

Background: GPR17 is a key player in oligodendrocyte differentiation. By regulating the availability of receptors at the cell surface, agonist-induced GPR17 trafficking may influence terminal cell fate.
Results: UDP-glucose and LTD4 induce GPR17 endocytosis and distribution in lysosomes or recycling compartments.
Conclusion: Agonist-activated GPR17 undergoes partial degradation and fast membrane recycling.
Significance: Understanding GPR17 trafficking may increase our knowledge of oligodendrocyte differentiation and myelination.
Keywords: Differentiation, Oligodendrocytes, Purinergic Receptor, Receptor Endocytosis, Trafficking
Abstract
GPR17 is a G-protein-coupled receptor that is activated by two classes of molecules: uracil-nucleotides and cysteinyl-leukotrienes. GPR17 is required for initiating the differentiation of oligodendrocyte precursors but has to be down-regulated to allow cells to undergo terminal maturation. Although a great deal has been learned about GPR17 expression and signaling, no information is currently available about the trafficking of native receptors after the exposure of differentiating oligodendrocytes to endogenous agonists. Here, we demonstrate that neuron-conditioned medium induces the transcriptionally mediated, time-regulated expression of GPR17 in Oli-neu, an oligodendrocyte precursor cell line, making these cells suitable for studying the endocytic traffic of the native receptor. Agonist-induced internalization, intracellular trafficking, and membrane recycling of GPR17 were analyzed by biochemical and immunofluorescence assays using an ad hoc-developed antibody against the extracellular N-terminal of GPR17. Both UDP-glucose and LTD4 increased GPR17 internalization, although with different efficiency. At early time points, internalized GPR17 co-localized with transferrin receptor, whereas at later times it partially co-localized with the lysosomal marker Lamp1, suggesting that a portion of GPR17 is targeted to lysosomes upon ligand binding. An analysis of receptor recycling and degradation demonstrated that a significant aliquot of GPR17 is recycled to the cell surface. Furthermore, internalized GPR17 displayed a co-localization with the marker of the “short loop” recycling endosomes, Rab4, while showing very minor co-localization with the “long loop” recycling marker, Rab11. Our results provide the first data on the agonist-induced trafficking of native GPR17 in oligodendroglial cells and may have implications for both physiological and pathological myelination.
Introduction
Recent work has led to the “deorphanization” of the G protein-coupled receptor (GPCR)5 referred to as GPR17, which is located at an intermediate phylogenetic position between the purinergic P2Y receptors and the CysLT1 and CysLT2 receptors for cysteinyl-leukotrienes (cysLTs; 1–6). Both recombinant and native GPR17 receptors respond to uracil nucleotides (e.g. UDP and UDP-glucose) and arachidonic acid-derived cysLTs (e.g. LTD4 and LTE4).
The physiological role of GPR17 has been deeply investigated in both in vivo and in vitro systems, and a number of studies have revealed its crucial role in oligodendrocyte precursor cell (OPC) differentiation (2, 7–11). Receptor expression, almost absent in early OPCs, gradually increases in more mature precursors, reaches a plateau in immature/pre-oligodendrocytes, and then gradually decreases during terminal differentiation. In line with these findings, GPR17 is co-expressed with the early oligodendrocyte marker NG2 and markers of pre/immature oligodendrocyte phenotype (such as O4 and DM-20) but is down-regulated in cells expressing myelin proteins such as myelin basic protein, which is highly synthesized in fully mature cells (7, 10, 11). Consistent with the role of GPR17 in oligodendrocyte ontogenesis, its activation by natural agonists promotes OPC differentiation under physiological conditions (2, 10). In contrast, the inhibition of GPR17 expression causes an impairment in oligodendrocyte differentiation and myelination in both in vivo (7) and in vitro systems (10). Altogether, these studies indicate that GPR17 is an integral signaling component controlling oligodendrocyte ontogenesis and suggest that the appropriate activation and deactivation of GPR17 are crucial steps in OPC maturation.
As it has been reported for many GPCRs, after ligand binding, GPR17 may undergo endocytosis and subsequent sorting into lysosomes for degradation and/or into recycling endosomes for re-incorporation into the plasma membrane. The balance of this dynamic intracellular trafficking is physiologically relevant because it modulates receptor levels at the cell surface. This process has important implications for the activation or silencing of GPR17-signaling pathway(s), and in turn, for OPC differentiation (12–16). It may even be hypothesized that GPR17 endocytosis may represent a key event necessary to allow OPCs to proceed to myelination. A similar process has been associated with the specification of other cell lineages, where the down-regulation of membrane receptors has been proposed to be necessary to allow cells to proceed toward terminal differentiation (17). Interestingly, the abnormal up-regulation of GPR17 has been associated with defective myelination during development and with multiple sclerosis (7). Thus, the characterization of the mechanisms involved in the expression of GPR17 in the plasma membrane may help us to better understand the molecular mechanisms of the contribution of GPR17 to oligodendrogenesis and may set the background for interpreting the consequences of GPR17 dysfunction in disease.
At present, however, there are very few studies available on the trafficking of GPR17 both under basal conditions and upon activation. In 1321N1 cells heterologously expressing hGPR17, it has been demonstrated that the GPR17 agonists UDP-glucose and LTD4 determine receptor desensitization/re-sensitization (6). On the other hand, a previous study has failed to demonstrate the direct activation of GPR17 by agonists, proposing that the receptor may function exclusively as a negative regulator for the cysLT1 receptor response to LTD4 treatment (18). Furthermore, Benned-Jensen and Rosenkilde (19) reported that mouse or human GPR17 is activated by uracil nucleotides but apparently not by LTD4 or LTC4 and showed that LTD4 did not significantly increase the internalization of FLAG-tagged hGPR17 in transiently transfected HEK293 cells. Moreover, despite the crucial role of GPR17 in OPCs (see above) and the evidence that the receptor is clearly down-regulated in cells achieving terminal maturation, no studies are yet available on the agonist-induced regulation of native GPR17 in cells of the oligodendroglial lineage.
In this study, therefore, we decided to analyze the endocytic trafficking of native GPR17 after activation with uracil nucleotides or cysLTs using a physiological expression system. Although OPC primary cultures would represent an ideal system, the necessity to isolate them from tissue for each experiment and the relatively low number of cells obtained from each preparation markedly limited their use in the detailed biochemical analysis planned for the present work. To avoid this problem, we selected Oli-neu cells, an OPC cell line immortalized from E16 mouse brains because these cells can be induced to recapitulate several features of differentiating OPCs in vitro (20, 21). Therefore, we first established the conditions for stimulating the endogenous expression of GPR17 in Oli-neu and then used these cells for the analysis of the endocytic trafficking of native GPR17. We found that Oli-neu cells express significant amounts of GPR17 after incubation with medium conditioned from neuronal primary cultures. The expression of the receptor was transient and depended on the maturational stages of Oli-neu toward a more differentiated phenotype; GPR17 synthesis was up-regulated during the early stages of differentiation and was down-regulated when the cells reached later stages of maturation, as shown in primary OPCs (2, 10, 11). Furthermore, the GPR17 gene promoter appeared to be highly activated by factors released in the medium by neurons and/or astrocytes. Finally, we demonstrate that UDP-glucose and LTD4, although with low efficiency, stimulated clathrin-mediated endocytosis of GPR17. After internalization, the receptor is delivered into early endosomes and then sorted either to lysosomes for degradation or recycled to the cell surface via the small G-protein Rab4-dependent pathway.
EXPERIMENTAL PROCEDURES
Materials, Antibodies, and Plasmids
The UDP-glucose, protease inhibitor cocktails, dithiothreitol (DTT), sodium 2-mercaptoethane sulfonate (MesNA), monoclonal antibodies against FLAG, anti-rabbit, and mouse IgG conjugated to horseradish peroxidase, and anti-rabbit IgG-light chain conjugated to horseradish peroxidase came from Sigma; LTD4 was from Cayman Europe. Montelukast was a gift from Merck, and cangrelor was a gift from The Medicines Co. (Parsippany, NJ). The sulfosuccinimidyl-2-(biotinamido)ethyl-1,3′dithiopropionate (EZ-LinkTM, Sulfo-NHS-SS-Biotin) was from Thermo Scientific (Milan, Italy), and streptavidin Plus UltraLink Resin was from Pierce. The antibodies against MAG came from Millipore (Billerica, MA), the anti-Lamp1 (lysosome-associated membrane protein-1) was from BD Biosciences, and the anti-transferrin receptor (TfR) was from Zymed Laboratories Inc. (San Francisco, CA). The polyclonal antibody against FoxO1 (C29H4, Cell Signaling Technology) was from Millipore. An antibody against a C-terminal region of GPR17 (anti-Ct-GPR17) was raised in rabbits and affinity-purified as previously described (1). The fluorescein-, rhodamine- or Cy3-conjugated anti-mouse, rabbit, or rat IgGs were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA). The siRNA against GPR17 were designed and synthesized by Qiagen (Milan, Italy), and the transfection reagent INTERFERinTM came from Polyplus-transfection (Illkirch, France).
Antibodies against the N-terminal Region of GPR17
An antibody was raised in rabbit using a synthetic peptide corresponding to the N-terminal sequence of mouse GPR17 (MNGLEAALPSLTDNSSLAYSEQC) coupled to keyhole-limpet hemocyanin (22). The antibodies (from here on referred to as anti-Nt-GPR17) were affinity-purified and tested for their specificity for in vivo immunolabeling.
Cloning of the GPR17 Promoter Region and the Generation of Luciferase Constructs
The human GPR17 promoter was analyzed with the Genomatix suite. A small fragment of 909 bp was hypothesized to be a regulatory region, and it was cloned into a pGL4.17 vector (Promega), upstream to the luc2 reporter gene, using the primers 5′-CGCTCGAGTTCCCATGTTGCTGGATGTA-3′ (forward) and 5′-CAAGCTTCGCTGAGTGTTTCCTCTGCT-3′ (reverse) containing a cassette for the restriction enzymes XhoI and HindIII, respectively. The generated vector was called pGL4-h909.
Cell Culture and Differentiation
The Oli-neu murine OPC cell line was kindly provided by Prof. J. Trotter (University of Mainz) and cultured in Sato medium containing 1% horse serum as previously described (20). The neuron-conditioned medium (CM) was obtained from primary cultured neurons prepared from the cerebral cortex of 18-day-old rat embryos and maintained in neurobasal medium supplemented with B27 as previously described (23). The CM was collected after 7–10 days and passed through a 22-μm filter. The Oli-neu cells were cultured in normal medium or normal medium plus CM in a 1:1 ratio. COS-7 cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum.
