

Background: The epoxygenase metabolites (EETs) inhibit ENaC by unknown mechanisms.
Results: 14,15-EET stimulates an ERK1/2-catalyzed inhibitory phosphorylation of the ENaC β and γ subunits.
Conclusion: A CYP2C44 epoxygenase/ERK1/2-mediated pathway for ENaC regulation has been characterized.
Significance: Roles for human CYP2C8 and CYP2C9 as antihypertensive epoxygenases and for the EETs as antihypertensive drug targets are proposed.
Keywords: Cytochrome P450, ENaC, Epidermal Growth Factor (EGF), Epoxygenase Pathway, ERK, Hypertension, Eicosanoid Function
Abstract
The epithelial sodium channel (ENaC) participates in the regulation of plasma sodium and volume, and gain of function mutations in the human channel cause salt-sensitive hypertension. Roles for the arachidonic acid epoxygenase metabolites, the epoxyeicosatrienoic acids (EETs), in ENaC activity have been identified; however, their mechanisms of action remain unknown. In polarized M1 cells, 14,15-EET inhibited amiloride-sensitive apical to basolateral sodium transport as effectively as epidermal growth factor (EGF). The EET effects were associated with increased threonine phosphorylation of the ENaC β and γ subunits and abolished by inhibitors of (a) mitogen-activated protein kinase/extracellular signal-regulated kinase kinase/extracellular signal regulated kinases 1 and 2 (MEK/ERK1/2) and (b) EGF receptor signaling. CYP2C44 epoxygenase knockdown blunted the sodium transport effects of EGF, and its 14,15-EET metabolite rescued the knockdown phenotype. The relevance of these findings is indicated by (a) the hypertension that results in mice administered cetuximab, an inhibitor of EGF receptor binding, and (b) immunological data showing an association between the pressure effects of cetuximab and reductions in ENaCγ phosphorylation. These studies (a) identify an ERK1/2-dependent mechanism for ENaC inhibition by 14,15-EET, (b) point to ENaC as a proximal target for EET-activated ERK1/2 mitogenic kinases, (c) characterize a mechanistic commonality between EGF and epoxygenase metabolites as ENaC inhibitors, and (d) suggest a CYP2C epoxygenase-mediated pathway for the regulation of distal sodium transport.
Introduction
Prevalence, complexity, and multiple medical consequences make chronic hypertension a major health challenge, and there is a growing need for new approaches for its diagnosis and treatment. The epithelial sodium channel (ENaC)2 plays an important role in renal sodium reabsorption, and gain or loss of function mutations in the human channel are associated with hypertensive or hypotensive phenotypes, respectively (1–3). ENaC is composed of three subunits (α, β, and γ) (4) that during inward sodium transport (JNa+) localize to the apical surface of the collecting duct (CD) principal cells (5–7). ENaC is regulated by hormones such as aldosterone and insulin (5, 8–10), and epidermal growth factor (EGF) inhibits its activity by an ERK1/2-catalyzed phosphorylation of its β (ENaCβ) and γ (ENaCγ) subunits (11–15).
The cytochrome P450 CYP2C (gene family 2, subfamily C) epoxygenases metabolize arachidonic acid to epoxyeicosatrienoic acids (EETs) (16). Electrophysiology studies in microdissected rat and mouse CDs (16–18) or cultured CD cells (19) identified the EETs as inhibitors of ENaC gating and pointed to the CYP2C epoxygenases as regulators of ENaC activity and distal sodium excretion (16). Furthermore, roles for the CYP2C epoxygenases and their EET metabolites in regulating blood pressure were suggested by reports that (a) epoxygenase inhibition causes salt-sensitive hypertension (20) and that (b) hypertensive salt-sensitive Dahl rats and Cyp4a10(−/−) mice show reduced renal epoxygenase activity (18, 20). However, the mechanisms by which the EETs regulate ENaC and ultimately blood pressure are presently unknown, and issues such as (a) direct effects resulting from EET binding to ENaC proteins and/or altering channel membrane density or microenvironments and/or (b) indirect effects such as EET-triggered alterations in ENaC proteolysis, glycosylation, and/or phosphorylation remain unanswered.
Similarities between EETs and EGF as activators of MEK/ERK1/2 kinases and inhibitors of ENaC gating (11–14, 17–19, 21, 22) suggest that their effects on ENaC activity proceeded by common, ERK1/2-mediated mechanisms. Here we characterize a hitherto unrecognized relationship between the seemingly unrelated mitogenic and transport properties of the CYP2C epoxygenase metabolites by showing that in polarized M1 cells (a) 14,15-EET inhibited amiloride-sensitive JNa+ by promoting the ERK1/2-catalyzed phosphorylation of ENaCβ and ENaCγ and (b) the CYP2C44 epoxygenase mediated the EGF effects on JNa+. Furthermore, inhibition of EGF receptor (EGFR) signaling abrogated the cellular effects of EGF and 14,15-EET on JNa+ and caused hypertension in mice.
EXPERIMENTAL PROCEDURES
Animal protocols were approved by the Institutional Animal Care and Use Committee. Adult male 129SeV mice (12–18 months old) were allowed free access to water and solid diets containing normal (5L0D, PMI Nutrition International) or high salt (TD92012, Harlan) (0.3 or 8% NaCl, respectively). Where indicated, mice were left untreated (controls) or administered cetuximab every 48 h for 8–10 days (12 mg/kg of body weight in ≤150 μl of 150 mm NaCl/mouse intraperitoneally). Blood pressure measurements were done utilizing the facilities and expertise of the Vanderbilt University Mouse Physiology Core. The systolic blood pressures (BPs) of conscious mice were measured between 9 and 11 a.m. by means of a Micro-Renthane tapered catheter (300–500 μm outer diameter) inserted into the right carotid artery of Nembutal-anesthetized mice (75 mg/kg of body weight intraperitoneally) and connected to a remote pressure sensor (Digi-Med Blood Pressure Analyzer, Micro-Med Inc.) (18). One and 2 days after surgery the animals were allowed to become familiar with the environment, and their BPs were monitored continuously for at least 40 min at an ambient temperature of 23 °C. BP data are reported as group averages ± S.E. calculated from ≥30 such measurements per animal.
