

Background: Trypanosoma brucei S-adenosylmethionine decarboxylase (AdoMetDC) is activated by heterodimerization with a catalytically dead paralog, prozyme.
Results: Trypanosomatid-specific residues in the AdoMetDC N terminus are essential for prozyme-mediated activation but not for heterodimerization.
Conclusion: AdoMetDC activation likely involves a conformational change of the N-terminal peptide.
Significance: Development of conformationally sensitive AdoMetDC inhibitors may provide a species-selective mechanism to inhibit trypanosomatid AdoMetDCs.
Keywords: Allosteric Regulation, Parasite Metabolism, Polyamines, S-Adenosylmethionine (SAM), Trypanosome, S-Adenosylmethionine Decarboxylase, Catalytically Dead Paralog
Abstract
Human African trypanosomiasis is caused by a single-celled protozoan parasite, Trypanosoma brucei. Polyamine biosynthesis is a clinically validated target for the treatment of human African trypanosomiasis. Metabolic differences between the parasite and the human polyamine pathway are thought to contribute to species selectivity of pathway inhibitors. S-adenosylmethionine decarboxylase (AdoMetDC) catalyzes a key step in the production of the polyamine spermidine. We previously showed that trypanosomatid AdoMetDC differs from other eukaryotic enzymes in that it is regulated by heterodimer formation with a catalytically dead paralog, designated prozyme, which binds with high affinity to the enzyme and increases its activity by up to 103-fold. Herein, we examine the role of specific residues involved in AdoMetDC activation by prozyme through deletion and site-directed mutagenesis. Results indicate that 12 key amino acids at the N terminus of AdoMetDC are essential for prozyme-mediated activation with Leu-8, Leu-10, Met-11, and Met-13 identified as the key residues. These N-terminal residues are fully conserved in the trypanosomatids but are absent from other eukaryotic homologs lacking the prozyme mechanism, suggesting co-evolution of these residues with the prozyme mechanism. Heterodimer formation between AdoMetDC and prozyme was not impaired by mutation of Leu-8 and Leu-10 to Ala, suggesting that these residues are involved in a conformational change that is essential for activation. Our findings provide the first insight into the mechanisms that influence catalytic regulation of AdoMetDC and may have potential implications for the development of new inhibitors against this enzyme.
Introduction
Human African trypanosomiasis (HAT),2 also known as sleeping sickness, is a vector-borne parasitic disease caused by the protozoan pathogen Trypanosoma brucei (1, 2). The World Health Organization estimates that up to 50 million people in Sub-Saharan Africa are at risk with reported yearly infections in the tens of thousands.3 HAT is characterized by severe neurological symptoms that lead to coma and eventual death in nearly all untreated individuals (4, 5). The World Health Organization has increased surveillance and control of African trypanosomiasis (6); however, one of the greatest remaining challenges to eliminating HAT is the lack of safe and effective therapies (7–9). The recent introduction of nifurtimox-eflornithine combination therapy has improved management of late stage disease (10), but it is a difficult therapy to administer and is not effective against all species of the parasite. Eflornithine, also known as α-difluoromethylornithine, is a suicide inhibitor of ornithine decarboxylase, implicating polyamine biosynthesis as a key target for HAT therapy.
Polyamines are small organic cations that are required for cell growth and that play important roles in diverse cellular processes such as replication, transcription, and translation (11–13). The key steps in polyamine biosynthesis are catalyzed by ornithine decarboxylase, which leads to formation of putrescine, and by the pyruvoyl-dependent S-adenosylmethionine decarboxylase (AdoMetDC), which catalyzes the decarboxylation of AdoMet to produce the aminopropyl group required for synthesis of spermidine from putrescine (Scheme 1) (14). Human AdoMetDC is a α2β2 homodimer, and the active sites that contain the covalently bound pyruvate cofactor sit in a cleft between β-sheets oriented away from the dimer interface (see Fig. 1A) (15, 16). The pyruvate is generated by an autoprocessing reaction that also leads to cleavage of the peptide bond into the α and β chains (17, 18). Putrescine stimulates both the processing and decarboxylation reactions of the mammalian enzyme (15, 18).
SCHEME 1.

Polyamine biosynthetic pathway in T. brucei. dcAdoMet, decarboxylated AdoMet; ODC, ornithine decarboxylase; SpdSyn, spermidine synthase; TrypSyn, trypanothione synthetase; γ-GCS, γ-glutamylcysteine synthetase; GS, glutathione synthetase; γ-GC, γ-glutamylcysteine.
FIGURE 1.

A, structure of human AdoMetDC (1I7M) bound to 4-amidinoindan-1-one-2′-amidinohydrazone (CGP 48664A; green) and to putrescine (Put; orange balls); α chains are displayed in purple, β chains are displayed in tan, and N-terminal restudies are shown as sticks. The N and C termini and part of the putrescine-binding site (Asp-174) are labeled. B, N-terminal sequence alignment of eukaryotic AdoMetDCs with prozyme (Pr). Highlighted in turquoise are residues essential for activity. Highlighted in yellow is the conserved 12-amino acid peptide found only in the trypanosomatid AdoMetDCs. T.br, T. brucei; T.cr, T. cruzi; L.ma, L. major.
Polyamine metabolism in trypanosomatids is unique, and differences with the human enzyme are thought to contribute to selective toxicity of inhibitors that target the pathway (19–22). These include differences in protein turnover rates and the finding that spermidine is conjugated to glutathione, forming a novel molecule termed trypanothione, which is required for redox chemistry. Genetic and chemical studies have demonstrated that polyamine and trypanothione biosynthetic enzymes are essential for T. brucei growth (20), and in addition to α-difluoromethylornithine, inhibitors of AdoMetDC and trypanothione synthetase showing good antitrypanosomal activity have also been described (23–25).
Polyamine levels are tightly controlled throughout the cell cycle in eukaryotes, but the regulatory mechanisms established in other eukaryotes are not found in T. brucei (26, 27). We showed previously that the trypanosomatid AdoMetDC is allosterically activated by heterodimer formation with a catalytically dead paralog, designated prozyme, which is present only in the trypanosomatids (28). Prozyme is neither processed to generate the pyruvate cofactor, nor does it display catalytic activity. However, upon binding, prozyme stimulates AdoMetDC activity by ∼103-fold. Putrescine, which stimulates the activity of human AdoMetDC, does not affect the activity of the T. brucei heterodimer (15); however, it is required for the Trypanosoma cruzi heterodimer to reach full activation (29). In T. brucei, prozyme expression levels appear to be translationally regulated in response to inhibition or knockdown of AdoMetDC, suggesting that the parasite controls polyamine synthesis at least in part through this mechanism (30).