Pulse-chase Experiments
Differentiated Oli-neu cells were preincubated for 1 h in methionine- and cysteine-free Oli-neu medium and labeled with 200 μCi/ml Express 35S-protein labeling mix ([35S]methionine/[35S]cysteine; PerkinElmer Life Sciences) for 90 min. They were then washed twice with complete medium containing a 2-fold excess of methionine/cysteine and chased in the absence or presence of the drugs for the times indicated under “Results.” At the end of the chase, the cells were lysed in ice-cold buffer A and centrifuged at 20,000 × g for 20 min. The extracts were immediately analyzed by gel electrophoresis or after immunoprecipitation (see above). The gels were exposed to phosphor screens and analyzed with a Storm PhosphorImager (Molecular Dynamics). Band intensity was quantified with ImageQuant software.
RNA Interference and Plasmid Transfections
For RNA interference, the Oli-neu cells were plated the day before and incubated in CM for 6 h before transfection. The cells were transfected using the transfection reagent INTERFERinTM without siRNA (mock transfection control), 2.5 nm scrambled siRNA (negative control), or a siRNA designed to silence mouse GPR17 (Qiagen; target sequences), in accordance with the manufacturer's instructions. Then, 24 or 48 h after transfection, the cells were fixed for immunofluorescence analysis or the cellular proteins extracted for immunoblotting. For forkhead box protein (Fox)O1 knock-down, Oli-neu cells were transfected with 50 nm siRNA designed to silence mouse FoxO1 (Cell Signaling Technology, Millipore). Thirty-six hours after transfection cells were incubated for 24 h with CM and then fixed as described above.
For cDNA transfection, the day before transfection, Oli-neu cells were seeded onto 24 × 24-mm glass coverslips (75,000 cells/coverslip) and incubated in CM. After 24 h, the cells were transfected with plasmids (0.5 μg/coverslip) encoding the N-terminally enhanced GFP-tagged Rab5a (24), Rab4a, or Rab11a (25), a generous gift of Dr. C. Bucci (Dept. of Science, University of Salento) and B. Chini (Institute of Neuroscience, Milan, Italy). cDNAs were mixed with jetPEI reagent (PolyplusTransfection) in a 1:2 ratio, incubated for 20 min at room temperature, and gently added to the cells. After 24 h of transfection, the medium was removed, and fresh CM was added. Analysis of GPR17 internalization was performed ∼48 h after transfection. For FoxO1 overexpression, Oli-neu were transfected with cDNAs encoding GFP-FoxO1 or FLAG-FoxO1-alanine-aspartic acid-alanine (ADA) obtained from Dr. D. Accili (Columbia University, New York) and provided by Addgene. After 48 h of incubation in Sato medium, cells were fixed and processed for immunofluorescence. For the analysis of promoter induction, Oli-neu cells were transfected with the normalizing vector pGL4-TK-Renilla (Promega) along with either the reporter construct pGL4-h909 or the corresponding empty vector (as a negative control) in a 1:50 ratio. A total of 250 ng of DNA was transfected with the JetPEI reagent. The day after transfection, the cells were incubated for 24 or 48 h with the selected treatments. The Dual Reporter Luminometer Assay (Promega) was performed according to the manufacturer's instructions.
Pharmacological Treatments and Cell Solubilization
Agonists were added to differentiating Oli-neu cells at the following concentrations: 100 μm UDP- glucose and 50 nm LTD4. Cells were then incubated at 37 °C for the indicated times. When required, cells were preincubated for 10 min at 4 °C with antagonists (10 μm cangrelor; 1 μm montelukast). Cells were lysed in buffer A (150 mm NaCl, 2 mm EGTA, 50 mm Tris-HCl, pH 7.5, and a Sigma protease inhibitor mixture diluted 1:1000) containing 1% Triton X-100 and then centrifuged 20,000 × g for 20 min at 4 °C.
Immunoprecipitation, Endoglycosidase Digestions, and Western Blotting
For the immunoprecipitation of GPR17, aliquots of the cell extracts (100–200 μg of protein) were incubated for 1 h with protein A- or G-Sepharose beads. The beads were removed by centrifugation, and the “precleared” supernatants were added to protein A or G beads that had been preincubated with the primary antibodies or non-immune IgG for 2 h at 4 °C. After overnight incubation, the beads were extensively washed with 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, and 0.3% (w/v) Triton X-100. For endoglycosidase digestions, proteins isolated by immunoprecipitation were eluted from protein A beads by heating at 65 °C in a solution containing 0.5% SDS and 1% β-mercaptoethanol. Aliquots of each sample were then diluted with Nonidet P-40 (1% final concentration) and concentrated endoglycosidase F or endoglycosidase H reaction buffers (G7 or G5, New England Biolabs) in accordance with the manufacturer's instructions and digested with 250 units of endoglycosidase F or H for 1 h at 37 or 0 °C. The reactions were stopped by the addition of Laemmli sample buffer and analyzed by Western blotting as previously described (22) using anti-rabbit IgG light chains or anti-mouse IgG conjugated to peroxidase (diluted 1:50,000) as secondary antibodies. The peroxidase was revealed using a chemiluminescent substrate (Pierce). For quantitative analysis, unsaturated autoradiograms were acquired using an ARCUS II scanner (Agfa-Gevaert, Mortsel, Germany), and the density of each band was quantified using NIH Image J software (National Technical Information Service, Springfield, VA).
RNA Extraction, Real-time and Reverse Transcription-PCR
Total RNA was purified from tissue, platelets, or cell cultures using an RNeasy Plus Mini kit (Qiagen) in accordance with the manufacturer's instructions. The target sequences for the quantitative, real-time PCR were amplified from 1 μg cDNA; for analysis of gene expression, we used the ABI Prism® 7000 Sequence Detection System, SDS software Version 1.2.3 (Applied Biosystems, CA). The target sequences were amplified using a pre-programmed thermal profile of enzyme activation at 50 °C followed by 40 identical cycles of denaturation at 95 °C for 15 s and annealing and amplification at 60 °C for 1 min; these conditions were previously determined to be optimal by Applied Biosystems. The TaqMan® primer and probe assays used were GPR17 (Mm02619401_s1) and the endogenous control glyceraldehyde-3-phosphate dehydrogenase (Mm99999915_g1). Control reactions that were performed in the absence of Quantiscript reverse transcriptase (Qiagen) excluded the possibility of genomic traces. The results were calculated using the 2−ΔΔCt method, allowing for the normalization to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) with the calibrator set to a value of 1 (26). For reverse transcription-PCR, 1 μg of RNA was transcribed to cDNA using the QuantiTect reverse transcription kit (Qiagen), and aliquots (1 μl) of the total cDNA were amplified in 40 cycles of PCR prepared using TopTaq Master Mix kit (Qiagen). The primers are indicated in Table 1. The DNA fragments were then analyzed by means of agarose gel electrophoresis.
TABLE 1.
Primers used in reverse transcription-PCR experiments
Fw, forward; Rv, reverse.
| Target gene | Primers | Product length |
|---|---|---|
| bp | ||
| P2Y1 | Fw 5′-CCTGCGAAGTTATTTCATCTA-3′ | 318 |
| Rv 5′-GTTGAGACTTGCTAGACCTCT-3′ | ||
| P2Y2 | Fw 5′-GCAGCATCCTCTTCCTCACCT-3′ | 499 |
| Rv 5′-CATGTTGATGGCGTTGAGGGT-3′ | ||
| P2Y4 | Fw 5′-GGCATTGTCAGACACCTTGTA-3′ | 550 |
| Rv 5′-AAGGCACGAAGCAGACAGCAA-3′ | ||
| P2Y6 | Fw 5′-CGCTTCCTCTTCTATGCCAA-3′ | 481 |
| Rv 5′-GTAGGCTGTCTTGGTGATGTG-3′ | ||
| P2Y12 | Fw 5′-CAGGTTCTCTTCCCATTGCT-3′ | 638 |
| Rv 5′-CAGCAATGATGATGAAAACC-3′ | ||
| P2Y14 | Fw 5′-TGTCTGCCGTGATCTTCT-3′ | 589 |
| Rv 5′-GGGTCCAGACACACATTG-3′ | ||
| GPR17 | Fw 5′-CTGCTTCTACCTTCTGGACTTCAT-3′ | 344 |
| Rv 5′-AGGTGAGCATAGAGAGGTCTTCGA-3′ | ||
| cysLT1 | Fw 5′-CTTGAACGTACTCTGACACTACAA-3′ | 241 |
| Rv 5′-GAGATGTCGTCAGATTTT-3′ | ||
| cysLT2 | Fw 5′-AGATAAGATGTCCATCATGT-3′ | 264 |
| Rv 5′-ACAGTTCCTGTTACTATAAC-3′ | ||
| GAPDH | Fw 5′-GCCATCAACGACCCCTTCATTG-3′ | 597 |
| Rv 5′-TGCCAGTGAGCTTCCCGTTC-3′ |
Biotinylation Assay
Cells were incubated in neuron-conditioned medium for 48 or 72 h and then biotinylated using 0.3 mg/ml of sulfo-NHS-SS-Biotin (Thermo Scientific) dissolved in PBS with 0.1 mm CaCl2 and 1 mm MgCl2 for 30 min at 4 °C. The labeled cells were washed 3 times for 10 min with 50 mm glycine in TBS (25 mm Tris, 85 mm NaCl, 5 mm KCl, 1 mm CaCl2, 1 mm MgCl2) to quench free biotin. When required, the cells were subsequently incubated in Oli-neu medium in the absence or presence of the drugs for the times indicated in the results. After a 4 °C wash with ice-cold PBS containing 0.1 mm CaCl2 and 1 mm MgCl2, the remaining surface biotin labeling was removed by incubating the cells twice with 50 mm DTT or MesNA, a membrane-impermeable reducing agent, at 4 °C. Both reducing agents were then neutralized with iodoacetamide (10 mm) in PBS with 0.1 mm CaCl2 and 1 mm MgCl2, and the cells were lysed in buffer A containing 1% Triton X-100 and a protease inhibitor mixture. After centrifugation (20,000 × g for 20 min at 4 °C), supernatants containing equal amounts of protein were incubated with streptavidin beads to isolate the biotinylated proteins. After extensive washes in extraction buffer, the proteins were eluted from the streptavidin beads and analyzed by SDS-PAGE followed by Western blotting. Unsaturated autoradiograms were acquired using an ARCUS II scanner (Agfa-Gevaert, Mortsel, Germany), and the density of each band was quantified using NIH Image J software (National Technical Information Service, Springfield, VA). Data were collected from at least three experiments.