Cell Culture
M1 cells (ATCC) were cultured in DMEM-Ham's F-12 medium (Invitrogen) containing 10% fetal bovine serum (Invitrogen) and used between passages 2 and 8. For CYP2C44 knockdown, cells were incubated with CYP2C44 coding (sh-2c44) or non-coding (mock) shRNA plasmids (SABiosciences) as described (23). Neomycin-resistant cells were cultured in neomycin-free medium and used after two passages. Transepithelial resistance (TER) and apical to basolateral sodium fluxes (JNa+) were measured in M1 cells (three to five passages) seeded (2.5 × 105 cells/well) onto 12-well Transwells (Corning Inc.) and cultured until confluence and TER was stable. Experimental TER measurements (in ohms × cm2) were done 2 h after the addition of tests compounds using an EVOM volt-ohm meter (World Precision Instruments). Conductances (G) were calculated from TER values and expressed as siemens/cm2. For JNa+ measurements, 1 nCi of 22Na in serum-free medium was added to the apical compartment of polarized M1 cells in serum-free medium. Following the addition of test compounds, cells were incubated for 60 min at 37 °C, and the 22Na was quantified by liquid scintillation. JNa+ in pmol/cm2/h was calculated based on the final specific activity of the 22Na tracer (Ci/mol of Na+). Where indicated, differences between total and amiloride-insensitive JNa+ were used as estimates of amiloride-sensitive, ENaC-mediated JNa+. Amiloride, EETs, PD98059, U0126, or EtOH as vehicle (≤0.2% final) was added to the apical compartment, and EGF was added to the basolateral compartment. CYP2C24 mRNAs levels were determined by quantitative real time PCR and normalized to the β-actin mRNA as reported (23). Eicosanoids in cell pellets or culture medium (15-mm plates) were extracted in the presence of 20-2H3-labeled 8,9-, 11,12-, and 14,15-EET (5 ng each) and 2H8-labeled 5,6-, 8,9-, 11,12-, and 14,15-dihydroxyeicosatrienoic acid (5 ng each) and quantified by ultrahigh pressure liquid chromatography-tandem mass spectrometry (23, 24). As is the case with most EETs present endogenously in biological samples (24, 25), greater than 90% of the EETs in M1 cell extracts are found as glycerophospholipid esters and must be released by alkaline hydrolysis prior to their mass spectral quantification (24).
Protein Expression and Phosphorylation
Western blots of kidney membranes or cell lysates (in radioimmune precipitation assay buffer; Bio-Rad) were done in 7% polyacrylamide gels by standard SDS-PAGE/blotting techniques using rabbit antibodies raised against β-actin (Sigma) or rat CYP2C11 (cross-reactive toward mouse CYP2C29 and CYP2C38) or peptides antibodies targeting CYP2C44 (GenScript); phospho-ERK1/2 (Tyr-202/204), ERK1/2, EGFR, phosphothreonine-containing peptides (Cell Signaling Technology), and goat anti-ENaCγ (Santa Cruz Biotechnology). Immunoreactive proteins were detected with an Immobilon Western Chemiluminescent HRP Substrate kit (Millipore). Membranes were isolated from kidneys homogenized in 0.1 m potassium phosphate buffer (pH 7.4) containing 0.25 m sucrose, 10 mm Na3VO4, protease and phosphatases inhibitor mixtures 2 and 3 (10 μl/ml each) (Sigma) in a T8-Ultra-Turrax (IKA, Germany) homogenizer. After sonication (six pulses, power 4) (Ultrasonic Homogenizer 4710, Cole Parmer Instruments, Chicago, IL), the homogenates were centrifuged for 15 min at 5,000 and 10,000 × g. Membrane fractions collected at 100,000 × g for 60 min were suspended in homogenization buffer and used within 24 h.
Immunoprecipitation studies were done in cell lysates from 48-h cultures and prepared in 0.1 m Tris-Cl buffer (pH 7.4) containing 1% Triton X-100 (v/v), and protease and phosphatase inhibitor mixtures 2 and 3 (10 μl/ml each). Lysates were clarified by centrifugation (10 min at 10,000 × g), incubated with protein A-coated Dynabeads (Invitrogen), and exposed to a magnetic field, and non-attached proteins were incubated (60 min at 4 °C) with anti-phosphothreonine antibodies. After capture with protein A-coated Dynabeads, immunoreactive proteins were eluted with Laemmli electrophoresis buffer (10 min at 95 °C) and analyzed by Western blot using anti-ENaCβ and -ENaCγ antibodies. For studies of 32P incorporation, plates of equivalent M1 cell density cultured for 48 h in medium containing 1 μm aldosterone were incubated for 30 min in phosphate-free high glucose minimum Eagle's medium containing okadaic acid (0.3 μm) and then for 3 h in [32P]orthophosphate (1 mCi/ml). After a 15-min exposure to vehicle, 11,12-EET, 14,15-EET (10 μm each), or EGF (10 ng/ml), the cells were washed with cold PBS containing 10 mm Na4P2O7, 10 mm Na3VO4, 1 mm EDTA, and protease and phosphatase inhibitor mixtures 2 and 3; lysed as above; and exposed to ENaCβ or ENaCγ antibodies; and protein A affinity-purified immunoreactive proteins were quantified by liquid scintillation. Short incubation times with agonists were used to minimize potential complications resulting from, among other things, changes in the levels of ENaC proteins and/or labile [γ-32P]ATP.
For immunofluorescence imaging studies, paraffin embedded sections (5 μm) from kidneys perfused in situ with 40 mm sodium phosphate (pH 7.4) containing 4% formaldehyde, 0.1 m NaCl, 50 mm NaF, 0.5 mm Na3VO4, 30 mm Na4P2O7, and 0.1% HOAc were exposed to white light for 48–72 h to reduce autofluorescence. Following microwave antigen retrieval (1× Citra Buffer, BioGenex) and blocking (in 10% goat serum), sections were incubated with biotinylated Dolichos biflorus agglutinin (Vector Labs) followed by rabbit anti-phosphothreonine antibodies and then exposed to mixtures of fluorescein isothiocyanate (FITC)-conjugated streptavidin (GE Healthcare) and rhodamine-conjugated anti-rabbit IgGs (Jackson ImmunoResearch Laboratories). To characterize the anti-phosphothreonine reactive material present in Dolichos biflorus-positive segments, samples of rabbit anti-phosphothreonine IgGs (Cell Signaling Technology catalog number 9381) (0.3 μg each) were incubated for 3–5 h at 4 °C with 15, 30, 60, 150, or 300 μg of a synthetic peptide containing the target ENaCγ phosphorylated threonine (bold and underlined) and flanking residues, PEAPVPG-(T-p)-PPPRYN. Kidney sections were blocked and exposed to biotinylated D. biflorus agglutinin followed by untreated anti-phosphothreonine or ENaCγ peptide-treated anti-phosphothreonine antibodies, and after removing unbound capture peptide, the immunofluorescence analyses proceeded as above.
Statistical Analyses
Data were analyzed using a two-tailed Student's t test (Excel). Error bars represent S.E. p values ≤0.05 were considered statistically significant.
RESULTS
To study mechanisms of ENaC regulation by EETs, we compared their effects on the transepithelial conductance and JNa+ of polarized M1 cells with those of EGF or amiloride, a selective ENaC inhibitor (4, 12). M1 cells are derived from mouse CDs (26), express a functional ENaC (27, 28), and metabolize endogenous arachidonic acid to EETs as shown by the presence of 11,12-, and 14,15-EET, and their corresponding dihydroxyeicosatrienoic acids in cell extracts (supplemental Fig. 1A). As with most cell preparations and organ tissues, greater than 90% of the EETs present in M1 cells are found as esters of cellular glycerophospholipids (25), and thus, the cellular concentration of free, non-esterified EETs represent a fraction of the total.