The structural basis for the activation of trypanosomatid AdoMetDC by prozyme remains an open question as no structural data are available for any of the trypanosomatid enzymes. To gain insight into this question, we evaluated the role of trypanosomatid-specific amino acid residues in the prozyme-mediated activation of T. brucei AdoMetDC through deletion and site-directed mutagenesis. Results indicate that the unique trypanosomatid N-terminal 16-amino acid peptide of AdoMetDC is essential for this process. We found that deletion of these residues led to a loss of prozyme activation. Subsequent site-directed mutagenesis identified two Leu residues (8 and 10) and two Met residues (11 and 13) necessary for prozyme activation. Interestingly, unlike the wild-type heterodimeric enzyme, the L8A and L10A mutant enzymes were activated by putrescine, although activity remained below that of the fully functional wild-type enzyme. Sedimentation velocity experiments demonstrated that mutation of Leu-8 and Leu-10 to Ala does not impair heterodimer formation, suggesting that these residues are involved in a conformational change that is essential for activation. The 16-amino acid unique N terminus is partially conserved in prozyme, but deletion of these residues in prozyme did not significantly affect activity. Our findings provide the first insight into the structural basis and mechanisms that control AdoMetDC activation by prozyme, demonstrating the importance of a unique N-terminal peptide in the process.
EXPERIMENTAL PROCEDURES
Multiple Sequence Alignment
AdoMetDC and prozyme sequences were obtained from PubMed (Homo sapiens gi:178518 and Solanum tuberosum gi:416883) and GeneDB (Tb927.6.4460, Tc00.1047053504257.30, LmjF30.3110, Tb927.6.4470, Tc00.1047053509167.110, and LmjF30.3120) databases and converted into a FASTA format. Alignment was performed using the ClustalW2 program.
Construction of Wild-type and Mutant Escherichia coli Expression Constructs
T. brucei AdoMetDC was amplified by PCR from the pET15b-AdoMetDC plasmid DNA (28) and cloned into the pE-SUMO (LifeSensors, Malvern, PA) vector. This construct was then used as a template to produce the Δ16 AdoMetDC mutant using complementary primer pairs encoding the sequence alteration. Point mutants were created using a QuikChange site-directed mutagenesis kit (Stratagene). T. brucei prozyme was previously cloned into the pT7-FLAG1 (Sigma) vector for expression of FLAG-tagged (amino acid sequence, MDYKDDDDK) protein in E. coli (28, 30). This construct was used as the PCR template to generate a Δ25 N-terminal deletion of prozyme, which was then cloned it into the pET28b vector engineered to generate a tagless protein. Cloning primers are provided in supplemental Table S1. E. coli TOP10 competent cells were transformed with each plasmid, and mutations were confirmed by DNA sequencing.
AdoMetDC·Prozyme Expression and Purification
E. coli BL21/DE3 cells were transformed with the construct of interest and grown at 37 °C until A600 reached between 0.6 and 0.8. Protein expression was induced by the addition of 0.2 mm isopropyl 1-thio-β-d-galactopyranoside, and cells were grown overnight at 20 °C. Cells were harvested by centrifugation and resuspended in lysis buffer (50 mm Hepes, 100 mm NaCl, 10 mm imidazole, 5 mm β-mercaptoethanol). Protease inhibitors were added directly to the cell lysate (Mixture 1 (1:1000), 1 mg/ml leupeptin, 2 mg/ml antipain, and 10 mg/ml benzamidine; Mixture 2 (1:1000), 1 mg/ml pepstatin, 1 mg/ml chymostatin; and 0.2 m phenylmethylsulfonyl fluoride (PMSF) in a 1:500 ratio) (Sigma). Bacterial cells were lysed using an EmulsiFlex-C5 homogenizer followed by centrifugation (Beckman Ti-45 rotor) at 15,000 rpm for 30 min. The soluble fraction was subjected to Ni2+-agarose (Qiagen, Valencia, CA) column chromatography (buffer A, 50 mm Hepes, pH 8.0, 100 mm NaCl, 1 mm β-mercaptoethanol, 10 mm imidazole; buffer B, 50 mm Hepes, pH 8.0, 100 mm NaCl, 1 mm β-mercaptoethanol, 800 mm imidazole) as described previously (28, 31). Heterodimeric enzymes were obtained by mixing the separate lysates and co-purifying the His6-SUMO-tagged AdoMetDC with the FLAG-tagged wild-type prozyme or tagless Δ25 prozyme. AdoMetDC-containing fractions were pooled together, concentrated, and diluted in buffer A to remove excess imidazole. The protein sample was then incubated with ULP1 (see below), also known as SUMO protease 1, which recognizes the tertiary structure of SUMO in the fusion protein and cleaves with high specificity. This resulted in the removal of the His6-SUMO tag, generating AdoMetDC such that no tag region remained. AdoMetDC (wild type or mutant) sample was incubated with ∼300 μg of ULP1 for every 20–35 mg of AdoMetDC for 2 h at 4 °C with gentle agitation. After cleavage, a second Ni2+-agarose purification step was performed in which the now tagless enzyme was collected in the flow-through fractions. Anion-exchange chromatography was subsequently performed as described previously (28, 31) for WT homodimer/heterodimer, Δ16 homodimer/heterodimer, and the AdoMetDC LSL-AAA heterodimer, all of which underwent careful binding analyses. Remaining mutants were sufficiently pure after the second Ni2+-agarose purification step. Extinction coefficients (supplemental Table S1) for the wild-type and mutant enzymes were obtained through the ExPASy-ProtParam program and used to quantify protein by measuring absorbance at 280 nm. These calculations were performed using the Beer's law equation (A = ϵlc where A is absorbance, ϵ is extinction coefficient, l is the path length, and c is concentration).
Expression and Purification of ULP-1
The pET15b-ULP1 plasmid was provided by Kim Orth. Expression of this N-terminal His-tagged protease was performed as above with the following modifications. Isopropyl 1-thio-β-d-galactopyranoside induction was performed at 30 °C overnight, and modified lysis (50 mm Tris-HCl, pH 7.5, 350 mm NaCl, 1 mm β-mercaptoethanol, 10 mm imidazole, 0.2% IGEPAL, 20% glycerol) and purification buffers (buffer A, 50 mm Tris-HCl, pH 7.5, 350 mm NaCl, 1 mm β-mercaptoethanol, 10 mm imidazole, 20% glycerol; buffer B, buffer A plus 800 mm imidazole) were used.
