

Background: Macrophage-specific apolipoprotein E (apoE) secretion has been implicated as protective in atherosclerosis.
Results: Protein kinase C (PKC) inhibition or knockdown in macrophages inhibits apoE secretion at a post-Golgi, vesicular transport level and is mediated by MARCKS.
Conclusion: Classical PKC isoforms regulate apoE secretion.
Significance: Current drug therapies targeting PKC may have an off-target effect on macrophage apoE secretion and atherosclerosis.
Keywords: Apolipoproteins, Atherosclerosis, Macrophages, Protein Kinase C (PKC), Protein Secretion
Abstract
Macrophage-specific apolipoprotein E (apoE) secretion plays an important protective role in atherosclerosis. However, the precise signaling mechanisms regulating apoE secretion from primary human monocyte-derived macrophages (HMDMs) remain unclear. Here we investigate the role of protein kinase C (PKC) in regulating basal and stimulated apoE secretion from HMDMs. Treatment of HMDMs with structurally distinct pan-PKC inhibitors (calphostin C, Ro-31-8220, Go6976) and a PKC inhibitory peptide all significantly decreased apoE secretion without significantly affecting apoE mRNA or apoE protein levels. The PKC activator phorbol 12-myristate 13-acetate (PMA) stimulated apoE secretion, and both PMA-induced and apoAI-induced apoE secretion were inhibited by PKC inhibitors. PKC regulation of apoE secretion was found to be independent of the ATP binding cassette transporter ABCA1. Live cell imaging demonstrated that PKC inhibitors inhibited vesicular transport of apoE to the plasma membrane. Pharmacological or peptide inhibitor and knockdown studies indicate that classical isoforms PKCα/β and not PKCδ, -ϵ, -θ, or -ι/ζ isoforms regulate apoE secretion from HMDMs. The activity of myristoylated alanine-rich protein kinase C substrate (MARCKS) correlated with modulation of PKC activity in these cells, and direct peptide inhibition of MARCKS inhibited apoE secretion, implicating MARCKS as a downstream effector of PKC in apoE secretion. Comparison with other secreted proteins indicated that PKC similarly regulated secretion of matrix metalloproteinase 9 and chitinase-3-like-1 protein but differentially affected the secretion of other proteins. In conclusion, PKC regulates the secretion of apoE from primary human macrophages.
Introduction
Apolipoprotein E (apoE) is a 34-kDa multifunctional protein that plays a critical role in processes relevant to atherosclerosis (1–3). ApoE mediates the clearance of triglyceride-rich lipoproteins, inhibits proliferation of smooth muscle cells and lymphocytes, contributes to antigen presentation, and promotes cholesterol efflux from foam cell macrophages (3–9). ApoE knock-out mice develop hyperlipidemia and severe atherosclerotic lesions (10, 11). Transplantation of apoE-expressing bone marrow into apoE knock-out mice results in a marked decrease in diet-induced atherosclerosis in these mice (12). In contrast, more recent studies demonstrate a strong association between elevated plasma apoE and increased risk of cardiovascular death in the elderly (13). These apparently disparate effects may relate to the differences in function between macrophage-derived apoE and apoE derived from other cellular sources (14) and highlight the importance of understanding the regulation of macrophage apoE secretion.
It is known that newly synthesized apoE follows the classical secretory pathway, whereby it is trafficked into the endoplasmic reticulum, post-translationally glycosylated and sialylated in the Golgi network, and transported in vesicles to the plasma membrane for secretion (1, 8, 15, 16) or reinternalization and resecretion or degradation (2, 17–19). ApoE secretion is a constitutive process controlled by the ATP-binding cassette transporter, ABCA1 (20, 21). In macrophages, apoE is synthesized in excess of cellular requirements (22). The balance between secretion and degradation of apoE depends on a range of factors, including the presence of secretory stimuli, such as high density lipoprotein (HDL) and apoAI (23–26).
Recently, our laboratory has demonstrated roles for phospholipase C, protein kinase A (PKA), intracellular calcium (Ca2+), and protein phosphatase 2B (PP2B)2 in regulating apoE secretion from macrophages (16, 27). In macrophages, apoE-containing vesicles are associated with the microtubular network, and perturbing this network inhibits apoE secretion (16). Stimulation of PKA activity did not enhance apoE secretion, suggesting that additional pathways were active. Because PKA and protein kinase C (PKC) interact in various cell signaling networks (28, 29) and PKC is known to be activated downstream of phospholipase C (30), it is likely that PKC plays a role in apoE secretion. A previous study indirectly implicated PKC in apoE secretion by demonstrating that phorbol ester inhibited apoE secretion in mouse macrophages secondary to decreased apoE synthesis (31). However, the PKC-independent effects of prolonged phorbol 12-myristate 13-acetate (PMA) exposure complicate interpretation of these data.
A direct role for PKC in protein secretion per se is supported by its established roles in mediating the secretion of various cargoes, such as glutamate and noradrenaline from neuronal cell lines (32–35), mucin from colonic tumor cell lines (36), histamine from rat basophilic leukemia mast cells (37), and insulin and glucagon from pancreatic cells (38–41). Furthermore, PKC has been reported to interact with a number of proteins associated with intracellular transport (e.g. actin, tubulin, β′-COP, p62-ZIP, and myristoylated alanine-rich protein kinase C substrate (MARCKS)) (42).
PKC is a member of the serine/threonine family of kinases with at least 11 isoforms classified into three groups: classical (α, β, γ), novel (δ, ϵ, θ, η), and atypical (ζ, ι, μ, λ) (30, 43). Macrophages express the α, β, δ, ϵ, θ, ζ, ι, and λ PKC isoforms (44). Clarifying the role of specific PKC isoform(s) in apoE secretion may be of particular clinical relevance because PKC activation has been observed in various diseases, and inhibition of PKC has been investigated for treatment of diabetic peripheral retinopathy (e.g. ruboxistaurin/LY333531), cancer (e.g. UCN-01, CGP41251), and psoriasis (e.g. AEB071) (45–48). The biological consequences of inhibition of PKC may be both diverse and clinically important. Given differences in the isoform expression of PKC in different cell types, data specific to primary human macrophages are important.
The present study has investigated the role of PKC in regulating the secretion of apoE from primary human macrophages. We identify for the first time likely roles for the classical PKC isoforms in this process, establish that PKC acts independently of ABCA1, and report a likely role for MARCKS as a downstream mediator of this process.
EXPERIMENTAL PROCEDURES
Materials
Calphostin C (CalpC), Ro-31-8220, bisindolylaimeide I (BisI), Gö6976, PMA, 4-α-phorbol, and PKC isoform-specific inhibitory peptides (to PKCϵ, -θ, and -ι/ζ) were purchased from Merck Australia. The broad PKC inhibitory peptide (fragment 19–36), BAPTA-AM, 2-aminoethoxydiphenylborate (2-APB), PD98059, and SB203580 were from Sigma. BIO-11000 was synthesized by GL Biochem (Shanghai). The LY379196 compound was provided by Lilly (Grant ExNCR: B7A-AYV003). Antibodies raised against PKCα/β, PKCβ, PKCδ, fibronectin, and HSP90 were from BD Biosciences. Phospho-MARCKS (Ser-152/156), MARCKS, phospho-ERK44/42 (Thr-202/Tyr-204), ERK44/42, phospho-p38 MAPK (Thr-180/Tyr-182), and p38 MAPK antibodies were from Cell Signal Technology. Stealth siRNA, non-silencing control, and RNAiMax were from Invitrogen. Human apoAI, acetylated LDL, and lipoprotein-deficient serum were all prepared as described previously (49). The apoE-green fluorescent protein (GFP) construct was generated as described previously (16).
Culture of Human Monocyte-derived Macrophages (HMDMs) and Inhibitor Treatment
Human monocytes were isolated through density gradient centrifugation from buffy coat preparations from healthy donors of the New South Wales Red Cross and differentiated for 7–9 days into HMDMs as described previously (26). For inhibitor treatment and pulse-chase experiments, HMDMs were enriched with cholesterol by incubating them with RPMI 1640 medium supplemented with 10% (v/v) lipoprotein-deficient serum and 50 μg/ml acetylated LDL for 2 days to maximize apoE synthesis (26, 50–54). For inhibitor experiments, HMDMs were incubated with the indicated concentrations of PKC inhibitors or corresponding vehicle (DMSO) control in RPMI medium containing 0.1% (w/v) BSA. Levels of secreted apoE in the medium were measured by ELISA and/or Western blot, and cellular apoE protein levels were analyzed by Western blot, as described previously (16). Western blots were quantified using ImageJ version 1.42b (21). Total RNA from HMDMs was isolated using TRISure and quantified by real-time RT-PCR as described previously (21). All experiments were conducted in triplicate cultures, and treatments were expressed relative to control. All experiments were repeated using multiple independent donors of primary HMDMs.