Based on studies of 11,12- and 14,15-EET concentration and time-dependent effects on transcellular G (supplemental Fig. 1, B and C), we chose to (a) utilize 14,15-EET for these studies because it is the major epoxygenase metabolite in cultured M1 cells (Table 1) and (b) perform all subsequent measurements 2 h after the addition of either vehicle (EtOH; ≤0.2%), 14,15-EET, EGF, or amiloride (5 μm, 10 ng/ml, and 10 μm final concentrations, respectively). 11,12-EET was identified as a selective ENaC inhibitor in microdissected rat and mouse CDs (17, 18). On the other hand, 11,12- and 14,15-EET were found to be equally effective ENaC inhibitors in cultures of mouse CD principal cells (19). It is of relevance to point out that because 14,15-EET is hydrated by cytosolic epoxide hydrolase at rates substantially higher than 11,12-EET (29), differences in rates of metabolic inactivation by cultured CD cells or freshly dissected CDs could account for the observed differences in EET biological activity.
TABLE 1.
The levels of epoxygenase metabolites in cultured M1 cells are EGF-sensitive
The concentrations of endogenous epoxygenase metabolites present in cultured M1 cells were quantified as described under “Experimental Procedures” and expressed as the sum of 11,12-EET + 11,12-dihydroxyeicosatrienoic acid and of 14,15-EET + 14,15-dihydroxyeicosatrienoic acid. Values (in ng/mg of cell protein) are averages ±S.E. calculated from three different experiments.
| Vehicle | EGF | |
|---|---|---|
| 11,12-Epoxygenase | 0.13 ± 0.11 | 0.30 ± 0.01 |
| 14,15-Epoxygenase | 0.56 ± 0.15 | 1.11 ± 0.02a |
| Total epoxygenase | 0.69 ± 0.26 | 1.50 ± 0.13a |
a Significantly different from vehicle-treated controls, p < 0.05.
Comparisons of the effects of 14,15-EET and amiloride on apical to basolateral sodium transport showed that both reduced JNa+ by about 27–29% (Fig. 1A) and that amiloride had no significant additional effects when added in the presence of 14,15-EET; i.e. JNa+ becomes essentially amiloride-insensitive in the presence of 14,15-EET (Fig. 1A). These data indicate that 14,15-EET and amiloride share a common target, i.e. ENaC-mediated transcellular sodium transport, and characterize (a) 14,15-EET as an inhibitor of cellular JNa+ and amiloride as its functional analog and (b) ENaC as a target of the effects of 14,15-EET on JNa+.
FIGURE 1.

The transport effects of 14,15-EET are amiloride-insensitive and, as with EGF, ERK1/2-mediated. A, changes in JNa+ induced by control, vehicle (black bars) or amiloride (10 μm) (white bars), or by 14,15-EET (5 μm) added in the presence of vehicle (black bars) or amiloride (10 μm) (white bars). Values (in pmol/h/cm2) are averages ± S.E. (error bars) calculated from five different cell samples. Differences from vehicle are indicated as follows: *, p < 0.0008; +, p < 0.0001; ≠, p < 0.002. The differences between amiloride and 14,15-EET or between amiloride added in the absence or presence of 14,15-EET were not significant (p > 0.05). The amiloride-sensitive components of the JNa+ responses to the agonists were 34, 26, and 37%, respectively, for amiloride, 14,15-EET, and a combination of both. B, cells incubated without (control) or with U0126 (apical; 10 μm) were exposed to vehicle, 14,15-EET (5 μm), or EGF (10 ng/ml) (black, gray, and white bars, respectively), and JNa+ was determined 2 h later. Values (in pmol/h/cm2) are averages ± S.E. (error bars) calculated from five different cell samples. Differences from controls are indicated as follows: *, p < 0.05; +, p < 0.0001; ≠, p < 0.01. The amiloride-sensitive components of the total cell JNa+ effects corresponded to 39, 30, and 25% for control untreated and to 46, 54, and 40% for U0126-treated cells for vehicle, 14,15-EET, and EGF, respectively. C and D, Western blots of cell lysates probed with anti-phospho-ERK1/2 (p-ERK) (upper panels) or -ERK1/2 (lower panels) antibodies. Shown are immunoreactive proteins with the mobilities of phospho-ERK1/2 and ERK1/2. C, lysates (50 μg of protein/lane) isolated from cells incubated for 10, 30, or 60 min with vehicle (lane 1) or 14,15-EET (5 μm) (lane 2). D, lysates from cells incubated without (control) or with U0126 (10 μm) for 1 h prior to a 30-min exposure to vehicle, 14,15-EET (5 μm), or EGF (10 ng/ml) (lanes 1, 2, and 3, respectively; 50–60 μg of protein/lane) probed with peptide antibodies.
14,15-EET and EGF Inhibit JNa+ by a Common, ERK1/2-mediated Mechanism
The effects of 14,15-EET and EGF (11, 12, 19) on amiloride-sensitive JNa+ (the difference between JNa+ before and after adding amiloride) were measured in the presence or absence of the MEK inhibitor U0126 (30). Although 14,15-EET and EGF reduced amiloride-sensitive JNa+ by similar extents (to 60–68% of vehicle controls), U0126 increased JNa+ in vehicle-, EGF-, and EET-treated cells by 1.8-, 2.6-, and 1.6-fold over control cells, respectively (Fig. 1B). Moreover, U0126 raised total JNa+ in control cells from 18.9 ± 1.8 to 30.1 ± 2.0 pmol/cm2/h (n > 5); however, the amiloride-sensitive component of JNa+ in control and U0126-treated cells was ∼34% of total JNa+. These results (a) show that under basal and stimulated conditions a portion of cellular JNa+ is MEK inhibitor-sensitive and (b) indicate a role for the MEK kinases in amiloride-sensitive basal JNa+.
The association between the effects of U0126 on the transport responses to 14,15-EET and EGF and its effects on the MEK/ERK1/2 pathway were investigated by Western blot analysis of ERK1/2 phosphorylation in U0126-treated and untreated cells. Control experiments showed that ERK1/2 activation by 14,15-EET reached its maximum within the first 10–30 min of treatment and decreased thereafter (Fig. 1C). This early activation of the MEK/ERK1/2 cascade is a known feature of growth factor and EET signaling and precedes their functional responses (21–23). A link between ERK1/2 activation and the functional effects of U0126 (Fig. 1B) was substantiated by the demonstration that the 14,15-EET- and EGF-mediated increases in ERK1/2 phosphorylation were blunted after preincubation with U0126 (Fig. 1D). In support of the above, 14,15-EET, EGF, and amiloride reduced transcellular G by ∼21, 27, and 32%, respectively (supplemental Fig. 2B), and PD98059, another MEK inhibitor (30), blunted the EET effects and reduced those of EGF and amiloride by ∼54 and 34%, respectively (supplemental Fig. 2B). As with the effect of U0126 and JNa+, the PD98059 effects on G were accompanied by reduced ERK1/2 phosphorylation (supplemental Fig. 2A). These data, consistent with the known effects of 14,15-EET and EGF on ERK1/2 activation and ENaC inhibition (17–19, 21, 22), suggest that (a) ENaC is but one component of the effects that MEK inhibitors have on cellular ion transport, and (b) although the MEK inhibitors and amiloride share common targets at least partially, their mechanisms of action are different. Additionally, they support the proposal that (a) under basal conditions JNa+ and G are under regulation by endogenous EETs and their effects on ERK1/2 phosphorylation (Fig. 1, B and D, and supplemental Figs. 1A and 2, A and B) and that (b) the responses to 14,15-EET and EGF are due to the amplification of basal, endogenous EET-mediated transport inhibition (Fig. 1B and supplemental Figs. 1A and 2B). These proposals are also supported by the demonstration that (a) under basal conditions ENaC oscillates between open and closed states in an epoxygenase-regulated fashion (17, 18), (b) epoxygenase inhibition blunts the effects of arachidonic acid on ENaC gating (17), and (c) EETs revert the effects of epoxygenase inhibition on ENaC gating (17).
