

Abstract
The pharmacology and regulation of Transient Receptor Potential Ankyrin 1 (TRPA1) ion channel activity is intricate due to the physiological function as an integrator of multiple chemical, mechanical, and temperature stimuli as well as differences in species pharmacology. In this study, we describe and compare the current inhibition efficacy of human TRPA1 on three different TRPA1 antagonists. We used a homology model of TRPA1 based on Kv1.2 to select pore vestibule residues available for interaction with ligands entering the vestibule. Site-directed mutation constructs were expressed in Xenopus oocytes and their functionality and pharmacology assessed to support and improve our homology model. Based on the functional pharmacology results we propose an antagonist-binding site in the vestibule of the TRPA1 ion channel. We use the results to describe the proposed intravestibular ligand-binding site in TRPA1 in detail. Based on the single site substitutions, we designed a human TRPA1 receptor by substituting several residues in the vestibule and adjacent regions from the rat receptor to address and explain observed species pharmacology differences. In parallel, the lack of effect on HC-030031 inhibition by the vestibule substitutions suggests that this molecule interacts with TRPA1 via a binding site not situated in the vestibule.
Introduction
The nonselective cation channel TRPA1 is the single member of the ankyrin repeat-rich branch of the transient receptor potential (TRP) channel family comprising 28 mammalian subunit genes (1). TRPA1 is expressed in peripheral and central termini of small diameter primary afferent neurons and the ganglia of these dorsal, trigeminal, and nodose neurons (2,3). Here it integrates the nociception of a large variety of different, potentially damaging and noxious stimuli: cold (3–5), electrophilic compounds (2,6,7), divalent ions (8,9), and mechanical stimulation (10). The involvement of TRPA1 in noceptive transduction and neurogenic inflammation (11,12) is supported by knockout experiments in mice (13,14) and a human gain-of-function mutation in TRPA1, which causes familial episodic pain syndrome (15). Despite the convincing target validation of TRPA1 as a target for analgesic therapy, there are only a few de novo designed TRPA1 antagonists described in literature and patents (seven different institutional applicants), and no program has started Phase II recruitment yet (according to www.clinicaltrial.gov).
TRPA1 activity is regulated by divalent cations, such as Ca2+ that interact with the EF-hand motive in the N-terminal region, while Zn2+ binds to single residues (cysteine and histidine) at both the N- and the C-terminal (8,9). The influx of Ca2+ ions through the TRPA1 ion channel increases the open channel probability (9) and this amplification by Ca2+ is followed by an equally Ca2+-dependent desensitization of TRPA1 (16). Site-directed mutagenesis studies have revealed the N-terminal segment as an important interaction site for electrophilic compounds, which covalently binds to cysteine or lysine residues in this region (17,18). Several studies have shown the species specific pharmacology between rodent and human TRPA1 by new antagonists (19,20) and recently a more complete species comparison has been published by Bianchi et al. (21). Chen et al. (19) show by substitution studies that their electrophilic, thioaminal-containing compounds, which covalently bind to the upper S6 domain of TRPA1, block human TRPA1 but activate rat TRPA1. These structurally analogous compounds are believed to interact with only a few residues in the S6 transmembrane domain: S943 and I946 in the human receptor, and the corresponding residues A946 and M949 in rats.
The functional TRPA1 ion channel is, like many other TRP family members, a homotetramer with fourfold symmetry around a central ion-conducting pore (22). Each subunit is suggested to have six transmembrane (S1–S6) segments with intracellular N- and C-termini and the S5 and S6 segments lining the pore (23,24). This overall topology is similar to the general potassium channel architecture based on multiple bacterial potassium channel structures (25). Support for the hypothesis of S5 and S6 lining the pore can also be drawn from high sequence similarity in S5 and S6 between different TRP channels (26).
The multiple potentiation and activation/inactivation modes of TRPA1 pose a challenge to the interpretation of mutagenesis-based structure function studies given the interdependencies of the various stimuli. Further, the peculiar rodent to human pharmacology species differences pose problems for drug discovery programs when trying to translate preclinical analgesia models to human pain conditions. At the same time, however, these species differences can yield clues to the ligand-regulated activity of TRPA1 and identify putative drugable binding sites. Therefore, drug discovery and structure-function studies on TRPA1 must go hand in hand.
In this article, we profile three structurally diverse TRPA1 antagonist compounds: the novel TRPA1 antagonist AZ868 (Compound 16 in Vallin et al. (27)) and the previously described antagonists A-967079 (28) and HC-030031 (11,29) on the wild-type human TRPA1 receptor, heterologously expressed in Xenopus oocytes using the two-electrode voltage-clamp method. A homology model of hTRPA1 was created using the atomic structure with an open pore activation gate of Kv1.2/Kv2.1 chimera (25) as structural scaffold. Information from the homology model was used to select putative compound interacting amino residues, spanning the upper S6 segment and the lower part of the pore mouth. Site-directed mutagenesis studies replaced the amino residues in hTRPA1 either to alanine (which changed the functionality of an amino-acid side chain) or to the corresponding set of amino residues in rTRPA1. The different TRPA1 constructs were then analyzed for functionality and pharmacology and compared with the wild-type hTRPA1 receptor. Based on the functional results and the final iteration of our homology model, we propose and describe a ligand binding site in the pore vestibule of hTRPA1.
Materials and Methods
Molecular biology
The hTRPA1 coding determining sequence (of NM_007332) was subcloned into an oocyte transcription vector containing the 5′ and 3′ untranslated regions of the Xenopus β-globin gene. hTRPA1 mutants were generated (M911A, M912A, M911A-M912A, Q940A, L941A, S943A, F944A, T945A, I946M, F947A, V948A, and I950F) using cassettes of synthetic DNA fragments (GeneArt, Regensburg, DE). Similarly, in an additional mutant, amino acids were substituted to the rat equivalents, respectively (S900T, I905L, E920D, S921A, Y926L, L927F, H933Y, S937T, A939G, V942I, S943A, I946M, and S985N). The constructs were sequenced, linearized, and in vitro transcribed (mMESSAGE mMACHINE T7 Kit; Ambion, Austin, TX). Gel electrophoresis and UV-spectrophotometry were used to confirm high purity and integrity of the mRNA preparations.