Steady-state Kinetic Analysis
The enzymatic activity of AdoMetDC was assessed using the 14CO2 trapping method (32). AdoMetDC (1–3 μm for homodimers and 0.1 and 1–6 μm for wild-type and mutant heterodimers, respectively) was titrated into reactions containing 25 μm 14CO2-AdoMet (American Radiolabeled Chemicals), a range of unlabeled AdoMet concentrations (25–1475 μm; Affymetrix), 100 mm Hepes, pH 8.0, 50 mm NaCl, 1 mm DTT. Putrescine (5 mm) was included as indicated. Test tubes were capped and placed in a water bath at 37 °C. The CO2 liberated from the substrate was trapped on a filter paper soaked in 80 μl of saturated Ba(OH)2. After 5 min, reactions were quenched using 6 m HCl. The filter paper was then transferred to a scintillation vial containing 5 ml of CytoScint. Samples were placed on the scintillation counter, and the counts per minute (cpm) obtained were converted into velocity (s−1) values using the specific activity of the substrate. Data were fitted to the Michaelis-Menten equation to determine the steady-state kinetic parameters (kcat and Km) using Prism (GraphPad, San Diego, CA). The kcat was calculated based on the molecular weight of the dimeric species.
Sedimentation Velocity
All analytical ultracentrifugation experiments were performed in a Beckman-Coulter (Indianapolis, IN) Optima XL-I ultracentrifuge using the Beckman An50Ti rotor. Studies were carried out at 20 °C and 50,000 rpm. Samples of co-purified wild-type or mutant AdoMetDC·prozyme were diluted to their final concentrations in analytical ultracentrifugation buffer (50 mm Hepes, pH 8.0, 50 mm NaCl, 1 mm β-mercaptoethanol) and incubated overnight at 4 °C before loading into dual sectored, charcoal-filled Epon centerpieces that had been sandwiched between sapphire windows in a centerpiece housing. The assembled housings were placed in the rotor and allowed to incubate at 20 °C for 2.5 h before centrifugation. Radially dependent concentration profiles were acquired at ∼10-min time intervals using the absorption optics tuned to 280 nm.
Data were analyzed using either SEDFIT or SEDPHAT, and c(s) distributions were used to fit the data (33, 34). Regularization levels of 0.68 were used. In the case of the Δ16 mutant, these distributions exhibited strong concentration dependence. Thus, these data were fitted to solutions of the Lamm equation with explicit consideration of the thermodynamics and kinetics of the AdoMetDC/prozyme interaction (35, 36). For wild-type AdoMetDC/prozyme mixture, a fixed fraction (23%) of the protein did not participate in the heterodimeric complex regardless of the concentration of the complex. Based on the method of purification, we assumed that this represented a fraction of AdoMetDC that was incompetent to form the complex and that the incompetent fraction was identical and constant in all wild-type and mutant AdoMetDC preparations. To carry out analysis of the association of Δ16 and prozyme, six sedimentation experiments were analyzed globally in SEDPHAT with several other assumptions and constraints as follows. 1) Monomeric AdoMetDC and monomeric prozyme behaved identically hydrodynamically. 2) The sedimentation coefficient of the complex was the same as that of the wild-type complex. 3) The material that sediments with an s20,w value of 4.9 S is all heterodimeric with no homodimers of either protein present. 4) No hyper- or hypochromicity accompanied the interaction. 5) All proteins and the complex have the same partial specific volume (0.73 cm3/g). 6) An observed 7 S species did not participate in the interaction and could be treated as “non-participating.” 7) The molar ratio of AdoMetDC and prozyme was constant (supplemental Fig. S1). The error interval for the association constant was calculated using the projection method (37) with the F-statistic described by Johnson (38). All analytical ultracentrifugation-derived figures were made using GUSSI. The same software was used to convert c(s) distributions to standard conditions. SEDNTERP (39) was used to calculate buffer viscosity, buffer density, and protein partial specific volumes.
Molecular Modeling
Human AdoMetDC (Protein Data Bank code 1I7B (15, 18)) was displayed using the graphics program PyMOL (40).
RESULTS
Sequence Analysis of Eukaryotic AdoMetDCs
To elucidate the structural requirements for AdoMetDC activation by prozyme, we performed a sequence alignment of representative AdoMetDCs to identify any sequence signatures that were unique to the trypanosomatid enzymes and thereby might be involved in prozyme activation. The analysis indicated that the N terminus contains a unique 16–20-amino acid peptide present in the trypanosomatid (T. brucei, T. cruzi, and Leishmania major) AdoMetDCs but absent from other eukaryotic homologs (e.g. human and plant; Fig. 1B). In this region, 12 specific amino acids are particularly conserved between the parasitic enzymes. Interestingly, although prozyme contains this stretch of residues as well, the degree of conservation with the respective AdoMetDC or prozyme from different species is lower. The human enzyme is fully active as a homodimer and does not contain this peptide. Thus, this conserved N-terminal extension is only present in the AdoMetDCs that are activated by prozyme, suggesting that it might play a role in the prozyme-mediated activation of the trypanosomatid enzymes.
Unique N terminus of T. brucei AdoMetDC Is Essential for Activation by Prozyme
A mutant version of T. brucei AdoMetDC was constructed lacking the N-terminal 16 amino acids to determine whether this region was required for activation. Both homodimeric (Δ16 AdoMetDC) and heterodimeric (Δ16 AdoMetDC·prozyme) enzymes were expressed and purified from E. coli. Like wild-type AdoMetDC, Δ16 AdoMetDC undergoes a self-processing reaction to generate the active enzyme consisting of two chains, α (32kDa) and β (8 kDa) (data not shown). In addition, Δ16 AdoMetDC, like the wild-type enzyme, forms a stable complex with wild-type prozyme that co-purified over Ni2+-agarose and ion-exchange chromatography. In the absence of putrescine, the Δ16 AdoMetDC homodimer has a kcat/Km of 16 m−1 s−1, which is similar to that of the wild-type homodimer, although the Km is increased significantly by this mutation (Table 1 and Fig. 2). However, prozyme is unable to activate the mutant enzyme, which displays a catalytic activity for the Δ16 AdoMetDC·prozyme heterodimer that is 100-fold lower than the wild-type heterodimer (Fig. 2). This strong decrease in catalytic efficiency is the consequence of an effect on both the apparent substrate binding constant (Km) and the catalytic rate (kcat) (Table 1). Putrescine stimulates the activity of the wild-type AdoMetDC homodimer and has no effect on the heterodimer. Activities of the truncated dimeric enzymes (Δ16 AdoMetDC and Δ16 AdoMetDC·prozyme) were not significantly affected by the addition of putrescine.