Mechanisms of ERK1/2 Activation by 14,15-EET
To study the role of the EGFR on the ERK1/2-mediated transport responses to 14,15-EET, we determined whether (a) the EET activated EGFR-associated tyrosine kinases and whether (b) inhibition of the receptor tyrosine kinases or ligand binding altered the functional responses to 14,15-EET. As seen in Fig. 2C, 14,15-EET was as effective as EGF in stimulating EGFR phosphorylation at tyrosine 1173, a residue involved in mitogenic signaling (31). Furthermore, Inhibitor III, an inhibitor of the EGFR tyrosine kinase (32), increased the G responses to 14,15-EET, amiloride, and EGF (by ∼1.5-, 1.4-, and 2.3-fold, respectively) (Fig. 2A) and blunted the increases in ERK1/2 phosphorylation induced by 14,15-EET and EGF (Fig. 2D). As with MEK inhibition (Fig. 1A and supplemental Fig. 2B), the effects of Inhibitor III on amiloride-sensitive G (Fig. 2A) suggest additional, non-ENaC-mediated transport effects. Likewise, inhibition of EGF binding to its receptor with cetuximab, an EGFR monoclonal antibody (33), had minimal effects on basal ERK1/2 activation but, reduced EGF- and 14,15-EET-stimulated ERK1/2 phosphorylation (Fig. 2E), and raised the G responses to 14,15-EET and EGF by 18–20% without changing that of amiloride (Fig. 2B). As shown in Fig. 2, A and B, although the effects of 14,15-EET, amiloride, and EGF on the conductance of cells exposed only to vehicle were more or less similar, their responses in the presence of Inhibitor III were significantly higher than those obtained in the presence of cetuximab. Although the potentiating effects of EGF on Inhibitor III efficacy have been reported (31), concentrations of cetuximab higher than 40 μg/ml resulted in progressive reductions in amiloride-insensitive TER likely due to increases in cell toxicity. To minimize the potential for unwanted side effects, cell studies were done at cetuximab concentrations ≤20 μg/ml. Based on these studies, it was concluded that EGFR mediates at least partially the transport and ERK1/2 activation responses to 14,15-EET and that these involve the EGFR tyrosine kinases and thus effects upstream of the MEK/ERK1/2 pathway. Roles for the EGFR tyrosine kinases in 14,15-EET-, ERK1/2-mediated mitogenesis have been published (21, 22).
FIGURE 2.

Inhibition of the EGFR tyrosine kinase or EGF binding blunts the effects of 14,15-EET and EGF on ERK1/2 activation and transcellular conductance. The transcellular conductance responses of cells preincubated for 1 h with vehicle or Inhibitor III (apical; 1 μm) (black and white bars, respectively) (A) or with vehicle or cetuximab (basolateral; 20 ng/ml) (black and white bars, respectively) (B) were calculated from TER measurements performed 2 h after the addition of 14,15-EET (5 μm), amiloride (10 μm), or EGF (10 ng/ml). Values are averages ± S.E. (error bars) calculated from three experiments, each performed in triplicates. A, different from controls lacking Inhibitor III: *, p < 10−5; +, p < 10−5; ≠, p < 10−4. B, different from controls lacking cetuximab: *, p < 10−3; ≠, p < 10−5. The differences between amiloride in the presence or absence of cetuximab were not significant (p > 0.05). Western blots of cell lysates probed with anti-EGFR tyrosine 1173 (C) or anti-ERK1/2 and -phospho-ERK1/2 (P-ERK1/2) peptide antibodies (D and E) show immunoreactive proteins with the mobilities of glycosylated EGFR (C) and ERK1/2 and phospho-ERK1/2 (D and E). C, cells (50 μg of protein/lane) were isolated 30 min after the addition of vehicle, 14,15-EET (5 μm), or EGF (10 ng/ml). D, cells were preincubated for 1 h without (lane 1) or with Inhibitor III (apical; 1 μm) (lane 2) and for 30 min in the presence of vehicle, 14,15-EET (5 μm), or EGF (10 ng/ml) (15–20 and 40–60 μg of protein/well for ERK1/2 and phospho-ERK1/2, respectively). D, cells were preincubated for 1 h without or with cetuximab (basolateral; 20 ng/ml) (lanes 1 and 2, respectively) and for 30 min with vehicle, 14,15-EET (5 μm), or EGF (10 ng/ml) (30–45 and 70–90 μg of protein/well for ERK1/2 and phospho-ERK1/2, respectively). P-Tyr, phosphotyrosine; S, siemens.
The Epoxygenase Metabolites Stimulate the Threonine Phosphorylation of ENaCβ and -γ
Two approaches were used to study whether 14,15-EET and/or 11,12-EET, a known inhibitor of ENaC gating (17), increase ENaCβ and/or ENaCγ threonine phosphorylation (13). In the first approach, lysates of cells incubated for 15 min (10, 34) with vehicle, EGF, 14,15-EET, or 11,12-EET were exposed to anti-phosphothreonine antibodies, and after protein G affinity purification, the immunoreactive proteins were analyzed by Western blot using anti-ENaCβ and -ENaCγ antibodies. As shown in Fig. 3, A and B, the EETs and EGF increased the yields of anti-phosphothreonine and anti-ENaCβ and -ENaCγ immunoreactive proteins with mobilities of ∼100 and 110 kDa and of 80–85 kDa, representing the glycosylated forms of ENaCβ and ENaCγ, respectively (35–38). In the second approach, cells incubated for 2 h in [32P]orthophosphate were exposed for 15 min (10, 34) to vehicle, 14,15-EET, or EGF, and after immunoprecipitation with either anti-ENaCβ or -ENaCγ antibodies, the 32P label of the protein G affinity-purified immunoreactive proteins was quantified by liquid scintillation. Fig. 3, C and D, show that (a) 14,15-EET and EGF increased the yield of the corresponding 32P-labeled immunoreactive proteins and that (b) the EET mimics EGF in its capacity to stimulate the threonine phosphorylation of the channel subunits.
FIGURE 3.
