

Abstract
Previous cysteine scanning studies of the cystic fibrosis transmembrane conductance regulator (CFTR) chloride channel have identified several transmembrane segments (TMs), including TM1, 3, 6, 9, and 12, as structural components of the pore. Some of these TMs such as TM6 and 12 may also be involved in gating conformational changes. However, recent results on TM1 seem puzzling in that the observed reactive pattern was quite different from those seen with TM6 and 12. In addition, whether TM1 also plays a role in gating motions remains largely unknown. Here, we investigated CFTR’s TM1 by applying methanethiosulfonate (MTS) reagents from both cytoplasmic and extracellular sides of the membrane. Our experiments identified four positive positions, E92, K95, Q98, and L102, when the negatively charged MTSES was applied from the cytoplasmic side. Intriguingly, these four residues reside in the extracellular half of TM1 in previously defined CFTR topology; we thus extended our scanning to residues located extracellularly to L102. We found that cysteines introduced into positions 106, 107, and 109 indeed react with extracellularly applied MTS probes, but not to intracellularly applied reagents. Interestingly, whole-cell A107C-CFTR currents were very sensitive to changes of bath pH as if the introduced cysteine assumes an altered pKa-like T338C in TM6. These findings lead us to propose a revised topology for CFTR’s TM1 that spans at least from E92 to Y109. Additionally, side-dependent modifications of these positions indicate a narrow region (L102-I106) that prevents MTS reagents from penetrating the pore, a picture similar to what has been reported for TM6. Moreover, modifications of K95C, Q98C, and L102C exhibit strong state dependency with negligible modification when the channel is closed, suggesting a significant rearrangement of TM1 during CFTR’s gating cycle. The structural implications of these findings are discussed in light of the crystal structures of ABC transporters and homology models of CFTR.
Introduction
Cystic fibrosis transmembrane conductance regulator (CFTR), an integral membrane protein whose dysfunction causes the inherited disease cystic fibrosis (1), is a unique member of the ABC transporter superfamily in that it serves as an anion channel that allows chloride ions to move passively across plasma membranes down their electrochemical gradient (1,2). Like many ABC exporters, CFTR harbors two nucleotide-binding domains (NBDs) and two transmembrane domains (TMDs), each of which comprises six transmembrane segments. It has long been established that CFTR, although belonging to the ABCC subfamily (3), has several unique molecular properties. CFTR distinguishes itself from other ABC transporters; first, because it possesses a regulatory domain (R domain) in which several consensus serines can be phosphorylated by protein kinase A (PKA). Phosphorylation of the R domain is a prerequisite for ATP to gate the channel (4–6). Second, although CFTR retains the conserved motion of dimerization and partial separation of its NBDs for gating conformational changes (7–10), the structural rearrangements of its TMDs must be somewhat different from those adopted by ABC transporters because the classical flip-flop movements of the TMDs in ABC transporters (11) that shuffle the conformation between inward-facing and outward-facing configurations do not afford a contiguous aqueous pathway for ion diffusion. Nevertheless, a recent report (12) provided evidence supporting the degraded transporter hypothesis: CFTR may employ similar conformational changes in its TMDs during gating but its cytoplasmic gate is degenerated (see (12) for details, but cf. (13)).
More than two decades since the CFTR gene was cloned (1), numerous structural and functional studies have amassed tremendous knowledge with respect to the gating and permeation mechanisms of CFTR (reviewed in (14)). Among many issues of interest, identifying the structural components for the anion permeation pathway is especially interesting in light of the apparent evolutionary relationship between CFTR, an ion channel, and ABC exporters carrying out the function of active transport. Based on hydropathy analysis, 12 transmembrane segments (TMs) were assigned to CFTR’s TMDs (Fig. 1 A, (1)). Substituted cysteine accessibility methods (SCAM) have been employed in examining targeted transmembrane segments in numerous transporters and channels (12,15–21). For CFTR, this technique has also been fruitful in recent years in identifying several pore-lining segments (12,16,18,22–25), some of which are intimately involved in gating motion (12,16,18,22,26). Finally, crystal structures of four ABC exporters, namely Sav1866 (27,28), MsbA (29), mouse P-glycoprotein (30), and TM287/288 (31), have been solved. Although the field is still waiting for the high-resolution structure of CFTR, these crystal structures of ABC exporters do provide the framework for building homology models that could potentially shed some light on the structural attributes of CFTR (15,32–34). Furthermore, although bearing some deficiencies (see Discussion for details), these models indeed provide a handful of predictions that have been verified by functional studies (12,16,18,22).
Figure 1.