Solutions and compounds
Buffers used for two-electrode voltage-clamp experiments were ND96 (NaCl 96 mM, CaCl2 2.4 mM, KCl 2.0 mM, MgCl2 1.0 mM and HEPES 5.0 mM), ND96 Ca2+-free (ND96 without CaCl2), and ND96, 150 μM Ca2+ (ND96 with the CaCl2 concentration reduced to 150 μM). The pH of the buffers was adjusted to 7.5 with NaOH and the solutions were sterile-filtered before use. Compounds were diluted from 10 mM DMSO stocks. Zn2+ and compounds to be tested were diluted from stock solutions in ND96 Ca2+-free, and then loaded on a 96-well compound plate. The samples were administered at a volume of 950 μL. For IonWorks (Molecular Devices, Sunnyvale, CA) current recordings, we used an intracellular solution (100 mM K-gluconate, 40 mM KCl, 3.2 mM MgCl2, 5 mM HEPES, and 3 mM EGTA pH 7.2, osmolarity: 280 mOsmol) and assay buffer (DPBS buffer supplemented with 0.55 mM D-Glucose and 0.05 mM MgCl2).
AZ868 (Compound 16 in Vallin et al. (27)) was synthesized as previously described in Vallin et al. (27). A-967079 was synthesized in two steps:
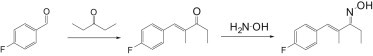
Step 1: (E)-1-(4-fluorophenyl)-2-methylpent-1-en-3-one
A solution of 4-fluorobenzaldehyde (1.70 mL, 16.1 mmol) and 3-pentanone (1.70 mL, 16.1 mmol) in 37% hydrochloric acid (4.0 mL) was heated to reflux for 3 h. After cooling to ambient temperature, the mixture was extracted with ether. The organic layer was washed with water, aqueous saturated sodium bicarbonate solution, and again with water. After drying over Na2SO4, the solution was concentrated. The product was purified using column chromatography (silica gel with an N-heptan/EtOAc gradient) The resulting product still contained impurities and was recrystallized from methanol/water 1.5:1 to yield 1.40 g (45%) E)-1-(4-fluorophenyl)-2-methylpent-1-en-3-one. 1H NMR (500 MHz, DMSO-d6) d ppm 1.03 (t, J = 7.25 Hz, 3 H), 1.96 (d, J = 1.26 Hz, 3 H), 2.87 (q, J = 7.25 Hz, 2 H), 7.29 (t, J = 8.83 Hz, 2 H), 7.54–7.61 (m, 2 H), and 7.65 (s, 1 H). MS: [M+H]+ = 193.2.
Step 2: (1E,3E)-1-(4-fluorophenyl)-2-methylpent-1-en-3-one oxime
Hydroxylamine hydrochloride (0.65 g, 9.4 mmol) was added in one portion to a solution of (E)-1-(4-fluorophenyl)-2-methylpent-1-en-3-one (1.2 g, 6.2 mmol) in pyridine (15 mL). The obtained reaction mixture was stirred at ambient temperature for 2 h. The solvent was evaporated and the residue partitioned between 1 M aqueous hydrochloric acid and ethyl acetate. The organic phase was washed with water, dried over Na2SO4, filtered, and concentrated. Two products with the same mass according to LC/MS were observed. The product mixture was purified using column chromatography (silica gel, elution with N-heptan/EtAOAc gradient) and yielded 1.03 g (79%) of the pure major product. 1H NMR (500 MHz, DMSO-d6) d ppm 1.04 (t, J = 7.57 Hz, 3 H), 1.98 (s, 3 H), 2.60 (q, J = 7.57 Hz, 2 H), 6.89 (s, 1 H), 7.15–7.27 (m, 2 H), 7.36–7.50 (m, 2 H), and 11.08 (s, 1 H). MS: [M+H]+ 208.1.
HC-030031 was purchased from TOSlab (Ekaterinburg, Russia). All solids were dissolved in DMSO, with a final concentration of 10 mM.
Xenopus oocyte preparation
Stage V-IV Xenopus laevis oocytes were obtained defolliculated and ready for injection from Ecocyte Bioscience (Castrop-Rauxel, Germany). The oocytes were injected with 3 ng mRNA encoding wild-type or mutant TRPA1 and then incubated at 18°C in ND96 supplemented with gentamicin (80 μg/mL), penicillin (100 U/mL), and streptomycin (100 μg/mL). Experiments were carried out 3–6 days after injection.
Two-electrode voltage-clamp
Experiments were performed using OpusXpress 6000A (Molecular Devices), a semiautomated system that uses the two-electrode voltage-clamp technique to record whole-cell currents from eight Xenopus oocytes in parallel. Glass electrodes were pulled from borosilicate glass capillaries with inner filament (TW150F-6; World Precision Instruments, Stevenage, UK) using the electrode puller PP-83 (Narishige, London, UK) and then filled with 3 M KCl solution. When immersed in perfusion buffer, the resistance of the current electrode measured 0.4–2 MΩ and the voltage electrode 0.4–10 MΩ. Oocytes were placed separately in the recording chambers with a volume of 150 μL and continuously perfused with ND96.
