

Abstract
Lipoproteins are a family of plasma nanoparticles responsible for the transportation of lipids throughout the body. High-density lipoprotein (HDL), the smallest of the lipoprotein family, measures 7–13 nm in diameter and consists of a cholesteryl ester and triglyceride core that is covered with a monolayer of phospholipids and apolipoproteins. We have developed an iron oxide core HDL nanoparticle (FeO-HDL), which has a lipid based fluorophore incorporated in the phospholipid layer. This nanoparticle provides contrast for optical imaging, magnetic resonance imaging (MRI) and transmission electron microscopy (TEM). Consequently, FeO-HDL can be visualized on the anatomical, cellular and sub-cellular level. In the current study we show that the biophysical features of FeO-HDL closely resemble those of native HDL and that FeO-HDL possess the ability to mimic HDL characteristics both in vitro as well as in vivo. We demonstrate that FeO-HDL can be applied to image HDL interactions and to investigate disease settings where HDL plays a key function. More generally, we have demonstrated a multimodal approach to study the behavior of biomaterials in vitro as well as in vivo. The approach allowed us to study nanoparticle dynamics in circulation, as well as nanoparticle targeting and uptake by tissues and cells of interest. Moreover, we were able to qualitatively assess nanoparticle excretion, critical for translating nanotechnologies to the clinic.
Keywords: High-density lipoprotein, Iron oxide nanoparticles, Transmission electron microscopy, Optical imaging, Nanoparticle excretion
1. Introduction
Lipoproteins are a family of plasma nanoparticles responsible for the transportation of lipids throughout the body [1]. They are divided into five main classes, high-density lipoprotein (HDL), low-density lipoprotein (LDL), intermediate-density lipoprotein (IDL), very low-density lipoprotein (VLDL), and chylomicrons, based on their density, and numerous subclasses also exist within the main classes [1]. The different lipoproteins share common physical features; they consist of a hydrophobic core, containing a different mix of cholesteryl esters and triglycerides, which is coated by an amphiphilic monolayer of phospholipids. Apolipoproteins are embedded in this lipidic corona to assure structural integrity and provide targeting functions [1].
High-density lipoprotein (HDL) is a crucially important vector and is the subject of intense study [2–4]. It is the smallest of the lipoprotein family, measures 7–13 nm in diameter and is initially synthesized in the liver and the intestine. HDL carries various apolipoproteins [1,3], and the most abundant protein components are apolipoprotein A-I (apoA-I) and apolipoprotein A-II (apoA-II) [5,6]. ApoA-1, like most apolipoproteins, is composed of an amphiphatic α-helical structure [7], with its hydrophobic region facing the core of the particle and the hydrophilic region facing the surrounding aqueous milieu. The apolipoprotein reduces the surface pressure on the lipoprotein, thereby stabilizing it [8,9]. Additionally, it activates the enzyme lecithin cholesterol acyltransferase (LCAT) [2,3], which catalyzes esterfication of free cholesterol into cholesteryl esters.
Most importantly, HDL is the main actor in the process known as reverse cholesterol transportation (RCT), in which HDL promotes the transportation of excess cholesterol from extra-hepatic or peripheral tissue to the liver for elimination through the biliary system [4,10]. Reverse efflux of cholesterol from plaque macrophages has an important protective effect in atherosclerosis [4], and HDL is believed to have other atheroprotective properties as well [3,4,11]. Hence serum HDL levels are negatively correlated with heart attack risk [12]. The two key proteins on the surface of plaque macrophages with which HDL interacts to accept cholesterol are ABCA1 and ABCG1, which are members of the ATP-binding cassette family and which transfer cholesterol to HDL species in an ATP-dependent manner [13]. HDL also interacts with the scavenger receptor-B1 (SR-B1) in the liver [14].
In vivo (MR) imaging is a powerful tool to investigate the dynamics, metabolism and trafficking of lipoproteins non-invasively. We recently published a study on a novel nanocrystal HDL-based nanoparticle platform [15], which allowed multimodal imaging of macrophages in atherosclerotic plaques of the apoE knockout (apoE KO) mouse model of atherosclerosis. For this platform, the natural core of triglycerides and cholesteryl esters of the HDL particles was substituted with a single gold, quantum dot or iron oxide nanocrystal. The biological features of this platform, including size, coating composition and macrophage specificity, were found to be similar to native HDL. Iron oxide core HDL (FeO-HDL), is schematically depicted in Fig. 1a. It has a lipid based fluorophore incorporated in the phospholipid layer, which enables optical imaging (Supplementary Fig. S1), while the iron oxide can be detected by magnetic resonance imaging (MRI) and transmission electron microscopy (TEM). Consequently FeO-HDL can be visualized on the anatomical, cellular and sub-cellular level. This allowed us to image macrophages in atherosclerotic lesions of apoE KO mice with MRI (Fig. 1b) in vivo and confocal laser microscopy (Fig. 1c) ex vivo. The in vivo MRI methodology and settings were described in detail previously [15].
Fig. 1.
