, which facilitates the formation of the VTV from the ER membranes. In this study, we tested our hypothesis and provide the evidence that supports the involvement of an additional protein, CideB, in VTV biogenesis. We found that CideB interacts with VLDL protein (apoB100) and COPII components.
Our recent study describing the VTV proteome suggested the presence of a small Mr protein, CideB, in VTVs (48). In this study, using biochemical methods, we confirmed that the VTVs contain CideB. CideB has been shown to be present in ER, Golgi, and lipid droplets. However, the presence of CideB in ER-derived VTV is of particular interest because: (i) the VTV exports nascent VLDL to the Golgi lumen; (ii) VLDL gets further lipidated in the Golgi; and (iii) CideB has been recently shown to promote VLDL lipidation and maturation in the Golgi lumen (50). Together, these observations led us to postulate that CideB might have an unidentified role in VLDL export from the ER to the Golgi.
The role of CideB in intracellular transport processes has not been reported previously; however, a number of findings related to CideB are consistent with the novel role proposed in this study. For example, the liver of CideB knock-out mice has been shown to secret reduced amounts of triglycerides (51). Moreover, knockdown or complete ablation of CideB leads to the secretion of triglyceride-poor VLDL particles from hepatocytes (49); in other words, CideB plays an important role in VLDL delivery to the Golgi where VLDL undergoes additional lipidation.
Although COPII proteins are sufficient to form vesicles from the ER membranes, they interact with other proteins to make specific COPII coats for distinct cargoes (23, 39–43). We contemplated the feasibility that CideB may interact with COPII components of the VTV to form a specialized larger COPII cage because CideB is present in COPII-coated VTVs, which are larger in their size than standard PTVs. Our results clearly indicate that CideB interacts with COPII components, Sar1 and Sec24. Binding of CideB with Sec24 is especially significant because the resulting CideB-Sec24 heterodimer might be involved in the formation of larger ER-derived vesicles that can accommodate VLDL-sized particles, which is consistent with data reported previously (39). Moreover, the presence of Sec23 in the VTV (21) raises the possibility that Sec24 forms two heterodimers, CideB/Sec24 and Sec23/Sec24, to facilitate the biogenesis of the VTV, and this supposition is consistent with other studies (39).
Does CideB interact with other secretory proteins and facilitate their ER exit? Co-immunoprecipitation analysis suggested that CideB does not associate with liver secretory protein, albumin, which raises another important question whether the classical COPII-coated PTVs contain CideB or not. Because a number of previous studies have revealed that albumin utilizes COPII-coated PTVs for its transport to the Golgi (15, 21), we speculated that CideB might not be present in PTVs. The data presented in this study demonstrate that PTVs indeed do not contain CideB, suggesting that typical secretory cargo proteins do not require proteins in addition to COPII components, which is consistent with previous unambiguous findings suggesting that COPII proteins are sufficient to recruit nascent proteins and generate PTVs from the ER membranes (22–31, 59). In contrast, it has been shown that special secretory cargo proteins control the formation and the size of ER-derived PTVs (39). Using a temperature-sensitive form of vesicular stomatitis viral glycoprotein (VSV-Gts), Aridor et al. (60) showed that the generation of PTVs from the ER membranes could be controlled by biosynthetic cargoes. The ability of secretory cargo proteins to modulate the size of ER-derived vesicles has been demonstrated by many groups (39, 61, 62). Recently, it has been shown that ubiquitination of Sec31 by ubiquitin ligase CUL3-KLHL12 regulates the size of the COPII coat (62). However, several studies suggest the requirement of additional protein(s) to form assorted COPII coats that lead to the generation of larger ER-derived vesicles (39). The formation of pre-chylomicron transport vesicle requires four proteins in addition to COPII proteins (43). Moreover, Shimoni et al. (39) showed that the transport of plasma membrane ATPase from the ER to Golgi requires Lst1 protein, which substitutes for Sec24 and forms the heterodimer with Sec23, leading to plasma membrane ATPase selection into PTVs. Interestingly, these vesicles were found to be larger than typical COPII-coated PTVs, supporting the thesis that cargo can influence the size of the vesicle. These observations support our findings that VLDL regulates the formation and size of specialized ER-derived VTVs.