The oocytes were impaled and then clamped at a holding voltage of – 60 mV (voltage-clamp gain, 2000; voltage-clamp lag, 220 μs; output gain, 1; filter, 100 Hz) before starting the protocol. Data series with Zn2+ responses larger than 2 μA or smaller than 0.15 μA were rejected, because large currents caused unstable responses and currents smaller than 0.15 μA had too low amplitude/noise ratio for reproducible assessment of antagonist efficacy. To quantify the effect of the antagonists on the current amplitude elicited by Zn2+, the current amplitude in the presence of antagonist was normalized to the current amplitude in the absence of antagonist. To get a stable and reproducible agonist response, Zn2+ activation was repeated three times before coaddition of Zn2+ and antagonist, followed by at least two repeated Zn2+ additions without antagonist to monitor current amplitude stability and reversibility of the tested compounds.
IonWorks QT System (Molecular Dynamics)
Whole-cell currents from CHO cells stably expressing human TRPA1 were measured on the IonWorks QT System (Molecular Devices). Fresh cell suspension was prepared immediately before use. Cells were detached and resuspended in 6–8 mL assay buffer. The final cell suspension volume was adjusted to ∼2,000,000 cells. Preweighed 25–27 mg samples of amphotericin B were dissolved in 0.58 mL of 100% DMSO and further diluted in 175 mL of intracellular solution to a final concentration of 0.1 mg/mL. Test compounds were serial-diluted in 100% DMSO and subsequently diluted in assay buffer to a final concentration between 25 μM and 7.9 nM in the patch plate. Currents were activated by 125 μM cinnamaldehyde. The final DMSO concentration did not exceed 0.4%. During experiments, the electronic head and fluidic head of the IonWorks unit were washed with DPBS between compound additions. Agonist first and then test compound were applied in a dual application protocol. A depolarizing ramp voltage protocol from −80 mV to +100 mV (500 ms) was applied before agonist addition (Prescan), after agonist addition (Postscan1), and after compound addition (Postscan2). Agonist activation time was 100 s, compound incubation time 60 s.
Data analysis
The variable studied in the two-electrode voltage-clamp recordings was the maximum peak amplitude of the current responses measured in μA. Leak subtraction was performed in the software CLAMPFIT 9.2 (Molecular Devices) before the data were transferred to the software GRAPHPAD PRISM 5 (GraphPad Software, La Jolla, CA) for further analysis. Unless otherwise indicated, calculated data for each compound are presented as mean blocking effect (%) mean ± SE. For the IonWorks experiments, the metric was set to find the mean peak current at −80 and +100 mV, respectively, in the voltage ramp. The mean offset current at 0 mV was subtracted from the mean peak currents yielding the absolute difference. The raw metric values at −80 and +100 mV were used to calculate % inhibitory effect at each compound concentration ratio as follows:
For concentration-response curves, data were analyzed by nonlinear regression analysis with variable slope using GRAPHPAD PRISM ver. 6.0 for Windows (GraphPad Software) using the equation
Statistical significance was evaluated using a one-way ANOVA nonparametric Krauskal-Wallis test, and p values < 0.05 (95% confidence level) were considered statistically significant.
Homology models
The amino-acid sequences of human and rat TRPA1 were retrieved from the SWISS-PROT Protein Sequence Data Bank (entries O75762 and Q6RI86). The Kv1.2 x-ray structure used as a template in modeling was retrieved from the Protein Data Bank (PDB:2R9R). The sequence alignment was performed in the software PRIME (Ver. 3.0; Schrödinger, New York, NY) with subsequent manual adjustments. The homology modeling and energy minimization were performed using MAESTRO molecular modeling software (ver. 9.2; Schrödinger). Energy minimization procedure was performed in a stepwise fashion and performed both with and without ligand present in the pore region. The calculations were performed ignoring solvent interactions, and the force field used was MMFF as implemented in MAESTRO. Homology modeling was performed using the PRIME module in MAESTRO. Rotamers from conserved residues were retained whereas other residues were built and energy-minimized. Evaluation of the model was performed by Ramachandran plot and protein reports from MAESTRO. Visualization and characterization of the pore region binding site was done using the SiteMap module in MAESTRO (SiteMap ver. 2.5; Schrödinger). Docking of ligands to the channel pore region was performed with the GLIDE module (ver. 5.7; Schrödinger). Selection of most possible ligand pose was done by a combination of docking score results and visual inspection.
Results
To examine which TRPA1 residues that are of importance for ligand binding, we selected amino-acid substitutions based on several initially prepared homology models. The homology models of the rat TRPA1 and human TRPA1 were constructed with a Kv1.2 structure as a template. Alignment and models were then revised according to the results of the mutation studies. The resulting alignment shows that Kv1.2 and TRPA1 share a sequence identity of only 11% in the S5–S6 region. Several possible alignments have been proposed (19,30), but referring to our substitution results, we suggest an alignment as shown (Fig. 1). After building the final rat and human homology models conforming to the highest degree with the mutation experiments, we energy-minimized those in a stepwise fashion. The overall conformations of the structures were maintained properly during this procedure. There were no obvious differences between structures with ligand docked before or after energy minimization, suggesting that the model pore region is stable.
Figure 1.
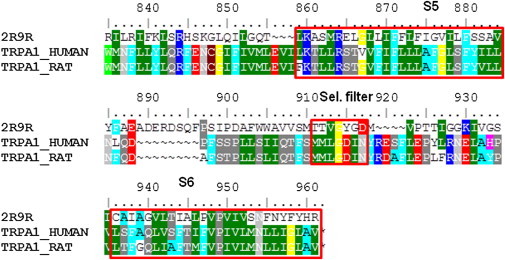
Sequence alignment of Kv1.2 (2R9R) with human and rat TRPA1. (Shading is according to similarity; amino acids with similar properties have identical tone.)