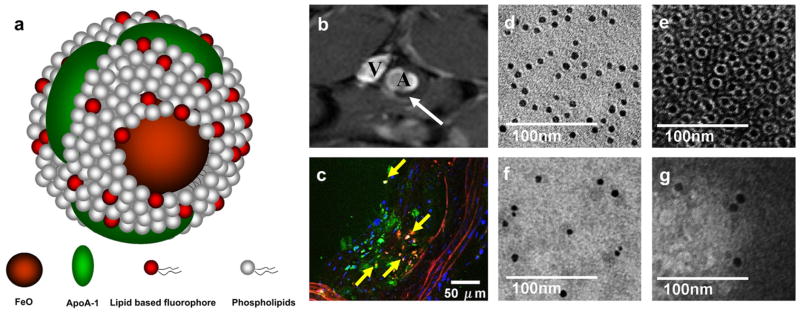
(a) schematic depiction of FeO-HDL. (b) in vivo T2*-weighted (TR = 10 ms, TE = 4 ms) MR image of abdominal region of an apoE KO mouse, 24 h post injection of FeO-HDL. The white arrow denotes the black rim (black spot) in the aortic wall observed after FeO-HDL administration. (A, aorta, V, inferior cava vein). (c) ex vivo confocal laser microscopy of excised aorta section. Nuclei are shown in blue, DAPI staining, FeO-HDL in red (rhodamine), Green colors marks macrophages, stained with Alexa 647-CD68 and yellow marks co-localization of FeO-HDL and macrophages. (d) In vitro TEM images of FeO-HDL in buffer. (e) In vitro negative staining TEM image of FeO-HDL, showing that each iron oxide (IO) particle is coated with a single monolayer of phospholipids. (f, g) TEM and negative staining TEM of blood samples 5 min after the administration of FeO-HDL revealed individually dispersed particles, i.e. no aggregation and that the lipid coating is intact.
Although FeO-HDL closely mimics the size and lipid/protein composition of native HDL it remains to be investigated if it is justifiable to extrapolate the biological properties, dynamics and trafficking of this HDL-like particle to that of natural HDL. We investigated, therefore, several key features of FeO-HDL and compared them with those of native HDL. First, we investigated the structure of FeO-HDL, both before and after injection into mice. HDL naturally binds to and interacts with macrophage cells and hepatocytes during RCT, so we probed the binding of FeO-HDL to these cells and the cholesterol efflux caused. Different lipoprotein fractions exist in a state of dynamic equilibrium with each other, so we explored lipid exchange dynamics in mice. Furthermore, we examined the deposition of FeO-HDL in atherosclerotic plaques and in the liver, and tracked the excretion of the iron cores via the bile into the feces.
2. Methods and materials
2.1. Materials
Myristoyl hydroxy phosphatidylcholine (MHPC) distearoyl phosphoethanol-amine-N-[methoxy(polyethylene glycol)-2000] ammonium salt (PEG-DSPE), dimyristoyl phosphoethanolamine (DMPE) and dimyristoyl phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) ammonium salt (Rhod-DMPE) were all purchased from Avanti Polar Lipids and used as received. The Cy5.5 NHS ester was purchased from Amersham Biosciences (Piscataway, NJ). Cell culture supplies were purchased from Invitrogen (Carlsbad, CA). Native HDL used was harvested from human plasma as per the method of Havel [16].
2.2. Synthesis
FeO-HDL was synthesized as reported previously [15]. Typically, 50 mg of MHPC lipids were solubilized in a chloroform methanol 20:1 solvent mixture. Fluorophore labeled lipid was added to the solution as 1% of total lipid mass and 10 mg of oleic acid coated iron oxide cores (NN Labs, Fayetteville, AR, 10 nm Fe3O4 formulation) were suspended in chloroform and slowly mixed with the lipid solution. The mixed solution was drop-wise added to heated deionized water under stirring. Apolipoprotein A-I was added after which the particles were purified. An extensive description of the synthesis can be found in the Supplementary data.
2.3. Dynamic light scattering
Dynamic light scattering was performed with ZetaPALS instrument with BI-MAS particle sizing option from Brookhaven Instruments Corporation, Holtsville, NY on 50 μl of FeO-HDL nanoparticles just after synthesis, diluted in 2 ml of PBS filtered with a 0.22 μm membrane and 24 h after incubation in 10% serum.
2.4. Cell culture
Murine macrophage cells J774A.1 (ATCC, Manassas, VA) and HepG2 hepatocytes (ATCC, Manassas, VA) were cultured in DMEM supplemented with 10% FBS and 1% streptomycin/penicillin. Mouse aortic endothelial cells (MAEC) were cultured with DMEM-F12 supplemented with 20% FBS and 1% penicillin/streptomycin plus bovine brain extracts and were a kind gift from Dr. Carlos Zaragoza and Dr. Beatriz Herranz (CNIC, Spain). After the fifth splitting, the cells were lifted either by scraping (macrophages) or using trypsin-edta (HepG2 hepatocytes and MAEC) and placed in wells plates for experiments. When the wells were approximately at 70–80% confluence they were incubated with the agent added into a relevant concentration or used as control cells.