The specific nature of the interaction between CideB and apoB100 raises the possibility that CideB plays a crucial role in VLDL sorting and the VTV biogenesis. Because previous published data suggested that VTVs do not carry albumin to the Golgi (15, 21), it is likely that CideB functions as VLDL-selecting protein for the VTV. However, the absence of CideB in the PTV and its interaction with COPII components support the thesis that it facilitates the process of VTV formation. Fig. 7 summarizes a novel role of CideB in VTV biogenesis. The potential role of CideB in VLDL selection as well as VTV formation is consistent with other studies in which Lst1 has been shown to mediate the selection of specific secretory cargo and the formation of larger ER-derived vesicles (39, 40).
FIGURE 7.

Schematic diagram summarizing the proposed novel role of CideB in the formation of the VTVs. CideB is localized to the ER membrane and VTVs. CideB binds to VLDL-apoB and may facilitate the VTV formation. CideB interacts with Sar1 and Sec24 to assemble an intricate COPII coat that may be necessary for the formation of large sized vesicles, the VTV, from hepatic ER membranes. CideB does not interact with nascent secretory proteins such as albumin and is not present in PTVs.
Interaction of CideB with apoB100, Sar1, and Sec24 and its presence in the VTV but not in other ER-derived vesicles (e.g. PTV or PCTV) indicated that it might play a functional role specifically in VTV generation. The data presented in this study clearly demonstrate that both blocking CideB with specific antibodies and CideB silencing using an siRNA approach significantly reduced the generation of the VTV, confirming its functional role in VTV genesis. Unlike Sar1, a COPII component that initiates VTV budding from hepatic ER membranes, knockdown of CideB is not lethal. Primary hepatocytes lacking CideB (hepatocytes isolated from CideB-null mice) continued to secrete VLDL particles; however, these particles were found to be smaller in size (49). Because CideB has been proposed to be a multifunctional protein, which is involved in various stages of VLDL lipidation in the ER as well as in the Golgi (49, 50), knockdown of CideB from hepatocytes would lead to incomplete VLDL lipidation in both the ER and the Golgi. Under these circumstances, immature VLDLs (smaller than normal VLDL particles) would not require larger ER-derived vesicles; instead, they can be transported to the Golgi in conventional COPII-coated PTVs. However under normal conditions, a larger vesicle (i.e. VTV) is required for exporting VLDL to the Golgi, and CideB plays an essential role in VTV biogenesis (Fig. 7).
In the present study, we have identified a new physiological role for CideB as we demonstrated that CideB facilitates the formation of ER-derived large vesicles. Using an siRNA approach, we were able to demonstrate that the knockdown of CideB significantly reduced the VTV generation. Our co-immunoprecipitation data show that CideB interacts with the COPII components that might be necessary to build a larger COPII cage and thus for the formation of VTV. We suggest that the interaction between CideB and apoB100 (a VTV cargo protein) triggers the process of VTV formation. These findings suggest that the size and the type of cargo regulate the assembly of assorted coat complexes, leading to the genesis of vesicles of various sizes.
This work was supported, in whole or in part, by National Institutes of Health Grant RO1 DK-81413 through the NIDDK (to S. A. S.).
- ER
- endoplasmic reticulum
- CideB
- cell death-inducing DFF45-like effector b
- VTV
- VLDL transport vesicle
- PTV
- protein transport vesicle
- PCTV
- pre-chylomicron transport vesicle
- COPII
- coat complex II
- apoB100
- apolipoprotein B100
- TAG
- triacylglycerol
- ANOVA
- analysis of variance.