Antibody Labeling of Cell Surface-exposed GPR17 in Living Cells
Cells that were cultured on coverslips as described above were chilled at 4 °C and washed once in ice-cold PBS containing 0.1 mm CaCl2, 1 mm MgCl2, and 1% horse serum. Cells were then incubated at 4 °C for 45 min with rabbit anti-Nt-GPR17 antibody (10 μg IgG/24 × 24-mm coverslips) diluted in ice-cold PBS with 0.1 mm CaCl2, 1 mm MgCl2, and 1% horse serum. After the incubation period, the cells were washed 5 times with PBS containing 0.1 mm CaCl2, 1 mm MgCl2, and 1% horse serum and either fixed with formaldehyde or incubated in Sato medium (controls) supplemented with agonists in the absence or presence of antagonists, 0.45 m sucrose, or 80 μm dynasore as reported under “Results.“ After an incubation at 37 °C for different times, the cells were chilled at 4 °C to block endocytosis and washed with PBS containing 0.1 mm CaCl2, 1 mm MgCl2, and when required, 50 mm glycine in Hanks' balanced salt solution (glycine buffer). Finally, the cells were fixed and processed for immunofluorescence.
Immunofluorescence
The cells were fixed for 8–10 min with 4% paraformaldehyde in phosphate buffer, pH 7.3, containing 4% sucrose at 37 °C and permeabilized for 5 min at room temperature in PBS containing 0.3% Triton X-100. After immunostaining as previously described (27), images were recorded using either a Zeiss LSM510 Meta or an MRC-1024 laser-scanning microscope (Bio-Rad) equipped with a 60× objective or an Axiovert 200M (Zeiss) confocal system equipped with a spinning disc (PerkinElmer Life Sciences) with a 63× or 40× objectives. To compare the double-stained patterns, images from the fluorescein or rhodamine channels were acquired separately and superimposed. The images were processed using Photoshop (Adobe Systems, Mountain View, CA).
Image Analyses and Quantitation
We quantified the internalization of GPR17 in Oli-neu cells by measuring the immunofluorescence intensity of anti-Nt-GPR17 in cells before (time 0) or after incubations with or without agonists at 37 °C followed by washes with glycine buffer to strip the antibody bound to the remaining cell surface-exposed receptors. At least three independent experiments were performed for each condition, and ∼180 cells were examined in each experiment. All images were recorded with an MRC-1024 laser-scanning microscope (Bio-Rad) using identical parameters. Serial sections were acquired for each field, and the images were exported as TIFF files for analysis with Image J software. Values are expressed as the -fold increase from the intensity detected in the cells at time 0 (background).
Statistical Analysis
We analyzed the data using GraphPad Prism software and expressed the results as mean values ±S.E. The significance of the differences was assessed using either a two-tailed, non-paired Student's t test or a two-way analysis of variance, and the customary threshold of p < 0.05 was used to declare statistical significance (confidence intervals 95%).
RESULTS
Endogenous Expression and Biochemical Characterization of GPR17 in Oli-neu
To investigate whether Oli-neu cells express GPR17, cell cultures maintained in normal conditions (Sato medium) were analyzed by confocal immunofluorescence microscopy after immunolabeling with antibodies directed against the C terminus of GPR17 (anti-Ct-GPR17; 1) or protein markers typical of different stages of oligodendrocyte differentiation. In line with previous data, Oli-neu cells showed very few cell processes and expressed classical markers of an immature oligodendrocyte phenotype (Olig2 and NG2, data not shown) when grown in Sato medium. At this stage very few (∼4%) GPR17-positive cells were detected, whereas the majority of cells did not show immunoreactivity for GPR17 or MAG (Fig. 1A). As a number of studies have reported that Oli-neu cells can be induced to acquire a more mature phenotype if they are placed under differentiating conditions (20, 28), we decided to incubate Oli-neu cells in selected conditions to promote their differentiation and possibly modulate their expression of GPR17. However, many of the tested treatments (e.g. ATP, leukemia inhibitory factor, and insulin-like growth factor administration, data not shown) were ineffective or induced low levels of GPR17 expression after 72 h (e.g. dibutyryl-cAMP, data not shown). Conversely, when the cells were cultured in the presence of medium collected from primary cultures of cortical neurons (from here on referred to as CM), a sustained increase in the levels of GPR17 expression was observed. Notably, after a 24-h incubation in CM, immunoreactivity for GPR17 was detected in a large number of Oli-neu cells. Twenty-four hours later (+CM 48 h), ∼80% of cells appeared intensively immunolabeled for GPR17. These GPR17-positive cells showed an increased number of processes, and some started to co-express the myelin protein, MAG (Fig. 1A). To confirm the specificity of the immunostaining for GPR17, 48-h differentiated cells were transfected with a small interfering (si) RNA specific to silence mouse GPR17. As shown in Fig. 1B, GPR17 labeling was largely reduced upon treatment with the specific siRNA but remained unchanged when the cells were transfected with a nonspecific (scrambled) siRNA.
FIGURE 1.

GPR17 expression in Oli-neu cells. Oli-neu cells were cultured in Sato medium ([minus]CM) or conditioned medium (+CM, A) or transfected with scrambled siRNA (+siRNAsc) or a specific siRNA targeted against GPR17 (+siRNAGPR17) and then incubated in CM (+CM, B). At selected time points, the cells were fixed and double-immunolabeled with the anti-Ct-GPR17 antibody (∼1 μg/100 μl; green) and a monoclonal antibody against MAG (1:400, red) followed by fluorochrome-conjugated secondary antibodies. Images were recorded using a confocal microscope; merged images selected from four independent experiments are shown. Note the morphological changes and the increase in GPR17-labeling in Oli-neu cells maintained in medium supplemented with CM. Immunoreactivity for GPR17 was largely reduced upon transfection with a specific siRNA. Scale bar = 20 μm. In C, total membrane extracts (30 μg proteins) of Oli-neu cells cultured in Sato medium or CM (indicated as CM − or +) were analyzed by Western blotting using the anti-Ct-GPR17 antibody (1 μg/ml); the arrow and arrowhead indicate bands of 46–48 and 38 kDa, respectively, specifically immunodetected in differentiated cells. In D, total membrane extracts (30 μg) of Oli-neu cells cultured for 48 h in CM with scrambled siRNA (siRNAsc) or siRNAGPR17 were immunoblotted using antibodies against TfR (1:1000 dilution) or GPR17. Note the specific reduction of the 38 and 46–48 kDa bands after GPR17 knockdown, whereas Tfr and the 57-kDa band are unchanged. E, quantitative analysis of the intensity of the 46–48-kDa bands detected in Western blots of control and silenced cells; the data are expressed as arbitrary units and represent the mean values ±S.E. of three independent experiments (*, p < 0.05).
The expression of GPR17 in CM-stimulated Oli-neu cells was also analyzed in cell extracts by Western blotting. As shown in Fig. 1C, a broad band of ∼46–48 kDa and a sharper band of 38 kDa were both detected with the anti-Ct-GPR17 antibody in differentiated but not in undifferentiated Oli-neu cells. The expression of these two polypeptides was largely reduced upon receptor knockdown with the specific siRNA (Fig. 1D). In contrast, a polypeptide of ∼57 kDa, which was also recognized by the antibody in undifferentiated and differentiated Oli-neu cells, was unaffected by GPR17 knockdown, suggesting that this band is not related to GPR17 but, rather, corresponds to a nonspecific signal. Altogether, these data further demonstrate the expression of GPR17 in differentiating Oli-neu cells and indicate that the fully mature, post-translationally modified receptor displays a molecular mass of 46–48 kDa, whereas the detected band of 38 kDa likely represents a precursor form. These conclusions are also supported by two additional observations (Fig. 2). First, pulse-chase experiments with [35S]Met/Cys followed by immunoprecipitation of 35S-labeled GPR17 from cell extracts indicated the presence of two major 35S-labeled polypeptides of 46–48 and 38 kDa, respectively, after labeling (pulse), whereas the 38-kDa polypeptide disappeared after the chase, suggesting that it indeed corresponds to a precursor form of the receptor that is converted to the mature receptor during the chase experiment (Fig. 2A). Second, endoglycosidase F digestion (to remove all N-linked carbohydrates) converted both polypeptides of 46–48 and 38 kDa into a single band of 33 kDa (Fig. 2B). In contrast, the 46–48-kDa band remained unchanged after endoglycosidase H digestion, which only removes high mannose N-linked carbohydrates, whereas the 38-kDa band was largely converted to the 33-kDa band. This result indicates that the 38-kDa polypeptide is a precursor form of GPR17 carrying high mannose oligosaccharide chains (Fig. 2B).
FIGURE 2.
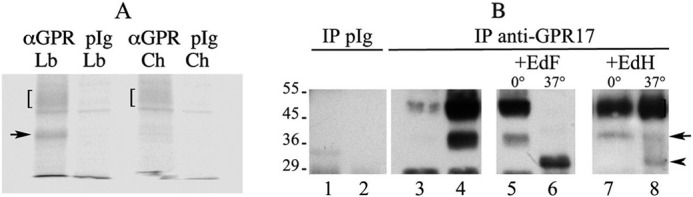
Synthesis and biochemical characterization of GPR17. A, immunoprecipitates with anti-Ct-GPR17 (αGPR) or preimmune rabbit IgG (pIg) from aliquots (100 μg of protein) of cell extracts from labeled (Lb) or 5 h “chased” (Ch) samples were analyzed by Western blotting followed by phosphorimaging analysis. Square brackets = 46–48-kDa bands; arrow = 38-kDa band. B, GPR17 glycosylation is shown. Protein (100 μg) from Triton X-100 extracts of undifferentiated (lanes 1 and 3) or 72 h-differentiated (lanes 2 and 4–8) Oli-neu cells were immunoprecipitated (IP) with anti-GPR17 or preimmune rabbit IgG (pIg). Before Western blot analysis, the samples in lanes 5–8 were incubated with endoglycosidase F (EdF) or H (EdH) at 37 or 0 °C. Square bracket = 46–48-kDa band, mature GPR17; arrow = 38-kDa band, immature GPR17; arrowhead = 33 kDa band un-glycosylated GPR17.