2.5. Cholesterol efflux measurements
This protocol was carried out as previously published [17]. J774A.1 macrophages (ATCC, Manassas VA, USA) were incubated overnight with 15 μg/ml cholesterol and 0.5 μCi/ml [3H]cholesterol in 0.2% BSA-DMEM. After an equilibration period of 3 h with 1 μg/ml of F-1394 (an acyl-CoA:cholesterol acyl-transferase inhibitor [18] in 0.2% BSA-DMEM, efflux was initiated by the addition of 50 μg/ml (by protein content) of acceptors (i.e. lipoprotein or BSA control). 100 μl aliquots of the medium were collected after 4 h, and the [3H]cholesterol measured by liquid scintillation counting (LSC). In order to measure the [3H] cholesterol present in the cells, cell lipids were then extracted by incubating the cell monolayers overnight in isopropanol. After lipid extraction, the total [3H] cholesterol present in lipid extract was measured by LSC and the efflux calculated as percentage of total.
2.6. Cell incubations
J774A.1 murine macrophage cells (ATCC, Manassas, VA), HepG2 hepatocytes (ATCC, Manassas, VA) and mouse aortic endothelial cells (MAEC) were cultured as described above. After the fifth splitting, the cells were put into 96 wells plates with 0.1 ml of media in each well. The cells were allowed to grow to 70–80% confluence and the media replaced with fresh, before FeO-HDL-Cy5.5 or FeO-PEG-Cy5.5 were added. After 1 h, the media in the wells was removed and the wells were extensively washed with PBS to get rid of unbound FeO-HDL in the media and subsequently fixed with 4% paraformaldehyde. Finally the wells were imaged using an IVIS Imaging System 200 (Xenogen, Alameda, CA) with Cy5.5 emission (615–665 nm) and excitation (695–770 nm) settings. An ROI was placed covering each well and a photon count obtained from each well. The data was analyzed using “Origin®” software.
For competitive inhibition experiments the cells were treated in a similar fashion as described above. However the cells were incubated with the same concentration of FeO-HDL-Cy5.5, but with three different concentrations of native HDL or FeO-HDL without any fluorophore. After incubation of 1 h the cells were washed and fixed as described above and imaged with the IVIS Imaging System 200.
2.7. Confocal laser microscopy
Imaging was performed on a Zeiss LSM 510 META microscope (Carl Zeiss, Oberkochen, Germany) in an inverted configuration. Objectives used were Plan-Apochromat 20×, Plan-Neofluar 40x (1.3 oil DIC) and Plan-Apochromat 63x (1.4 oil DIC) lenses. Data were captured and analyzed using Zeiss LSM 510 Meta and Image Browser software (Zeiss). J774A.1 macrophage cells and HepG2 cells for confocal microscopy were cultured as described above and grown on coverslips placed at the bottom of six wells plates. After 1 h of incubation with FeO-HDL the media was discarded, and 1 ml of 4% paraformaldehyde was added. After 5 min the cover slip, with the now fixed cells, was stained with DAPI, placed on a glass slide and sealed.
2.8. Transmission electron microscopy of cells
The instrument used was a Hitachi H7650 system linked to a SIA (Scientific Instruments and Applications) digital camera controlled by Maxim CCD software. TEM was performed on FeO-HDL samples suspended in ammonium acetate buffer, made of 0.125 M ammonium acetate, 2.6 mM ammonium carbonate, and 0.26 mM tetrasodium EDTA at pH 7.4. The nickel TEM-grids were cleaned in acetone before one drop of the ammonium acetate-nanoparticle solution was applied to the grid. After drying one drop of 2% sodium phosphotungstate was applied to the grid as described by Forte and Nordhausen [19] J774A.1 murine macrophage cells were cultured and incubated as described above. After an incubation of 15 min the media in the wells was removed and the cells were washed 3 times with PBS, scraped from the bottom of the wells and centrifuged to cell pellets. The supernatant of PBS was discarded and the cell pellets were re-suspended and fixed in 2.5% glutaraldehyde. The resulting cell pellets, were further fixed in osmium tetroxide, dehydrated and samples were cut in ultrathin sections (~60 nm), stained with 4% uranyl acetate and Reynold’s lead citrate [20] and placed on grids prior to imaging.
2.9. Transmission electron microscopy of tissue sections
ApoE KO mice were anesthetized using isoflurane. FeO-HDL was administrated through a tail vein injection. At 15 min, 30 min, 1 h and 2 h, 24 h after injection liver biopsies were obtained. The biopsies were immediately placed in 2.5% glutaraldehyde. After 2 h and 24 h the mice were sacrificed and perfused through the heart with saline. Liver and aortas were harvested and these organs were placed in 2.5% glutaraldehyde and prepared along with the biopsies for TEM.
2.10. Transmission electron microscopy of blood
ApoE KO and wild type mice were anesthetized using isoflurane. FeO-HDL-Cy5.5 was administrated through a tail vein injection. After 5 min blood samples were collected from the femoral vein. TEM was performed on the blood samples suspended in ammonium acetate buffer as described above. The nickel TEM-grids were cleaned in acetone before one drop of the ammonium acetate-nanoparticle solution was applied to the grid. After drying one drop of 2% sodium phosphotungstate was applied to the grid as described by Forte and Nordhausen [19].