AZ868 and A-967079 are docked in the human TRPA1 model, showing possible interactions with side chains of S6 amino-acid residues and residues corresponding to the selectivity filter (Fig. 2). For clarity, only two of the four subunits are displayed, and only the substituted residues are included. Even though the sequence identities are quite high between species (∼80% between human and rat), several studies and observations in-house suggest that TRPA1 pharmacology is not conserved between species (21). Specifically, a number of nonconserved residues have been suggested important for activation and inhibition pharmacology (19,20).
Figure 2.
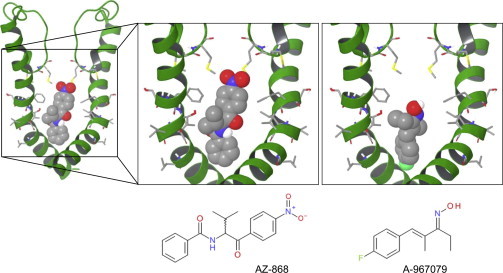
Docking poses of AZ868 and A-967079 in the pore region of TRPA1 according to the homology model prediction. Structures of AZ868 and A-967079 are shown in the panels below the docking poses.
To test the homology model and examine the importance of specific amino acids for ligand binding in the proposed S6 region and selectivity filter, we constructed a number of hTRPA1 constructs with one or more amino-acid substitutions (see Fig. 3 and the Materials and Methods for a complete list of mutations made). To explore the putative species difference, we also constructed a rat TRPA1 homology model from the human TRPA1 model. We then substituted all residues which differed between the rat and human receptor to construct a human receptor with the differing rat residues in the S5–S6 linker, the selectivity filter, and S6 (Fig. 3, right).
Figure 3.
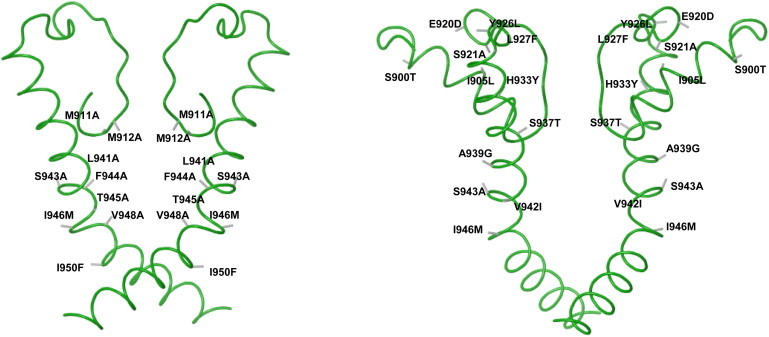
(Left) Illustrating the selectivity filter, S5–S6 linker, and S6 fold according to the homology model. The single and double substitutions studied are shown at predicted positions on the backbone fold (solid strand). (Right) Illustrating the complete construct with the 12 rat residues substituted in the human receptor replacing human residues with the corresponding rat residues at predicted positions according to the homology model.
To assess the pharmacological effect of the substitutions (shown in Fig. 3), we developed a functional assay using two-electrode voltage-clamping of Xenopus oocytes injected with mRNA coding for the substituted and wild-type TRPA1 channels. We used Zn2+ as agonist, as it has been shown to be potent and activate TRPA1 channels by a noncovalent and fully reversible fashion (8). Further Zn2+ alone does not evoke currents in uninjected oocytes (L. Eisele, G. Klement, and M. Dabrowski, unpublished). To avoid desensitization of the TRPA1 current, Ca2+ was excluded during antagonist application and from the wash buffer used for perfusion immediately before and after antagonist application. However, between ligand additions, the oocytes were perfused with a Ca2+-containing buffer (ND96, 150 μM Ca2+), because a complete exclusion of Ca2+ proved to cause unstable current responses and rapidly deteriorating oocyte integrity. The complete assay protocol, with details of perfusion and buffers, is illustrated in Fig. 4, A and B.
Figure 4.
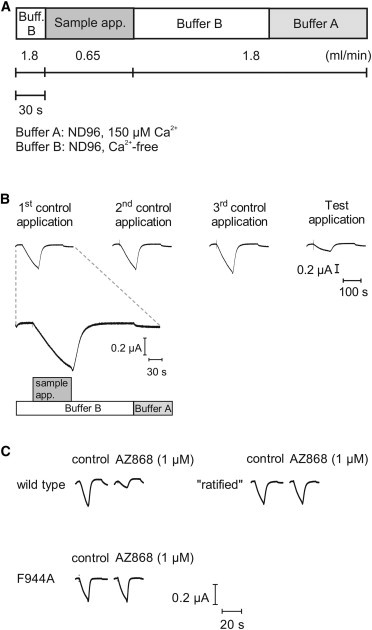
(A) One application cycle of the assay illustrating type of application: buffer type or sample. Flow rate and length of application as indicated (horizontal bars). (B) Examples of corresponding current traces from an application cycle and a test application using an antagonist. First application scale showing buffer changes as in panel A. (Vertical scale bar) Current amplitude. (Horizontal bar) Timescale for all traces. (C) Example current traces from control and test application obtained using the application cycle shown in panels A and B. The current traces are recorded from, and comparing, TRPA1 WT; rat residues substituted in to the human TRPA1 receptor, ratified; and the TRPA1 F944A substitution in control and with 1 μM AZ868 applied as indicated above the traces. The scale bars show current amplitude and timecourse for all three receptor constructs.
During characterization of the substituted and wild-type channels, we noted that assay stability was compromised by current amplitudes above 1 μA. Despite the careful application protocol, avoiding Ca2+ during agonist presence, it was not possible to obtain concentration response curves for Zn2+ due to desensitization and destabilization of the oocytes seen at larger current amplitudes. Thus, in lieu of using a fixed Zn2+ concentration based on a concentration response current activation curve, the Zn2+ concentration was adjusted in each experiment to suit the level of expression, oocyte condition, and the difference in Zn2+ sensitivity between substituted channels. Typical Zn2+ concentration ranged between 2 and 7 μM. Representative current traces from wild-type and substituted TRPA1 channels are shown in Fig. 4 C.