Next, we evaluated the expression of GPR17 in Oli-neu cells at different time points after the administration of CM. Previous data have demonstrated that in OPCs, GPR17 expression increases during differentiation from the precursor stage to pre-oligodendrocytes and subsequently decreases during the transition from pre-oligodendrocytes to mature oligodendrocytes (2, 7, 10, 11). Importantly, in Oli-neu cells incubated in CM, the protein levels of GPR17 showed a similarly transient expression. As shown in Fig. 3, after 48–72 h incubation in CM, a strong immunoreactivity for GPR17 was detected in ∼80–90% of cells. This signal declined at later times, and few GPR17 highly positive cells were detected after 96–120 h. In contrast, the number of cells expressing detectable levels of myelin proteins increased strongly after 72 h in CM, and almost 80% of the cells were highly positive for myelin proteins after 96 h (Fig. 3). These differences in GPR17 and MAG expression were also confirmed by Western blotting followed by densitometric analysis. As shown in Fig. 3, B and C, the peak of GPR17 protein expression preceded that of MAG and then rapidly declined, whereas the levels of the myelin protein continued to increase for up to 96 h. Changes in GPR17 protein expression after incubation with CM were consistent with the changes in the level of GPR17 mRNA during Oli-neu cell differentiation. As revealed by real-time PCR (Fig. 3D), the mRNA coding for GPR17 was up-regulated during differentiation and increased 10-fold after a 48-h incubation in CM. In accordance with the protein expression levels, the levels of GPR17 mRNA declined rapidly and returned to a lower level after 96 h. In contrast, the expression of most of the other members of the purinergic or cysLT receptor families detected in Oli-neu cells were almost unchanged, with the only exception being P2Y2, whose mRNA levels were increased upon incubation with CM (Fig. 4). Interestingly, P2Y12 and cysLT1 receptor mRNAs were undetected at any time.
FIGURE 3.
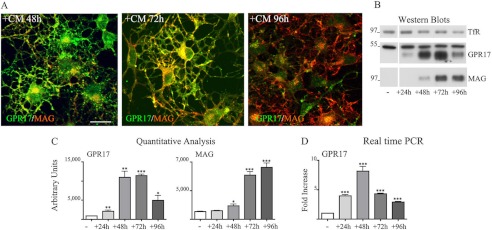
Transient expression of GPR17 in Oli-neu cells incubated in neuron-conditioned medium. In A, Oli-neu cells were cultured in CM for 48 h, 72 h, or 96 h (+CM) and then fixed and double-immunolabeled with antibodies against GPR17 or MAG (A′–C′). Images were recorded using a confocal microscope, and superimposed images (merges) are shown. Selected images from four independent experiments are shown. Scale bar = 20 μm. In B, cell extracts from Oli-neu cells cultured either in Sato medium (−) or in medium supplemented with CM (+) for 24, 48, 72, or 96 h were analyzed by Western blotting using antibodies against TfR, GPR17, or MAG (1:2,000 dilution). C, quantitative analysis of the Western blots from three independent experiments is shown; the intensity of the bands as measured by the ImageJ program is given as arbitrary units (*, p < 0.05; **, p < 0.01; ***, p < 0.0001). D, shown is real time PCR analysis of GPR17 gene expression in Oli-neu cells cultured in Sato medium (−) or exposed to CM (+) for the selected time points. GPR17 mRNA levels are relative to the housekeeping gene GAPDH and are expressed as -fold increase with respect to the mRNA levels (=1) detected in Oli-neu cells maintained in Sato medium. Data are from three independent experiments and represent the mean ± S.E. Statistical analysis was performed using a two-tailed, non-paired Student's t test (*, p < 0.05; **, p < 0.01; ***, p < 0.0001).
FIGURE 4.
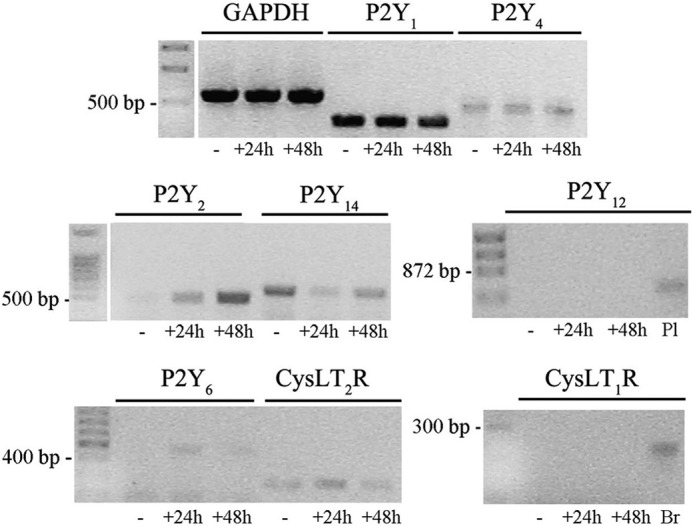
Analysis of purinergic and CysLT receptors in Oli-neu cells. Reverse transcription-PCR amplification was performed using specific primers (see “Experimental Procedures”) and 1 μg of RNA extracted from Oli-neu cells cultured in Sato medium alone (−) or in medium supplemented with CD (+) for 24 or 48 h. RNA from mouse brain (Br) and mouse platelets (Pl) was used as positive controls as indicated under “Experimental Procedures.” GAPDH was used as an internal standard. The final products were separated on an agarose gel (5 μl of the reaction mixtures were loaded on each lane). The bands revealed by ethidium bromide staining corresponded to the predicted sizes of the amplified DNA fragments.
To evaluate whether the increase in GPR17 expression in Oli-neu cells during differentiation occurred via a mechanism involving gene activation, one of the putative promoter regions identified on the human GPR17 gene (h909) and corresponding to highly conserved orthologous sequences on the mouse and rat genes (Fig. 5A) was cloned into a luciferase reporter vector and transfected in Oli-neu cells to measure promoter activity upon the exposure of cells to various stimuli. As reported in Fig. 5B, a statistically significant stimulation of the GPR17 promoter and luciferase activity was observed after a 48-h exposure of Oli-neu cells to CM. A slight, but not statistically significant stimulation of GPR17 promoter activity was detected after the exposure of Oli-neu cells to neurobasal medium supplemented with B27 (which is normally used to culture cortical neurons, indicated as +NB in Fig. 5B). In accordance with these results, Western blot analysis of cell extracts from parallel cultures revealed a slight increase in the GPR17 protein in Oli-neu cells after incubation in neurobasal medium supplemented with B27 (+NB in Fig. 5C) at later times (72–96 h), whereas receptor protein levels were strongly increased after a 48-h incubation in CM, suggesting for the first time that the GPR17 promoter is highly activated by factors released by neurons and/or astrocytes. To shed light on the mechanisms underlying the induction of GPR17 in Oli-neu, we turned our attention to recent data demonstrating that the transcription factor FoxO1 binds to the GPR17 promoter and increases receptor expression in a subset of hypothalamic neurons critical for initiating food intake (29). To investigate whether FoxO1 is also implicated in the expression of GPR17 in differentiating Oli-neu, we first examined the levels of FoxO1 in Oli-neu incubated in Sato medium or after administration of CM. Western blotting analysis demonstrated the presence of a faint band of ∼78 kDa in extracts of cells incubated for 24 h, suggesting that very low levels of FoxO1 are normally expressed in Oli-neu (Fig. 5D).
FIGURE 5.

The GPR17 promoter is selectively activated by CM. A, shown is a schematic representation of the human (h), mouse (m), and rat (r) GPR17 gene. The region upstream to the coding sequence has been analyzed with the Genomatix suite. Small blocks (from 600 bp to 1 kb) have been identified as putative promoter regions. Five regions in human, two in mouse, and two in rat were found (in gray). Only the region 1 is highly conserved among the three species. The human sequence 1 (909 bp) has been cloned in a pGL4 vector for the reporter assay. B, results of dual reporter luciferase assays show the GPR17 promoter activity in Oli-neu cells after transfection with the reporter construct pGL4-h909 or the corresponding empty vector. This was followed by incubation in Sato medium (control), neurobasal medium plus B27 (+NB), or CM for 24–48 h. Data are from three independent experiments and represent the mean ± S.E. Statistical analysis was performed using a two-way analysis of variance (***, p < 0.0001). C, cell extracts (30 μg protein) from Oli-neu cells incubated with either medium alone (−) or CM or neurobasal medium plus B27 (+NB) for the selected time points were analyzed by Western blotting using antibodies against GPR17. D, cell extracts from COS-7 (30 μg) or Oli-neu cells incubated with either Sato medium alone (−) or supplemented with CM for the selected time points were analyzed by Western blotting using antibodies against FoxO1 or GPR17. E, Oli-neu cells transiently transfected with cDNAs encoding for GFP-FoxO1 (FoxO1) or FLAG-FoxO1ADA were cultured in Sato medium for 48, fixed, and then labeled with anti-Ct-GPR17 antibodies and anti-FLAG antibody followed by secondary antibodies. Cells were examined using an Axiovert 200M confocal system equipped with a spinning disc. Arrows indicate FoxO1-transfected cells. Note that wild type protein is accumulated mainly in cytoplasm, whereas the constitutively active form (FoxO1ADA) is accumulated in the nucleus. The images are representatives from three independent experiments. Bar = 20 μm.
We then investigated the possible role of FoxO1 on GPR17 expression by mRNA silencing as well as by protein overexpression. After transfection with either a siRNA targeting mouse FoxO1 or scrambled siRNA, Oli-neu were incubated for 36 h in CM to induce GPR17 expression. Confocal images revealed no differences between specific or negative siRNA-transfected cells (data not shown). Next, we transfected Oli-neu with either wild type GFP-FoxO1 or a constitutively active FoxO1 (FLAG-FoxO1ADA) in which phosphorylation sites have been mutated to prevent FoxO1 inactivation (29). After a 48-h incubation of cells in Sato medium, high levels of FoxO1 were detected in the cytoplasm or in the nucleus of ∼10% of the cells, but no increases in GPR17 expression were observed in transfected compared with untransfected cells, thus suggesting that FoxO1 does not regulate GPR17, at least in Oli-neu oligodendroglial cells and under the tested experimental conditions (Fig. 5E). Altogether, these data demonstrate that GPR17 expression in Oli-neu cells can be modulated during differentiation, as observed in OPC primary cultures or in vivo (2, 7, 10, 11), and support the conclusion that these cells are an appropriate model for investigating GPR17 trafficking.