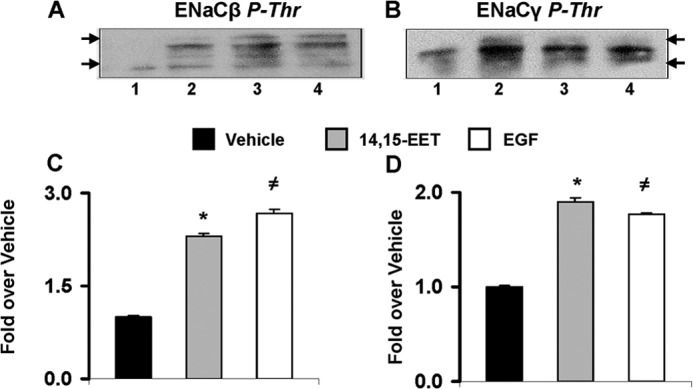
EGF and EETs stimulate threonine phosphorylation of ENaCβ and -γ. A and B, equivalent volumes of lysates from cells (at 80–85% confluence) treated for 15 min with vehicle, 11,12-EET (10 μm), 14,15-EET (5 μm), or EGF (lanes 1, 2, 3, and 4, respectively) (10 ng of protein/lane) were exposed to anti-phosphothreonine (P-Thr) antibodies, and after protein G affinity purification, the anti-phosphothreonine immunoreactive proteins were analyzed by Western blot using anti-ENaCβ (A) or -ENaCγ (B) antibodies. Arrows show mobilities for 75- and 125-kDa proteins. C and D, lysates from 32P-labeled cells incubated for 15 min with vehicle, 14,15-EET (5 μm), or EGF (10 ng/ml) (black, gray, and white bars, respectively) were exposed to anti-ENaCβ (C) or -ENaCγ (D) antibodies, and the 32P contents of affinity-purified immunoreactive proteins were determined by β-counting. Values are averages ± S.E. (error bars) calculated from two different experiments, each done in duplicates. C, different from vehicle: *, p < 0.001; ≠, p < 0.0003. D, different from vehicle: *, p < 0.0004; ≠, p < 0.0004.
The CYP2C44 Epoxygenase Mediates the Effects of EGF on Sodium Transport
The role of the CYP2C44 epoxygenase on the inhibition of J22Na+ by EGF was studied in cells expressing a scrambled (mock) or a CYP2C44-selective silencing RNA. Compared with mock-transfected cells, CYP2C44 knockdown cells (shRNA cells) showed reduced levels of (a) CYP2C44 transcripts (Fig. 4A); (b) anti-CYP2C44 immunoreactive proteins with mobilities of CYP2C44 (56.7 kDa) (39) and a 65-kDa protein (Fig. 4A, inset) that is also down-regulated in shRNA cells, suggesting that it corresponds to a post-translationally modified CYP2C44 epoxygenase; and (c) endogenous EETs and dihydroxyeicosatrienoic acids (Fig. 4B). Importantly, whereas polarized mock and shRNA cells showed no differences in amiloride-sensitive JNa+ (Fig. 4C), EGF reduced JNa+ in mock but not in CYP2C44 knockdown cells (Fig. 4C), and the knockdown phenotype was reversed upon the addition of 14,15-EET, a CYP2C44 epoxygenase metabolite (39) (Fig. 4C). These results indicate that (a) the CYP2C44 epoxygenase mediates the inhibition by EGF of amiloride-sensitive, ENaC-dependent JNa+, and (b) although CYP2C44 is involved in the JNa+ responses to EGF under basal, steady state conditions, it appears to make limited contributions to the regulation of cellular JNa+ (Fig. 4C) (17, 18). This and the stimulation of 11,12- and 14,15-EET biosynthesis by EGF (Table 1) point to CYP2C44 as a component of the EGF-stimulated, ERK1/2-mediated inhibitory phosphorylation of ENaCβ and ENaCγ.
FIGURE 4.

CYP2C44 knockdown reduces epoxygenase expression and blunts the effects of EGF on sodium transport. A, quantitative real time PCR analysis of mRNAs present in cells expressing non-coding (mock) (black bars) or CYP2C44-coding silencing RNAs (shRNA) (gray bars) using CYP2C44-selective primers (23). Values, normalized to β-actin mRNA levels, are averages ± S.E. (error bars) calculated from three different cell samples, each analyzed in triplicates. Difference from mock cells is indicated as follows: *, p < 0.004. Inset, Western blots of lysates from mock and shRNA cells probed with anti-CYP2C44 antibodies (upper panel) and normalized to the levels of anti-β-actin immunoreactive protein (lower panel). The arrows indicate approximate mobilities for 56- and 65-kDa proteins. B, the sum of EETs and dihydroxyeicosatrienoic acids (DHETs) present in mock and shRNA cells was extracted and quantified using ultrahigh pressure liquid chromatography-tandem mass spectrometric techniques as described under “Experimental Procedures.” Values are -fold change averages calculated from three different experiments. Differences from mock controls are indicated as follows: *, p < 0.02; ≠, p < 0.04. C, amiloride-sensitive JNa+ responses for mock (black bars) and shRNA cells (gray bars) exposed to vehicle or EGF (10 ng/ml) in the absence or presence of 14,15-EET (5 μm). Values (in pmol/h/cm2) are averages ± S.E. (error bars) calculated from five cell samples. Differences are indicates as follows: from vehicle-treated mock cells: *, p < 0.004; +, p < 0.001; from EGF-treated mock cells: ≠, p < 0.04. The differences between vehicle- and EGF-treated shRNA cells, EGF- and EGF plus 14,15-EET- treated mock cells, and EGF plus 14,15-EET-treated mock and shRNA cells were not significant (p > 0.05).
Cetuximab Alters ENaCγ Phosphorylation and Raises Blood Pressure
The biological significance of EGFR-dependent, EET-mediated ENaC inhibition was explored by administering cetuximab to mice and measuring its effects on (a) systemic BP and (b) kidney levels of phosphorylated ENaCγ. Cetuximab was chosen because it is in use for the treatment of certain types of human cancers (33, 40), and thus the study could have clinical implications. Compared with untreated controls, cetuximab raised the BP of mice on normal salt diets by 25 mm Hg (120 ± 3 versus 145 ± 2 mm Hg) and that of those on high salt diets by 34 mm Hg (127 ± 3 versus 161 ± 6 mm Hg) (Fig. 5A), suggesting a dietary salt component of its pressure effects. Western blots of kidney membranes from untreated and cetuximab-treated mice on normal or high salt diets showed similar levels of anti-ENaCγ reactive proteins corresponding to ENaCγ (75–80 kDa) and its serine protease product (about 50 kDa) (36, 37, 41) (Fig. 5B). Western blots of these samples identified the presence of anti-phosphothreonine reactive proteins with the mobility of ENaCγ (∼75 kDa) (Fig. 5C). The common origin of these anti-ENaCγ and -phosphothreonine immunoreactive proteins (Fig. 5, B and C) was further suggested by stripping and exposing both membranes to the alternate antibodies (not shown). An association between the prohypertensive effects of cetuximab and reduced ENaCγ threonine phosphorylation was indicated by the fact that cetuximab-treated mice showed reduced kidney levels of an anti-phosphothreonine immunoreactive protein with the mobility of ENaCγ (38) (Fig. 5C). Finally, as shown in supplemental Fig. 3, cetuximab had little or no effect on the levels of plasma-circulating EETs, kidney CYP2C44 and murine CYP2C cross-reactivity toward rat anti-CYP2C11 (CYP2C29 and CYP2C38), or microsomal arachidonic acid metabolism in mice fed a high salt diet.
FIGURE 5.