Live Cell Imaging and Analysis
Cultured HMDMs (2 × 105 cells/ml) were transiently transfected with 3–5 μg of apoE-GFP cDNA using the Amaxa transfection system according to the manufacturer's instructions and incubated overnight. HMDMs were treated with 200 nm CalpC or 5 μm Ro-31-8220 for 10–20 min prior to imaging. Live cell imaging of apoE-GFP-positive cells for 3–5 min was performed using a Leica Microsystems TCS SP5 confocal laser-scanning microscope equipped with a ×63 water immersion lens and a heated stage. The vesicular speed of 100 representative apoE-GFP-containing vesicles from at least two independent buffy coat preparations of HMDMs was quantified using Imaris software (Bitplane AG).
siRNA Knockdown Experiments
HMDMs were transfected with Stealth siRNA (Invitrogen) targeting PKCα/β or PKCδ isoforms using RNAiMax transfection reagent (Invitrogen) according to the manufacturer's instructions and incubated for 2 days. Cells were cultured in RPMI + 10% (v/v) human serum for a further 2–4 days and then washed and incubated with RPMI medium containing 0.1% (w/v) BSA for 1 h prior to harvesting medium and cells. Levels of PKC isoform expression were determined by Western blot analysis and real-time PCR. Cellular apoE and secreted apoE were detected using Western blot analysis.
Cell Viability
Cell viability was routinely assessed by measuring the leakage of lactate dehydrogenase into the medium as previously described (21) and was greater than 80% for all experiments.
PKC Activity Assays
In preliminary experiments, PKC activity was determined using the PKC assay kit (Millipore) as per the manufacturer's instructions in order to confirm that concentrations of inhibitors were effective in HMDMs. The PKC inhibitors BisI, CalpC, Ro-31-8220, and Gö6976 inhibited PKC activity by between 28 and 95%, depending on the specific inhibitor and concentration, whereas acute exposure to PMA (40 min at 250 nm) increased PKC activity (by a mean of 295%) (data not shown).
Statistical Analysis
Data are presented as the mean ± S.E. of the indicated number of independent experiments or the mean ± S.D. of one experiment representative of at least two separate experiments, each experiment using independent HMDM donors. Data were analyzed by non-parametric or parametric tests as appropriate. For non-parametric data, significant difference between control and multiple treatment groups was assessed by the Kruskal-Wallis test, and comparison of two groups was performed by the Mann-Whitney U test. Time course studies were analyzed using two-way analysis of variance with treatment as the between-group effect and time as the within-group effect. Analyses were performed using GraphPad Prism version 6.0. A two-tailed p < 0.05 was considered statistically significant.
RESULTS
Structurally Distinct Pan-PKC Inhibitors Inhibit Basal ApoE Secretion from HMDMs
To investigate whether PKC plays a role in apoE secretion from HMDMs, the effect of several pan-PKC inhibitors with differing sites of action was investigated. CalpC is a light-sensitive perylene quinone PKC inhibitor that targets the PKC regulatory domain (55, 56). Both Ro-31-8220 and BisI target the PKC catalytic domain containing the ATP binding site (45, 55). The PKC peptide fragment 19–36 inhibits PKC by binding to its pseudosubstrate domain. BisI (data not shown), CalpC (Fig. 1A), and Ro-31-8220 (Fig. 1B) all caused a rapid (within 1 h) dose-dependent decrease in apoE secretion from cholesterol-loaded HMDMs. The PKC inhibitory peptide also significantly inhibited apoE secretion, albeit less effectively (Fig. 1C). The effect of CalpC and Ro-31-8220 was similar in HMDMs that were not cholesterol-enriched (supplemental Fig. 1). Although CalpC, Ro-31-8220 and PKC inhibitory peptide significantly inhibited apoE secretion, they only minimally affected cellular levels of apoE protein (supplemental Fig. 2 A– C, respectively) and mRNA (supplemental Fig. 2, D–F, respectively), suggesting a direct effect on secretion rather than an effect secondary to decreased synthesis.
FIGURE 1.

Structurally distinct pan-PKC inhibitors inhibit apoE secretion from HMDMs in a dose-dependent manner. Primary HMDMs were differentiated and enriched with cholesterol prior to treatment with PKC inhibitors (CalpC (A), Ro-31-82220 (B), and PKC inhibitory peptide (C)) at specified concentrations or vehicle (DMSO) control. ApoE released into the culture medium was measured after 1 h by ELISA and is expressed relative to control. Western blots of representative experiments are depicted above the corresponding bar graphs. Data represent mean ± S.E. (error bars) of at least three independent experiments from different donors, each containing triplicate cultures. *, p < 0.0001, Kruskal-Wallis test.
To determine whether PKC inhibition exerted a direct effect on apoE secretion independent of ongoing apoE synthesis, pulse-chase experiments were performed as described previously (16, 21). In brief, cholesterol-loaded HMDMs were incubated with [35S]methionine for 3 h in methionine/cysteine-free medium, followed by a chase in methionine/cysteine-replete medium not containing [35S]methionine, and levels of cell-associated and secreted [35S]apoE were determined and expressed relative to starting material (Fig. 2). Despite donor-specific differences in the rates of apoE secretion (compare controls in Fig. 2, A and D), both CalpC and Ro-31-8220 consistently and directly inhibited secretion of [35S]apoE within 1 h by 58% (from 20.7 ± 1.2 to 8.7 ± 2.5% secretion; Fig. 2A; p < 0.001) and 51% (from 10.6 ± 0.4 to 5.1 ± 0.5% secretion; Fig. 2D; p < 0.001), respectively. There were no detectable effects on levels of cell-associated apoE (Fig. 2, B and E) or calculated degradation during PKC inhibition (Fig. 2, C and F).
FIGURE 2.

Pulse-chase metabolic labeling studies demonstrate that CalpC and Ro-31-8220 directly inhibit secretion of preformed 35S-labeled apoE. HMDMs were differentiated, cholesterol-enriched, and labeled with [35S]methionine/cysteine, as described under “Experimental Procedures,” and chased in medium containing 0.1% BSA for 3 h. Cell-associated and secreted [35S]apoE was immunoprecipitated and quantified using phosphorimaging after SDS-PAGE, and net degradation of apoE was calculated by deducting both secreted and cellular apoE for each time point from the original apoE pool available at time 0 (t0) (before chase). Data are mean ± S.D. (error bars) of triplicate cultures from one experiment representative of two different donors. A and D, secreted [35S]apoE, percentage of starting material; B and E, cell-associated apoE; C and F, net degradation of apoE. *, p < 0.001 for difference between treatment and control at respective time points (two-way analysis of variance with post hoc Bonferroni correction).
PKC Inhibition Immobilizes ApoE-containing Vesicles in Human Macrophages
ApoE is extensively glycosylated, and the presence of multiple glycoforms on two-dimensional electrophoresis is an indicator of normal transport and processing through the Golgi and trans-Golgi network (15, 57). Two-dimensional electrophoresis analysis of cellular apoE after PKC inhibition showed no significant change in the glycoform profile (supplemental Fig. 3), suggesting that PKC inhibition effects are post-Golgi.
We have previously reported that in murine RAW macrophages, apoE is transported to the plasma membrane in discrete vesicles and that inhibition of PKA leads to immobilization of the movement of these vesicles (16), whereas PP2B inhibition had no effect (16, 27). To investigate the effect of PKC inhibition on such vesicular transport, human macrophages were transiently transfected with apoE-GFP cDNA. Live cell imaging of control HMDMs demonstrated rapid, multidirectional movement of apoE-containing vesicles, including movement to the plasma membrane (supplemental Movies 1 and 3 and Fig. 3A). This movement was arrested in the presence of 200 nm CalpC (supplemental Movie 2 and Fig. 3B) and 5 μm Ro-31-8220 (supplemental Movie 4). Quantification of the speed of vesicular movement in control versus CalpC- or Ro-31-8220-treated cells demonstrated a marked decrease in the average speed of apoE-containing vesicles from 0.42 μm/s in control cells to 0.14 μm/s (CalpC; p < 0.0001) and 0.15 μm/s (Ro-31-8220; p < 0.0001) in cells treated with PKC inhibitors (Fig. 3C). This result supports a direct effect of PKC inhibition on apoE vesicular traffic and, in this respect, indicates that PKC is similar to PKA and dissimilar to PP2B in its site of regulation of apoE secretion in macrophages.
FIGURE 3.