TABLE 1.
Steady-state kinetic analysis of wild-type and mutant T. brucei AdoMetDC enzymes
kcat/Km (m−1 s−1) values were determined through linear regression (slope) of the velocity versus substrate plots, and the highest substrate concentration tested was used to determine a lower estimate of Km and kcat. The error represents the S.D. for n = 3 replicates.
| Enzyme | 5 mm putrescine |
No putrescine |
||||
|---|---|---|---|---|---|---|
| kcat | Km | kcat/Km | kcat | Km | kcat/Km | |
| s−1 | mm | m−1 s−1 | s−1 | mm | m−1 s−1 | |
| WT AdoMetDC homodimer | 0.0065 ± 0.0004 | 0.14 ± 0.04 | 46 ± 12 | 0.0058 ± 0.0003 | 0.40 ± 0.06 | 15 ± 2 |
| WT AdoMetDC·prozyme | 0.21 ± 0.01 | 0.13 ± 0.02 | (1.7 ± 0.3) × 103 | 0.25 ± 0.02 | 0.14 ± 0.02 | (1.8 ± 0.5) × 103 |
| Δ16 AdoMetDC homodimer | >0.03 | >2 | 16 ± 3a | >0.032 | >2 | 16 ± 11a |
| Δ16 AdoMetDC·prozyme | 0.017 ± 0.001 | 0.60 ± 0.1 | 28 ± 5 | 0.014 ± 0.001 | 0.80 ± 0.1 | 18 ± 3 |
| AdoMetDC(KDS-AAA)·prozyme | 0.33 ± 0.07 | 0.60 ± 0.3 | (5.7 ± 0.3) × 102 | 0.071 ± 0.005 | 0.37 ± 0.07 | (1.9 ± 0.4) × 102 |
| AdoMetDC(LSL-AAA)·prozyme | 0.011 ± 0.001 | 1.2 ± 0.2 | 9.0 ± 1.0 | >0.01 | >2 | 5.1 ± 0.5a |
| AdoMetDC(MAM-AAA)·prozyme | 0.026 ± 0.002 | 0.31 ± 0.08 | 85 ± 22 | >0.01 | >2 | 4.8 ± 0.1a |
| AdoMetDC(WGS-AAA)·prozyme | 0.10 ± 0.01 | 0.30 ± 0.1 | (2.8 ± 0.1) × 102 | 0.18 ± 0.04 | 1.0 ± 0.5 | (1.7 ± 0.8) × 102 |
| AdoMetDC(L8A)·prozyme | 0.12 ± 0.003 | 0.45 ± 0.03 | (2.5 ± 0.2) × 102 | >0.03 | >2 | 16.6 ± 0.5a |
| AdoMetDC(S9A)·prozyme | 0.29 ± 0.02 | 0.90 ± 0.1 | (3.4 ± 0.5) × 102 | 0.23 ± 0.01 | 1.4 ± 0.1 | (1.8 ± 0.2) × 102 |
| AdoMetDC(L10A)·prozyme | 0.084 ± 0.009 | 0.22 ± 0.08 | (3.8 ± 0.1) × 102 | >0.10 | >2 | 50 ± 9a |
| WT AdoMetDC·Δ25 prozyme | 0.15 ± 0.01 | 0.20 ± 0.04 | (7.7 ± 0.2) × 102 | 0.23 ± 0.04 | 0.60 ± 0.20 | (4 ± 1) × 102 |
| Δ16 AdoMetDC·Δ25 prozyme | >0.004 | >2 | 2.2 ± 0.1a | >0.006 | >2 | 3.1 ± 0.3a |
a Kinetic experiments in which substrate saturation was not observed.
FIGURE 2.

Catalytic efficiency (kcat/Km) of wild-type and mutant T. brucei (T.br) AdoMetDCs in the presence (purple) and absence (blue) of 5 mm putrescine (Put). The inset represents the N-terminal region with the conserved trypanosomatid residues underlined. HM, AdoMetDC homodimer; HT, AdoMetDC·prozyme heterodimer. Error bars represent the S.D. for n = 3 replicates.
Identification of Residues within the N-terminal Peptide That Are Essential for Prozyme Activation
To identify specific residues within the N-terminal peptide that were involved in activation by prozyme, Ala-scanning mutagenesis was performed. The key residues were initially narrowed down by construction of four mutants, each containing three consecutive amino acids substituted with Ala unless the native residue was Ala or Gly in which case it was unaltered. Similar to the Δ16 homodimer and heterodimer, these mutants were all capable of self-processing, and the heterodimeric complexes held together through all purification steps. Kinetic analysis of each mutant was performed on the AdoMetDC·prozyme complex. Results indicate that AdoMetDC triple mutant L8A/S9A/L10A had the strongest effect; the catalytic efficiency was 360-fold lower than for the wild-type enzyme both in the presence and absence of putrescine (Table 1 and Fig. 2). AdoMetDC M11A/M13A showed similarly reduced enzyme activity in the absence of putrescine, but in this case, activity was stimulated 20-fold by putrescine to within ∼20-fold of wild-type heterodimer levels. The remaining two triple and double mutants (K5A/D6A/S7A and W14A/S16A) were only minimally impaired (3–6-fold) in their ability to be activated by prozyme. To further study the contributions of Leu-8, Ser-9, and Leu-10, individual point mutations to Ala were constructed for each of these residues (Fig. 2). Ser-9 was not essential because activity within 10-fold of wild-type heterodimer levels was observed upon mutation to Ala. However, when Leu-8 or Leu-10 were replaced by Ala, the activity was similar to that of the truncated Δ16 AdoMetDC·prozyme complex, although the activity of these mutants was stimulated by putrescine to within 10-fold of wild-type levels.