Cetuximab causes hypertension in mice and reduces ENaCγ threonine phosphorylation. Mice on normal salt (NS) or high salt (HS) diets were left untreated or administered cetuximab every other day for 10 days. A, systolic BPs of untreated or cetuximab-treated mice (white and black bars, respectively). Values are averages ± S.E. (error bars) calculated from ≥30 measurements/mouse performed for groups of untreated mice on normal or high salt diets (five and six animals, respectively) or for cetuximab-treated mice on normal or high salt diets (six and nine mice, respectively). Differences are indicated as follows: *, untreated on high salt diet, p < 10−3; cetuximab on normal diet, p < 10−5; cetuximab on high salt diet, p < 10−6; ≠, cetuximab on high salt diet, p < 10−4; untreated on high salt diet, p < 10−5. B and C, Western blots of kidney membranes from cetuximab-treated and untreated mice fed normal salt or high salt diets and probed with ENaCγ (B) or phosphothreonine (P-Thr) (C) antibodies. Loadings (60–40 and 30–20 μg of protein/lane for B and C, respectively) were normalized by comparisons with Coomassie Blue-stained membranes. Lanes 1 and 3, untreated normal salt and high salt diets, respectively. Lanes 2 and 4, cetuximab-treated normal salt and high salt diets, respectively. D, paraffin-embedded kidney sections from mice fed normal salt diets and exposed to D. biflorus agglutinin followed by anti-phosphothreonine antibodies (p-Thr) were incubated in the absence or presence of a threonine phosphorylated peptide coding for the ENaCγ target threonine (Control and Peptide panels, respectively), and the immunofluorescence analyses proceeded as described under “Experimental Procedures.” Shown are 400× images of green D. biflorus (DB), red (p-Thr), and overlay fluorescence (DB + p-Thr) emissions showing D. biflorus-positive signals in CDs and of anti-phosphothreonine-positive signals in CDs and non-CD tubular segments as well as the presence of anti-phosphothreonine immunoreactive proteins in the CD (white arrows) only in those sections exposed to IgGs incubated in the absence of the ENaCγ peptide. The white scale bars correspond to 0.5 μm.
Immunofluorescence imaging of kidney sections using biotinylated D. biflorus agglutinin, a CD marker (42), and anti-phosphothreonine antibodies showed the presence of immunoreactive proteins in (a) the CD as illustrated by co-localization of green and red fluorescence from the CD marker and anti-phosphothreonine immunoreactive proteins, respectively (Fig. 5D, Control panels), and (b) a few non-CD tubular sections (Fig. 5D, Control panels). Next, similar sections were exposed to D. biflorus and anti-phosphothreonine antibodies preincubated with a threonine phosphorylated peptide coding for the ENaCγ target threonine and flanking residues (13–15) (see “Experimental Procedures” for details). The ENaCγ selectivity of the anti-phosphothreonine-derived red fluorescence in the CD is indicated by the fact that at a capture peptide/antibody ratio of 100 (w/w), the anti-phosphothreonine signals were quenched in D. biflorus-positive structures but not in the remaining, non-CD tubular segment (Fig. 5D, Peptide panels). The results in Fig. 5 support the proposition that (a) the anti-phosphothreonine-positive proteins present in the CDs correspond to phosphorylated ENaCγ and that (b) ENaCγ undergoes threonine phosphorylation in vivo.
DISCUSSION
Salt-sensitive hypertension is associated with sodium retention and compensatory increases in plasma volume with sodium reabsorption by ENaC serving as a rate-limiting step for this important kidney function (5). The identification of 11,12- and 14,15-EET as inhibitors of ENaC activity (17–19) and associations between blood pressure and kidney CYP2C44 epoxygenase expression (18) support a role for EETs in ENaC-mediated sodium reabsorption and ultimately blood pressure (18). As with many ion channels, ENaC is regulated by factors that alter gating, expression, subunit assembly, membrane translocation, and/or residence time. Multiple mechanisms can alter one or more of these factors, including proteolysis (8, 35–38); hormonal effects on translocation and membrane assembly (6, 7, 36, 41, 43); changes in ubiquitination, retrieval, and degradation (37, 38, 44, 45); and protein kinase-mediated negative or positive effects (8, 10, 11–15, 44, 46). The earlier identification of 11,12-EET and 14,15-EET as ENaC inhibitors (17–19) and the present characterization of 14,15-EET as an inhibitor of amiloride-sensitive JNa+ in M1 cells can be added to this list. The present study expands on earlier demonstrations of ENaC inhibition by EGF and EETs and identifies a common mechanism to explain their effects on ENaC activity. The mechanism proposed (a) involves an EET-mediated, ERK1/2-catalyzed phosphorylation of the channel β and γ subunits, (b) is consistent with the known roles of EETs in EGF signaling and ERK1/2 phosphorylation (21, 22), and (c) identifies ENaC as a proximal functional target for the CYP2C44 epoxygenase and MEK/ERK1/2 kinases. Importantly, this characterization of a mechanistic commonality between sodium transport and mitogenic responses described for the EETs provides a mechanistic platform that could serve to rationalize the many, apparently unrelated biological activities reported for these lipids.
Pathways by which 14,15-EET could regulate MEK/ERK1/2 kinases and ENaC activity include (a) transmembrane signaling by EET receptor(s) (Fig. 6, 1), (b) effects on EGFR signaling and/or ligand binding (Fig. 6, 2), (c) activation of kinases upstream of ERK1/2 (Fig. 6, 2), and (d) non-receptor-mediated, direct effects on ERK1/2 phosphorylation (Fig. 6, 3) or ENaC gating (Fig. 6, 4). With few exceptions (47), the identification of EET receptors capable of transmembrane signaling has proven inconclusive, and support for direct EET effects on protein kinases or ENaC activity is lacking. Studies in epithelial and endothelial cells identified EET roles in the activation of kinases such as MEK, Ras, and Akt (21–23), and the demonstration that 14,15-EET mediates heparin-binding EGF-like growth factor processing and binding provided a cogent explanation for its pleiotropic effects on these kinases (22). Based on these precedents, we propose a similar mechanism for the 14,15-EET effects on the EGFR and MEK/ERK1/2 pathway of M1 cells. The fact that CYP2C44 knockdown abrogated the in vitro effects of EGF on JNa+ and that inhibition of EGFR ligand binding reduced ENaCγ phosphorylation and caused hypertension illustrates the potential significance of these findings to the in vivo regulation of ENaC activity and blood pressure and points to a hitherto unrecognized, concerted role for CYP2C44, EGF, and EGFR in blood pressure regulation.
FIGURE 6.

Regulation of ENaC and sodium transport by up-regulation of the kidney. CYP2C44 epoxygenase and EET biosynthesis (for example, by increased salt intake) activates an EGFR-mediated signaling cascade, leading to increased ERK1/2-mediated ENaCβ and ENaCγ phosphorylation, channel inactivation, and increased sodium excretion. Alterations in EGFR-mediated signaling and/or CYP2C44 activity and/or expression reduce ERK1/2-mediated ENaC regulatory inhibition, leading to sodium retention and increased blood pressure. Upward or downward arrows denote increases or decreases in concentration or enzymatic activity, respectively. Red squares 1, 2, 3, and 4 depict potential sites of EET action: a membrane-bound EET receptor (EET-R) (1), direct actions on EGFR ligand binding or signaling (2), and direct effects on MEK1/2 (3) or ENaC (4) activity.