REFERENCES
- 1. Ginsberg H. N. (2002) New perspectives on atherogenesis: role of abnormal triglyceride-rich lipoprotein metabolism. Circulation 106, 2137–2142 [DOI] [PubMed] [Google Scholar]
- 2. Sehayek E., Eisenberg S. (1994) The role of native apolipoprotein B-containing lipoproteins in atherosclerosis: cellular mechanisms. Curr. Opin. Lipidol. 5, 350–353 [DOI] [PubMed] [Google Scholar]
- 3. Olofsson S. O., Asp L., Borén J. (1999) The assembly and secretion of apolipoprotein B-containing lipoproteins. Curr. Opin. Lipidol. 10, 341–346 [DOI] [PubMed] [Google Scholar]
- 4. Hussain M. M., Bakillah A., Nayak N., Shelness G. S. (1998) Amino acids 430–570 in apolipoprotein B are critical for its binding to microsomal triglyceride transfer protein. J. Biol. Chem. 273, 25612–25615 [DOI] [PubMed] [Google Scholar]
- 5. Hussain M. M., Shi J., Dreizen P. (2003) Microsomal triglyceride transfer protein and its role in apoB-lipoprotein assembly. J. Lipid Res. 44, 22–32 [DOI] [PubMed] [Google Scholar]
- 6. Shelness G. S., Ingram M. F., Huang X. F., DeLozier J. A. (1999) Apolipoprotein B in the rough endoplasmic reticulum: translation, translocation, and the initiation of lipoprotein assembly. J. Nutr. 129, 456S–462S [DOI] [PubMed] [Google Scholar]
- 7. Jamil H., Gordon D. A., Eustice D. C., Brooks C. M., Dickson J. K., Jr., Chen Y., Ricci B., Chu C. H., Harrity T. W., Ciosek C. P., Jr., Biller S. A., Gregg R. E., Wetterau J. R. (1996) An inhibitor of the microsomal triglyceride transfer protein inhibits apoB secretion from HepG2 cells. Proc. Natl. Acad. Sci. U.S.A. 93, 11991–11995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bakillah A., Nayak N., Saxena U., Medford R. M., Hussain M. M. (2000) Decreased secretion of ApoB follows inhibition of ApoB-MTP binding by a novel antagonist. Biochemistry. 39, 4892–4899 [DOI] [PubMed] [Google Scholar]
- 9. Kulinski A., Rustaeus S., Vance J. E. (2002) Microsomal triacylglycerol transfer protein is required for lumenal accretion of triacylglycerol not associated with ApoB, as well as for ApoB lipidation. J. Biol. Chem. 277, 31516–31525 [DOI] [PubMed] [Google Scholar]
- 10. Tiwari S., Siddiqi S. A. (2012) Intracellular trafficking and secretion of VLDL. Arterioscler. Thromb. Vasc. Biol. 32, 1079–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gusarova V., Seo J., Sullivan M. L., Watkins S. C., Brodsky J. L., Fisher E. A. (2007) Golgi-associated maturation of very low density lipoproteins involves conformational changes in apolipoprotein B, but is not dependent on apolipoprotein E. J. Biol. Chem. 282, 19453–19462 [DOI] [PubMed] [Google Scholar]
- 12. Tran K., Thorne-Tjomsland G., DeLong C. J., Cui Z., Shan J., Burton L., Jamieson J. C., Yao Z. (2002) Intracellular assembly of very low density lipoproteins containing apolipoprotein B100 in rat hepatoma McA-RH7777 cells. J. Biol. Chem. 277, 31187–31200 [DOI] [PubMed] [Google Scholar]
- 13. Swift L. L. (1996) Role of the Golgi apparatus in the phosphorylation of apolipoprotein B. J. Biol. Chem. 271, 31491–31495 [PubMed] [Google Scholar]
- 14. Olofsson S. O., Bjursell G., Boström K., Carlsson P., Elovson J., Protter A. A., Reuben M. A., Bondjers G. (1987) Apolipoprotein B: structure, biosynthesis and role in the lipoprotein assembly process. Atherosclerosis 68, 1–17 [DOI] [PubMed] [Google Scholar]
- 15. Gusarova V., Brodsky J. L., Fisher E. A. (2003) Apolipoprotein B100 exit from the endoplasmic reticulum (ER) is COPII-dependent, and its lipidation to very low density lipoprotein occurs post-ER. J. Biol. Chem. 278, 48051–48058 [DOI] [PubMed] [Google Scholar]