Inhibition of PKC arrests vesicular movement of apoE-containing vesicles. HMDMs were transiently transfected with apoE-GFP cDNA and cultured for 24 h prior to performing microscopy in RPMI containing 0.1% BSA. Individual cells expressing apoE-GFP were identified using confocal Leica SP5 microscope on a heated stage, and GFP-positive vesicles were tracked and average speed was determined after exposure to CalpC (200 nm) or Ro-31-8220 (5 μm) for 10–20 min prior to imaging. A and B show a representative sequence of still images tracking apoE-GFP vesicles of the same cell before (A) and after (B) the addition of CalpC. Average speed (C) of vesicles was obtained from 100 representative vesicles from at least two independent macrophage preparations from different donors. Statistical analysis was performed using the Mann-Whitney U test with p < 0.0001 relative to control.
Stimulation of ApoE Secretion by ApoAI Is PKC-dependent and ABCA1-independent
ApoAI stimulates apoE secretion by redirecting apoE away from degradation toward secretion (21, 25). Previous studies indicated that apoAI activates phospholipase C, which in turn activates PKCα, and that HDL stimulates PKC during its stimulation of cholesterol efflux from cells (58–60). However, it is unclear whether PKC regulates apoAI-mediated apoE secretion. In order to investigate this, HMDMs were pretreated with PKC inhibitor CalpC (Fig. 4A) or Ro-31-8220 (data not shown) prior to the addition of apoAI. The stimulation of apoE secretion by apoAI was markedly inhibited by pretreatment with CalpC. CalpC decreased basal and apoAI-stimulated apoE secretion to similar extents (−52.2% versus −53.5%, respectively, n = 3 experiments). ApoAI did not significantly increase apoE secretion when cells were pretreated with CalpC (p = 0.2, n = 3 experiments). Overall, these data indicate that PKC activity is required for effective stimulation of both basal and apoAI-stimulated apoE secretion.
FIGURE 4.

ApoAI-mediated apoE secretion is PKC-dependent, and this process is ABCA1-independent. HMDMs isolated from healthy donors (A) and Tangier disease (TD) patients (B) were acutely exposed to ApoAI (25 μg/ml) in the presence or absence of 200 nm CalpC. Levels of secreted apoE were detected by Western blot analysis and quantified relative to control. Results shown represent mean ± S.E. (error bars) of three different donors, each containing triplicate cultures. Statistical analysis was performed using the Mann-Whitney U test. *, p < 0.01, control versus ApoAI, ApoAI versus ApoAI + CalpC, and control versus CalpC.
From previous studies, it is known that apoAI-induced apoE secretion is ABCA1-independent (21, 61). However, because PKC activity has been implicated in ABCA1-mediated cholesterol efflux to apoAI, we investigated whether PKC-dependent apoAI-mediated apoE secretion was dependent on ABCA1 activity. HMDMs were isolated from three separate Tangier disease subjects, who have documented extremely low HDL cholesterol, with homozygous and compound heterozygous mutations in the ABCA1 gene (Patient 1, homozygous for c.4121C>T (R1270X) in exon 27 (27, 62); Patients 2 and 3, c.1759C>T (p.Arg587Trp) in exon 14 and c.4957_4961del (p.Val1653CysfsX48) in exon 37 (63)). In these studies, CalpC inhibited apoE secretion in HMDMs from patients with Tangier disease (Fig. 4B) in a similar manner as in HMDMs from a healthy individual (Fig. 4A), confirming that apoAI-mediated apoE secretion is regulated independently of ABCA1.
PKC Activation by PMA Increases ApoE Secretion
Our previous studies identified that stimulation of PKA activity failed to stimulate apoE secretion above basal levels (16). The effect of PKC stimulation was investigated using brief exposure to PMA, which activates classical and novel PKC isoforms by binding to their diacyl glycerol binding or regulatory sites (55). To minimize transcriptional and long term off target effects of PMA, HMDM exposure to PMA was limited to 40 min. Under these conditions, PMA induced a dose-dependent increase in apoE secretion (Fig. 5A). This was inhibited by CalpC (Fig. 5C) and Ro-31-8220 (data not shown), supporting the PKC dependence of the effect. The increased apoE secretion stimulated by PMA was independent of changes in cellular levels of apoE protein (Fig. 5B) and mRNA (data not shown). In addition, the PMA analog, 4-α-phorbol, which does not affect PKC activity, also had no effect on apoE secretion (data not shown). To investigate whether PKA, PP2B, and Ca2+ were involved in PMA-stimulated apoE secretion, we preincubated HMDMs with the PKA inhibitor H89, the PP2B inhibitor cyclosporin A, the intracellular Ca2+ chelator BAPTA-AM, and the inositol 1,4,5-trisphosphate receptor antagonist 2-APB prior to stimulation with PMA. PMA-mediated apoE secretion was PKA-dependent (Fig. 5D), PP2B-dependent (Fig. 5E), and Ca2+-dependent (Fig. 5F), suggesting that PKC acts upstream of PKA and PP2B and is dependent on intracellular Ca2+.
FIGURE 5.

Acute exposure to PMA stimulates apoE secretion from HMDMs. HMDMs were incubated with PMA (250–1000 nm) for 40 min, during which time apoE secreted into the medium was determined by ELISA (A), and cellular apoE was determined by Western blot analysis (B). HMDMs were incubated with PMA (500 nm) in the presence or absence of CalpC (C), H89 (D), cyclosporin A (E), 2-ABP (F), or BAPTA-AM (F), and levels of apoE secretion were detected using ELISA. Graphs in A and B depict mean ± S.D. (error bars) of triplicate culture from one representative experiment. Graphs in C–E represent mean ± S.E. (error bars) of three different donors, each containing triplicate cultures. The graph in F depicts mean ± S.D. (error bars) of triplicate cultures from one experiment representative of three different donors. A, *, p < 0.001, Kruskal-Wallis test. C–F, *, p < 0.01, Mann-Whitney U test.
Because secreted apoE is lipidated (61), it is possible that PKC activity regulates apoE secretion qualitatively by affecting the lipidation of secreted apoE. ApoE lipidation was measured indirectly by measuring the densities of secreted apoE using a sucrose gradient as described previously (27). Stimulation of apoE secretion using PMA or inhibition of apoE secretion using Ro-31-8220 or CalpC did not significantly affect the density of secreted apoE (1.02–1.20 g/ml) (supplemental Fig. 4). Thus, the regulation of apoE secretion by PKC is probably distinct from the processes regulating its lipidation. In other experiments, acute inhibition of PKC using Ro-31-8220 or CalpC and its acute stimulation with PMA had no demonstrable effects on the hydrolysis of VLDL triglyceride by primary macrophages or on the release of cholesterol from cholesterol-enriched primary macrophages (data not shown).
Classical PKC Isoforms, Most Likely PKCα/β, Are Involved in Basal ApoE Secretion
Macrophages express the classical PKC isoforms α and β; the novel PKC isoforms δ, ϵ, and θ; and the atypical PKC isoforms ζ, ι, and λ (44). The pan-PKC inhibitors used in Fig. 1 (CalpC, BisI, and Ro-31-8220) target both classical and novel PKC isoforms. To distinguish between classical and novel isoforms, we investigated the effect of the relatively specific classical isoform (PKCα, -β, and -γ) inhibitor Gö6976 (55, 64) on basal apoE secretion. Similar to the pan-PKC inhibitors, Gö6976 induced a dose-dependent decrease in apoE secretion (Fig. 6A) from HMDMs but exerted no significant effect on cellular apoE mRNA or protein levels (not shown). Because PKCγ is not expressed in human macrophages (44), these data indicate a role for PKCα and/or PKCβ in apoE secretion. Thus, we also explored the PKCβ inhibitor, LY379196 (Lilly). Treatment of HMDMs with the PKCβ inhibitor LY379196 (10 μm) decreased apoE secretion by 22.9 ± 2.3% (n = 4 different donors) (Fig. 6B). Although the concentration of LY379196 used here may potentially target other PKC isoforms, human macrophages often require a higher concentration of pharmacological inhibitors than many cell lines for effect, and LY379196 has been used previously in primary human macrophages at concentrations up to 10 μm (65).
FIGURE 6.

The classical PKC isoforms, PKCα and PKCβ, regulate apoE secretion from HMDMs. HMDMs were treated with the specific classical PKC inhibitor Gö6976 (A) or the PKCβ inhibitor LY379196 (B) for 1 h, and secreted apoE was detected by ELISA. Western blots of representative experiments are depicted above the corresponding bar graphs. HMDMs were transfected with Stealth siRNA oligonucleotides targeting either the classical PKC isoforms PKCα and PKCβ, PKCδ, or non-silencing control (NS control). Cells were then further cultured for 2–4 days in RPMI containing 10% human serum. HMDMs were harvested, and levels of PKCα/β (C), PKCδ (E), and secreted apoE were detected by Western blot analysis (D) and ELISA (F). HMDMs were also treated with PKCϵ translocation inhibitory peptide and PKCθ and PKCι/ζ pseudosubstrate inhibitory peptides for 1 h, and levels of apoE secretion were determined by ELISA (G) and Western blot analysis (H). A–D, mean ± S.E. (error bars) of at least four independent experiments from different donors, each containing triplicate cultures. #, p < 0.005, Mann-Whitney U test; *, p < 0.001, Kruskal-Wallis. E–H, representative experiments conducted in triplicate of at least two independent donors (mean ± S.D. (error bars)).