Agonist-induced Internalization of Native GPR17
In light of these results, we investigated the internalization of GPR17 in differentiated Oli-neu cells after the administration of either agonists or antagonists. Cells were incubated with sulfo-NHS-SS-Biotin under conditions that prevent plasma membrane internalization (4 °C). Preliminary experiments revealed that GPR17 and TfRs exposed to the cell surface were efficiently biotinylated, whereas, as expected, cytosolic proteins such as actin were not. Furthermore, biotin labeling of GPR17 and TfR was abolished by stripping with disulfide reducing reagents (such as DTT or MesNA), further indicating that no membrane internalization occurred at 4 °C. Conversely, when biotinylated Oli-neu cells were incubated for 15 min at 37 °C, a portion of biotinylated receptors (10.82% ± 6.63% of total labeling, n = 3 independent experiments; Fig. 6C) was protected from stripping with reducing agents, suggesting that GPR17, similar to many other GPCRs, undergoes constitutive internalization. To investigate agonist-induced endocytosis, biotinylated Oli-neu cells were incubated with micromolar concentrations of uracil nucleotides (100 μm UDP-glucose) and nanomolar concentrations of cysLTs (50 nm LTD4); these agonist concentrations were shown previously to activate the receptors in OPC primary cultures (10). Cells were exposed to the agonists for 5, 15, and 30 min at 37 °C and at the end of each time point incubated with a reducing agent as described above. The results of Western blotting indicated that the agonists activated the endocytosis of native GPR17 in a time-dependent manner (Fig. 6D) as has been previously reported for the receptor in a heterologous expression system (6). Quantitative analysis of the internalization of biotinylated receptors after a 15-min incubation with the agonists demonstrated that UDP-glucose was more efficient at stimulating the endocytosis of GPR17 compared with LTD4. The amount of internalized receptor was indeed increased to 392.9 ± 97.2% (n = 3; p = 0.0241) or to 157.9 ± 12.21% (p < 0.0091) compared with the constitutively internalized receptor after an incubation with UDP-glucose or LTD4, respectively (Fig. 6, F and G). Moreover, the effects of the two agonists were counteracted by the addition of cangrelor or montelukast, two antagonists previously shown to inhibit receptor activation mediated by UDP-glucose or LTD4, respectively (1, 2, 10).
FIGURE 6.

Agonist-induced internalization of GPR17. Internalization of GPR17 was measured in differentiated Oli-neu cells after cell surface labeling at 4 °C with the thiol-cleavable sulfo-NHS-SS-biotin. A, after labeling, cells were washed without (−) or with (+) DTT to remove the biotin from surface, and cell extracts (250 μg) were incubated with streptavidin. The proteins bound to streptavidin beads (B) and 5% of the cell extracts used for streptavidin-bead incubation (C.Ex) and also 5% of the supernatants from the streptavidin incubation (unbound proteins (Ub)) were analyzed by Western blotting with antibodies against TfR, GPR17, and actin. B, after labeling, Oli-neu cells were incubated for 15 min at 37 °C to allow for receptor internalization and then incubated without (−) or with (+) DTT. The cell extracts were analyzed as described above. In C, quantitative analysis of Western blots revealed that 10.82 ± 6.63% of the total biotinylated GPR17 underwent internalization under basal conditions. Data are from three independent experiments (the mean ± S.E.). ***, p < 0.0001. In D and E, after labeling cells were extracted or incubated with either UDP-glucose (100 μm, UDPg) or LTD4 (50 nm). At selected time points, the cells were cooled at 4 °C, incubated with the membrane-impermeable reducing agent MesNA to remove biotin from the remaining cell surface receptors, and solubilized. Aliquots (250 μg) of cell extracts were incubated with streptavidin beads and proteins bound to streptavidin (E) and also 10 μg of total cell extracts (D) were analyzed by Western blotting with antibodies against GPR17. F, after biotin labeling, Oli-neu cells were incubated at 37 °C in the absence (−) or presence of UDP-glucose, UDP-glucose (UDPg) and cangrelor (10 μm, Cng), LTD4, or LTD4 and montelukast (1 μm, MTL). After 15 min, cells were cooled at 4 °C, treated with reducing agents, and solubilized. Cell extracts (200 μg) were incubated with streptavidin beads, and bound proteins were analyzed by Western blotting with anti-GPR17 antibodies. The image shows representative blots from three independent experiments. G, graphs represent the quantitative analysis of blots, and the data are expressed as the percentage of biotinylated GPR17 internalized in the absence of agonists (−) and represent the mean ± S.E. (*, p < 0.05; **, p < 0.01, two-tailed, non-paired Student's t test).
The Distribution of GPR17 in the Endocytic Compartments after Agonist Administration
To analyze the trafficking of endocytosed GPR17 after the administration of UDP-glucose or LTD4, we employed protocols for tracking cell surface receptors by using antibodies directed against extracellular epitopes (30). To this end we raised antibodies directed against the extracellular N-terminal (Nt) domain of GPR17 and tested their ability to recognize the receptor on the plasma membranes of living cells. An anti-Nt-GPR17 antibody was found to primarily label the plasma membrane of differentiated Oli-neu cells after incubation at 4 °C (Fig. 7A). The specificity of the antibody was demonstrated by (i) the absence of labeling on the plasma membrane of undifferentiated cells (data not shown) and (ii) the abolishment of immunostaining after antibody preadsorption with the antigen (Fig. 7B).
FIGURE 7.

Labeling of cell surface-exposed GPR17 with an antibody raised against the N terminus of GPR17. Cells were cultured with CM for 48 h and then incubated for 45 min at 4 °C with the anti-Nt-GPR17 antibody without or with preincubation with the antigen. After washing, the cells were fixed and labeled with a monoclonal antibody against MAG followed by an incubation with anti-mouse IgG conjugated to Cy3 (MAG, red) and an anti-rabbit IgG conjugated to fluorescein to reveal the distribution of the anti-GPR17 antibody (GPR17, green). Note that anti-Nt-GPR17 labels the surface of cells that do not express or express small amounts of MAG. Immunostaining for GPR17 was completely abolished by preincubating the antiserum with the peptide used for rabbit immunization. The merged images are shown. Bars = 10 μm.
This antibody was then used to follow the endocytic pathways taken by GPR17. Receptors exposed to the cell surface were labeled with the anti-Nt-GPR17 antibody at 4 °C and then incubated at 37 °C with or without agonists for different times. To allow for a better localization of the internalized receptor, after an incubation at 37 °C, the cells were washed with glycine buffer, pH 2.8, to strip the antibodies bound to the receptors remaining at the cell surface. Cells were then fixed, permeabilized, and labeled with anti-rabbit IgG conjugated to Cy3 or double-labeled using monoclonal antibodies directed against TfR or Lamp1 followed by species-specific secondary antibodies. As a first step, we examined whether labeling of cells with anti-Nt-GPR17 antibody could per se increase the basal internalization of the receptor. Quantitative analysis of the intensity of immunofluorescence detected on Oli-neu after labeling at 4 °C or after labeling followed by a 15-min incubation at 37 °C without agonists indicated that the antibody increased the internalization of GPR17 only slightly compared with biotin (18.30% ± 1.29% of total labeling, n = 158 cells from two independent experiments), thus suggesting that it has little effect on GPR17.
After 2 min of incubation at 37 °C with agonists, immunostaining for the anti-Nt-GPR17 was still largely detected at the cell surface, although the labeling appeared more clustered, suggesting that receptor stimulation results in a more efficient recruitment of the receptor to endocytic patches and vesicles. At later times (8 and 12 min at 37 °C), an increasing number of dot-like structures that were immunolabeled for GPR17 appeared concentrated in the cytoplasm of cells following the administration of agonists. We traced the endocytic route of GPR17 by double-labeling with antibodies against protein markers of early/recycling endosomes (TfR) or lysosomes (Lamp1). As shown in Fig. 8, after 8 min of endocytosis, GPR17 partially co-localized with TfR in small vesicles that were dispersed throughout the cytoplasm, whereas at later times (15 and 30 min), both GPR17 and TfR were found to co-localize in larger dot-like structures located in the perinuclear region, presumably recycling endosomes (Fig. 9). In addition, after 30 min of LTD4 or UDP-glucose administration, GPR17 immunoreactivity was accumulated in a number of Lamp1-positive vesicles (Fig. 9). These data suggest that GPR17 follows the same intracellular route after the administration of LTD4 or UDP-glucose and that it may be degraded in lysosomal compartments or recycled to the cell surface.
FIGURE 8.

Endocytosis of surface-labeled GPR17. Differentiated Oli-neu cells that were surface-labeled with the affinity-purified anti-Nt-GPR17 antibody were incubated without (control (CTR)) or with agonists (50 nm LTD4 or 100 μm UDP-glucose (UDPglc)). After an 8-min incubation at 37 °C, cells were washed with glycine buffer to remove the antibodies bound to the remaining cell surface-exposed receptors, fixed, and immunostained with a monoclonal antibody against TfR followed by incubation with anti-mouse IgG conjugated to fluorescein (TfR) and anti-rabbit IgG conjugated to Cy3 (GPR17). Confocal microscopy images show immunostaining for GPR17 was accumulated in small structures dispersed within the cytoplasm. Arrows indicate vesicles labeled for GPR17 and TfR. Images are representative of four independent experiments. The merged images are shown in A, B, and C. Bars = 10 μm.
FIGURE 9.

GPR17 is accumulated in TfR- or Lamp1-positive compartments after 15 and 30 min of exposure to agonists. Differentiated Oli-neu cells were labeled with anti-Nt-GPR17 antibodies and incubated for 15 and 30 min at 37 °C with agonists (LTD4 or UDP-glucose (UDPglc)). After glycine washing, cells were fixed and labeled with monoclonal antibodies against TfR or Lamp1 followed by incubation with anti-rabbit IgG conjugated to Cy3 (GPR, red) and anti-mouse or anti-rat IgG conjugated to fluorescein (TfR or LAMP, green). Merged images represent individual confocal sections (0.5 μm) and are representative of four independent experiments. Note a subset of cytoplasmic structures co-labeled (yellow) for GPR17 and TfR or Lamp1. Bar = 10 μm.
GPR17 Endocytosis Is Mediated by Clathrin
Before analyzing the degradation and/or recycling of GPR17 receptors, we examined whether GPR17 undergoes clathrin-dependent or -independent endocytosis (31). To this aim, after labeling with the anti Nt-GPR17-antibody, Oli-neu cells were incubated for 12 min at 37 °C with the agonists in the presence of hypertonic sucrose to prevent the assembly of clathrin lattices. As shown in Fig. 10, the intracellular accumulation of cell surface-labeled GPR17 was largely inhibited by treatment with 0.45 m sucrose, which is also demonstrated by the quantitative analysis of the intensity of receptor immunoreactivity recovered in cells after stimulation in the presence or absence of hypertonic sucrose (Fig. 10). This analysis demonstrates that the internalization of GPR17 was increased 2.4- or 3.5-fold with respect to background after incubation with LTD4 or UDP-glucose, respectively; incubation in hypertonic sucrose largely reduced this agonist-activated receptor endocytosis. These data are in line with the biochemical analysis of biotinylated receptors (Fig. 6), confirming that the intracellular accumulation of GPR17 is greater in UDP-glucose than in LTD4-stimulated cells and demonstrating that the endocytosis of GPR17 is clathrin-mediated. In line with these data, we found that dynasore, an inhibitor of dynamin polymerization, largely reduced the internalization of GPR17, further supporting the conclusion that GPR17-endocytosis is a clathrin- and dynamin-dependent process (Fig. 11).