2.11. In vivo lipid exchange
ApoE KO and wild type mice were anesthetized using isoflurane. FeO-HDL-Cy5.5 was administrated through a tail vein injection. After 2 h the mice were injected with a mixture of ketamine and xylazine and sacrificed by heart puncture. A minimum of 500 μl blood was obtained from the heart. Plasma was isolated by centrifugation with 10.000 rpm for 5 min. A minimum of 220 μl plasma was run through FPLC and 4 plasma fractions were obtained. The 4 plasma fractions, VLDL, LDL, HDL and plasma proteins, were imaged with IVIS Imaging System 200 (Xenogen, Alameda, CA).
2.12. TEM of bile and feces
ApoE KO mice were injected with FeO-HDL and sacrificed 2 h later, as described above. Non-injected apoE KO mice were used as controls. The gall bladder was drained and the bile thus obtained was dropped onto a grid and imaged without further processing. The intestines were cut into four sections and feces were taken from each section before fixing, staining and imaging as detailed above for tissue.
3. Results and discussion
3.1. Nanoparticle morphology after intravenous administration
We previously reported a nanocrystal core HDL particle platform similar to native HDL [15]. TEM and negative stain TEM images of FeO-HDL are depicted in Fig. 1d and e, revealing a monodisperse core size, that the FeO-HDL particles were within the size range of native HDL (7–13 nm) and that each individual particle was coated with a single monolayer of phospholipids. Subsequent to our work other groups have also developed lipoprotein like and nanocrystal containing nanoparticles, but in one case the lipoprotein template was 250 nm chylomicrons [21], and in the other case gold core HDL was synthesized and was investigated as a therapeutic, not as a contrast agent [22]. Here, we provide further evidence that our contrast agent platform closely mimics native HDL. We investigated the nanoparticle morphology under physiological conditions by applying the following approach: TEM and negative staining TEM was performed on plasma of an apoE KO mouse obtained 5 min after the injection of FeO-HDL (Fig. 1f and g). This time point was used in order to ensure a reasonable concentration of nanoparticles in the blood. For investigating the stability of FeO-HDL over longer time periods, FeO-HDL was exposed to plasma for 24 h at 37 °C and examined for aggregation with dynamic light scattering. The sizes of the particles were found not to change significantly after the 24 h incubation in plasma as the sizes before and after incubation were 8.3 nm ± 0.3 nm and 8.6 nm ± 0.6 nm, respectively.
We demonstrated that in circulation and for 24 h incubation in plasma FeO-HDL nanoparticles remained individually dispersed, i.e. no aggregation, and that the phospholipid corona remained intact, confirming the physical structure remains as native HDL after intravenous administration.
3.2. Confocal and transmission electron microscopy of FeO-HDL uptake by macrophages and hepatocytes
When cholesterol is removed from cells in the atherosclerotic plaque and transported back to the liver, HDL interacts with macrophages and hepatocytes. To demonstrate the affinity of FeO-HDL for these cell types, we incubated murine J774A.1 macrophages and HepG2 cells with rhodamine labeled FeO-HDL. As a control particle we used PEGylated iron oxide particles (FeO-PEG) that were labeled with rhodamine. Fig. 2 illustrates the qualitative preferential uptake of FeO-HDL by macrophages visualized with confocal microscopy (2a) and TEM (2b). Confocal microscopy clearly showed high quantities of fluorescence from FeO-HDL present in the cytoplasm.
Fig. 2.

(a) Confocal microscopy of the uptake of FeO-HDL and FeO-PEG in macrophages and hepatocytes. (b) TEM of FeO-HDL uptake in macrophages (top panels) and hepatocytes (lower panels). Single iron oxide nanoparticles were found in close proximity to the cell surface in vitro, or just inside (or on) the cell surface, demonstrating the uptake of single nanoparticles on a sub-cellular level in vitro. (c) endosome containing iron oxides, in this case inside a macrophage cell. (d) Cell viability after incubation with FeO-HDL in macrophages and Hep2G cells for 4 h and 8 h.
To visualize the uptake of the FeO-HDL at the sub-cellular level we examined fixed macrophage cells that were incubated for 15 min and looked for particles close to or just inside the cells with TEM. Interestingly we found single particles in close proximity of the cells, indicating that the particles were taken up individually and not as clusters/aggregates (Fig. 2b top panel). However, it has to be noted that these images are not necessarily representative for the whole cell population. Inside some cells the iron oxide nanoparticles were aggregated in endosomes (Fig. 2c), but from a qualitative assessment we found most of the nanoparticles individually taken up by the cells and only subsequently clustered in endosomes. The same trend was found when FeO-HDL was applied to hepatocytes, as can be seen in Fig. 2a and b (lower panels).
3.3. Cell viability
As a measure of the viability of cells that were incubated with FeO-HDL we determined their ATP content. J774A.1 murine macrophages and HepG2 hepatocytes, two cell types that naturally interact with HDL in vivo, were incubated with FeO-HDL for 1, 4 and 8 h at a concentration of 4 μg/ml. We based this number on what we found to cause signal attenuation in MRI of cell pellets in our previous study[15], which is comparable to the iron oxide content accumulated in the atherosclerotic plaques of mice injected with FeO-HDL. The results after incubation for 4 and 8 h are portrayed in Fig. 2d and demonstrate no significant decrease of cell vitality.