In summary, this characterization of a role for the murine CYP2C44 epoxygenase and 14,15-EET in the ERK1/2-mediated regulation of amiloride-sensitive JNa+ as well as published studies of CYP2C epoxygenase and EET effects on ENaC gating, distal sodium excretion, and blood pressure control (16–18, 20) offers new approaches for the understanding of plasma sodium homeostasis and suggests that maneuvers designed to (a) up-regulate the expression or activity of the human functional homologues of the CYP2C44 epoxygenase (CYP2C8 and/or CYP2C9) or (b) increase kidney EET concentrations could serve as a basis for the development of new antihypertensive therapies as well as strategies for the early diagnosis and detection of the disease. Finally, the demonstration that cetuximab causes hypertension in mice raises a need for careful studies of its blood pressure effects in humans.
Acknowledgment
The contributions of the Vanderbilt Mouse Physiology Core and O'Brien Mouse Kidney Physiology Center are acknowledged.
This work was supported, in whole or in part, by National Institutes of Health Grants DK038226 (to J. H. C. and R. C. H.), DK51265 (to R. H. C.), and HL34300 (to W. H. W.) from the USPHS.

This article contains supplemental Figs. 1–3.
- ENaC
- epithelial sodium channel
- BP
- systolic blood pressure
- CD
- collecting duct
- EET
- epoxyeicosatrienoic acid
- EGFR
- EGF receptor
- G
- conductance
- TER
- transepithelial resistance.
REFERENCES
- 1. Schild L. (1996) The ENaC channel as the primary determinant of two human diseases: Liddle syndrome and pseudohypoaldosteronism. Nephrologie 17, 395–400 [PubMed] [Google Scholar]
- 2. Warnock D. G. (2001) Liddle syndrome: genetics and mechanism of Na+ channel defects. Am. J. Med. Sci. 322, 302–307 [DOI] [PubMed] [Google Scholar]
- 3. Rossier B. C., Schild L. (2008) Epithelial sodium channel. Mendelian versus essential hypertension. Hypertension 52, 595–600 [DOI] [PubMed] [Google Scholar]
- 4. Canessa C. M., Schild L., Buell G., Thorens B., Gautschi I., Horisberger J. D., Rossier B. C. (1994) Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature 367, 463–467 [DOI] [PubMed] [Google Scholar]
- 5. Hamm L. L., Feng Z., Hering-Smith K. S. (2010) Regulation of sodium transport by ENaC in the kidney. Curr. Opin. Nephrol. Hypertens. 19, 98–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Snyder P. M. (2002) The epithelial sodium channel: cell surface insertion and retrieval in Na+ homeostasis and hypertension. Endocr. Rev. 23, 258–275 [DOI] [PubMed] [Google Scholar]
- 7. Butterworth M. B., Edinger R. S., Frizzell R. A., Johnson J. P. (2009) Regulation of epithelial sodium channel by membrane trafficking. Am. J. Physiol. Renal Physiol. 296, F10–F24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Soundararajan R., Pearce D., Hughey R. P., Kleyman T. R. (2010) Role of epithelial sodium channels and their regulators in hypertension. J. Biol. Chem. 285, 30363–30369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bubien J. K. (2010) Epithelial Na+ channel (ENaC), hormones, and hypertension. J. Biol. Chem. 285, 23527–23531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang Y. H., Alvarez de la Rosa D., Canessa C. M., Hayslett J. P. (2005) Insulin-induced phosphorylation of ENaC correlates with increased sodium channel function in A6 cells. Am. J. Physiol. Cell Physiol. 288, C141–C147 [DOI] [PubMed] [Google Scholar]
- 11. Vehaskari V. M., Hering-Smith K. S., Moskowitz D. W., Weiner I. D., Hamm L. L. (1989) Effect of epidermal growth factor on sodium transport in the cortical collecting tubule. Am. J. Physiol. Renal Physiol. 256, F803–F809 [DOI] [PubMed] [Google Scholar]
- 12. Shen J. P., Cotton C. U. (2003) Epidermal growth factor inhibits amiloride-sensitive sodium absorption in renal collecting duct cells. Am. J. Physiol. Renal Physiol. 284, F57–F64 [DOI] [PubMed] [Google Scholar]
- 13. Shi H., Asher C., Chigaev A., Yung Y., Reuveny E., Seger R., Garty H. (2002) Interactions of β and γ ENaC with Nedd4 can be facilitated by an ERK-mediated phosphorylation. J. Biol. Chem. 277, 13539–13547 [DOI] [PubMed] [Google Scholar]
- 14. Falin R., Veizis I. E., Cotton C. U. (2005) A role for ERK1/2 in EGF- and ATP-dependent regulation of amiloride-sensitive sodium absorption. Am. J. Physiol. Cell Physiol. 288, C1003–C1011 [DOI] [PubMed] [Google Scholar]
- 15. Falin R. A., Cotton C. U. (2007) Acute downregulation of ENaC by EGF involves the PY motif and putative ERK phosphorylation site. J. Gen. Physiol. 130, 313–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Capdevila J. H. (2007) Regulation of ion transport and blood pressure by cytochrome P450 monooxygenases. Curr. Opin. Nephrol. Hypertens. 16, 465–470 [DOI] [PubMed] [Google Scholar]
- 17. Wei Y., Lin D.-H., Kemp R., Yaddanapudi G. S., Nasjletti A., Falck J. R., Wang W. H. (2004) Arachidonic acid inhibits epithelial Na channel via cytochrome P450 (CYP) epoxygenase-dependent metabolic pathways. J. Gen. Physiol. 124, 719–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nakagawa K., Holla V. R., Wei Y., Wang W. H., Gatica A., Wei S., Mei S., Miller C. M., Cha D. R., Price E., Jr., Zent R., Pozzi A., Breyer M. D., Guan Y., Falck J. R., Waterman M. R., Capdevila J. H. (2006) Salt-sensitive hypertension is associated with dysfunctional Cyp4a10 gene and kidney epithelial sodium channel. J. Clin. Investig. 116, 1696–1702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pavlov T. S., Ilatovskaya D. V., Levchenko V., Mattson D. L., Roman R. J., Staruschenko A. (2011) Effects of cytochrome P450 metabolites of arachidonic acid on the epithelial sodium channel (ENaC). Am. J. Physiol. Renal Physiol. 301, F672–F681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Makita K., Takahashi K., Karara A., Jacobson H. R., Falck J. R., Capdevila J. H. (1994) Experimental and/or genetically controlled alterations of the renal microsomal cytochrome P450 epoxygenase induce hypertension in rats fed a high salt diet. J. Clin. Investig. 94, 2414–2420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen J. K., Wang D.-W., Falck J. R., Capdevila J., Harris R. C. (1999) Transfection of an active cytochrome P450 arachidonic acid epoxygenase indicated that 14,15-epoxyeicosatrienoic acid functions as an intracellular second messenger in response to epidermal growth factor. J. Biol. Chem. 274, 4764–4769 [DOI] [PubMed] [Google Scholar]