Interestingly all substituted channels proved to be functional, as current responses were evoked by application of Zn2+. However, some of the substituted constructs yielded altered characteristics of the ion channel with respect to current amplitudes, Ca2+ sensitivity, and background activity (as determined by leak current at −60 mV). Two substitutions, M911A-M912A and F944A, resulted in noticeably smaller current responses and a lower background activity compared to wild-type channels. Oocytes expressing L941A or I950F substituted channels displayed markedly larger currents upon Zn2+ activation and more depolarized resting membrane potentials compared to wild-type, suggesting an increased background activity of the substituted channels. L941A- and I950F-expressing oocytes also responded strongly to Ca2+ in the wash buffer. The rat-substituted construct (S900T, I905L, E920D, S921A, Y926L, L927F, H933Y, S937T, A939G, V942I, S943A, I946M, and S985N), together with six of the other substitutions (M911A, Q940A, S943A, T945A, I946M, and V948A), did not seem to affect the channel properties in these respects.
The profile of the three tested TRPA1 antagonists on substituted and human wild-type TRPA1 receptors are shown in Fig. 5. Results are expressed as % remaining current normalized to the control activation by Zn2+ alone. The concentration of the three TRPA1 antagonists was guided by their potencies obtained in the IonWorks assay (Table 1). We selected concentrations slightly higher than the IC50 values to generate a large but not complete inhibition percentage on the wild-type channel (60–80%). For clarity and statistical significance testing, the results from the oocyte experiments are summarized in Table 2. It is evident that the efficacy of AZ868 is largely dependent on the presence of the methionines at position 911 and 912 in contrast to the A-967079 and HC-030031 compounds.
Figure 5.
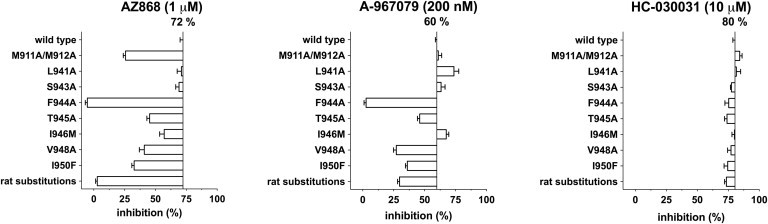
Box plot of the mean inhibition percentage of the maximal current amplitude for AZ868 (1 μM), A-967079 (0.2 μM), and HC-030031 (10 μM) as indicated above each panel. The TRPA1 construct type is listed on the y axis and the level of inhibition scale is shown on the x axis. Wild-type inhibition percentage is written above the vertical line and the inhibition percentage on the individual substitutions is shown as boxes drawn from the WT level. n = 2–16 (see Table 2 for tabulated list of inhibition and significance).
Table 1.
Potencies of the tested compounds measured on IonWorks (Molecular Dynamics) current-clamped CHO cells expressing human TRPA1 preactivated by 125 μM cinnamaldehyde
| CHO cell | AZ868 | A-967079 | HC-030031 |
|---|---|---|---|
| Clamped @ −80 mV | 0.7 ± 0.2 | 0.08 ± 0.02 | 6.7 ± 1.6 |
| Clamped @ +100 mV | 0.9 ± 0.2 | 0.1 ± 0.02 | 8.1 ± 0.6 |
Half-maximum inhibition values (IC50) in μM. Data represents mean ± SE for n = 4–20 concentration response curves.
Table 2.
Inhibition of the maximal current amplitude of the three compounds tested on all substitution constructs made
| Constructs | AZ868 (1 μM) | A-967079 (0.2 μM) | HC-030031 (10 μM) |
|---|---|---|---|
| Wild-type | 72 ± 2 (n = 7) | 60 ± 1 (n = 12) | 80 ± 2 (n = 14)) |
| M911A/M912A | 26 ± 2a (n = 10) | 61 ± 3 (n = 5) | 84 ± 2 (n = 10) |
| L941A | 71 ± 3 (n = 5) | 74 ± 4 (n = 3) | 81 ± 4 (n = 2) |
| S943A | 69 ± 3 (n = 10) | 63 ± 3 (n = 7) | 78 ± 1 (n = 6) |
| F944A | −5 ± 2a (n = 8) | 3 ± 2a (n = 16) | 75 ± 3 (n = 11) |
| T945A | 45 ± 2 (n = 8) | 46 ± 2 (n = 12) | 74 ± 2 (n = 10) |
| I946M | 57 ± 4 (n = 10) | 67 ± 2 (n = 7) | 81 ± 2 (n = 5) |
| V948A | 41 ± 4b (n = 11) | 27 ± 2c (n = 10) | 77 ± 3 (n = 15) |
| I950F | 33 ± 2a (n = 13) | 36 ± 1 (n = 5) | 75 ± 3 (n = 16) |
| Rat substitutions | 3 ± 1a (n = 5) | 30 ± 2c (n = 11) | 73 ± 1 (n = 9) |
Values are mean ± SE and statistical significance is based on a one-way ANOVA test versus the WT inhibition and significance is given by the p values as stated.
p < 0.001.
p = 0.05–0.01.
p = 0.01–0.001.
Reducing the size of the amino-acid side chain by replacing methionine with the smaller side-chain alanine is less efficacious in coordinating and stabilizing the AZ868 molecule in the vestibule. The rest of the pattern on the substituted channels is mostly shared between A-967079 and AZ868 except for the L941A and I946M substitutions. L941A has no effect on AZ868 inhibition but increase the inhibition efficacy by A-967079 slightly. I946M also improves A-967079 inhibition but reduces the inhibition by AZ868. All tested substitutions were without significant effect on the HC-030031 inhibition efficacy. The largest effect is seen when we remove the benzene ring of F944, completely abolishing both AZ868 and A-967079 inhibition of TRPA1 but having no effect on HC-030031 inhibition.