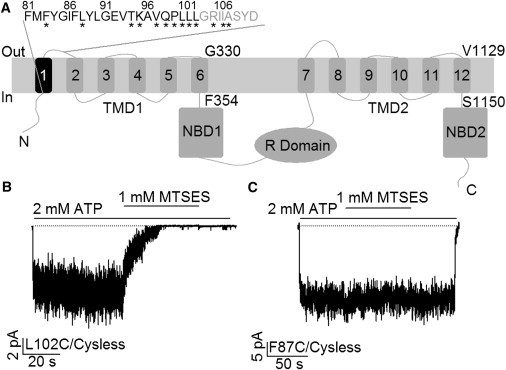
Traditional topology of CFTR and effects of MTSES on cysteine-substituted CFTR mutant channels. (A) Topological map of CFTR based on hydropathy analysis shows the domain structure of CFTR: two TMDs, two NBDs, and an R domain. The first transmembrane segment (TM1) is colored black with its amino acid sequence shown above. Eight residues previously assigned as the first extracellular Loop (ECL1) were labeled in gray. The innermost and outermost residues of TM6 and TM12 were also marked. (B) Cytoplasmic application of MTSES dramatically reduced ATP-gated L102C/Cysless channel currents in an excised inside-out membrane patch. (C) Macroscopic current trace showing a lack of effect of MTSES on F87C/Cysless channels.
A general picture emerges from our previous SCAM studies on TM6 and 12: there is an accessibility limit in the middle of these two TMs when thiol-reactive, channel impermeant probes were applied from the cytoplasmic side of the membrane (12,16). Together with data from whole-cell experiments where the engineered cysteines were probed from the extracellular end, Alexander et al. (15) and Norimatsu et al. (23) proposed a bottleneck region in the pore that is composed of amino acids 338–341 in TM6 and 1131–1140 in TM12. By contrast, recent data on TM1 (35), depict a very different picture as this barrier to channel impermeant thiol-reagents seems located close to the extracellular end of this TM defined originally in Riordan et al. (1). This puzzling observation raises at least two possibilities. First, TM1 may not line the pore and the reported macroscopic current response to methanethiosulfonate (MTS) reagents is a result of effects on gating. Second, the assigned topology of TM1 (1) may not be accurate so that the pore-lining segment of TM1 actually extends into the first extracellular loop. To resolve this issue, we decided to carry out more extensive SCAM studies on TM1.
In the current study, SCAM experiments were performed with both inside-out membrane patches, to which MTS probes were applied from the cytoplasmic side, and whole-cell recordings, which allow testing reactivity with extracellular-applied MTS reagents. Overall, seven reactive sites (E92, K95, Q98, L102, I106, A107, Y109) were identified. The periodicity of these positive hits suggests that TM1, like the internal part of TM6 and TM12 (12,16), also assumes an α-helical structure. More importantly, the reactivity patterns toward intracellularly and extracellularly applied MTS probes indeed suggest a 4-amino-acid bottleneck (102–106) similar to what has been reported for TM6 (15,16,23). However, as positions 103–109 were classified as the first extracellular loop in the traditional topological map of CFTR’s TMDs (1), our findings suggest a different membrane-spanning topology for TM1: instead of being part of a disordered extracellular loop, we propose that amino acids 103–109 constitute part of a more structured ion permeation pathway. Another unique feature of TM1 is the observation that modifications of engineered cysteines at positions 95, 98, and 102 are strictly state dependent—modifications occur nearly exclusively in the open state. Structural implications of this finding will be discussed.
Materials and Methods
Channel expression
To avoid effects of MTS reagents on endogenous cysteines in CFTR, a construct with all endogenous cysteines altered (i.e., cysless background) was used. All mutations were confirmed with DNA sequence (DNA Core, University of Missouri). Chinese hamster ovary cells grown at 37° in Dulbecco’s modified Eagle’s medium plus 10% fetal bovine serum were used to express the channels. The pcDNA of all mutants were transfected into cells together with pEGFP-C3 (Takara Bio), which encodes green fluorescent protein, using PolyFect reagent (QIAGEN, Venlo, Netherlands) according to manufacturer’s instructions. After transfection, cells were transferred to 35 mm tissue culture dishes and incubated under 27° for 2–7 days before experiments were performed.
Electrophysiology
Inside-out mode
Glass electrodes were prepared with a two-stage vertical puller (Narishige, Tokyo, Japan), and polished with a homemade microforge to yield a resistance between 2 and 5 MΩ when placed in the bath solutions containing (in mM): 145 NaCl, 5 KCl, 2 MgCl2, 1 CaCl2, 5 glucose, 5 HEPES, 20 sucrose, with pH adjusted to 7.4 using NaOH. After the patch was excised into an inside-out configuration, the perfusion solution was changed to one containing (in mM): 150 NMDG-Cl, 10 EGTA, 10 HEPES, 8 Tris, 2 MgCl2, with pH adjusted to 7.4 using NMDG. The pipette solution contained 140 NMDG-Cl, 2 MgCl2, 5 CaCl2, 10 HEPES, with pH adjusted to 7.4 using NMDG. To achieve fast solution changes, the pipette tip was moved to the outlets of a three-barrel glass tubing under the control of a fast solution exchange system (SF-77B; Warner Instruments, Hamden, CT). The current signal was recorded with a patch clamp amplifier (EPC10; HEKA), filtered at 100 Hz with a Bessel filter and digitized at 500 Hz using Pulse (V8.80; HEKA). All the experiments were performed at room temperature.