To confirm involvement of PKCα and/or PKCβ, HMDMs were incubated with siRNA against either PKCα or PKCβ. However, most likely due to the strong sequence homology between these PKC isoforms, siRNAs designed against either PKCα or PKCβ also targeted the opposite isoform. Treatment of HMDMs with PKCα/β siRNAs 228 and 429 resulted in a marked reduction in PKCα/β mRNA (data not shown) and protein levels (64.4 ± 8.3 and 71.8 ± 2.4%, respectively) (Fig. 6C). Both siRNAs 228 and 429 resulted in an inhibition of apoE secretion (39.1 ± 7.4 and 39.2 ± 8.0%, respectively) (Fig. 6D). There was a minor decrease in cellular apoE levels (data not shown). In contrast, knockdown of PKCδ (59.7 ± 1.5 to 68.7 ± 1.3%) in human macrophages (Fig. 6E) had no effect on apoE secretion (Fig. 6F) or cellular apoE levels (data not shown). Inhibitory peptides specific for PKCϵ (Fig. 6G), PKCθ, and PKCι/ζ (Fig. 6H) also had no effect on apoE secretion. Taken together, our data strongly support a role for classical isoforms PKCα and -β, but not PKC isoform δ, ϵ, θ, or ι/ζ, in macrophage-mediated apoE secretion.
PKC Regulates ApoE Secretion via MARCKS
To determine potential downstream targets or other signaling proteins involved in regulating PKC-mediated apoE secretion, we investigated proteins known to be involved in PKC-regulated protein secretion in other cells (66–71). HMDMs were treated with PMA, Ro-31-8220, or Gö6976 for 5 min, and the ratios of phosphoprotein/total protein were determined using Western blot analysis. PMA treatment caused a 3.3-fold increase in phospho-MARCKS, whereas Ro-31-8220 and Gö6976 resulted in a decrease by 5- and 2-fold, respectively (Fig. 7A). To further confirm a role for MARCKS, HMDMs were treated with its synthetic peptide inhibitor, BIO-11000 (72), which decreased apoE secretion (Fig. 7B). In contrast, PMA, Ro-31-8220, and Gö6976 had no effect on p38 MAPK phosphorylation (supplemental Fig. 5A), whereas its inhibitor, SB203580, had no significant effect on apoE secretion (supplemental Fig. 5B), thereby ruling out a role for p38 MAPK. Other experiments examining phosphorylation of ERK44/42 upon PKC inhibition demonstrated minimal change, and the ERK44/42 inhibitor, PD98059, did not identify any substantial effects on apoE secretion (data not shown). These results point to a prominent role for MARCKS relative to other downstream PKC targets in regulating apoE secretion.
FIGURE 7.
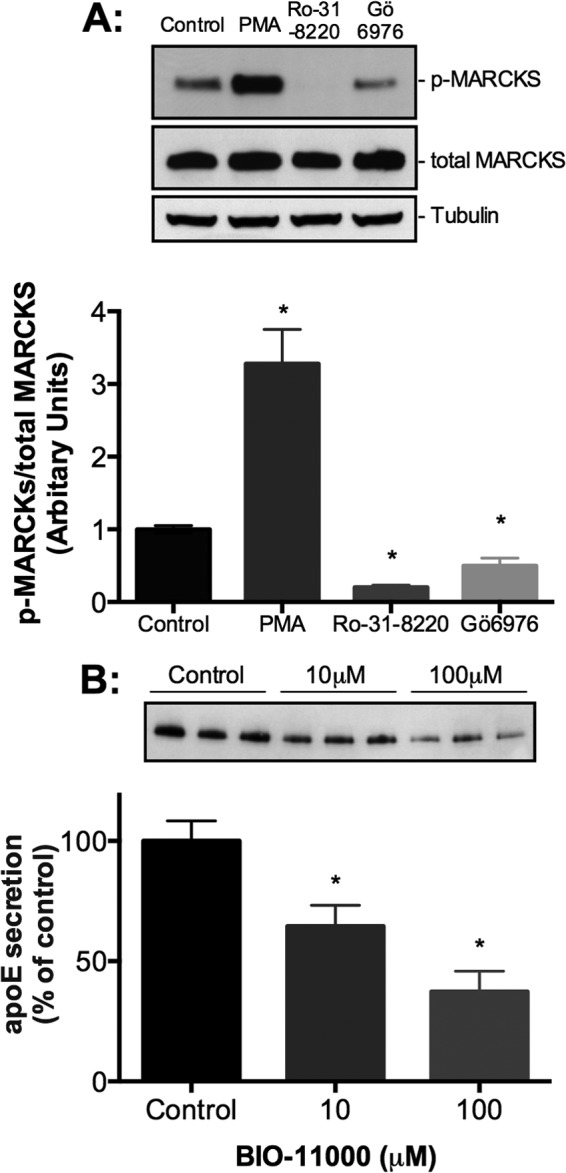
A role for MARCKS in regulating apoE secretion from HMDMs. HMDMs were incubated 500 nm PMA, 5 μm Ro-31-8220, or 500 nm Gö6976 for 5 min prior to lysis. A, levels of phospho-MARCKS (p-MARCKS), total MARCKS, and tubulin detected by Western blot analysis. Graphs demonstrate a ratio of phosphorylated protein to total protein of three independent experiments. Statistical analysis was performed using the Mann-Whitney U test. *, p < 0.01, PMA versus control, Ro-31-8220 versus control, and Gö6976 versus control. Representative Western blots from the same donor are depicted above the respective graphs. HMDMs were treated with the MARCKS inhibitor BIO-11000 (B) for 1 h, and levels of secreted apoE were determined by Western blot analysis and ELISA (data not shown). Graphs represent mean ± S.E. (error bars) of four independent donors. *, p < 0.05, Kruskal-Wallis.
Differential Regulation of Macrophage Protein Secretion by PKC
To determine whether our studies of apoE secretion from human macrophages had general implications for the secretion of other proteins, we reviewed the literature for other known constitutively secreted proteins from human macrophages and surveyed their presence in our media in preliminary experiments. We identified six other proteins that were consistently secreted by HMDMs from multiple independent donors: matrix metalloproteinase 9 (MMP9) (73), chitinase-3-like-1 protein (CHI3L1; also known as HC-gp-39) (74, 75), lysozyme (76), fibronectin (76, 77), cyclophilin A (CypA) (78), and heat shock protein (HSP) 90 (79). As summarized in Table 1, we found three distinct response profiles. MMP9 and CHI3L1 behaved similarly to apoE in demonstrating both increased secretion in response to PMA and decreased secretion with PKC inhibition. In contrast, lysozyme and fibronectin release were not stimulated by PMA, but their secretion was decreased by PKC inhibition. Last, CypA and HSP90 were unaffected by PMA or PKC inhibition. Thus, PKC modulation demonstrates a degree of specificity for the secretion of different cargoes in human macrophages.
TABLE 1.
Regulation of macrophage protein secretion by PKC activation or inhibition
Cholesterol-enriched primary HMDMs were treated with 500 nm PMA, 5 μm Ro-31-8220, or 200 nm CalpC for 1 h, and levels of the indicated proteins were determined by Western blot analysis and expressed as percentage change relative to control. Data are representative of experiments performed in triplicate cultures from at least three independent donors (mean ± S.E.). Statistical analysis was performed by Mann-Whitney U test. ↑, increase in percentage change; ↓, decrease in percentage change.
| Secreted protein | Change relative to control |
||
|---|---|---|---|
| PMA | Ro-31-8220 | CalpC | |
| % | % | % | |
| ApoE | ↑ 61 ± 19a | ↓ 48 ± 12b | ↓ 56 ± 6b |
| MMP9 | ↑ 32 ± 8a | ↓ 44 ± 13b | ↓ 58 ± 8b |
| CHI3L1 | ↑ 43 ± 17a | ↓ 56 ± 10b | ↓ 53 ± 9b |
| Lysozyme | 10 ± 9 | ↓ 29 ± 5b | ↓ 42 ± 20b |
| Fibronectin | −6 ± 15 | ↓ 51 ± 15b | ↓ 67 ± 12a |
| CypA | −12 ± 18 | −29 ± 29 | −38 ± 76 |
| HSP90 | −3 ± 7 | 7 ± 8 | −0.13 ± 54 |
a p < 0.01.
b p < 0.05.