Analysis of the Dimer Formation by Sedimentation Velocity
Although all mutant enzymes could be co-purified as heterodimers, to assess the relative strengths of the associations between the prozyme and various AdoMetDC constructs, we performed analytical ultracentrifugation experiments in the sedimentation velocity mode at three different protein concentrations (Fig. 3). Wild-type heterodimer binding affinity (Kd) was compared with those mutations that led to the largest decrease in activity. If the dissociation constant for the heterodimers was near the concentrations used, a concentration dependence would be expected for the observed sedimentation coefficients or for the populations of the resolved species, whereas if the Kd was below the lowest tested concentration no dependence would be observed (41, 42).
FIGURE 3.
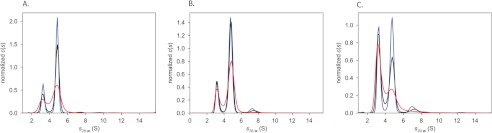
Sedimentation velocity analysis. A, size distributions for the wild-type AdoMetDC·prozyme heterodimer. Distributions have been normalized by the total signal present between 2 and 7 S in the respective distributions. Material sedimenting at <1 S (probably resulting from a buffer mismatch) has been excluded from the plot. AdoMetDC·prozyme concentrations were 0.3 (red), 1.7 (black), and 3.8 μm (blue). B, size distributions of the L8A/S9A/L10A AdoMetDC·prozyme heterodimer. Distributions have been normalized by the total amount of signal present. Concentrations of the complex were 0.7 (red), 2.2 (black), and 3.7 μm (blue). C, size distributions of the Δ16 AdoMetDC·prozyme heterodimer. Distributions were normalized by the total amount of signal present between 2 and 6 S, and slowly sedimenting material of <1 S has been excluded. The concentrations of the (potential) complex were 0.7 (red), 1.9 (black), and 3.3 μm (blue).
For the wild-type and L8A/S9A/L10A mutant AdoMetDC·prozyme complexes, the c(s) distributions, which relate the signal populations of sedimenting species directly to the sedimentation velocity data, revealed two well resolved peaks (Fig. 3A). The peaks occurred at s20,w values of 3.3 and 4.8 S, and these values were not dependent on the concentration of the complex. The estimated molar masses associated with the two species were 40 and 70 kg/mol, consistent with the expected masses for the monomeric (3.3 S) and heterodimeric (4.8 S) species. Significantly, the relative signal populations of the two species did not change over the concentration range studied (0.3–3.8 μm), demonstrating that these species were not in equilibrium. The lack of relative population variation shows that the dissociation constants were below the lowest complex concentration studied (Kd <0.3 μm for wild-type and Kd <0.7 μm for the L8A/S9A/L10A mutant). The result for the wild-type enzyme is similar to our prior sedimentation equilibrium analysis where we found the dissociation constant between AdoMetDC and prozyme to be below 0.5 μm (28). The data also suggest that the 3.3 S material (∼23% of the total) contains protein that was no longer competent to form a dimer. SDS-PAGE analysis of the AdoMetDC·prozyme preparations showed that they contain approximately equal concentrations of the two proteins, consistent with the observation that the incompetent material does not represent a major fraction of the protein (supplemental Fig. S1). Based on the method of purification, excess incompetent prozyme could not be carried through the purification because it lacked a His tag. This suggests that either the 3.3 S peak contained largely incompetent AdoMetDC or that incompetence occurred after the Ni2+ affinity purification step and that the 3.3 S material is a mixture of incompetent AdoMetDC and prozyme. Two conclusions may be drawn from these data. First, the LSL mutations did not increase the dissociation constant of AdoMetDC and prozyme to above the limit of detection (Kd <0.7 μm), and given that this value is well below the concentration used in the enzyme assays, these data provide a clear indication that the reduced activity of this mutant does not result from loss of dimerization. Second, the amount of incompetent AdoMetDC in the mutant preparation (∼21%) was similar to the wild-type enzyme, suggesting that its relative abundance does not strongly vary from preparation to preparation. The cause of this phenomenon is unknown.
In contrast to the wild-type and L8A/S9A/L10A protein complexes, Δ16 AdoMetDC·prozyme displayed robust concentration dependence in the sedimentation velocity studies (Fig. 3C). Specifically, when going from low to high concentrations, the signal population (the areas of the peaks in Fig. 3C) of the 3.3 S species decreases, whereas that of the 4.8 S species increases, consistent with mass action law. This observation and our experiences with the wild-type AdoMetDC allowed us to construct a constrained Δ16 + prozyme ↔ Δ16·prozyme interaction model and use it to fit the sedimentation velocity data using the Lamm equation with explicitly considered thermodynamics and kinetics (see “Experimental Procedures” for the assumptions and constraints built into the model). Given the assumptions and constraints, we found that the dissociation constant was 1.0 (0.9–1.4) μm (the numbers in parentheses indicate the 68.3% error interval). The quality of the fits to the data were high based on the low overall root mean square of the differences between the data and the fits (0.0065 arbitrary unit) and the non-systematic nature of the residuals (supplemental Fig. S2). Our ability to resolve the complex from the uncomplexed material in the c(s) distributions indicates a slow off-rate for the interaction. Thus, although wild-type and the triple mutant heterodimers bind tightly, there is decreased affinity between subunits in the Δ16 heterodimer. This implies that truncation at the N terminus weakens the dimer interface but that it does not block dimer formation.
To further confirm that poor dimer formation did not contribute to the lack of activation of the Δ16 heterodimer, an enzyme concentration titration (1–32 μm) was performed using the kinetic activity assay. Results demonstrate that as the concentration of Δ16 AdoMetDC·prozyme was doubled the enzyme velocity also doubled, and there was no indication of a greater than proportional increase in rate as would be expected if monomers were contributing to the lowered rate of the reaction. Thus, based on the sedimentation velocity results, the Δ16 truncation at the N terminus weakens the dimer interface, but the dimer was fully formed under the conditions of the enzyme assay. These data suggest that the presence of substrate may impact the Kd for dimerization of the Δ16 mutant.
N Terminus of T. brucei Prozyme Is Not Involved in AdoMetDC Activation
Because T. brucei prozyme also contains residues at the N terminus with similarity, although not full conservation, to the trypanosomatid AdoMetDC N-terminal signature, we evaluated the effects of truncation of the prozyme N terminus as well. Results demonstrate that when wild-type AdoMetDC is co-purified with Δ25 prozyme, a catalytically efficient enzyme was still obtained (Table 1 and Fig. 4). Similarly to wild-type prozyme, Δ25 prozyme was not able to stimulate Δ16 AdoMetDC activity, demonstrating once again the essentiality of the AdoMetDC N terminus for AdoMetDC activation. Putrescine had no effect on the activity of these complexes.