FIGURE 10.

GPR17 is internalized via clathrin-dependent endocytosis. In A, differentiated Oli-neu cells were surface-labeled with anti-Nt-GPR17 antibodies and then incubated for 12 min at 37 °C without (control (CTR)) or with agonists (LTD4 or UDP-glucose (UDPglc)) in Sato medium minus (−) or supplemented (+) with 450 mm sucrose. After glycine washing, cells were fixed and labeled with monoclonal antibodies against Lamp1 followed by incubation with anti-rat IgG conjugated to fluorescein (LAMP) and anti-rabbit IgG conjugated to Cy3 (GPR17). Confocal microscopy images show no accumulation of GPR17 immunostaining in the cells incubated in hypertonic sucrose. Images are representative of three independent experiments. The merged images are shown. Bar = 10 μm. B, to quantify the effects of clathrin lattice inhibition on the endocytosis of GPR17, confocal images were collected from Oli-neu cells incubated with or without hypertonic sucrose (+), and the pixel intensity was determined by ImageJ software. The graph represents the increase of GPR17 immunoreactivity in the cells over background (background = the immunofluorescence detected in the cells after labeling at 4 °C followed by acid stripping). The values are the mean (± S.E.) of three independent experiments. *, p > 0.05; **, p < 0.01; ***, p < 0.0001 (two-tailed, non-paired Student's t test).
FIGURE 11.

Dynamin inhibition affects GPR17 endocytosis. Oli-neu cells were labeled with anti-Nt-GPR17 antibodies, pretreated at 4 °C with or without the dynamin inhibitor dynasore, and then incubated at 37 °C for 12 min in Sato medium (CTR) or medium supplemented with dynasore and UDP-glucose or LTD4. After fixation, cells were double-immunolabeled with monoclonal antibodies against Lamp1 followed by incubation with anti-rat IgG conjugated to fluorescein (LAMP, green) and anti-rabbit IgG conjugated to Cy3 (GPR17, red). Note the large reduction of GPR17 internalization in Oli-neu incubated with the inhibitor of dynamin (Bar = 20 μm).
A Portion of GPR17 Is Re-routed to the Plasma Membrane via Rab4-positive Recycling Endosomes
We next asked whether receptors underwent recycling and/or degradation after agonist-activated internalization. To test this, agonist-activated receptor internalization was measured after the biotinylation of the cell surface, stimulation at 37 °C for 15 min, and biotin stripping as described above. To reveal the re-routing of receptors to the cell surface, parallel samples of Oli-neu cells were reincubated at 37 °C in normal medium for 15 and 30 min at 37 °C to allow for the sorting of internalized receptors to the cell surface or degradative compartments. At selected time points, cells were lysed without or after biotin cleavage. Biotinylated receptors were then isolated from cell extracts by streptavidin beads and analyzed by Western blotting as described above. Recycling was measured as the amount of biotinylated receptors that were not protected by MesNA cleavage and thus reappeared at the cell surface. Degradation was calculated by subtracting the amount of receptors recycled to the cell surface and the amount of receptors that were still present intracellularly from the total amount of internalized receptors after agonist administration (time 0). The results clearly indicate that a significant amount of GPR17 is recycled to the plasma membrane (Fig. 12). Densitometric analysis of Western blots from three independent experiments demonstrated that although ∼50–45% of endocytosed GPR17 underwent degradation after agonist administration, a consistent portion of the receptor (∼25 and ∼30% after LTD4 or UDP-glucose, respectively) was also recycled to the cell surface (Fig. 12). The amounts of recycled or degraded receptors were not significantly different after stimulation with either agonist.
FIGURE 12.
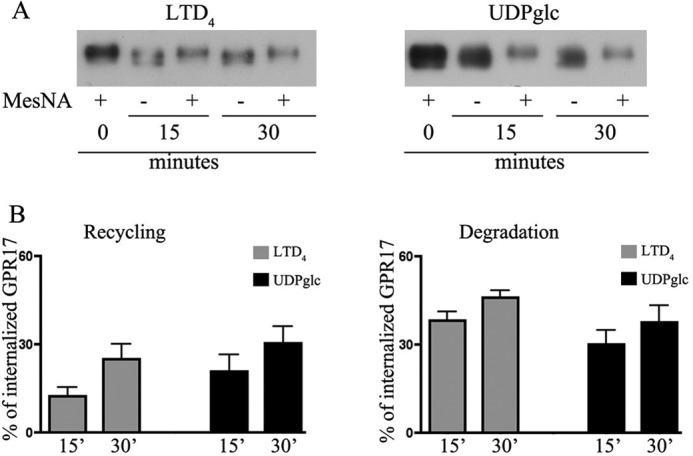
Recycling and degradation of GPR17 after agonist-induced endocytosis. Oli-neu cells after cell surface labeling with sulfo-NHS-SS-biotin at 4 °C were incubated at 37 °C with UDP-glucose (100 μm, UDPglc) or LTD4 (50 nm). After 15 min, cells were cooled at 4 °C, incubated with MesNA to remove biotin from the remaining cell surface receptors, and solubilized (time 0) or further incubated in normal medium. At selected times, cells were either solubilized (−) or re-incubated with MesNA to remove biotinylated receptors re-exposed to the cell surface. In A aliquots (200 μg) of cell extracts were incubated with streptavidin beads, and the proteins bound to streptavidin were analyzed by Western blotting with antibodies against GPR17. In B, graphs represent the quantitative analysis of GPR17 recycling and degradation, which is reported as a percentage of the amount of receptor internalized after 15 min at 37 °C with LTD4 or UDP-glucose; the values are the mean of four independent experiments ±S.E.
To further characterize the recycling pathways taken by GPR17, we compared its distribution to that of the small G-proteins Rab4, Rab5, and Rab11 at different times after the administration of agonists. A number of important studies have demonstrated the relevance of these Rab proteins as regulators of the recycling/endosomal compartments. It has also been demonstrated that early/sorting endosomes in the cell periphery contain primarily Rab5 and Rab4, whereas recycling endosomes in the perinuclear area contain primarily Rab4 or Rab11 (32–35). To investigate the distribution of GPR17 and Rab proteins, we transfected Oli-neu cells with cDNAs (0.5–0.2 μg) encoding for Rab4a, Rab5a, or Rab11a bearing a green fluorescent protein EGFP tag at the N terminus. After transfection, the cells were differentiated with CM for 48 h and then processed as described above to investigate the internalization and intracellular distribution of GPR17. Cells expressing moderate levels of GFP-Rabs (∼5% of total cells) were selected for analysis. Under this condition, Rab5, Rab4, and Rab11 show an intracellular distribution similar to that previously reported (32). Based on the confocal images (Fig. 13), GPR17 largely co-localized with GFP-Rab5 and Rab4 in small vesicles that were primarily scattered in the cytoplasm or located at the periphery of the cells at early times of internalization. At later times, GPR17 partially co-localized with Rab4 primarily in large, dot-like structures that were distributed in the perinuclear region (Fig. 14). A very minor co-localization of GPR17 with Rab11a could also be detected, as reported in Fig. 15. Altogether, these data support the conclusion that a portion of GPR17 is also recycled to the plasma membrane after internalization upon agonist stimulation through a fast recycling loop as revealed by co-localization with Rab4.
FIGURE 13.

At early time points endocytosed GPR17 accumulates in Rab5- or Rab4-positive vesicles. Oli-neu cells transiently transfected with cDNA encoding for GFP-Rab5a (Rab5) or GFP-Rab4a (Rab4) were labeled with anti-Nt-GPR17 antibodies and then stimulated with LTD4 or UDP-glucose (UDPglc) for 15 min. After glycine washing, the cells were fixed, and the endocytosed GPR17 was visualized by labeling with Cy3-conjugated antibodies (red). The cells were then examined using an Axiovert 200M confocal system equipped with a spinning disc. Note the co-localization of Rab proteins with GPR17 (arrows) in small vesicles scattered throughout the cytoplasm. The images are representatives from three independent experiments. Bar = 5 μm.
FIGURE 14.

At later time points endocytosed GPR17 accumulates in Rab4-positive structures distributed in the perinuclear region. Oli-neu cells transiently transfected with cDNA encoding for GFP-Rab4a (Rab4) were labeled with anti-Nt-GPR17 antibodies and then stimulated with LTD4 or UDP-glucose (UDPglc) for 30 min. After glycine washing, cells were fixed, and the endocytosed GPR17 was visualized by labeling with Cy3-conjugated antibodies (GPR). The cells were examined using an Axiovert 200M confocal system equipped with a spinning disc. Note the co-localization of GPR17 with Rab4 (arrows) in dot-like structures distributed in the perinuclear region. The images are representatives from three independent experiments. Bar = 5 μm.
FIGURE 15.

GPR17 exhibited a very minor degree of localization in Rab11-positive compartments. Oli-neu cells transiently transfected with cDNA encoding for GFP-Rab11a (Rab11) were labeled with anti-Nt-GPR17 antibodies and then stimulated with LTD4 or UDP-glucose (UDPglc) for 30 min. After glycine washing, the cells were fixed, and the endocytosed GPR17 was visualized by labeling with Cy3-conjugated antibodies (red, GPR). The cells were examined using an Axiovert 200M confocal system equipped with a spinning disc. The merged and single channel images shown are representatives from three independent experiments. Bar = 5 μm.
DISCUSSION
To investigate the intracellular trafficking of GPR17 in a native expression model, we have first characterized its expression in Oli-neu cells, an immortalized OPC cell line, and showed that medium derived from cortical neuron primary cultures stimulates the transient expression of GPR17, as previously observed in maturing cultured OPCs (2, 7, 10, 11). Using Oli-neu cells, we were then able to investigate the endocytic trafficking of the receptor after the administration of agonists and to demonstrate that (i) GPR17 undergoes clathrin-mediated endocytosis and (ii) the internalized receptor is partially recycled to the cell surface and partially sorted to lysosomes for degradation. Furthermore, although UDP-glucose and LTD4 bind to distinct sites on GPR17 (36), we found that both agonists mediate the trafficking of GPR17 to similar endocytic compartments, and no obvious differences were observed between LTD4 or UDP-glucose stimulation; the only exception is that the latter agonist causes a more efficient internalization of the receptor.