DISCUSSION
This is the first report of a role for the classical PKCα/β isoforms and MARCKS in regulating apoE secretion from any cell type and the first to demonstrate a specific role for PKC in regulating vesicular protein transport in HMDMs. Importantly, these results also confirm PKC as a signaling target capable of enhancing apoE secretion from HMDMs.
A range of structurally diverse chemical PKC inhibitors, a PKC inhibitory peptide, and PKCα/β isoform siRNA knockdown all inhibited apoE secretion from HMDMs and clearly indicate a role for PKC in this process. Our results show that PKC inhibition directly regulates apoE secretion. PKC inhibition prevented the secretion of recently synthesized [35S]apoE and inhibited transport of apoE-GFP-containing vesicles but did not affect apoE glycoform distribution, suggesting that PKC acts on the transport of apoE after completion of its glycosylation and sialylation in the Golgi and trans-Golgi networks. Levels of cell-associated apoE were minimally affected by PKC inhibition. This may be due to efficient degradation of unsecreted apoE or the relatively small size of the retained pool relative to cellular apoE, which limits the sensitivity with which small degrees of accumulation could be detected under our experimental conditions.
A role for PKA, PP2B, and phospholipase C signaling pathways in regulation of apoE secretion has only recently been elucidated (16, 27). PKC and PKA share a number of similarities in their effects on apoE secretion. Inhibition of PKA and PKC similarly directly inhibit apoE secretion without materially affecting apoE mRNA or cell apoE protein levels, and both profoundly inhibit the movement of apoE-containing vesicles in macrophages. These results clearly distinguish the effects of PKC from those of PP2B, whose inhibition with cyclosporin A reduced apoE secretion and degradation, resulting in substantial apoE accumulation in the cell. In addition, PP2B inhibition did not affect the transport of apoE-containing vesicles. PKC, however, differs from PKA in that stimulation of PKC activity with PMA rapidly increased apoE secretion, whereas stimulation of PKA activity did not (16). These distinctions indicate that PKC may have a unique role in the stimulation of apoE secretion in human macrophages, providing a signaling pathway by which secretion of apoE can be enhanced. Furthermore, the stimulation of apoE secretion by PMA is decreased by both PKA and PP2B inhibition (Fig. 5, D and E), suggesting that PKC is likely to be upstream of PKA and PP2B in the signaling cascade regulating apoE secretion. Further, chelation of Ca2+ by BAPTA-AM and blocking Ca2+ release from the endoplasmic reticulum using the inositol 1,4,5-trisphosphate receptor antagonist 2-APB both decreased PMA-mediated apoE secretion (Fig. 5F), confirming the importance of Ca2+ in regulating this process.
The stimulation of apoE secretion by PMA implies that classical and novel PKC isoforms, but not atypical isoforms, are involved in stimulated apoE secretion. This is also supported by our data demonstrating that CalpC, Ro-31-8220, BisI, Gö6976, LY379196, and PKC inhibitory peptide all inhibited apoE secretion based on their reported specificities (45, 55, 65). The effects of Gö6976 (inhibitor of classical PKC isoforms) and LY379196 (PKCβ inhibitor) on apoE secretion (decrease in apoE secretion) imply that PKCα and/or PKCβ are most likely to regulate apoE secretion in HMDMs because these cells do not express PKCγ (44). The use of pharmacological inhibitors to study cell signaling molecules, such as PKC, can be complicated by off-target effects, particularly at higher concentrations. This problem is reduced, but not eliminated, by the use of multiple inhibitors at the lowest effective concentrations, as we have done in this study. The role of PKCα/β was further confirmed by our siRNA studies. Because both PKCα and PKCβ share ∼80% homology, designing specific siRNA oligonucleotides that exclusively target one of the isoforms is technically challenging, and we found that all oligonucleotides, although specifically designed to affect only one isoform, also inhibited the other. Thus, although we cannot be conclusive about the relative importance of PKCα and PKCβ in regulating apoE secretion or the possibility of functional redundancy of the two isoforms, a role for one or both of these isoforms is clear. siRNA knockdown of the novel PKC isoform, PKCδ, and peptides inhibitory to PKC isoform δ, θ, or ι/ζ had no effect on apoE secretion from human macrophages (Fig. 6, E–H). Future studies using cells from PKCα or PKCβ knock-out mice may be useful to elucidate the relative importance of either or both of these isoforms in regulating apoE secretion.
The expression and function of specific PKC isoforms varies between cells, tissues, and species (30). For example, PKCα is expressed in abundance in the human leukemia cell line (HL-60), whereas it has relatively minor expression in rat brain cells (80) and in primary macrophages.3 Thus, studies using primary human macrophages, although technically challenging, appear to be essential for clarifying PKC and isoform specificity in relation to apoE secretion.
There are very few studies of vesicular transport in primary human macrophages. Here, we transfected primary human macrophages with apoE-GFP to track apoE-containing vesicles. Inhibition of PKC with pharmacological inhibitors arrested apoE-containing vesicles rapidly, identifying the site of action at the vesicular transport level. A number of vesicular transport proteins have been linked to PKC activation in cell systems other than macrophages, including ADP-ribosylation factor, β′-COP, and MARCKS. Our data indicate that the PKC activation or inhibition corresponds directly with the degree of MARCKS phosphorylation (Fig. 7A) and is supported by inhibited apoE secretion by the peptide inhibitor, BIO-11000. MARCKS regulates the hypersecretion of mucus in respiratory diseases, such as asthma and cystic fibrosis (70, 81). Phosphorylation of MARCKS allows movement to the cytoplasm, binding to chaperone HSP70, which engages the cysteine string protein located on the surface of the secretory vesicles containing mucin (69, 82). Because apoE is structurally a mucin, we hypothesize a similar model for apoE secretion, and future research will delineate the exact chaperone molecules and mechanisms through which MARCKS regulates both basal and stimulated apoE secretion.
PKC has effects on the secretion of other proteins released from human macrophages but demonstrates some selectivity. We confirmed that MMP9 secretion from human macrophages is PKC-dependent as previously reported in other cell types (83, 84). Furthermore, consistent with previous reports in macrophage cell lines, PMA did not increase lysozyme secretion (85). However, PMA did not increase fibronectin secretion from primary human macrophages as previously reported in other cell types (86, 87). The effects of PKC activation and inhibition on CHI3L1 secretion from human macrophages observed in this study are very similar to that of apoE secretion, and these findings are novel. A potential role for CHI3L1 has been identified in Gaucher disease and atherosclerosis, where distinct subsets of macrophages express CHI3L1 and its homolog, chitotriosidase (88). However, the mechanisms through which CHI3L1 mediates these disease processes remain to be elucidated.
PKC plays a role in various disease states, such as bipolar disorders, diabetes, arteriosclerosis, Alzheimer disease, cardiac hypertrophy, and various types of cancer (30, 43, 64). A number of animal models and clinical trials have been conducted with pharmacological inhibitors of PKC to treat disorders, such as diabetic peripheral retinopathy (e.g. LY333531/ruboxistaurin), cancer (e.g. UCN-01 and CGP41251), and autoimmune diseases, such as psoriasis (e.g. AEB071) (45–48). Based on our results, the effects of such inhibition on protein secretion will vary with the protein cargo and the PKC isoforms affected. The inhibition of apoE secretion by PKC inhibitors could have complex effects on atherosclerosis and immune function.
In summary, this is the first study to identify a role for PKC and MARCKS in regulating both basal and stimulated apoE secretion from primary human macrophages. Pharmacological inhibition of specific PKC isoform(s) and siRNA knockdown of PKCα/β suggest that the classical PKC isoforms are involved in mediating apoE secretion.
Acknowledgments
We thank Dr. Donna Dinnes, Kathlen Huang, Dr. Mathew Traini, and Dr. Carmel Quinn for helpful discussions and Dr. Youra Lee, Thuan Thai, and Dr. Macarena Rodriguez for technical assistance.
This work was supported by a Program Grant 222722 (to L.K and W.J) from the National Health and Medical Research Council, Australia, a Heart Foundation Postdoctoral Fellowship PF 10S 5362 (to D.K) from the National Heart Foundation, Australia, and a Practitioner Fellowship (to J.R.B.) from the Royal Perth Hospital Medical Research Foundation. Eli Lilly Australia provided the LY379196 compound as part of their grant (ExNCR: B7A-AY-V003).

This article contains supplemental Figs. 1–5 and Movies 1–4.
D. Karunakaran, M. Kockx, and L. Kritharides, unpublished data.
- PP2B
- protein phosphatase 2B
- BAPTA-AM
- 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetate-acetoxymethyl ester
- PMA
- phorbol 12-myristate 13-acetate
- MARCKS
- myristoylated alanine-rich protein kinase C substrate
- CalpC
- calphostin C
- BisI
- bisindolylaimeide I
- 2-APB
- 2-aminoethoxydiphenylborate
- HMDM
- human monocyte-derived macrophage.