FIGURE 4.

Catalytic efficiency (kcat/Km) of T. brucei AdoMetDC·truncated prozyme heterodimers. WT, wild-type AdoMetDC·wild-type prozyme complex; Δ16, Δ16 AdoMetDC·wild-type prozyme; Δ25 proz, wild-type AdoMetDC·Δ25 prozyme; and, Δ16/Δ25, Δ16 AdoMetDC·Δ25 prozyme. Data were collected in the presence (purple) and absence (blue) of 5 mm putrescine (Put). Error bars represent the S.D. for n = 3 replicates.
DISCUSSION
Regulation of polyamine biosynthesis is a key cellular function, and eukaryotic cells have evolved a wide range of transcriptional, translational, and posttranslational mechanisms to control pathway flux including several mechanisms that are unique to the polyamine pathway (11). Although trypanosomatids do not utilize the mechanisms demonstrated for mammals, yeast, and plants, they too have evolved a novel species-specific mechanism for regulation that relies on enzyme activation of a catalytically impaired AdoMetDC subunit with an inactive paralog, termed prozyme (20). The structural basis for the substantial activation (∼103-fold) that is observed upon heterodimer formation has been partially elucidated by our current studies. We found that the trypanosomatid AdoMetDCs contain a conserved N-terminal peptide of 12 nearly invariant residues that are not present in other eukaryotic AdoMetDCs, none of which are regulated by a prozyme mechanism. Truncation of these residues or point mutation of Leu-8, Leu-10, or Met-11 and Met-13 in combination led to a loss of T. brucei AdoMetDC prozyme activation despite the finding that the heterodimer was still formed. These data demonstrate that the conserved N-terminal peptide is essential for the activation process.
The N terminus of T. brucei AdoMetDC could potentially have played a role in AdoMetDC activation by prozyme either by promoting dimer formation or through an allosteric regulatory mechanism. Our results are consistent with the second possibility. First, although a number of the mutant enzymes that contain either truncations or point mutations in the N terminus of AdoMetDC are no longer activated by prozyme, they all form heterodimers that co-purify over multiple steps of column chromatography. Second, sedimentation velocity experiments on the AdoMetDC L8A/S9A/L10A mutant show that despite the lack of activation this protein forms a dimer with prozyme that is apparently as strong as the wild-type heterodimer. Third, although the Δ16 heterodimer does form a weaker dimer, we found no evidence that this dissociation occurs under the conditions of the enzyme assay. Thus, the data suggest that these N-terminal residues influence catalysis through an allosteric mechanism. Structural knowledge about AdoMetDC relies on the human homodimeric enzyme, which is activated by putrescine and does not have an N-terminal extension. T. brucei AdoMetDC shares 58% sequence identity with human AdoMetDC, and although the expected tertiary structure will be similar, the position of the N terminus of T. brucei AdoMetDC cannot be predicted with existing data. However, the N terminus of human AdoMetDC (residues 5–7) (Fig. 1) lines the substrate-binding pocket, suggesting that the more extensive N-terminal sequence of T. brucei could potentially fold back to interact with the active site. Thus, our data suggest a model in which binding of prozyme results in a conformational change that rearranges the N terminus closer to the active site residues, allowing new interactions that enhance catalysis (Fig. 5). Such a mechanism is reminiscent of the conversion of trypsinogen into its active form trypsin that is achieved after hydrolysis of the Lys15-Ile16 bond by enterokinase (43). This cleavage leads to the formation of a new N terminus, which rearranges and places key amino acids closer to the residues that form the oxyanion hole (Gly-193 and Ser-195). The AdoMetDC N terminus is not near the dimer interface so the mechanism by which heterodimerization impacts the conformation of the N-terminal 16 amino acids likely involves long range interactions as has been observed in other proteins (44).
FIGURE 5.

Hypothetical model for T. brucei AdoMetDC activation. A, WT, wild-type AdoMetDC ±putrescine (Put) showing that binding to putrescine leads to partial activation, whereas formation of the heterodimer with prozyme leads to full activation. The N terminus is shown undergoing a hypothetical conformational change as part of the activation process. B, LSL to Ala triple mutant; MAM to Ala double mutant, and the Δ16 truncation mutant ±putrescine showing that the heterodimeric enzymes exist in a low activity conformation(s), although binding to putrescine leads to partial activation of some of the mutants. Multiple low activity and partially active conformations may be present; thus, not all low activity or partially active forms may be structurally equivalent.
The role of putrescine in activating trypanosomatid AdoMetDCs differs between wild-type T. brucei AdoMetDC and N-terminal AdoMetDC mutants and between the trypanosomatid enzymes from different species. Although the T. brucei AdoMetDC homodimer is marginally activated by putrescine, the heterodimer is not affected (Fig. 5A). In contrast, for the T. cruzi enzyme, putrescine activates both the homodimer and the heterodimer, and both are required for full activation (29). In this study, we found that the M11A/M13A, L8A, and L10A mutant heterodimers, but not the Δ16 heterodimer, have become sensitive to putrescine, and these enzymes are now partially activated by the diamine. Our data suggest that mutation of the T. brucei AdoMetDC N-terminal residues leads to population of additional conformational states that are responsive to putrescine activation, leading to partially active conformations that are not observed in the wild-type enzyme (Fig. 5B). The wild-type T. cruzi heterodimeric enzyme must also populate additional partially active conformations that are not typically observed for the wild-type T. brucei enzyme. Our data suggest that there is an inner dependence between putrescine and prozyme binding and that both have the potential to influence the equilibrium between active and inactive conformers.
In the human enzyme, the putrescine-binding site is composed of acidic residues (Asp-174, Glu-178, and Glu-256) distant from the active site and closer to the dimer interface. Structural studies demonstrate that putrescine influences the orientation of essential catalytic residues through a hydrogen-bonding network mediated by Lys-80, His-243, Glu-11, and Ser-229 (45). However, although Asp-174 and Glu-256 in addition to some of the residues involved in connecting the putrescine-binding pocket to the active site are conserved between the trypanosomatid and human enzymes, they do not seem to play the same roles. We previously showed that, similarly to the human enzyme, Asp-174 is essential for putrescine to bind T. cruzi AdoMetDC and stimulate its activity, whereas the remaining amino acids known to be important in the human enzyme were not, supporting an alternative binding mode for putrescine to the trypanosomatid enzymes (46).