Interestingly, some effect is seen by substituting the two neighboring residues at positions 945 and 946 as well. However, the reduction in inhibition was consistent on both AZ868 and A-967079 but not significant when tested in one-way ANOVA. Thus, we observe effect on AZ868 and A-967079 interaction by primarily the side-chain positions of F944, suggesting access to this position from the inner vestibule. The last two substitutions, V948A and I950F, affect AZ868 and A-967079 inhibition in a similar fashion, reducing the TRPA1 current inhibition by half to 30–40% of the control response.
The observation that HC-030031 inhibition is only marginally affected by all the substitutions tested leads us to suggest that this compound inhibits TRPA1 by interacting with a different site than AZ868 and A-967079. Finally, we tested the set of 12 combined substitutions replacing the human TRPA1 receptor residue with the corresponding residue from the rat receptor in the proposed extracellular loop before S6 and along the pore segment of S6. This rat substitution construct was functionally like the other described substituted channels, but efficacy of AZ868 current inhibition was completely abolished, verifying the lack of species translation for this compound. Even A-967079 inhibition efficacy was markedly reduced on the rat substitution construct, whereas the HC-030031 effect was unaltered.
Homology models of rat and human TRPA1 were constructed with the Kv1.2 structure as a template. Whereas the sequence identities between the TRP ion channel family sequences allow for a quite straightforward alignment, the identity between Kv1.2 and TRPA1 is only 11% in the S5-S6 region, enabling several different and initially equally good suggestions for the alignment. However, referring to our substitution results reported here, we propose the final alignment as shown in Fig. 1. In favor of this alignment is the preservation of the positions of P949 and N954 relative to the template sequence as well as the results from the substitution functionality test. Interestingly, our homology model can only dock the active enatiomer of the racemic compound AZ868 ((27) and M. Svensson, G. Terp, and D. Weigelt, unpublished), further supporting the homology based on the alignment in Fig. 1.
Discussion
Substitution of individual amino-acid residues can directly affect the drug binding by changing the affinity due to changes in the available acceptor and donor pattern or even larger structural/geometric changes of the binding site. It could also indicate an interference of the access pathway of the compound, an allosteric effect, or a modification of the channel gating. In this study, we assess the macroscopic whole-cell current amplitudes and can thus not dissect the individual contributions to the efficacy of the antagonists. However, by the methodic substitutions and comparison with the proposed homology model using three different ligands, we argue that a ligand binding site exits in the vestibule of the channel pore of TRPA1.
By carefully adjusting assay conditions to optimize stability, avoid desensitization and other nonequilibrium artifacts, and by using a reversible ligand, we conclude that the differential observations of the compound characteristics on the mutant and wild-type receptors are indeed consequences of changes in direct interaction with the specific residues in the pore vestibule of the TRPA1 channel. The limitation of this argument is that we model a static picture of a pore region that we know is very flexible. Until crystal structures of TRP channels are solved, we are limited to this interpretation on how the ligands interact in the pore region, being in best agreement with our mutation results.
The AstraZeneca proprietary compound AZ868 shows an effect of the M911A and M912A substitutions in contrast to the lack of effect on A-967079 inhibition. According to our homology model, these methionines are positioned close to the selectivity filter and can be reached by AZ868. The SiteMap module (Schrödinger) evaluation, aiming to describe the size and properties of the pore binding site, shows possibilities for more elongated ligands to bind in the pore (exemplified by the AZ868 structure). Our results show that a hydrophobic part of the AZ868 structure interacts with the top of the inner vestibule defined by the methionines at positions 911 and 912. This is in accordance with the SiteMap results, showing a preference for hydrophobicity at this position. Extending the length of the A-967079 ligand and thereby allowing the structure to pick up the methionine interaction could possibly cause an increase in current inhibition efficacy.
The single most dramatic effect on current inhibition is seen by removing the benzene ring of F944. Substitution to the smaller alanine completely abolishes A-967079 and AZ868 current inhibition of TRPA1. Our hypothesis is that the channel adapts to ligand binding, leading to π-π stacking of the aromatic rings. In addition, the adaption to ligand binding could enable a hydrogen bond between the ligand and the threonine side chain. Results from the substitution study can then be explained by A-967079 and AZ868 interacting with both T945 and F944 as visualized in Fig. 3. The observation that lengthening the side chain from isoleucine to methionine at position 946 produces opposite effects between AZ868 and A-967079, is interesting; it also indicates that the vestibule segment of S6 will adapt to ligand binding. Similarly, the lack of effect by AZ868 and the modest increase in efficacy of A-967079 on L941A, further strengthen the hypothesis of smaller ligand induced structural changes in the vestibule.
The effects on V948A and I950F are similar between AZ868 and A-967079, suggesting a similar interaction pattern by these ligands in this end of the vestibule. We also confirm the human species selectivity of AZ868 is defined by the S6 residues because substitution of the rat residues into the human receptor results in complete abolishment of inhibition. In comparison, the reduction in inhibition efficacy for A-967079 is only 50% (from 60 to 30%), which suggests that A-967079 binds similarly, but not exactly like AZ868 to this set of residues and does indeed retain some activity in rats. Finally, we only observe a very limited effect on the inhibition by HC-030031 suggesting a good species crossover by this compound and entirely different binding mode compared to A-967079 and AZ868. As none of the substitutions influence the binding of HC-030031, we conclude that this ligand does not bind to TRPA1 in the pore region, whereas AZ868 and A-967079 efficacy are determined by direct interactions with amino acids in the pore vestibule.