REFERENCES
- 1. Basu S. K., Goldstein J. L., Brown M. S. (1983) Independent pathways for secretion of cholesterol and apolipoprotein E by macrophages. Science 219, 871–873 [DOI] [PubMed] [Google Scholar]
- 2. Werb Z., Chin J. R., Takemura R., Oropeza R. L., Bainton D. F., Stenberg P., Taylor J. M., Reardon C. (1986) The cell and molecular biology of apolipoprotein E synthesis by macrophages. Ciba Found. Symp. 118, 155–171 [DOI] [PubMed] [Google Scholar]
- 3. Mahley R. W. (1988) Apolipoprotein E. Cholesterol transport protein with expanding role in cell biology. Science 240, 622–630 [DOI] [PubMed] [Google Scholar]
- 4. Zhang W. Y., Gaynor P. M., Kruth H. S. (1996) Apolipoprotein E produced by human monocyte-derived macrophages mediates cholesterol efflux that occurs in the absence of added cholesterol acceptors. J. Biol. Chem. 271, 28641–28646 [DOI] [PubMed] [Google Scholar]
- 5. Huang Z. H., Lin C. Y., Oram J. F., Mazzone T. (2001) Sterol efflux mediated by endogenous macrophage apoE expression is independent of ABCA1. Arterioscler. Thromb. Vasc. Biol. 21, 2019–2025 [DOI] [PubMed] [Google Scholar]
- 6. Greenow K., Pearce N. J., Ramji D. P. (2005) The key role of apolipoprotein E in atherosclerosis. J. Mol. Med. 83, 329–342 [DOI] [PubMed] [Google Scholar]
- 7. van den Elzen P., Garg S., León L., Brigl M., Leadbetter E. A., Gumperz J. E., Dascher C. C., Cheng T. Y., Sacks F. M., Illarionov P. A., Besra G. S., Kent S. C., Moody D. B., Brenner M. B. (2005) Apolipoprotein-mediated pathways of lipid antigen presentation. Nature 437, 906–910 [DOI] [PubMed] [Google Scholar]
- 8. Kockx M., Jessup W., Kritharides L. (2008) Regulation of endogenous apolipoprotein E secretion by macrophages. Arterioscler. Thromb. Vasc. Biol. 28, 1060–1067 [DOI] [PubMed] [Google Scholar]
- 9. Allan L. L., Hoefl K., Zheng D. J., Chung B. K., Kozak F. K., Tan R., van den Elzen P. (2009) Apolipoprotein-mediated lipid antigen presentation in B cells provides a pathway for innate help by NKT cells. Blood 114, 2411–2416 [DOI] [PubMed] [Google Scholar]
- 10. Nakashima Y., Plump A. S., Raines E. W., Breslow J. L., Ross R. (1994) ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler. Thromb. 14, 133–140 [DOI] [PubMed] [Google Scholar]
- 11. Reddick R. L., Zhang S. H., Maeda N. (1994) Atherosclerosis in mice lacking apoE. Evaluation of lesional development and progression. Arterioscler. Thromb. 14, 141–147 [DOI] [PubMed] [Google Scholar]
- 12. Linton M. F., Atkinson J. B., Fazio S. (1995) Prevention of atherosclerosis in apolipoprotein E-deficient mice by bone marrow transplantation. Science 267, 1034–1037 [DOI] [PubMed] [Google Scholar]
- 13. Mooijaart S. P., Berbée J. F., van Heemst D., Havekes L. M., de Craen A. J., Slagboom P. E., Rensen P. C., Westendorp R. G. (2006) ApoE plasma levels and risk of cardiovascular mortality in old age. PLoS Med. 3, e176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huang Z. H., Reardon C. A., Subbaiah P. V., Getz G. S., Mazzone T. (2013) ApoE derived from adipose tissue does not suppress atherosclerosis or correct hyperlipidemia in EKO mice. J. Lipid Res. 54, 202–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zannis V. I., McPherson J., Goldberger G., Karathanasis S. K., Breslow J. L. (1984) Synthesis, intracellular processing, and signal peptide of human apolipoprotein E. J. Biol. Chem. 259, 5495–5499 [PubMed] [Google Scholar]
- 16. Kockx M., Guo D. L., Huby T., Lesnik P., Kay J., Sabaretnam T., Jary E., Hill M., Gaus K., Chapman J., Stow J. L., Jessup W., Kritharides L. (2007) Secretion of apolipoprotein E from macrophages occurs via a protein kinase A and calcium-dependent pathway along the microtubule network. Circ. Res. 101, 607–616 [DOI] [PubMed] [Google Scholar]
- 17. Fazio S., Linton M. F., Hasty A. H., Swift L. L. (1999) Recycling of apolipoprotein E in mouse liver. J. Biol. Chem. 274, 8247–8253 [DOI] [PubMed] [Google Scholar]
- 18. Zhao Y., Yue L., Gu D., Mazzone T. (2002) Regulation of macrophage apoE expression and processing by extracellular matrix. J. Biol. Chem. 277, 29477–29483 [DOI] [PubMed] [Google Scholar]
- 19. Hasty A. H., Plummer M. R., Weisgraber K. H., Linton M. F., Fazio S., Swift L. L. (2005) The recycling of apolipoprotein E in macrophages. Influence of HDL and apolipoprotein A-I. J. Lipid Res. 46, 1433–1439 [DOI] [PubMed] [Google Scholar]
- 20. Von Eckardstein A., Langer C., Engel T., Schaukal I., Cignarella A., Reinhardt J., Lorkowski S., Li Z., Zhou X., Cullen P., Assmann G. (2001) ATP binding cassette transporter ABCA1 modulates the secretion of apolipoprotein E from human monocyte-derived macrophages. FASEB J. 15, 1555–1561 [DOI] [PubMed] [Google Scholar]
- 21. Kockx M., Rye K. A., Gaus K., Quinn C. M., Wright J., Sloane T., Sviridov D., Fu Y., Sullivan D., Burnett J. R., Rust S., Assmann G., Anantharamaiah G. M., Palgunachari M. N., Katz S. L., Phillips M. C., Dean R. T., Jessup W., Kritharides L. (2004) Apolipoprotein A-I-stimulated apolipoprotein E secretion from human macrophages is independent of cholesterol efflux. J. Biol. Chem. 279, 25966–25977 [DOI] [PubMed] [Google Scholar]
- 22. Deng J., Rudick V., Dory L. (1995) Lysosomal degradation and sorting of apolipoprotein E in macrophages. J. Lipid Res. 36, 2129–2140 [PubMed] [Google Scholar]
- 23. Basheeruddin K., Rechtoris C., Mazzone T. (1992) Transcriptional and post-transcriptional control of apolipoprotein E gene expression in differentiating human monocytes. J. Biol. Chem. 267, 1219–1224 [PubMed] [Google Scholar]
- 24. Dory L. (1991) Regulation of apolipoprotein E secretion by high density lipoprotein 3 in mouse macrophages. J. Lipid Res. 32, 783–792 [PubMed] [Google Scholar]
- 25. Bielicki J. K., McCall M. R., Forte T. M. (1999) Apolipoprotein A-I promotes cholesterol release and apolipoprotein E recruitment from THP-1 macrophage-like foam cells. J. Lipid Res. 40, 85–92 [PubMed] [Google Scholar]
- 26. Rees D., Sloane T., Jessup W., Dean R. T., Kritharides L. (1999) Apolipoprotein A-I stimulates secretion of apolipoprotein E by foam cell macrophages. J. Biol. Chem. 274, 27925–27933 [DOI] [PubMed] [Google Scholar]
- 27. Kockx M., Guo D. L., Traini M., Gaus K., Kay J., Wimmer-Kleikamp S., Rentero C., Burnett J. R., Le Goff W., Van Eck M., Stow J. L., Jessup W., Kritharides L. (2009) Cyclosporin A decreases apolipoprotein E secretion from human macrophages via a protein phosphatase 2B-dependent and ATP-binding cassette transporter A1 (ABCA1)-independent pathway. J. Biol. Chem. 284, 24144–24154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Faux M. C., Rollins E. N., Edwards A. S., Langeberg L. K., Newton A. C., Scott J. D. (1999) Mechanism of A-kinase-anchoring protein 79 (AKAP79) and protein kinase C interaction. Biochem. J. 343, 443–452 [PMC free article] [PubMed] [Google Scholar]