T. brucei AdoMetDC has been both genetically and chemically validated as a potential drug target for the treatment of HAT (20). The finding that AdoMetDC is regulated by a catalytically dead homolog provides alternative mechanisms to active site targets for the design of species-selective enzyme inhibitors. The discovery that the N terminus of AdoMetDC is involved in an allosteric mechanism for prozyme activation indeed suggests several such approaches. Small molecules could be designed to block interaction between the peptide and key residues during catalysis, or compounds that block the conformational change that accompanies activation could also be sought. Gleevec, a drug used for the treatment of certain cancers including chronic myelogenous leukemia, is an inhibitor of the constitutively active Bcr-Abl tyrosine kinase (47, 48). Its mechanism of action represents a good example of a ligand exploiting the inactive conformation of a protein kinase. Another example for this type of mechanism is the development of biaryl urea inhibitors of p38 mitogen-activated protein kinase (3). More detailed structural information on the T. brucei AdoMetDC·prozyme heterodimeric enzyme is needed to fully elucidate the allosteric mechanism of prozyme activation and allow these approaches to be fully explored.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 AI34432 and R37AI034432 (to M. A. P.). This work was also supported by Welch Foundation Grant I-1257 (to M. A. P.).

This article contains supplemental Figs. S1 and S2 and Table S1.
World Health Organization, African Trypanosomiasis Fact Sheet, www.who.int/mediacentre/factsheets/fs259/en/index.html.
- HAT
- human African trypanosomiasis
- AdoMet
- S-adenosylmethionine
- AdoMetDC
- S-adenosylmethionine decarboxylase
- SUMO
- small ubiquitin-like modifier.
REFERENCES
- 1. Brun R., Blum J., Chappuis F., Burri C. (2010) Human African trypanosomiasis. Lancet 375, 148–159 [DOI] [PubMed] [Google Scholar]
- 2. Stuart K., Brun R., Croft S., Fairlamb A., Gürtler R. E., McKerrow J., Reed S., Tarleton R. (2008) Kinetoplastids: related protozoan pathogens, different diseases. J. Clin. Investig. 118, 1301–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Regan J., Breitfelder S., Cirillo P., Gilmore T., Graham A. G., Hickey E., Klaus B., Madwed J., Moriak M., Moss N., Pargellis C., Pav S., Proto A., Swinamer A., Tong L., Torcellini C. (2002) Pyrazole urea-based inhibitors of p38 MAP kinase: from lead compound to clinical candidate. J. Med. Chem. 45, 2994–3008 [DOI] [PubMed] [Google Scholar]
- 4. Barrett M. P., Vincent I. M., Burchmore R. J., Kazibwe A. J., Matovu E. (2011) Drug resistance in human African trypanosomiasis. Future Microbiol. 6, 1037–1047 [DOI] [PubMed] [Google Scholar]
- 5. Kennedy P. G. (2008) The continuing problem of human African trypanosomiasis (sleeping sickness). Ann. Neurol. 64, 116–126 [DOI] [PubMed] [Google Scholar]
- 6. Simarro P. P., Diarra A., Ruiz Postigo J. A., Franco J. R., Jannin J. G. (2011) The human African trypanosomiasis control and surveillance programme of the World Health Organization 2000–2009: the way forward. PLoS Negl. Trop. Dis. 5, e1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brun R., Don R., Jacobs R. T., Wang M. Z., Barrett M. P. (2011) Development of novel drugs for human African trypanosomiasis. Future Microbiol. 6, 677–691 [DOI] [PubMed] [Google Scholar]
- 8. Jacobs R. T., Nare B., Phillips M. A. (2011) State of the art in African trypanosome drug discovery. Curr. Top. Med. Chem. 11, 1255–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Phillips M. A. (2012) Stoking the drug target pipeline for human African trypanosomiasis. Mol. Microbiol. 86, 10–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yun O., Priotto G., Tong J., Flevaud L., Chappuis F. (2010) NECT is next: implementing the new drug combination therapy for Trypanosoma brucei gambiense sleeping sickness. PLoS Negl. Trop. Dis. 4, e720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pegg A. E. (2009) Mammalian polyamine metabolism and function. IUBMB Life 61, 880–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Childs A. C., Mehta D. J., Gerner E. W. (2003) Polyamine-dependent gene expression. Cell. Mol. Life Sci. 60, 1394–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tabor C. W., Tabor H. (1984) Polyamines. Annu. Rev. Biochem. 53, 749–790 [DOI] [PubMed] [Google Scholar]
- 14. Pegg A. E. (2009) S-Adenosylmethionine decarboxylase. Essays Biochem. 46, 25–45 [DOI] [PubMed] [Google Scholar]
- 15. Ekstrom J. L., Mathews I. I., Stanley B. A., Pegg A. E., Ealick S. E. (1999) The crystal structure of human S-adenosylmethionine decarboxylase at 2.25 Å resolution reveals a novel fold. Structure 7, 583–595 [DOI] [PubMed] [Google Scholar]
- 16. Pegg A. E., Xiong H., Feith D. J., Shantz L. M. (1998) S-Adenosylmethionine decarboxylase: structure, function and regulation by polyamines. Biochem. Soc. Trans. 26, 580–586 [DOI] [PubMed] [Google Scholar]
- 17. Tolbert W. D., Zhang Y., Cottet S. E., Bennett E. M., Ekstrom J. L., Pegg A. E., Ealick S. E. (2003) Mechanism of human S-adenosylmethionine decarboxylase proenzyme processing as revealed by the structure of the S68A mutant. Biochemistry 42, 2386–2395 [DOI] [PubMed] [Google Scholar]
- 18. Bale S., Ealick S. E. (2010) Structural biology of S-adenosylmethionine decarboxylase. Amino Acids 38, 451–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fairlamb A. H., Blackburn P., Ulrich P., Chait B. T., Cerami A. (1985) Trypanothione: a novel bis(glutathionyl)spermidine cofactor for glutathione reductase in trypanosomatids. Science 227, 1485–1487 [DOI] [PubMed] [Google Scholar]
- 20. Willert E., Phillips M. A. (2012) Regulation and function of polyamines in African trypanosomes. Trends Parasitol. 28, 66–72 [DOI] [PubMed] [Google Scholar]
- 21. Casero R. A., Jr., Marton L. J. (2007) Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat. Rev. Drug Discov. 6, 373–390 [DOI] [PubMed] [Google Scholar]