3.4. Cholesterol efflux properties
As mentioned in the introduction, HDL is pivotal for reverse cholesterol transport (RCT). Therefore we compared the macrophage cholesterol efflux capacity of FeO-HDL with native HDL. Macrophage cholesterol efflux is an established surrogate measure for RCT of lipoproteins [23] and has been used to probe the mechanism of RCT [24] as well as a method to evaluate synthetic HDL replacement therapies [25]. The level of macrophage cholesterol efflux capacity of FeO-HDL (0.91 ± 0.09) was found to be similar to that produced by native HDL (1 ± 0.03), as shown in Fig. 3a. As RCT is a key feature of HDL this result strongly contributes to the notion that FeO-HDL can be perceived as HDL-like.
Fig. 3.

(a) Normalized cholesterol efflux of BSA, native HDL and FeO-HDL. (b) Uptake curve of FeO-HDL in macrophage and in hepatocyte cells as function of incubation concentration. It can be seen that the uptake of FeO-HDL was saturable for both cell types. (c) Competitive inhibition of FeO-HDL-Cy5.5, using native HDL or non-labeled FeO-HDL (no fluorophore). (d) Normalized uptake of FeO-HDL-Cy5.5 and FeO-PEG-Cy5.5 in macrophages, hepatocytes and endothelial cells. (e) Normalized fluorescence profile of lipoprotein fractions from apoE KO mice. It is notable that fluorescence is found in all 4 fractions. (f) Fluorescence profiles from wild type mice. Lipoprotein fractions were separated based on size using FPLC.
3.5. Quantification of FeO-HDL uptake under different conditions
As previously mentioned, HDL interacts with the ABCA1 protein, ABCG-I or SR-BI receptors to perform reverse cholesterol transport (RCT). To investigate if FeO-HDL’s interaction with macrophages occurs in a receptor-like manner and to investigate nanoparticle uptake in detail, we carried out in vitro cell experiments where the FeO-HDL concentration was varied. To this aim, FeO-HDL was labeled with a lipid conjugated to a near infrared fluorophore Cy5.5 (FeO-HDL-Cy5.5). Macrophages and hepatocytes were incubated with different concentrations of FeO-HDL in a 96 wells plate (a representative fluorescent image is shown in Supplementary Fig. S2). The wells were imaged with a Xenogen IVIS 200 fluorescence imaging system and the uptake curve is depicted in Fig. 3b. We observed that the signal of FeO-HDL was saturable with increasing concentration, indicative of a receptor-like uptake. To further elaborate on this we performed competitive inhibition experiments where FeO-HDL-Cy5.5 was co-incubated with either native HDL or FeO-HDL without a fluorophore. We used concentrations of the competing species of 1.5–9 times the concentration of Cy5.5 labeled FeO-HDL. We found that the fluorescence signal of the Cy5.5 labeled particles decreased when the concentration of native HDL or FeO-HDL without a fluorophore was increased (Fig. 3c). Since the uptake of the FeO-HDL-Cy5.5 particle can be inhibited via competition with native HDL and unlabeled FeO-HDL the uptake mechanism most likely occurs in a receptor-mediated fashion, possibly via one of the proteins native HDL is known to interact with, i.e. the ABCA-I protein, ABCG-I or SR-BI. As a control cell for the uptake of FeO-HDL we used an endothelial cell line (MAEC), as these cells are the cell type primarily exposed to HDL as it circulates. When incubating macrophages, hepatocytes and endothelial cells with FeO-HDL-Cy5.5 and FeO-PEG-Cy5.5 we found a significantly higher uptake in macrophages and hepatocytes with FeO-HDL-Cy5.5, while the uptake in endothelial cells and the uptake of FeO-PEG-Cy5.5 were found to be substantially lower (Fig. 3d).
3.6. In vivo lipid dynamics
Lipoproteins are highly dynamic in the circulation. Interactions between different lipoprotein fractions occurs at high rates and lipids are exchanged between the different lipoprotein particles [26]. Therefore, FeO-HDL-Cy5.5 was injected intravenously in the tail vein of wild type mice (n = 2) and apoE KO mice (n = 3), a well recognized animal model of atherosclerosis [27]. After 2 h the mice were sacrificed, blood was collected via a heart puncture and the plasma subsequently isolated. The plasma was separated into 4 fractions: VLDL, LDL, HDL and plasma proteins with fast protein liquid chromatography(FPLC). These fractions were analyzed for fluorescence with the aforementioned IVIS fluorescence imaging system. Fluorescence in any other fraction than the HDL fraction would occur only if the near infrared fluorophore is transferred to another fraction in circulation. For the apoE KO mice, the fluorescence was almost equally distributed among all 4 fractions (Fig. 3e), indicating transfer of lipids between FeO-HDL and the different lipoprotein fractions.