- 29. Maioli E., Torricelli C., Fortino V. (2006) Functional interactions of protein kinase A and C in signalling networks. A recapitulation. Cell Mol. Life Sci. 63, 637–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Steinberg S. F. (2008) Structural basis of protein kinase C isoform function. Physiol. Rev. 88, 1341–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dory L. (1993) Post-transcriptional regulation of apolipoprotein E expression in mouse macrophages by phorbol ester. Biochem. J. 292, 105–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Coffey E. T., Herrero I., Sihra T. S., Sánchez-Prieto J., Nicholls D. G. (1994) Glutamate exocytosis and MARCKS phosphorylation are enhanced by a metabotropic glutamate receptor coupled to a protein kinase C synergistically activated by diacylglycerol and arachidonic acid. J. Neurochem. 63, 1303–1310 [DOI] [PubMed] [Google Scholar]
- 33. Turner N. A., Walker J. H., Ball S. G., Vaughan P. F. (1996) Phorbol ester-enhanced noradrenaline secretion correlates with the presence and activity of protein kinase C-α in human SH-SY5Y neuroblastoma cells. J. Neurochem. 66, 2381–2389 [DOI] [PubMed] [Google Scholar]
- 34. Goodall A. R., Turner N. A., Walker J. H., Ball S. G., Vaughan P. F. (1997) Activation of protein kinase C-alpha and translocation of the myristoylated alanine-rich C-kinase substrate correlate with phorbol ester-enhanced noradrenaline release from SH-SY5Y human neuroblastoma cells. J. Neurochem. 68, 392–401 [DOI] [PubMed] [Google Scholar]
- 35. Vaughan P. F., Walker J. H., Peers C. (1998) The regulation of neurotransmitter secretion by protein kinase C. Mol. Neurobiol. 18, 125–155 [DOI] [PubMed] [Google Scholar]
- 36. Hong D. H., Forstner J. F., Forstner G. G. (1997) Protein kinase C-ϵ is the likely mediator of mucin exocytosis in human colonic cell lines. Am. J. Physiol. 272, G31–G37 [DOI] [PubMed] [Google Scholar]
- 37. Ludowyke R. I., Scurr L. L., McNally C. M. (1996) Calcium ionophore-induced secretion from mast cells correlates with myosin light chain phosphorylation by protein kinase C. J. Immunol. 157, 5130–5138 [PubMed] [Google Scholar]
- 38. Zawalich W., Brown C., Rasmussen H. (1983) Insulin secretion. Combined effects of phorbol ester and A23187. Biochem. Biophys. Res. Commun. 117, 448–455 [DOI] [PubMed] [Google Scholar]
- 39. Deeney J. T., Cunningham B. A., Chheda S., Bokvist K., Juntti-Berggren L., Lam K., Korchak H. M., Corkey B. E., Berggren P. O. (1996) Reversible Ca2+-dependent translocation of protein kinase C and glucose-induced insulin release. J. Biol. Chem. 271, 18154–18160 [DOI] [PubMed] [Google Scholar]
- 40. Efanov A. M., Zaitsev S. V., Berggren P. O. (1997) Inositol hexakisphosphate stimulates non-Ca2+-mediated and primes Ca2+-mediated exocytosis of insulin by activation of protein kinase C. Proc. Natl. Acad. Sci. U.S.A. 94, 4435–4439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. De Marinis Y. Z., Zhang E., Amisten S., Taneera J., Renström E., Rorsman P., Eliasson L. (2010) Enhancement of glucagon secretion in mouse and human pancreatic α cells by protein kinase C (PKC) involves intracellular trafficking of PKCα and PKCδ. Diabetologia 53, 717–729 [DOI] [PubMed] [Google Scholar]
- 42. Jaken S., Parker P. J. (2000) Protein kinase C binding partners. BioEssays 22, 245–254 [DOI] [PubMed] [Google Scholar]
- 43. Newton A. C. (2001) Protein kinase C. Structural and spatial regulation by phosphorylation, cofactors, and macromolecular interactions. Chem. Rev. 101, 2353–2364 [DOI] [PubMed] [Google Scholar]
- 44. Ma H. T., Lin W. W., Zhao B., Wu W. T., Huang W., Li Y., Jones N. L., Kruth H. S. (2006) Protein kinase C β and δ isoenzymes mediate cholesterol accumulation in PMA-activated macrophages. Biochem. Biophys. Res. Commun. 349, 214–220 [DOI] [PubMed] [Google Scholar]
- 45. Way K. J., Chou E., King G. L. (2000) Identification of PKC-isoform-specific biological actions using pharmacological approaches. Trends Pharmacol. Sci. 21, 181–187 [DOI] [PubMed] [Google Scholar]
- 46. Hofmann J. (2004) Protein kinase C isozymes as potential targets for anticancer therapy. Curr. Cancer Drug Targets 4, 125–146 [DOI] [PubMed] [Google Scholar]
- 47. Skvara H., Dawid M., Kleyn E., Wolff B., Meingassner J. G., Knight H., Dumortier T., Kopp T., Fallahi N., Stary G., Burkhart C., Grenet O., Wagner J., Hijazi Y., Morris R. E., McGeown C., Rordorf C., Griffiths C. E., Stingl G., Jung T. (2008) The PKC inhibitor AEB071 may be a therapeutic option for psoriasis. J. Clin. Invest. 118, 3151–3159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Danis R. P., Sheetz M. J. (2009) Ruboxistaurin. PKC-β inhibition for complications of diabetes. Expert Opin. Pharmacother. 10, 2913–2925 [DOI] [PubMed] [Google Scholar]
- 49. Kritharides L., Jessup W., Mander E. L., Dean R. T. (1995) Apolipoprotein A-I-mediated efflux of sterols from oxidized LDL-loaded macrophages. Arterioscler. Thromb. Vasc. Biol. 15, 276–289 [DOI] [PubMed] [Google Scholar]
- 50. Basu S. K., Brown M. S., Ho Y. K., Havel R. J., Goldstein J. L. (1981) Mouse macrophages synthesize and secrete a protein resembling apolipoprotein E. Proc. Natl. Acad. Sci. U.S.A. 78, 7545–7549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Basu S. K., Ho Y. K., Brown M. S., Bilheimer D. W., Anderson R. G., Goldstein J. L. (1982) Biochemical and genetic studies of the apoprotein E secreted by mouse macrophages and human monocytes. J. Biol. Chem. 257, 9788–9795 [PubMed] [Google Scholar]
- 52. Kayden H. J., Maschio F., Traber M. G. (1985) The secretion of apolipoprotein E by human monocyte-derived macrophages. Arch. Biochem. Biophys. 239, 388–395 [DOI] [PubMed] [Google Scholar]
- 53. Mazzone T., Gump H., Diller P., Getz G. S. (1987) Macrophage free cholesterol content regulates apolipoprotein E synthesis. J. Biol. Chem. 262, 11657–11662 [PubMed] [Google Scholar]
- 54. Rouis M., Nigon F., Eggerman T. L., Brewer H. B., Jr., Chapman M. J. (1990) Apolipoprotein E expression by human-monocyte-derived macrophages. Modulation by opsonised zymosan and cholesterol. Eur. J. Biochem. 189, 447–453 [DOI] [PubMed] [Google Scholar]
- 55. Hofmann J. (1997) The potential for isoenzyme-selective modulation of protein kinase C. FASEB J. 11, 649–669 [DOI] [PubMed] [Google Scholar]
- 56. Pollack I. F., Kawecki S. (1997) The effect of calphostin C, a potent photodependent protein kinase C inhibitor, on the proliferation of glioma cells in vitro. J. Neurooncol. 31, 255–266 [DOI] [PubMed] [Google Scholar]
- 57. Lee Y., Kockx M., Raftery M. J., Jessup W., Griffith R., Kritharides L. (2010) Glycosylation and sialylation of macrophage-derived human apolipoprotein E analyzed by SDS-PAGE and mass spectrometry. Evidence for a novel site of glycosylation on Ser-290. Mol. Cell Proteomics 9, 1968–1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Garver W. S., Deeg M. A., Bowen R. F., Culala M. M., Bierman E. L., Oram J. F. (1997) Phosphoproteins regulated by the interaction of high density lipoprotein with human skin fibroblasts. Arterioscler. Thromb. Vasc. Biol. 17, 2698–2706 [DOI] [PubMed] [Google Scholar]
- 59. Deeg M. A., Bowen R. F., Oram J. F., Bierman E. L. (1997) High density lipoproteins stimulate mitogen-activated protein kinases in human skin fibroblasts. Arterioscler. Thromb. Vasc. Biol. 17, 1667–1674 [DOI] [PubMed] [Google Scholar]
- 60. Kanda Y., Richards R. G., Handwerger S. (1998) Apolipoprotein A-I stimulates human placental lactogen release by activation of MAP kinase. Mol. Cell. Endocrinol. 143, 125–131 [DOI] [PubMed] [Google Scholar]
- 61. Yancey P. G., Yu H., Linton M. F., Fazio S. (2007) A pathway-dependent on apoE, ApoAI, and ABCA1 determines formation of buoyant high-density lipoprotein by macrophage foam cells. Arterioscler. Thromb. Vasc. Biol. 27, 1123–1131 [DOI] [PubMed] [Google Scholar]