- 22. Chen J. K., Capdevila J., Harris R. C. (2002) Heparin-binding EGF-like growth factor mediates the biological effects of P450 arachidonate epoxygenase metabolites in epithelial cells. Proc. Natl. Acad. Sci. U.S.A. 99, 6029–6034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yang S., Wei S., Pozzi A., Capdevila J. H. (2009) The arachidonic acid epoxygenase is a component of the signaling mechanisms responsible for VEGF-stimulated angiogenesis. Arch. Biochem. Biophys. 489, 82–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Capdevila J. H., Dishman E., Karara A., Falck J. R. (1991) Cytochrome P450 arachidonic acid epoxygenase. Stereochemical characterization of epoxyeicosatrienoic acids. Methods Enzymol. 206, 441–453 [DOI] [PubMed] [Google Scholar]
- 25. Capdevila J. H., Falck J. R., Harris R. C. (2000) Cytochrome P450 and arachidonic acid bioactivation: molecular an functional properties of the arachidonate monooxygenase. J. Lipid Res. 41, 163–181 [PubMed] [Google Scholar]
- 26. Stoos B. A., Náray-Fejes-Tóth A., Carretero O. A., Ito S., Fejes-Tóth G. (1991) Characterization of a mouse cortical collecting duct cell line. Kidney Int. 39, 1168–1175 [DOI] [PubMed] [Google Scholar]
- 27. Korbmacher C., Segal A. S., Fejes-Tóth G., Giebisch G., Boulpaep E. L. (1993) Whole-cell currents in single and confluent M1 mouse cortical collecting duct cells. J. Gen. Physiol. 102, 761–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nakhoul N. L., Hering-Smith K. S., Gambala C. T., Hamm L. L. (1998) Regulation of sodium transport in M-1 cells. Am. J. Physiol. Renal Physiol. 275, F998–F1007 [DOI] [PubMed] [Google Scholar]
- 29. Zeldin D. C., Kobayashi J., Falck J. R., Winder B. S., Hammock B. D., Snapper J. R., Capdevila J. H. (1993) Regio- and enantiofacial selectivity of epoxyeicosatrienoic acid hydration by cytosolic epoxide hydrolase. J. Biol. Chem. 268, 6402–6407 [PubMed] [Google Scholar]
- 30. Hotokezaka H., Sakai E., Kanaoka K., Saito K., Matsuo K., Kitaura H., Yoshida N., Nakayama K. (2002) U0126 and PD98059, specific inhibitors of MEK, accelerate differentiation of RAW246.7 cells into osteoclast-like cells. J. Biol. Chem. 277, 47366–47372 [DOI] [PubMed] [Google Scholar]
- 31. Shushan A., Ben-Bassat H., Mishani E., Laufer N., Klein B. Y., Rojansky N. (2007) Inhibition of leiomyoma cell proliferation by genistein and the protein tyrosine kinase inhibitor TSKS050. Fertil. Steril. 87, 127–135 [DOI] [PubMed] [Google Scholar]
- 32. Chen F., Hancock C. N., Macias A. T., Joh J., Still K., Zhong S., MacKerell A. D., Jr., Shapiro P. (2006) Characterization of ATP-independent ERK inhibitors identified through in silico analysis of active ERK2 structure. Bioorg. Med. Chem. Lett. 16, 6281–6287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Harding J., Burtness B. (2005) Cetuximab: an epidermal growth factor receptor chimeric-human-murine monoclonal antibody. Drugs Today 41, 107–127 [DOI] [PubMed] [Google Scholar]
- 34. Shimkets R. A., Lifton R., Canessa C. M. (1998) In vivo phosphorylation of the epithelial sodium channel. Proc. Natl. Acad. Sci. U.S.A. 95, 3301–3305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ergonul Z., Frindt G., Palmer L. G. (2006) Regulation of maturation and processing of ENaC subunits in the rat kidney. Am. J. Physiol. Renal Physiol. 291, F683–F693 [DOI] [PubMed] [Google Scholar]
- 36. Hughey R. P., Bruns J. B., Kinlough C. L., Kleyman T. R. (2004) Distinct pools of epithelial sodium channel are expressed at the plasma membrane. J. Biol. Chem. 279, 48491–48494 [DOI] [PubMed] [Google Scholar]
- 37. Hu J. C., Bengrine A., Lis A., Awayda M. S. (2009) Alternative mechanisms of activation of the epithelial Na+ channel by cleavage. J. Biol. Chem. 284, 36334–36345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rossier B. C., Stutts M. J. (2009) Activation of the epithelial sodium channel (ENac) by serine proteases. Annu. Rev. Physiol. 71, 361–379 [DOI] [PubMed] [Google Scholar]
- 39. DeLozier T. C., Tsao C. C., Coulter S. J., Foley J., Bradbury J. A., Zeldin D. C., Goldstein J. A. (2004) CYP2C44, a new murine CYP2C that metabolizes arachidonic acid to unique stereospecific products. J. Pharmacol. Exp. Ther. 310, 845–854 [DOI] [PubMed] [Google Scholar]
- 40. Liu L., Cao Y., Tan A., Liao C., Mo Z., Gao F. (2010) Cetuximab-based therapy vs. noncetuximab therapy in advanced or metastatic colorectal cancer: a meta-analysis of seven randomized controlled trails. Colorectal Dis. 12, 399–406 [DOI] [PubMed] [Google Scholar]
- 41. Masilamani S., Kim G. H., Mitchell C., Wade J. B., Knepper M. A. (1999) Aldosterone-mediated regulation of ENaC α, β, and γ subunit proteins in rat kidney. J. Clin. Investig. 104, R19–R23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Holthöfer H., Schulte B. A., Spicer S. S. (1987) Expression of binding sites for Dolichos biflorus agglutinin at the apical aspect of collecting duct cells in rat kidney. Cell Tissue Res. 249, 481–485 [DOI] [PubMed] [Google Scholar]
- 43. Rotin D., Kanelis V., Schild L. (2001) Trafficking and cell surface stability of ENaC. Am. J. Physiol. Renal Physiol. 281, F391–F399 [DOI] [PubMed] [Google Scholar]
- 44. Thomas C. P., Itani O. A. (2004) New insights into epithelial sodium channel function in the kidney: site of action, regulation by ubiquitin ligases, serum- and glucocorticoid-inducible kinases and proteolysis. Curr. Opin. Nephrol. Hypertens 13, 541–548 [DOI] [PubMed] [Google Scholar]
- 45. Butterworth M. B., Weisz O. A., Johnson J. P. (2008) Some assembly required: putting the epithelial sodium channel together. J. Biol. Chem. 283, 35305–35309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fejes-Tóth G., Frindt G., Náray-Fejes-Tóth A., Palmer L. G. (2008) Epithelial Na+ channel activation and processing in mice lacking SGK1. Am. J. Physiol. Renal Physiol. 294, F1298–F1305 [DOI] [PubMed] [Google Scholar]
- 47. Yang W., Tuniki V. R., Anjaiah S., Falck J. R., Hillard C. J., Campbell W. B. (2008) Characterization of epoxyeicosatrienoic acid binding site in U937 membranes using a novel radiolabeled agonist 20-125I-14,15-epoxyeicosa-8(Z)-enoic acid. J. Pharmacol. Exp. Ther. 324, 1019–1027 [DOI] [PubMed] [Google Scholar]