- 16. Bamberger M. J., Lane M. D. (1990) Possible role of the Golgi apparatus in the assembly of very low density lipoprotein. Proc. Natl. Acad. Sci. U.S.A. 87, 2390–2394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Higgins J. A. (1988) Evidence that during very low density lipoprotein assembly in rat hepatocytes most of the triacylglycerol and phospholipid are packaged with apolipoprotein B in the Golgi complex. FEBS Lett. 232, 405–408 [DOI] [PubMed] [Google Scholar]
- 18. Rusiñol A., Verkade H., Vance J. E. (1993) Assembly of rat hepatic very low density lipoproteins in the endoplasmic reticulum. J. Biol. Chem. 268, 3555–3562 [PubMed] [Google Scholar]
- 19. Yamaguchi J., Gamble M. V., Conlon D., Liang J. S., Ginsberg H. N. (2003) The conversion of apoB100 low density lipoprotein/high density lipoprotein particles to apoB100 very low density lipoproteins in response to oleic acid occurs in the endoplasmic reticulum and not in the Golgi in McA RH7777 cells. J. Biol. Chem. 278, 42643–42651 [DOI] [PubMed] [Google Scholar]
- 20. Borchardt R. A., Davis R. A. (1987) Intrahepatic assembly of very low density lipoproteins: rate of transport out of the endoplasmic reticulum determines rate of secretion. J. Biol. Chem. 262, 16394–16402 [PubMed] [Google Scholar]
- 21. Siddiqi S. A. (2008) VLDL exits from the endoplasmic reticulum in a specialized vesicle, the VLDL transport vesicle, in rat primary hepatocytes. Biochem. J. 413, 333–342 [DOI] [PubMed] [Google Scholar]
- 22. Barlowe C. (1998) COPII and selective export from the endoplasmic reticulum. Biochim. Biophys. Acta 1404, 67–76 [DOI] [PubMed] [Google Scholar]
- 23. Barlowe C., Schekman R. (1993) SEC12 encodes a guanine-nucleotide-exchange factor essential for transport vesicle budding from the ER. Nature 365, 347–349 [DOI] [PubMed] [Google Scholar]
- 24. Barlowe C., d'Enfert C., Schekman R. (1993) Purification and characterization of SAR1p, a small GTP-binding protein required for transport vesicle formation from the endoplasmic reticulum. J. Biol. Chem. 268, 873–879 [PubMed] [Google Scholar]
- 25. Fromme J. C., Orci L., Schekman R. (2008) Coordination of COPII vesicle trafficking by Sec23. Trends. Cell Biol. 18, 330–336 [DOI] [PubMed] [Google Scholar]
- 26. Gürkan C., Stagg S. M., Lapointe P., Balch W. E. (2006) The COPII cage: unifying principles of vesicle coat assembly. Nat. Rev. Mol. Cell Biol. 7, 727–738 [DOI] [PubMed] [Google Scholar]
- 27. Tang B. L., Wang Y., Ong Y. S., Hong W. (2005) COPII and exit from the endoplasmic reticulum. Biochim. Biophys. Acta 1744, 293–303 [DOI] [PubMed] [Google Scholar]
- 28. Hughes H., Stephens D. J. (2008) Assembly, organization, and function of the COPII coat. Histochem. Cell Biol. 129, 129–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Barlowe C., Orci L., Yeung T., Hosobuchi M., Hamamoto S., Salama N., Rexach M. F., Ravazzola M., Amherdt M., Schekman R. (1994) COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell 77, 895–907 [DOI] [PubMed] [Google Scholar]
- 30. Jensen D., Schekman R. (2011) COPII-mediated vesicle formation at a glance. J. Cell Sci. 124, 1–4 [DOI] [PubMed] [Google Scholar]
- 31. Kuge O., Dascher C., Orci L., Rowe T., Amherdt M., Plutner H., Ravazzola M., Tanigawa G., Rothman J. E., Balch W. E. (1994) Sar1 promotes vesicle budding from the endoplasmic reticulum but not Golgi compartments. J. Cell Biol. 125, 51–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shoulders C. C., Stephens D. J., Jones B. (2004) The intracellular transport of chylomicrons requires the small GTPase, Sar1b. Curr. Opin. Lipidol. 15, 191–197 [DOI] [PubMed] [Google Scholar]
- 33. Levy E., Harmel E., Laville M., Sanchez R., Emonnot L., Sinnett D., Ziv E., Delvin E., Couture P., Marcil V., Sane A. T. (2011) Expression of Sar1b enhances chylomicron assembly and key components of the coat protein complex II system driving vesicle budding. Arterioscler. Thromb. Vasc. Biol. 31, 2692–2699 [DOI] [PubMed] [Google Scholar]