- 62. Hooper A. J., Robertson K., Ng L., Kattampallil J. S., Latchem D., Willsher P. C., Thom J., Baker R. I., Burnett J. R. (2009) A novel ABCA1 nonsense mutation, R1270X, in Tangier disease associated with an unrecognised bleeding tendency. Clin. Chim. Acta 409, 136–139 [DOI] [PubMed] [Google Scholar]
- 63. Tanaka A. R., Abe-Dohmae S., Ohnishi T., Aoki R., Morinaga G., Okuhira K., Ikeda Y., Kano F., Matsuo M., Kioka N., Amachi T., Murata M., Yokoyama S., Ueda K. (2003) Effects of mutations of ABCA1 in the first extracellular domain on subcellular trafficking and ATP binding/hydrolysis. J. Biol. Chem. 278, 8815–8819 [DOI] [PubMed] [Google Scholar]
- 64. Mackay H. J., Twelves C. J. (2007) Targeting the protein kinase C family. Are we there yet? Nat. Rev. Cancer 7, 554–562 [DOI] [PubMed] [Google Scholar]
- 65. Osto E., Kouroedov A., Mocharla P., Akhmedov A., Besler C., Rohrer L., von Eckardstein A., Iliceto S., Volpe M., Lüscher T. F., Cosentino F. (2008) Inhibition of protein kinase Cβ prevents foam cell formation by reducing scavenger receptor A expression in human macrophages. Circulation 118, 2174–2182 [DOI] [PubMed] [Google Scholar]
- 66. Ward R. A., Nakamura M., McLeish K. R. (2000) Priming of the neutrophil respiratory burst involves p38 mitogen-activated protein kinase-dependent exocytosis of flavocytochrome b558-containing granules. J. Biol. Chem. 275, 36713–36719 [DOI] [PubMed] [Google Scholar]
- 67. Chattopadhyay N., Tfelt-Hansen J., Brown E. M. (2002) PKC, p42/44 MAPK and p38 MAPK regulate hepatocyte growth factor secretion from human astrocytoma cells. Brain Res. Mol. Brain Res. 102, 73–82 [DOI] [PubMed] [Google Scholar]
- 68. Mócsai A., Jakus Z., Vántus T., Berton G., Lowell C. A., Ligeti E. (2000) Kinase pathways in chemoattractant-induced degranulation of neutrophils. The role of p38 mitogen-activated protein kinase activated by Src family kinases. J. Immunol. 164, 4321–4331 [DOI] [PubMed] [Google Scholar]
- 69. Green T. D., Crews A. L., Park J., Fang S., Adler K. B. (2011) Regulation of mucin secretion and inflammation in asthma. A role for MARCKS protein? Biochim. Biophys. Acta 1810, 1110–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Li Y., Martin L. D., Spizz G., Adler K. B. (2001) MARCKS protein is a key molecule regulating mucin secretion by human airway epithelial cells in vitro. J. Biol. Chem. 276, 40982–40990 [DOI] [PubMed] [Google Scholar]
- 71. Raufman J. P., Malhotra R., Xie Q., Raffaniello R. D. (1997) Expression and phosphorylation of a MARCKS-like protein in gastric chief cells. Further evidence for modulation of pepsinogen secretion by interaction of Ca2+/calmodulin with protein kinase C. J. Cell. Biochem. 64, 514–523 [PubMed] [Google Scholar]
- 72. Damera G., Jester W. F., Jiang M., Zhao H., Fogle H. W., Mittelman M., Haczku A., Murphy E., Parikh I., Panettieri R. A., Jr. (2010) Inhibition of myristoylated alanine-rich C kinase substrate (MARCKS) protein inhibits ozone-induced airway neutrophilia and inflammation. Exp. Lung Res. 36, 75–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Welgus H. G., Campbell E. J., Cury J. D., Eisen A. Z., Senior R. M., Wilhelm S. M., Goldberg G. I. (1990) Neutral metalloproteinases produced by human mononuclear phagocytes. Enzyme profile, regulation, and expression during cellular development. J. Clin. Invest. 86, 1496–1502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Coffman F. D. (2008) Chitinase 3-Like-1 (CHI3L1). A putative disease marker at the interface of proteomics and glycomics. Crit. Rev. Clin. Lab. Sci. 45, 531–562 [DOI] [PubMed] [Google Scholar]
- 75. Renkema G. H., Boot R. G., Au F. L., Donker-Koopman W. E., Strijland A., Muijsers A. O., Hrebicek M., Aerts J. M. (1998) Chitotriosidase, a chitinase, and the 39-kDa human cartilage glycoprotein, a chitin-binding lectin, are homologues of family 18 glycosyl hydrolases secreted by human macrophages. Eur. J. Biochem. 251, 504–509 [DOI] [PubMed] [Google Scholar]
- 76. Takemura R., Werb Z. (1984) Secretory products of macrophages and their physiological functions. Am. J. Physiol. 246, C1–C9 [DOI] [PubMed] [Google Scholar]
- 77. Alitalo K., Hovi T., Vaheri A. (1980) Fibronectin is produced by human macrophages. J. Exp. Med. 151, 602–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sherry B., Yarlett N., Strupp A., Cerami A. (1992) Identification of cyclophilin as a proinflammatory secretory product of lipopolysaccharide-activated macrophages. Proc. Natl. Acad. Sci. U.S.A. 89, 3511–3515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Février B., Raposo G. (2004) Exosomes. Endosomal-derived vesicles shipping extracellular messages. Curr. Opin Cell Biol. 16, 415–421 [DOI] [PubMed] [Google Scholar]
- 80. Makowske M., Ballester R., Cayre Y., Rosen O. M. (1988) Immunochemical evidence that three protein kinase C isozymes increase in abundance during HL-60 differentiation induced by dimethyl sulfoxide and retinoic acid. J. Biol. Chem. 263, 3402–3410 [PubMed] [Google Scholar]
- 81. Singer M., Martin L. D., Vargaftig B. B., Park J., Gruber A. D., Li Y., Adler K. B. (2004) A MARCKS-related peptide blocks mucus hypersecretion in a mouse model of asthma. Nat. Med. 10, 193–196 [DOI] [PubMed] [Google Scholar]
- 82. Park J., Fang S., Crews A. L., Lin K. W., Adler K. B. (2008) MARCKS regulation of mucin secretion by airway epithelium in vitro. Interaction with chaperones. Am. J. Respir. Cell Mol. Biol. 39, 68–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Liu J. F., Crépin M., Liu J. M., Barritault D., Ledoux D. (2002) FGF-2 and TPA induce matrix metalloproteinase-9 secretion in MCF-7 cells through PKC activation of the Ras/ERK pathway. Biochem. Biophys. Res. Commun. 293, 1174–1182 [DOI] [PubMed] [Google Scholar]
- 84. Park M. J., Park I. C., Lee H. C., Woo S. H., Lee J. Y., Hong Y. J., Rhee C. H., Lee Y. S., Lee S. H., Shim B. S., Kuroki T., Hong S. I. (2003) Protein kinase C-α activation by phorbol ester induces secretion of gelatinase B/MMP-9 through ERK 1/2 pathway in capillary endothelial cells. Int. J. Oncol. 22, 137–143 [PubMed] [Google Scholar]
- 85. Helal R., Melzig M. F. (2010) New aspects in the synthesis and secretion of lysozyme by cultured human monocyte cell lines. In Vitro Cell. Dev. Biol. Anim. 46, 492–496 [DOI] [PubMed] [Google Scholar]
- 86. Ha H., Yu M. R., Lee H. B. (2001) High glucose-induced PKC activation mediates TGF-β 1 and fibronectin synthesis by peritoneal mesothelial cells. Kidney Int. 59, 463–470 [DOI] [PubMed] [Google Scholar]
- 87. Arici M., Brown J., Williams M., Harris K. P., Walls J., Brunskill N. J. (2002) Fatty acids carried on albumin modulate proximal tubular cell fibronectin production. A role for protein kinase C. Nephrol. Dial. Transplant. 17, 1751–1757 [DOI] [PubMed] [Google Scholar]
- 88. Boot R. G., van Achterberg T. A., van Aken B. E., Renkema G. H., Jacobs M. J., Aerts J. M., de Vries C. J. (1999) Strong induction of members of the chitinase family of proteins in atherosclerosis. Chitotriosidase and human cartilage gp-39 expressed in lesion macrophages. Arterioscler. Thromb. Vasc. Biol. 19, 687–694 [DOI] [PubMed] [Google Scholar]