The multiple modes of regulation of TRPA1 activity pose a challenge for structure-function studies in general and antagonist ligand interaction in particular because antagonist potency varies pending the mode of activation. Therefore, when comparing inhibitors of TRPA1 activity, it is necessary to weigh-in the mode of activation. The S6 region was identified as critical for AITC and voltage gating of TRPA1 using site-directed mutagenesis and a homology model based on an alignment somewhat different than ours (30). The alignment is shifted by nine residues relative to our alignment, and the effect of this is that other residues are predicted to be exposed to the pore, and thereby suggested available for ligand interactions (30).
Menthol activates human TRPA1 as a full agonist but is only a partial agonist on murine TRPA1. By comparing menthol efficacy on chimeras between human and murine TRPA1 receptors, Xiao et al. (31) find that S5 is critical in determining the modulation of TRPA1 by menthol and several other modulators. One should, however, be careful when directly comparing molecular ligand- and Zn2+ activated pharmacology on TRPA1 for antagonists dependent on the pore regions. These results and ours further strengthen the suggestion of a flexible pore region but also the intricate interplay between molecular agonists and antagonist pharmacology.
In an elegant series of experiments using de novo designed TRPA1 antagonists, Chen et al. (19) identified a number of residues in the S5–S6 linker and the S6 region as determining the rat/human species difference, site, and type of effect (activation/inhibition). When comparing these data to our findings and homology model, they are somewhat different, particularly the S943 and, to some degree, I946 results. It is important to note the difference in activation type: covalent binding at cysteine residues versus Zn2+ charge-charge interaction and the different functional readout and Ca2+ influx in nonvoltage-controlled cells versus voltage-clamp current recordings. Finally, the Chen study also employs a different set of antagonists than ours in comparing three structurally different antagonists.
However, it is certainly interesting and somewhat surprising that the S943 and I946 substitutions abolishes inhibition by the profiled antagonist CMP1 (19), whereas we observe no change for A-967079 and AZ868 and only modest changes of the AZ868 and A-967079 inhibition on the I946M channel (Fig. 5). This could reflect the different experimental conditions as stated above, or the fact that different ligands induce different fits in the flexible pore vestibule. We favor the interpretation that all these residues can form part of a somewhat flexible binding site in the pore vestibule. A further observation supporting a flexible ligand binding site hypothesis is the reversible pore dilation of TRPA1 under prolonged activation observed by two groups (32,33). Both groups show that long AITC applications (tens of seconds) abolish rectification and uptake of the 629-Da large organic cationic dyes YoPro in a concentration-dependent fashion. This further stresses a high degree of structural flexibility in the extracellular loop between S5 and S6, and the selectivity filter for S5 and S6.
Conclusion
We propose the existence of a drugable binding site in the pore of TRPA1 ion channel vestibule defined by two consecutive methionines M911 and M912 from the inner mouth of the selectivity filter and a stretch of residues in S6 from L941 to I950. If compounds, which can enter the vestibule and pick up several of these proposed interactions, can be designed and synthesized, then it may be possible to design TRPA1 antagonists with high affinity and selectivity for the human TRPA1 ion channel. Such ligands hold the promise of becoming a new analgesic treatment in man.
Acknowledgments
We thank Fernando Sehgelmeble for the synthesis of A-967079. Further, we thank Zara Sands for inspiring us to develop this TRPA1 homology model.
We thank Åsa Malmberg, who sponsored the AstraZeneca Global Ion Channel initiative enabling these experiments.
References
- 1.Nilius B., Owsianik G. The transient receptor potential family of ion channels. Genome Biol. 2011;12:218. doi: 10.1186/gb-2011-12-3-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bandell M., Story G.M., Patapoutian A. Noxious cold ion channel TRPA1 is activated by pungent compounds and bradykinin. Neuron. 2004;41:849–857. doi: 10.1016/s0896-6273(04)00150-3. [DOI] [PubMed] [Google Scholar]
- 3.Story G.M., Peier A.M., Patapoutian A. ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell. 2003;112:819–829. doi: 10.1016/s0092-8674(03)00158-2. [DOI] [PubMed] [Google Scholar]
- 4.Fajardo O., Meseguer V., Viana F. TRPA1 channels mediate cold temperature sensing in mammalian vagal sensory neurons: pharmacological and genetic evidence. J. Neurosci. 2008;28:7863–7875. doi: 10.1523/JNEUROSCI.1696-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karashima Y., Talavera K., Voets T. TRPA1 acts as a cold sensor in vitro and in vivo. Proc. Natl. Acad. Sci. USA. 2009;106:1273–1278. doi: 10.1073/pnas.0808487106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jordt S.E., Bautista D.M., Julius D. Mustard oils and cannabinoids excite sensory nerve fibers through the TRP channel ANKTM1. Nature. 2004;427:260–265. doi: 10.1038/nature02282. [DOI] [PubMed] [Google Scholar]
- 7.Macpherson L.J., Geierstanger B.H., Patapoutian A. The pungency of garlic: activation of TRPA1 and TRPV1 in response to allicin. Curr. Biol. 2005;15:929–934. doi: 10.1016/j.cub.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 8.Hu H., Bandell M., Patapoutian A. Zinc activates damage-sensing TRPA1 ion channels. Nat. Chem. Biol. 2009;5:183–190. doi: 10.1038/nchembio.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zurborg S., Yurgionas B., Heppenstall P.A. Direct activation of the ion channel TRPA1 by Ca2+ Nat. Neurosci. 2007;10:277–279. doi: 10.1038/nn1843. [DOI] [PubMed] [Google Scholar]