- 34. Charcosset M., Sassolas A., Peretti N., Roy C. C., Deslandres C., Sinnett D., Levy E., Lachaux A. (2008) Anderson or chylomicron retention disease: molecular impact of five mutations in the SAR1B gene on the structure and the functionality of Sar1b protein. Mol. Genet. Metab. 93, 74–84 [DOI] [PubMed] [Google Scholar]
- 35. Georges A., Bonneau J., Bonnefont-Rousselot D., Champigneulle J., Rabès J. P., Abifadel M., Aparicio T., Guenedet J. C., Bruckert E., Boileau C., Morali A., Varret M., Aggerbeck L. P., Samson-Bouma M. E. (2011) Molecular analysis and intestinal expression of SAR1 genes and proteins in Anderson's disease (Chylomicron retention disease). Orphanet. J. Rare. Dis. 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Okada T., Miyashita M., Fukuhara J., Sugitani M., Ueno T., Samson-Bouma M. E., Aggerbeck L. P. (2011) Anderson's disease/chylomicron retention disease in a Japanese patient with uniparental disomy 7 and a normal SAR1B gene protein coding sequence. Orphanet. J. Rare. Dis. 6, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Saito K., Chen M., Bard F., Chen S., Zhou H., Woodley D., Polischuk R., Schekman R., Malhotra V. (2009) TANGO1 facilitates cargo loading at endoplasmic reticulum exit sites. Cell 136, 891–902 [DOI] [PubMed] [Google Scholar]
- 38. Malhotra V., Erlmann P. (2011) Protein export at the ER: loading big collagens into COPII carriers. EMBO J. 30, 3475–3480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shimoni Y., Kurihara T., Ravazzola M., Amherdt M., Orci L., Schekman R. (2000) Lst1p and Sec24p cooperate in sorting of the plasma membrane ATPase into COPII vesicles in Saccharomyces cerevisiae. J. Cell Biol. 151, 973–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Roberg K. J., Crotwell M., Espenshade P., Gimeno R., Kaiser C. A. (1999) LST1 is a SEC24 homologue used for selective export of the plasma membrane ATPase from the endoplasmic reticulum. J. Cell Biol. 145, 659–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mansbach C. M., Siddiqi S. A. (2010) The biogenesis of chylomicrons. Annu. Rev. Physiol. 72, 315–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Siddiqi S. A., Gorelick F. S., Mahan J. T., Mansbach C. M., 2nd (2003) COPII proteins are required for Golgi fusion but not for endoplasmic reticulum budding of the pre-chylomicron transport vesicle. J. Cell Sci. 116, 415–427 [DOI] [PubMed] [Google Scholar]
- 43. Siddiqi S., Saleem U., Abumrad N. A., Davidson N. O., Storch J., Siddiqi S. A., Mansbach C. M., 2nd. (2010) A novel multiprotein complex is required to generate the prechylomicron transport vesicle from intestinal ER. J. Lipid Res. 51, 1918–1928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Neeli I., Siddiqi S. A., Siddiqi S., Mahan J., Lagakos W. S., Binas B., Gheyi T., Storch J., Mansbach C. M., 2nd. (2007) Liver fatty acid-binding protein initiates budding of pre-chylomicron transport vesicles from intestinal endoplasmic reticulum. J. Biol. Chem. 282, 17974–17984 [DOI] [PubMed] [Google Scholar]
- 45. Siddiqi S. A., Mansbach C. M., 2nd. (2008) PKCζ-mediated phosphorylation controls budding of the pre-chylomicron transport vesicle. J. Cell Sci. 121, 2327–2338 [DOI] [PubMed] [Google Scholar]
- 46. Siddiqi S. A., Mahan J., Siddiqi S., Gorelick F. S., Mansbach C. M., 2nd. (2006) Vesicle-associated membrane protein 7 is expressed in intestinal ER. J. Cell Sci. 119, 943–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kim J., Hamamoto S., Ravazzola M., Orci L., Schekman R. (2005) Uncoupled packaging of amyloid precursor protein and presenilin 1 into coat protein complex II vesicles. J. Biol. Chem. 280, 7758–7768 [DOI] [PubMed] [Google Scholar]