HDL comprises the lipoprotein fraction with the smallest particle size, and it could be argued that fluorescence in other fractions merely was a result of FeO-HDL aggregation. However, when FeO-HDL was administrated to WT mice where the plasma levels of VLDL and LDL lipoproteins are substantially lower than in apoE KO mice [28], and therefore a lower probability of lipid exchange was expected, it was observed that the fluorescence primarily remained in the HDL fraction (Fig. 3f). This corroborated our TEM findings that FeO-HDL does not aggregate, and remains intact and within the size of native HDL after applied intravenously. The lipid transfer, reflecting a biological dynamic/active system, further confirms the similarities between native HDL and our iron oxide modified HDL.
3.7. Transmission electron microscopy of atherosclerotic plaque tissue
Excised abdominal aortas of apoE KO mice injected with FeO-HDL were extensively examined on sub-cellular level with TEM. The iron oxide cores were found in large quantities within atherosclerotic plaques of aortas. Importantly, we found the iron oxide monodispersely distributed within the plaque tissue (Fig. 4). This result corresponded well with our TEM examination of blood samples, where individual dispersed FEO-HDL particles were observed. Therefore, we conclude that FeO-HDL particles circulate as single entities and enter the plaque sites individually, similar to native HDL. Occasional aggregates inside endosomes were observed as well, Supplementary Fig. S3. In untreated apoE KO mice no iron oxide particles were found in the plaque tissue (Supplementary Fig. S4).
Fig. 4.

(a, d) Light microcopy of Azur B/Methylene Blue stained aortic section from an apoE KO mouse. (b, c, e, f) TEM images at two different magnifications of two plaque areas show monodisperse iron oxide particles within the atherosclerotic plaque (N, nucleus and L, lipid vacuole).
3.8. Trafficking and excretion of FeO-HDL
It is very important to consider biodegradability, excretion pathways and pharmacokinetics when nanoparticles are administrated in vivo [29]. In addition, one of the functions of HDL is to deliver cholesterol to the liver for excretion.
To determine the pharmacokinetic profile, we injected FeO-HDL-Cy5.5 into apoE KO mice and collected blood samples up to 24 h post injection. Analysis of the blood samples with IVIS fluorescence imaging (not shown) revealed a half life of approximately 90 min. To accompany the plasma half life measurements we used TEM to examine liver sections at different time points (15, 30, 45, 60 min) after FeO-HDL injection to investigate its trafficking through the liver. Furthermore, we studied the contents of the bile and feces of mice that were injected with FeO-HDL with TEM.
In the liver we found iron oxide to be present in the Kupffer cells (Fig. 5a, top), but to a large extent also in endosomes around or near the bile canaliculi within the hepatocytes (Fig. 5a, middle). The accumulation of iron oxide near the bile canaliculi indicated the route of excretion. We did not find any difference in the number of endosomes containing iron oxide around the bile canaliculi, in any of the time points examined. We believe the reason for lack of difference in iron oxide containing endosomes at the examined time points can be explained by that the metabolism and excretion starts almost immediately after injection. As a control for the liver examination we used untreated apoE KO mice. In the untreated apoE KO mice we found structures similar to the iron oxide containing endosomes. (Fig. 5a, bottom). These structures were found around the bile canaliculi as well. Thus, we were not able to firmly distinguish the two structures from one and other. We believe the structures found in the untreated mice likely originate from breakdown products of hemoglobin or metabolized iron from the diet or an artifact of section staining. As described by Choi et al. there is a clear cutoff size for structures excreted by the renal system [30]. Considering the core size of FeO-HDL (10 nm) we are encouraged to hypothesize that the observations in the liver sections, of the mice injected with FeO-HDL, indeed are metabolized iron cores from FeO-HDL. To confirm the route of excretion of FeO-HDL we examined the bile and the feces with TEM. As anticipated we found iron oxide particles both within the bile (Fig. 5b, top, middle) and the feces (Fig. 5c, top, middle) of mice injected with FeO-HDL, while this was not the case for the bile (Fig. 5b, bottom) and feces (Fig. 5c, bottom) from untreated mice. This indicates the route of excretion of part of the dose, with these iron oxide cores being excreted in a similar manner to cholesterol, HDL’s natural cargo. To conclusively establish overall excretion a more quantitative approach will be pursued in further studies.
Fig. 5.

(a), top TEM of Kupffer cells 45 min after injection of FeO-HDL in an apoE KO mouse (N, Nucleus. E, erythrocyte) Arrows indicate endosomes containing iron oxide. (a), middle TEM of hepatocyte 45 min after injection of FeO-HDL in an apoE KO mouse. Numerous endosomes containing iron oxide around the bile canaliculi (BC) were found, as indicated by black arrows. (a), bottom TEM of hepatocytes of an untreated apoE KO mouse (M, mitochondria), arrows marks structures similar to the iron oxide containing endosomes (a) middle. b Iron oxide particles were found with TEM examinations of the bile from an apoE KO mouse injected with FeO-HDL (top, middle), while TEM of the bile from an untreated mice revealed no presence of iron oxide particles (bottom). (c) TEM of feces from apoE KO mice treated with FeO-HDL revealed the presence of iron oxide particles (top, middle), while these were not present in untreated mice (bottom).