- 10.Corey D.P., García-Añoveros J., Zhang D.S. TRPA1 is a candidate for the mechanosensitive transduction channel of vertebrate hair cells. Nature. 2004;432:723–730. doi: 10.1038/nature03066. [DOI] [PubMed] [Google Scholar]
- 11.McNamara C.R., Mandel-Brehm J., Fanger C.M. TRPA1 mediates formalin-induced pain. Proc. Natl. Acad. Sci. USA. 2007;104:13525–13530. doi: 10.1073/pnas.0705924104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trevisani M., Siemens J., Geppetti P. 4-Hydroxynonenal, an endogenous aldehyde, causes pain and neurogenic inflammation through activation of the irritant receptor TRPA1. Proc. Natl. Acad. Sci. USA. 2007;104:13519–13524. doi: 10.1073/pnas.0705923104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bautista D.M., Jordt S.E., Julius D. TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell. 2006;124:1269–1282. doi: 10.1016/j.cell.2006.02.023. [DOI] [PubMed] [Google Scholar]
- 14.Kwan K.Y., Allchorne A.J., Corey D.P. TRPA1 contributes to cold, mechanical, and chemical nociception but is not essential for hair-cell transduction. Neuron. 2006;50:277–289. doi: 10.1016/j.neuron.2006.03.042. [DOI] [PubMed] [Google Scholar]
- 15.Kremeyer B., Lopera F., Ruiz-Linares A. A gain-of-function mutation in TRPA1 causes familial episodic pain syndrome. Neuron. 2010;66:671–680. doi: 10.1016/j.neuron.2010.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y.Y., Chang R.B., Liman E.R. The nociceptor ion channel TRPA1 is potentiated and inactivated by permeating calcium ions. J. Biol. Chem. 2008;283:32691–32703. doi: 10.1074/jbc.M803568200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hinman A., Chuang H.H., Julius D. TRP channel activation by reversible covalent modification. Proc. Natl. Acad. Sci. USA. 2006;103:19564–19568. doi: 10.1073/pnas.0609598103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.MacPherson L.J., Dubin A.E., Patapoutian A. Noxious compounds activate TRPA1 ion channels through covalent modification of cysteines. Nature. 2007;445:541–545. doi: 10.1038/nature05544. [DOI] [PubMed] [Google Scholar]
- 19.Chen J., Zhang X.F., Faltynek C.R. Molecular determinants of species-specific activation or blockade of TRPA1 channels. J. Neurosci. 2008;28:5063–5071. doi: 10.1523/JNEUROSCI.0047-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klionsky L., Tamir R., Gavva N.R. Species-specific pharmacology of trichloro(sulfanyl)ethyl benzamides as transient receptor potential ankyrin 1 (TRPA1) antagonists. Mol. Pain. 2007;3:39. doi: 10.1186/1744-8069-3-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bianchi B.R., Zhang X.F., Chen J. Species comparison and pharmacological characterization of human, monkey, rat, and mouse TRPA1 channels. J. Pharmacol. Exp. Ther. 2012;341:360–368. doi: 10.1124/jpet.111.189902. [DOI] [PubMed] [Google Scholar]
- 22.Gaudet R. TRP channels entering the structural era. J. Physiol. 2008;586:3565–3575. doi: 10.1113/jphysiol.2008.155812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoenderop J.G., Voets T., Bindels R.J. Homo- and heterotetrameric architecture of the epithelial Ca2+ channels TRPV5 and TRPV6. EMBO J. 2003;22:776–785. doi: 10.1093/emboj/cdg080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vannier B., Zhu X., Birnbaumer L. The membrane topology of human transient receptor potential 3 as inferred from glycosylation-scanning mutagenesis and epitope immunocytochemistry. J. Biol. Chem. 1998;273:8675–8679. doi: 10.1074/jbc.273.15.8675. [DOI] [PubMed] [Google Scholar]
- 25.Long S.B., Campbell E.B., MacKinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- 26.Ramsey I.S., Delling M., Clapham D.E. An introduction to TRP channels. Annu. Rev. Physiol. 2006;68:619–647. doi: 10.1146/annurev.physiol.68.040204.100431. [DOI] [PubMed] [Google Scholar]
- 27.Vallin K.S., Sterky K.J., Weigelt D. N-1-Alkyl-2-oxo-2-aryl amides as novel antagonists of the TRPA1 receptor. Bioorg. Med. Chem. Lett. 2012;22:5485–5492. doi: 10.1016/j.bmcl.2012.07.032. [DOI] [PubMed] [Google Scholar]
- 28.Chen J., Joshi S.K., Kym P.R. Selective blockade of TRPA1 channel attenuates pathological pain without altering noxious cold sensation or body temperature regulation. Pain. 2011;152:1165–1172. doi: 10.1016/j.pain.2011.01.049. [DOI] [PubMed] [Google Scholar]
- 29.Eid S.R., Crown E.D., Urban M.O. HC-030031, a TRPA1 selective antagonist, attenuates inflammatory- and neuropathy-induced mechanical hypersensitivity. Mol. Pain. 2008;4:48. doi: 10.1186/1744-8069-4-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Benedikt J., Samad A., Vlachova V. Essential role for the putative S6 inner pore region in the activation gating of the human TRPA1 channel. Biochim. Biophys. Acta. 2009;1793:1279–1288. doi: 10.1016/j.bbamcr.2009.04.014. [DOI] [PubMed] [Google Scholar]
- 31.Xiao B., Dubin A.E., Patapoutian A. Identification of transmembrane domain 5 as a critical molecular determinant of menthol sensitivity in mammalian TRPA1 channels. J. Neurosci. 2008;28:9640–9651. doi: 10.1523/JNEUROSCI.2772-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Banke T.G., Chaplan S.R., Wickenden A.D. Dynamic changes in the TRPA1 selectivity filter lead to progressive but reversible pore dilation. Am. J. Physiol. Cell Physiol. 2010;298:C1457–C1468. doi: 10.1152/ajpcell.00489.2009. [DOI] [PubMed] [Google Scholar]
- 33.Chen J., Kim D., Reilly R.M. Pore dilation occurs in TRPA1 but not in TRPM8 channels. Mol. Pain. 2009;5:3. doi: 10.1186/1744-8069-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]