- 22. Bacchi C. J., Nathan H. C., Hutner S. H., McCann P. P., Sjoerdsma A. (1980) Polyamine metabolism: a potential therapeutic target in trypanosomes. Science 210, 332–334 [DOI] [PubMed] [Google Scholar]
- 23. Spinks D., Torrie L. S., Thompson S., Harrison J. R., Frearson J. A., Read K. D., Fairlamb A. H., Wyatt P. G., Gilbert I. H. (2012) Design, synthesis and biological evaluation of Trypanosoma brucei trypanothione synthetase inhibitors. ChemMedChem 7, 95–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Barker R. H., Jr., Liu H., Hirth B., Celatka C. A., Fitzpatrick R., Xiang Y., Willert E. K., Phillips M. A., Kaiser M., Bacchi C. J., Rodriguez A., Yarlett N., Klinger J. D., Sybertz E. (2009) Novel S-adenosylmethionine decarboxylase inhibitors for the treatment of human African trypanosomiasis. Antimicrob. Agents Chemother. 53, 2052–2058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bacchi C. J., Nathan H. C., Yarlett N., Goldberg B., McCann P. P., Bitonti A. J., Sjoerdsma A. (1992) Cure of murine Trypanosoma brucei rhodesiense infections with an S-adenosylmethionine decarboxylase inhibitor. Antimicrob. Agents Chemother. 36, 2736–2740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hanfrey C., Elliott K. A., Franceschetti M., Mayer M. J., Illingworth C., Michael A. J. (2005) A dual upstream open reading frame-based autoregulatory circuit controlling polyamine-responsive translation. J. Biol. Chem. 280, 39229–39237 [DOI] [PubMed] [Google Scholar]
- 27. Pegg A. E. (2006) Regulation of ornithine decarboxylase. J. Biol. Chem. 281, 14529–14532 [DOI] [PubMed] [Google Scholar]
- 28. Willert E. K., Fitzpatrick R., Phillips M. A. (2007) Allosteric regulation of an essential trypanosome polyamine biosynthetic enzyme by a catalytically dead homolog. Proc. Natl. Acad. Sci. U.S.A. 104, 8275–8280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Willert E. K., Phillips M. A. (2009) Cross-species activation of trypanosome S-adenosylmethionine decarboxylase by the regulatory subunit prozyme. Mol. Biochem. Parasitol. 168, 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Willert E. K., Phillips M. A. (2008) Regulated expression of an essential allosteric activator of polyamine biosynthesis in African trypanosomes. PLoS Pathog. 4, e1000183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Osterman A., Grishin N. V., Kinch L. N., Phillips M. A. (1994) Formation of functional cross-species heterodimers of ornithine decarboxylase. Biochemistry 33, 13662–13667 [DOI] [PubMed] [Google Scholar]
- 32. Pegg A. E., Pösö H. (1983) S-Adenosylmethionine decarboxylase (rat liver). Methods Enzymol. 94, 234–239 [DOI] [PubMed] [Google Scholar]
- 33. Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schuck P., Perugini M. A., Gonzales N. R., Howlett G. J., Schubert D. (2002) Size-distribution analysis of proteins by analytical ultracentrifugation: strategies and application to model systems. Biophys. J. 82, 1096–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Brautigam C. A. (2011) Using Lamm-equation modeling of sedimentation velocity data to determine the kinetic and thermodynamic properties of macromolecular interactions. Methods 54, 4–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dam J., Velikovsky C. A., Mariuzza R. A., Urbanke C., Schuck P. (2005) Sedimentation velocity analysis of heterogeneous protein-protein interactions: Lamm equation modeling and sedimentation coefficient distributions c(s). Biophys. J. 89, 619–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bevington P. R., Robinson D. K. (1992) Data Reduction and Error Analysis for the Physical Sciences, 2nd Ed., McGraw-Hill Science/Engineering/Math, Columbus, OH [Google Scholar]
- 38. Johnson M. L. (1992) Why, when, and how biochemists should use least squares. Anal. Biochem. 206, 215–225 [DOI] [PubMed] [Google Scholar]
- 39. Laue T. M., Shah B. D., Ridgeway R. M., Pelletier S. L. (1992) in Analytical Ultracentrifugation in Biochemistry and Polymer Science (Harding S. E., Rowe A. J., Horton J. C., eds) pp. 90–125, The Royal Society of Chemistry, Cambridge, UK [Google Scholar]
- 40. DeLano W. L. (2002) The PyMOL Molecular Graphics System, Schrödinger, LLC, New York [Google Scholar]
- 41. Gilbert G. A., Jenkins R. C. (1956) Boundary problems in the sedimentation and electrophoresis of complex systems in rapid reversible equilibrium. Nature 177, 853–854 [DOI] [PubMed] [Google Scholar]
- 42. Brown P. H., Balbo A., Schuck P. (2008) Characterizing protein-protein interactions by sedimentation velocity analytical ultracentrifugation. Curr. Protoc. Immunol. Chapter 18, Unit 18.15 [DOI] [PubMed] [Google Scholar]
- 43. Kossiakoff A. A., Chambers J. L., Kay L. M., Stroud R. M. (1977) Structure of bovine trypsinogen at 1.9 Å resolution. Biochemistry 16, 654–664 [DOI] [PubMed] [Google Scholar]
- 44. Reynolds K. A., McLaughlin R. N., Ranganathan R. (2011) Hot spots for allosteric regulation on protein surfaces. Cell 147, 1564–1575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bale S., Lopez M. M., Makhatadze G. I., Fang Q., Pegg A. E., Ealick S. E. (2008) Structural basis for putrescine activation of human S-adenosylmethionine decarboxylase. Biochemistry 47, 13404–13417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Clyne T., Kinch L. N., Phillips M. A. (2002) Putrescine activation of Trypanosoma cruzi S-adenosylmethionine decarboxylase. Biochemistry 41, 13207–13216 [DOI] [PubMed] [Google Scholar]
- 47. Cherry M., Williams D. H. (2004) Recent kinase and kinase inhibitor x-ray structures: mechanisms of inhibition and selectivity insights. Curr. Med. Chem. 11, 663–673 [DOI] [PubMed] [Google Scholar]
- 48. Nagar B., Bornmann W. G., Pellicena P., Schindler T., Veach D. R., Miller W. T., Clarkson B., Kuriyan J. (2002) Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571). Cancer Res. 62, 4236–4243 [PubMed] [Google Scholar]