4. Conclusions
In the present study we investigated the biological characteristics, and uptake mechanism of FeO-HDL, an iron oxide based high-density lipoprotein mimicking nanoparticle. We observed this HDL nanoparticle platform to behave very similarly to native HDL. Moreover, we showed the stability of FeO-HDL in circulation, its individual receptor-like interaction with cells and cholesterol efflux capacity, as well as the dynamic lipid exchange behavior in vivo. We propose FeO-HDL as a probe to investigate HDL biology and as an imaging agent or vector to deliver drugs, proteins, etc. in disease settings where HDL is key.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
Partial support provided by NIH grants R01 HL71021, R01 HL78667 and R01 EB009638 (ZAF) and R01 HL084312 (EAF). We thank the AHA Founder’s Affiliate for Postdoctoral Fellowship 09POST2220194 (DPC) and the Danish Heart Association for studentship 07-10-A1655-22406 (TS). We thank CSL Ltd, Parkville, Australia for their kind gift of apolipoprotein A-I. We would like to acknowledge the invaluable help and assistance of Heather Bell of the Mount Sinai Pathology EM core. Confocal microscopy was performed at the MSSM–Microscopy Shared Resource Facility and (supported by NIH–National Cancer Institute Grant 5R24 CA095823-04, National Science Foundation Grant DBI-9724504, and NIH Grant 1 S10 RR0 9145-01).
Appendix. Supplementary data
The supplementary data associated with this article can be found in the on-line version at doi:10.1016/j.biomaterials.2010.08.078.
Appendix
Figures with essential color discrimination. Figs. 1–4 in this article are difficult to interpret in black and white. The full color images can be found in the on-line version, at doi:10.1016/j.biomaterials.2010.08.078.
References
- 1.Mahley RW, Innerarity TL, Rall SC, Weisgraber KH. Plasma-lipoproteins-apolipoprotein structure and function. J Lipid Res. 1984;25(12):1277–94. [PubMed] [Google Scholar]
- 2.Feig JE, Shamir R, Fisher EA. Atheroprotective effects of HDL: beyond reverse cholesterol transport. Curr Drug Targ. 2008;9(3):196–203. doi: 10.2174/138945008783755557. [DOI] [PubMed] [Google Scholar]
- 3.Barter P, Kastelein J, Nunn A, Hobbs R. High density lipoproteins (HDLs) and atherosclerosis; the unanswered questions. Atherosclerosis. 2003;168(2):195–211. doi: 10.1016/s0021-9150(03)00006-6. [DOI] [PubMed] [Google Scholar]
- 4.Viles-Gonzalez JF, Fuster V, Corti R, Badimon JJ. Emerging importance of HDL cholesterol in developing high-risk coronary plaques in acute coronary syndromes. Curr Opin Cardiol. 2003;18(4):286–94. doi: 10.1097/00001573-200307000-00008. [DOI] [PubMed] [Google Scholar]
- 5.Li H-h, Lyles DS, Pan W, Alexander E, Thomas MJ, Sorci-Thomas MG. ApoA-I structure on discs and spheres. Variable helix registry and conformational states. J Biol Chem. 2002;277(42):39093–101. doi: 10.1074/jbc.M206770200. [DOI] [PubMed] [Google Scholar]
- 6.Datta G, Chaddha M, Hama S, Navab M, Fogelman AM, Garber DW, et al. Effects of increasing hydrophobicity on the physical-chemical and biological properties of a class A amphipathic helical peptide. J Lipid Res. 2001;42(7):1096–104. [PubMed] [Google Scholar]
- 7.Silva RAGD, Huang R, Morris J, Fang J, Gracheva EO, Ren G, et al. Structure of apolipoprotein A-I in spherical high density lipoproteins of different sizes. Proc Nat Acad Sci. 2008;105(34):12176–81. doi: 10.1073/pnas.0803626105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thomas MJ, Bhat S, Sorci-Thomas MG. Three-dimensional models of HDL apoA-I: implications for its assembly and function. J Lipid Res. 2008;49(9):1875–83. doi: 10.1194/jlr.R800010-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Segrest JP, Jones MK, Klon AE, Sheldahl CJ, Hellinger M, De Loof H, et al. A detailed molecular belt model for apolipoprotein A-I in discoidal high density lipoprotein. J Biol Chem. 1999;274(45):31755–8. doi: 10.1074/jbc.274.45.31755. [DOI] [PubMed] [Google Scholar]
- 10.Lusis AJ. Atherosclerosis. Nature. 2000;407(6801):233–41. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Florentin M, Liberopoulos EN, Wierzbicki AS, Mikhailidis DP. Multiple actions of high-density lipoprotein. Curr Opin Cardiol. 2008;23(4):370–8. doi: 10.1097/HCO.0b013e3283043806. [DOI] [PubMed] [Google Scholar]
- 12.Naghavi M, Libby P, Falk E, Casscells SW, Litovsky S, Rumberger J, et al. From vulnerable plaque to vulnerable patient-a call for new definitions and risk assessment strategies: part II. Circulation. 2003;108(15):1772–8. doi: 10.1161/01.CIR.0000087481.55887.C9. [DOI] [PubMed] [Google Scholar]