GPR17 Expression in Oli-neu Cells Is Highly Regulated by Neuron-conditioned Medium
The Oli-neu cell line is derived from brain cultures enriched in OPCs after immortalization with the neu oncogene, and when maintained in normal Sato medium, the majority of cells express markers of immature oligodendrocyte phenotypes such as the O4 antigen and NG2 proteoglycan (20 and 21)6; at this stage, only 4% of the cells express detectable amounts of GPR17. Because our previous study demonstrated that a subpopulation of NG2-positive precursor cells up-regulates GPR17 when induced to differentiate to post-mitotic pre-myelinating oligodendrocytes (11), we tested the conditions necessary to promote Oli-neu cell differentiation and GPR17 expression. Our immunofluorescence and biochemical data show that medium from neuronal primary cultures, although fostering Oli-neu cells toward a more differentiated phenotype, up-regulates the expression of GPR17. According to previous studies in OPCs and also in Oli-neu cells, GPR17 is up-regulated at early stages of differentiation and is then turned down in cells with a more mature phenotype that synthesizes high levels of myelin proteins (10, 11). Furthermore, our data demonstrate that this is a transcriptionally mediated effect that is induced by factors released by neurons and/or astrocytes (which are also present, although in a minor amount, in cortical neuronal primary cultures) because neuron-conditioned (but not neurobasal unconditioned medium) efficiently activated the GPR17 gene promoter. A number of extrinsic signals (in cooperation with intrinsic mechanisms) are indeed expected to regulate oligodendroglial cell differentiation (and possibly modulate the expression of GPR17), including axonal surface ligands and secreted molecules as well as axonal activity and glutamate released from axons (37). In line with evidence indicating GPR17 down-regulation after the O4 stage (Ref. 10 and present data) and according to recent findings that have clearly coupled glutamate release with axonal myelination (38), an event known to occur at quite late stages of oligodendrocytes differentiation, we did not observe any activation of the GPR17 gene promoter or any increase in GPR17 synthesis in Oli-neu cells probed with glutamate or glutamate-receptor antagonists (data not shown). Alternatively, other factors may be involved. In this respect, we have also tested the action of ATP, platelet-derived growth factor, leukemia inhibitory factor, and insulin-like growth factor. These molecules are known to play important roles in OPC survival, differentiation, and myelination, but they were unable to up-regulate the expression of GPR17 in Oli-neu cells. Recently, Accili and co-workers (29) demonstrated that in a subset of hypothalamic neurons secreting Agouti-related peptide, GPR17 expression is regulated by FoxO1. Microarray analysis indicated that GPR17 is highly decreased in FoxO1-deficient AgRP neurons, whereas FoxO1 overexpression increased the expression of GPR17 mRNA in neuro2A neuroblastoma cells. Our present results, however, indicate that the mechanism(s) underlying GPR17 expression in Oli-neu is different from that reported for AgRP neurons and FoxO1 does not appear to be involved. This conclusion comes from three observations: (i) the amount of FoxO1 in Oli-neu is very low even after incubation with CM, (ii) its knockdown does not reduce GPR17-expression, and (iii) its overexpression (even of the constitutively active forms) does not induce expression of GPR17. Although FoxO1 does not regulate GPR17 in Oli-neu, FoxO proteins may have an important function in OPC proliferation and oligodendrogenesis. Recent studies have indeed demonstrated that after hypoxia these transcription factors, mainly FoxO1, can affect the expression of p27Kip1, a key regulator of oligodendrogenesis (39).
Concerning Oli-neu, we cannot exclude the possibility that factors present in the CM could counteract the signaling of the neu-oncogene, which has been used to immortalize the precursor cells isolated from E16 mouse brains and foster their proliferation (20). Exit from the mitotic cycle may trigger the expression of GPR17 and promote differentiation as has been reported for OPCs (10, 11). This hypothesis is supported by the observation that an erbB inhibitor (PD174,265) induced the expression of GPR17 and the rapid (24 h) differentiation of Oli-neu cells (data not shown). Further work is required to identify additional molecules that may promote GPR17 gene activation and receptor protein synthesis.
Agonist-dependent GPR17 Trafficking
Our previous work has shown that GPR17 is a “dualistic” receptor that is activated by uracil nucleotides and cysLTs (1, 2, 10). Moreover, studies in heterologously transfected cells demonstrated that, similar to other GPCRs, recombinant GPR17 can undergo a loss of function upon prolonged exposure to its endogenous agonists as shown by desensitization, i.e. the inability to further respond to agonist stimulation, followed by re-sensitization upon agonist removal (6). However, the relevance of these findings to the regulation of native GPR17 in physiological systems has not been investigated previously. Moreover, in our previous study, the role of endocytic trafficking in agonist-induced GPR17 desensitization/re-sensitization was only marginally analyzed. The dynamics of intracellular trafficking of the receptor is, nevertheless, physiologically relevant, as it modulates receptor levels at the cell surface (12, 14–16, 40). The internalization and subsequent sorting of the receptor into degradative or recycling compartments may have important implications for the activation/silencing of signaling pathway(s). In the case of GPR17, it may represent a key event necessary to achieve GPR17 down-regulation and in turn to allow immature oligodendroglial cells to proceed to a more mature phenotype. A similar process has been associated with the differentiation of erythrocytes, where the down-regulation of erythropoietin receptors via lysosomal degradation has been proposed to be necessary to allow cells to proceed toward terminal maturation (17). Therefore, it was important to investigate in detail the trafficking of GPR17 in a more physiological model such as differentiating Oli-neu cells. Furthermore, given the dualistic nature of GPR17 and the likely presence of two distinct binding sites, one for uracil nucleotides and one for cysLTs, in its structure (36), it was important to verify whether the two distinct classes of natural agonists modulate the endocytic pathways taken by the receptor in different ways.
In this study we demonstrate that native GPR17 in Oli-neu cells is internalized in a time- and dose-dependent manner after the administration of agonists, although the efficacy of LTD4 appeared to be less than that of UDP-glucose. This conclusion is supported by quantitative analysis performed by using biochemical (biotinylation of cell surface receptors) and immunocytochemical (immunolabeling of cell surface receptors with a specific antibody) approaches aimed at revealing the internalization of GPR17. Moreover, we demonstrate that, at least in our cell system, LTD4 acts by directly activating GPR17 and not via the CysLT1 receptor; this conclusion is based on the finding that this receptor is not expressed in Oli-neu cells. These data are in line with our previous data obtained in a heterologous expression system in which CysLTR1 is also not expressed (6), but we cannot exclude the possibility that GPR17 may be differentially regulated and, therefore, act as a negative regulator of CysLTR1 when both receptors are co-expressed in the same system as previously reported (18). However, this possibility will never occur in OPCs and pre-oligodendrocytes where CyLTR1 has not been detected (10), thus further confirming that in oligodendroglial cells, cysLTs act by directly promoting the activation of GPR17. Finally, the mechanisms and functional consequences of this difference in the internalization of GPR17 after exposure to either uracil nucleotides or cysLTs are not yet known. Differences in the efficiency of receptor internalization may be due to differences in binding to arrestins and/or the activation of different intracellular signals.
Although we did observe some quantitative differences in the efficiency of internalization, we did not observe any significant differences in the pathway taken by internalized receptors after UDP or LTD4 stimulation. Using a protocol that has allowed us to visualize the fate of internalized receptors in endocytic compartments by means of confocal microscopy analysis, we demonstrated that GPR17 is internalized via clathrin-mediated endocytosis after both LTD4 and UDP-glucose administration. At early times after internalization, the receptor reaches early endosomes independent of the agonist used for stimulation. At later times, it accumulated in dot-like structures and partially co-localized with a marker of lysosomes, thus suggesting that both agonists stimulated receptor degradation. This conclusion is also supported by the quantification of the amounts of biotinylated receptor recovered in cells after stimulation and agonist retrieval. In addition, these experiments indicate that although an aliquot of the receptor is degraded, a consistent portion of it is still recycled to the cell surface. Furthermore, using an immunocytochemical approach, we investigated whether internalized GPR17 is recycled via a rapid or slow recycling loop by comparing the distribution of GPR17 with that of the Rab proteins involved in endocytic traffic. After transfection in Oli-neu cells, Rab proteins exhibited a localization similar to that reported previously (32). Rab5 was mainly concentrated in vesicles that were dispersed in the cytoplasm, and Rab4 was located in small vesicles and in dotted structures at the perinuclear region, whereas given its distribution and cycling in the trans-Golgi-network, Rab11 was mainly detected in the perinuclear region. Our results demonstrate that GPR17 is transported into the early/sorting endosomes and can be recycled to the membrane via the fast recycling loop. This conclusion is supported by the observation that soon after internalization, GPR17 co-localized with Rab5 and Rab4 in a compartment at the periphery of the cell. At later times, GPR17 also accumulated in a Rab4-positive compartment known to be involved in the rapid recycling loop. Only a very small amount of co-localization was observed with Rab11, a marker of the slow recycling loop.
In conclusion, we demonstrate that after agonist activation, GPR17 undergoes degradation and recycling. The balance of receptor sorting into recycling and/or degradative compartments may have important implications in the down-regulation of GPR17 signaling pathway(s) in differentiating OPCs.
Acknowledgments
We are most grateful to Jacqueline Trotter for the Oli-neu cells and Cecilia Bucci and Bice Chini for the Rab constructs. Special thanks to the Monzino Foundation, Milan, Italy, for the gift of the Zeiss LSM510 Meta confocal microscope and the Axiovert 200M confocal system equipped with a spinning disc (PerkinElmer Life Sciences).
This work was supported by Progetti di Ricerca di Interesse Nazionale COFIN-PRIN 2008, Ministero dell'Istruzione dell' Università e Della Ricerca “Purinoceptors and neuroprotection: focus on the new purinergic receptor GPR17,” Regione Lombardia Project NUTEC ID 30263049 and Cariplo Fuondation rif.no. 2012-0546 (to P. R.). This work was supported in part by FISM Fondazione Italiana Sclerosi Multipla Grant 2010/R2 (to M. P. A.) and CNR-Progetto Invecchiamento, CUP B51J11000820005.
A. Fratangeli, E. Parmigiani, M. Fumagalli, D. Lecca, R. Benfante, M. Passafaro, A. Buffo, M. P. Abbracchio, and P. Rosa, unpublished results.
- GPCR
- G protein-coupled receptor
- LT
- leukotriene
- cysLT
- cysteinyl-leukotriene
- CM
- conditioned medium
- OPCs
- oligodendrocyte precursor cells
- Lamp1
- lysosomal-associated membrane protein 1
- MAG
- myelin-associated glycoprotein
- TfR
- transferrin receptor
- FoxO
- Forkhead box protein O
- MesNA
- sodium 2-mercaptoethane sulfonate
- Nt
- N-terminal.