- 48. Rahim A., Nafi-valencia E., Siddiqi S., Basha R., Runyon C. C., Siddiqi S. A. (2012) Proteomic analysis of the very low density lipoprotein (VLDL) transport vesicles. J. Proteomics 75, 2225–2235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ye J., Li J. Z., Liu Y., Li X., Yang T., Ma X., Li Q., Yao Z., Li P. (2009) Cideb, an ER- and lipid droplet-associated protein, mediates VLDL lipidation and maturation by interacting with apolipoprotein B. Cell. Metab. 9, 177–190 [DOI] [PubMed] [Google Scholar]
- 50. Li X., Ye J., Zhou L., Gu W., Fisher E. A., Li P. (2012) Opposing roles of cell death-inducing DFF45-like effector B and perilipin 2 in controlling hepatic VLDL lipidation. J. Lipid Res. 53, 1877–1889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen Z., Norris J. Y., Finck B. N. (2010) Peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) stimulates VLDL assembly through activation of cell death-inducing DFFA-like effector B (CideB). J. Biol. Chem. 285, 25996–26004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gong J., Sun Z., Li P. (2009) CIDE proteins and metabolic disorders. Curr. Opin. Lipidol. 20, 121–126 [DOI] [PubMed] [Google Scholar]
- 53. Siddiqi S. A., Siddiqi S., Mahan J., Peggs K., Gorelick F. S., Mansbach C. M., 2nd (2006) The identification of a novel endoplasmic reticulum to Golgi SNARE complex used by the prechylomicron transport vesicle. J. Biol. Chem. 281, 20974–20982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Thorngate F. E., Raghow R., Wilcox H. G., Werner C. S., Heimberg M., Elam M. B. (1994) Insulin promotes the biosynthesis and secretion of apolipoprotein B-48 by altering apolipoprotein B mRNA editing. Proc. Natl. Acad. Sci. U.S.A. 91, 5392–5396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yellaturu C. R., Deng X., Cagen L. M., Wilcox H. G., Mansbach C. M., 2nd, Siddiqi S. A., Park E. A., Raghow R., Elam M. B. (2009) Insulin enhances post-translational processing of nascent SREBP-1c by promoting its phosphorylation and association with COPII vesicles. J. Biol. Chem. 284, 7518–7532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Siddiqi S., Mani A. M., Siddiqi S. A. (2010) The identification of the SNARE complex required for the fusion of VLDL-transport vesicle with hepatic cis-Golgi. Biochem. J. 429, 391–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Siddiqi S., Siddiqi S. A., Mansbach C. M., 2nd. (2010) Sec24C is required for docking the prechylomicron transport vesicle with the Golgi. J. Lipid Res. 51, 1093–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Oh H. Y., Namkoong S., Lee S. J., Por E., Kim C. K., Billiar T. R., Han J. A., Ha K. S., Chung H. T., Kwon Y. G., Lee H., Kim Y. M. (2006) Dexamethasone protects primary cultured hepatocytes from death receptor-mediated apoptosis by upregulation of cFLIP. Cell. Death Differ. 13, 512–523 [DOI] [PubMed] [Google Scholar]
- 59. Stagg S. M., LaPointe P., Razvi A., Gürkan C., Potter C. S., Carragher B., Balch W. E. (2008) Structural basis for cargo regulation of COPII coat assembly. Cell 134, 474–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Aridor M., Bannykh S. I., Rowe T., Balch W. E. (1999) Cargo can modulate COPII vesicle formation from the endoplasmic reticulum. J. Biol. Chem. 274, 4389–4399 [DOI] [PubMed] [Google Scholar]
- 61. Kim S. D., Pahuja K. B., Ravazzola M., Yoon J., Boyadjiev S. A., Hammamoto S., Schekman R., Orci L., Kim J. (2012) SEC23-SEC31 the interface plays critical role for export of procollagen from the endoplasmic reticulum. J. Biol. Chem. 287, 10134–10144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jin L., Pahuja K. B., Wickliffe K. E., Gorur A., Baumgärtel C., Schekman R., Rape M. (2012) Ubiquitin-dependent regulation of COPII coat size and function. Nature 482, 495–500 [DOI] [PMC free article] [PubMed] [Google Scholar]