- 13.Tall AR. Cholesterol efflux pathways and other potential mechanisms involved in the athero-protective effect of high density lipoproteins. J Int Med. 2008;263(3):256–73. doi: 10.1111/j.1365-2796.2007.01898.x. [DOI] [PubMed] [Google Scholar]
- 14.Acton S, Rigotti A, Landschulz KT, Xu SZ, Hobbs HH, Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science. 1996;271(5248):518–20. doi: 10.1126/science.271.5248.518. [DOI] [PubMed] [Google Scholar]
- 15.Cormode DP, Skajaa T, van Schooneveld MM, Koole R, Jarzyna P, Lobatto ME, et al. Nanocrystal core high-density lipoproteins: a multimodality contrast agent platform. Nano Lett. 2008;8(11):3715–23. doi: 10.1021/nl801958b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Havel RJ, Eder HA, Bragdon JH. Distribution and chemical composition of ultracentrifugally separated lipoproteins in human serum. J Clin Invest. 1955;34(9):1345–53. doi: 10.1172/JCI103182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cormode DP, Briley-Saebo KC, Mulder WJM, Aguinaldo JGS, Barazza A, Ma Y, et al. An apoA-I mimetic peptide high-density-lipoprotein-based MRI contrast agent for atherosclerotic plaque composition detection. Small. 2008;4(9):1437–44. doi: 10.1002/smll.200701285. [DOI] [PubMed] [Google Scholar]
- 18.Rong JX, Kusunoki J, Oelkers P, Sturley SL, Fisher EA. Acyl-coenzymeA (CoA):cholesterol acyltransferase inhibition in rat and human aortic smooth muscle cells is nontoxic and retards foam cell formation. Arterioscler Thromb Vasc Biol. 2005;25(1):122–7. doi: 10.1161/01.ATV.0000148202.49842.3b. [DOI] [PubMed] [Google Scholar]
- 19.Forte TM, Nordhausen RW. Electron-microscopy of negatively stained lipoproteins. Meth Enzymol. 1986;128:442–57. doi: 10.1016/0076-6879(86)28086-6. [DOI] [PubMed] [Google Scholar]
- 20.Horak D, Babic M, Jendelova P, Herynek V, Trchova M, Pientka Z, et al. D-Mannose-modified iron oxide nanoparticles for stem cell labeling. Bioconjug Chem. 2007;18(3):635–44. doi: 10.1021/bc060186c. [DOI] [PubMed] [Google Scholar]
- 21.Bruns OT, Ittrich H, Peldschus K, Kaul MG, Tromsdorf UI, Lauterwasser J, et al. Real-time magnetic resonance imaging and quantification of lipoprotein metabolism in vivo using nanocrystals. Nat Nano. 2009;4(3):193–201. doi: 10.1038/nnano.2008.405. [DOI] [PubMed] [Google Scholar]
- 22.Thaxton CS, Daniel WL, Giljohann DA, Thomas AD, Mirkin CA. Templated spherical high density lipoprotein nanoparticles. J Am Chem Soc. 2009;131 (4):1384–5. doi: 10.1021/ja808856z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rothblat GH, de la Llera-Moya M, Atger V, Kellner-Weibel G, Williams DL, Phillips MC. Cell cholesterol efflux: integration of old and new observations provides new insights. J Lipid Res. 1999;40(5):781–96. [PubMed] [Google Scholar]
- 24.Remaley AT, Thomas F, Stonik JA, Demosky SJ, Bark SE, Neufeld EB, et al. Synthetic amphipathic helical peptides promote lipid efflux from cells by an ABCA1-dependent and an ABCA1-independent pathway. J Lipid Res. 2003;44 (4):828–36. doi: 10.1194/jlr.M200475-JLR200. [DOI] [PubMed] [Google Scholar]
- 25.Davidson WS, Lund-Katz S, Johnson WJ, Anantharamaiah GM, Palgunachari MN, Segrest JP, et al. The influence of apolipoprotein structure on the efflux of cellular free cholesterol to high density lipoprotein. J Biol Chem. 1994;269(37):22975–82. [PubMed] [Google Scholar]
- 26.Jonas A. Interaction of bovine serum high density lipoprotein with mixed vesicles of phosphatidylcholine and cholesterol. J Lipid Res. 1979;20(7):817–24. [PubMed] [Google Scholar]
- 27.Breslow JL. Mouse models of atherosclerosis. Science. 1996;272:685–8. doi: 10.1126/science.272.5262.685. [DOI] [PubMed] [Google Scholar]
- 28.Zadelaar S, Kleemann R, Verschuren L, de Vries-Van der Weij J, van der Hoorn J, Princen HM, et al. Mouse models for atherosclerosis and pharmaceutical modifiers. Arterioscler Thromb Vasc Biol. 2007;27(8):1706–21. doi: 10.1161/ATVBAHA.107.142570. [DOI] [PubMed] [Google Scholar]
- 29.Sanhai WR, Sakamoto JH, Canady R, Ferrari M. Seven challenges for nanomedicine. Nat Nano. 2008;3(5):242–4. doi: 10.1038/nnano.2008.114. [DOI] [PubMed] [Google Scholar]
- 30.Choi HS, Liu W, Misra P, Tanaka E, Zimmer JP, Ipe BI, et al. Renal clearance of quantum dots. Nat Biotech. 2007;25:1165–70. doi: 10.1038/nbt1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.