REFERENCES
- 1. Ciana P., Fumagalli M., Trincavelli M. L., Verderio C., Rosa P., Lecca D., Ferrario S., Parravicini C., Capra V., Gelosa P., Guerrini U., Belcredito S., Cimino M., Sironi L., Tremoli E., Rovati G. E., Martini C., Abbracchio M. P. (2006) The orphan receptor GPR17 identified as a new dual uracil nucleotides/cysteinyl-leukotrienes receptor. EMBO J. 25, 4615–4627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lecca D., Trincavelli M. L., Gelosa P., Sironi L., Ciana P., Fumagalli M., Villa G., Verderio C., Grumelli C., Guerrini U., Tremoli E., Rosa P., Cuboni S., Martini C., Buffo A., Cimino M., Abbracchio M. P. (2008) The recently identified P2Y-like receptor GPR17 is a sensor of brain damage and a new target for brain repair. PLoS One 3, e3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Parravicini C., Ranghino G., Abbracchio M. P., Fantucci P. (2008) GPR17. Molecular modeling and dynamics studies of the 3-D structure and purinergic ligand binding features in comparison with P2Y receptors. BMC Bioinformatics 9, 263–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pugliese A. M., Trincavelli M. L., Lecca D., Coppi E., Fumagalli M., Ferrario S., Failli P., Daniele S., Martini C., Pedata F., Abbracchio M. P. (2009) Functional characterization of two isoforms of the P2Y-like receptor GPR17. [35S]GTPγS binding and electrophysiological studies in 1321N1 cells. Am. J. Physiol. Cell. Physiol. 297, C1028–C1040 [DOI] [PubMed] [Google Scholar]
- 5. Daniele S., Lecca D., Trincavelli M. L., Ciampi O., Abbracchio M. P., Martini C. (2010) Regulation of PC12 cell survival and differentiation by the new P2Y-like receptor GPR17. Cell. Signal. 22, 697–706 [DOI] [PubMed] [Google Scholar]
- 6. Daniele S., Trincavelli M. L., Gabelloni P., Lecca D., Rosa P., Abbracchio M. P., Martini C. (2011) Agonist-induced desensitization/resensitization of human G protein-coupled receptor 17. A functional cross-talk between purinergic and cysteinyl-leukotriene ligands. J. Pharmacol. Exp. Ther. 338, 559–567 [DOI] [PubMed] [Google Scholar]
- 7. Chen Y., Wu H., Wang S., Koito H., Li J., Ye F., Hoang J., Escobar S. S., Gow A., Arnett H. A., Trapp B. D., Karandikar N. J., Hsieh J., Lu Q. R. (2009) The oligodendrocyte-specific G protein-coupled receptor GPR17 is a cell-intrinsic timer of myelination. Nat. Neurosci. 12, 1398–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ceruti S., Villa G., Genovese T., Mazzon E., Longhi R., Rosa P., Bramanti P., Cuzzocrea S., Abbracchio M. P. (2009) The P2Y-like receptor GPR17 as a sensor of damage and a new potential target in spinal cord injury. Brain 132, 2206–2218 [DOI] [PubMed] [Google Scholar]
- 9. Ceruti S., Viganò F., Boda E., Ferrario S., Magni G., Boccazzi M., Rosa P., Buffo A., Abbracchio M. P. (2011) Expression of the new P2Y-like receptor GPR17 during oligodendrocyte precursor cell maturation regulates sensitivity to ATP-induced death. Glia 59, 363–378 [DOI] [PubMed] [Google Scholar]
- 10. Fumagalli M., Daniele S., Lecca D., Lee P. R., Parravicini C., Fields R. D., Rosa P., Antonucci F., Verderio C., Trincavelli M. L., Bramanti P., Martini C., Abbracchio M. P. (2011) Phenotypic changes, signaling pathway, and functional correlates of GPR17-expressing neural precursor cells during oligodendrocyte differentiation. J. Biol. Chem. 286, 10593–10604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Boda E., Viganò F., Rosa P., Fumagalli M., Labat-Gest V., Tempia F., Abbracchio M. P., Dimou L., Buffo A. (2011) The GPR17 receptor in NG2 expressing cells. Focus on in vivo cell maturation and participation in acute trauma and chronic damage. Glia 59, 1958–1973 [DOI] [PubMed] [Google Scholar]
- 12. Ferguson S. S. (2001) Evolving concepts in G protein-coupled receptor endocytosis. The role in receptor desensitization and signaling. Pharmacol. Rev. 53, 1–24 [PubMed] [Google Scholar]
- 13. Marchese A., Paing M. M., Temple B. R., Trejo J. (2008) G protein-coupled receptor sorting to endosomes and lysosomes. Annu. Rev. Pharmacol. Toxicol. 48, 601–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dunham J. H., Hall R. A. (2009) Enhancement of the surface expression of G protein-coupled receptors. Trends Biotechnol. 27, 541–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ritter S. L., Hall R. A. (2009) Fine-tuning of GPCR activity by receptor-interacting proteins. Nat. Rev. Mol. Cell Biol. 10, 819–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sorkin A., von Zastrow M. (2009) Endocytosis and signaling. Intertwining molecular networks. Nat. Rev. Mol. Cell Biol. 10, 609–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Walrafen P., Verdier F., Kadri Z., Chrétien S., Lacombe C., Mayeux P. (2005) Both proteasomes and lysosomes degrade the activated erythropoietin receptor. Blood 105, 600–608 [DOI] [PubMed] [Google Scholar]
- 18. Maekawa A., Balestrieri B., Austen K. F., Kanaoka Y. (2009) GPR17 is a negative regulator of the cysteinyl leukotriene 1 receptor response to leukotriene D4. Proc. Natl. Acad. Sci. U.S.A. 106, 11685–11690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Benned-Jensen T., Rosenkilde M. M. (2010) Distinct expression and ligand binding profiles of two constitutively active GPR17 splice variants. Br. J. Pharmacol. 159, 1092–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jung M., Krämer E., Grzenkowski M., Tang K., Blakemore W., Aguzzi A., Khazaie K., Chlichlia K., von Blankenfeld G., Kettenmann H. (1995) Lines of murine oligodendroglial precursor cells immortalized by an activated neu tyrosine kinase show distinct degrees of interaction with axons in vitro and in vivo. Eur. J. Neurosci. 7, 1245–1265 [DOI] [PubMed] [Google Scholar]
- 21. Winterstein C., Trotter J., Krämer-Albers E. M. (2008) Distinct endocytic recycling of myelin proteins promotes oligodendroglial membrane remodeling. J. Cell Sci. 121, 834–842 [DOI] [PubMed] [Google Scholar]
- 22. Taverna E., Saba E., Linetti A., Longhi R., Jeromin A., Righi M., Clementi F., Rosa P. (2007) Localization of synaptic proteins involved in neurosecretion in different membrane microdomains. J. Neurochem. 100, 664–677 [DOI] [PubMed] [Google Scholar]
- 23. Passafaro M., Piëch V., Sheng M. (2001) Subunit-specific temporal and spatial patterns of AMPA receptor exocytosis in hippocampal neurons. Nat. Neurosci. 4, 917–926 [DOI] [PubMed] [Google Scholar]
- 24. Bucci C., Lütcke A., Steele-Mortimer O., Olkkonen V. M., Dupree P., Chiariello M., Bruni C. B., Simons K., Zerial M. (1995) Co-operative regulation of endocytosis by three Rab5 isoforms. FEBS Lett. 366, 65–71 [DOI] [PubMed] [Google Scholar]
- 25. Conti F., Sertic S., Reversi A., Chini B. (2009) Intracellular trafficking of the human oxytocin receptor. Evidence of receptor recycling via a Rab4/Rab5 “short cycle.” Am. J. Physiol. Endocrinol. Metab. 296, E532–E542 [DOI] [PubMed] [Google Scholar]
- 26. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔCT) method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 27. Linetti A., Fratangeli A., Taverna E., Valnegri P., Francolini M., Cappello V., Matteoli M., Passafaro M., Rosa P. (2010) Cholesterol reduction impairs exocytosis of synaptic vesicles. J. Cell Sci. 123, 595–605 [DOI] [PubMed] [Google Scholar]
- 28. Kippert A., Trajkovic K., Fitzner D., Opitz L., Simons M. (2008) Identification of Tmem10/Opalin as a novel marker for oligodendrocytes using gene expression profiling. BMC Neurosci. 9, 40–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ren H., Orozco I. J., Su Y., Suyama S., Gutiérrez-Juárez R., Horvath T. L., Wardlaw S. L., Plum L., Arancio O., Accili D. (2012) FoxO1 target Gpr17 activates AgRP neurons to regulate food intake. Cell 149, 1314–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Klein U., von Zastrow M. (2000) in Regulation of G-protein-coupled Receptor Function and Expression (Benovic J. L., ed.) pp. 4–13, New York, Wiley-Liss, Inc. [Google Scholar]
- 31. Doherty G. J., McMahon H. T. (2009) Mechanisms of endocytosis. Annu. Rev. Biochem. 78, 857–902 [DOI] [PubMed] [Google Scholar]
- 32. Sönnichsen B., De Renzis S., Nielsen E., Rietdorf J., Zerial M. (2000) Distinct membrane domains on endosomes in the recycling pathway visualized by multicolor imaging of Rab4, Rab5, and Rab11. J. Cell Biol. 149, 901–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Seachrist J. L., Ferguson S. S. (2003) Regulation of G protein-coupled receptor endocytosis and trafficking by Rab GTPases. Life Sci. 74, 225–235 [DOI] [PubMed] [Google Scholar]
- 34. Platta H. W., Stenmark H. (2011) Endocytosis and signaling. Curr. Opin. Cell Biol. 23, 393–403 [DOI] [PubMed] [Google Scholar]
- 35. Zerial M., McBride H. (2001) Rab proteins as membrane organizers. Nat. Rev. Mol. Cell Biol. 2, 107–117 [DOI] [PubMed] [Google Scholar]
- 36. Parravicini C., Abbracchio M. P., Fantucci P., Ranghino G. (2010) Forced unbinding of GPR17 ligands from wild type and R255I mutant receptor models through a computational approach. BMC Struct. Biol. 10, 8–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Emery B. (2010) Regulation of oligodendrocyte differentiation and myelination. Science 330, 779–782 [DOI] [PubMed] [Google Scholar]
- 38. Wake H., Lee P. R., Fields R. D. (2011) Control of local protein synthesis and initial events in myelination by action potentials. Science 333, 1647–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jablonska B., Scafidi J., Aguirre A., Vaccarino F., Nguyen V., Borok E., Horvath T. L., Rowitch D. H., Gallo V. (2012) Oligodendrocyte regeneration after neonatal hypoxia requires FoxO1-mediated p27Kip1 expression. J. Neurosci. 32, 14775–14793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hanyaloglu A. C., von Zastrow M. (2008) Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu. Rev. Pharmacol. Toxicol. 48, 537–568 [DOI] [PubMed] [Google Scholar]