Whole-cell mode
Pipettes used in whole-cell experiments were made with the same puller described previously, but not polished to yield a resistance of ∼1.5–2.5 MΩ when filled with whole-cell pipette solution containing (in mM): 10 EGTA, 10 HEPES, 20 TEACl, 10 MgATP, 2 MgCl2, 85 aspartate, 16 pyruvate, 5.8 glucose, with pH adjusted to 7.4 using CsOH. Bath solutions of various pH values were adjusted with H2SO4 and cacodylic acid, respectively. A voltage ramp of ±100 mV over 200 ms was applied every 5 s. The current was acquired with a patch clamp amplifier (EPC10; HEKA), filtered at 1 kHz and digitized at 2 kHz with Pulse software (V8.80; HEKA). Experiments were also conducted at room temperature.
Reagents and chemical modification
MTS reagents (Toronto Research Chemicals, Toronto, Ontario, Canada) were stored at −70° as 100 mM stock solutions. Each aliquot was diluted into 1 mM (inside-out) or 0.5 mM (whole-cell) immediately before application. Due to possible spontaneous oxidation of the introduced cysteines (36,37), dithiothreitol (DTT; Sigma-Aldrich, St. Louis, MO) was added into the cocktail containing PKA and ATP to ensure that the engineered cysteines are mostly in the reduced form before applications of the MTS reagents. CFTRinh-172 was kindly provided by Dr. Robert Bridges with support from the Cystic Fibrosis Foundation Therapeutics. ATP and PKA were bought from Sigma-Aldrich.
Data analysis
To calculate the modification rate by MTS reagents, we first fitted the current decay phase in each recording with a single exponential function with the Igor program (V4.07; WaveMetrics, Portland, OR) to obtain the time constant, τ. The modification rate constant was computed as 1/(τ∗ [MTSES]) where [MTSES] is the concentration of MTSES. Single-channel kinetics analysis was conducted using the program developed by L. Csanády (2000) (38), where C-O-B mode was chosen to extract the kinetics parameters. Student’s paired and equal variance t-test (two-tailed) was conducted with Excel (Microsoft). P < 0.05 was considered significant. Data in the text were presented as means ± SE. n represents the number of data points for each experiment.
Results
Cysteine scanning of TM1 using both internal and external thiol-specific probes
To investigate the role of TM1 in CFTR’s pore-forming constituents, individual cysteines were introduced into each position (81–102) along previously assigned TM1 (1) under the cysless background as described before (12,16). After the channels were expressed in the cell membrane, inside-out patches containing cysteine-substituted channels were made to test their reactivity toward 2-sulfonatoethyl MTS reagents. In all the inside-out recordings, a cocktail containing PKA and ATP was first used to maximally activate the channels. Subsequently, chloride currents were induced by ATP to a steady-state level before the channels were exposed to intracellularly applied MTS reagents in the continuous presence of ATP for at least 1 min or until the current reaches a steady level. We then switched the perfusion solution back to one with ATP alone to ensure that any reactivity is not reversed by removal of the reagent. Fig. 1 B shows a representative recording of L102C mutant channels in response to the application of MTSES. This observed decrease of macroscopic currents is due to covalent modification of the engineered cysteines by the reagent as the effect persisted even after a complete removal of MTSES. Similar observations were made for K95C- and Q98C-CFTR. We could only obtain microscopic current with E92C-CFTR probably due to a poor expression, but the application of MTSES decreased the single-channel amplitude of this construct (see below). Strikingly, cysteines placed in all other 18 positions did not respond to MTSES. For instance, in the case of the F87C mutant channel, no obvious changes in the mean current were seen during a 1-min application of MTSES (Fig. 1 C). However, one cannot rule out the possibility that these channels were modified but without apparent changes in gating or permeation.
Fig. 2 summarizes our results described previously. Data on TM6 and 12 from our previous reports (12,16) are presented for comparison. Two differences were noted. First, fewer positions show positive response to MTSES (4 in TM1 vs. 6 in TM6 and 8 in TM12). Nonetheless, the apparent periodicity with the four reactive positions on TM1 is consistent with the notion that this part (E92 to L102) of TM1 assumes a secondary structure of an α-helix, just like the intracellular half of TM6 and TM12 (12,16). Second, most surprisingly, unlike TM6 and TM12, all four reactive positions on TM1 reside in the extracellular half of this segment according to the traditional topology built on hydropathy analysis (Figs. 1 A and 2). Thus, although the accessibility limits for TM6 (position 341) and TM12 (position 1140) are located approximately in the middle of these TMs, L102 is the outermost residue in the classical topological map of TM1 (1). We therefore extended our cysteine scanning of TM1 to residues beyond the L102 position. The results were also summarized in Fig. 2. Although we failed to detect any currents from G103C-CFTR, all other seven cysteine-substituted constructs did not respond to cytoplasmic application of MTSES to a significant extent (Fig. 2). We conclude that the accessibility limit to intracellular MTSES is indeed at position 102 as reported by Wang et al. (35).
Figure 2.
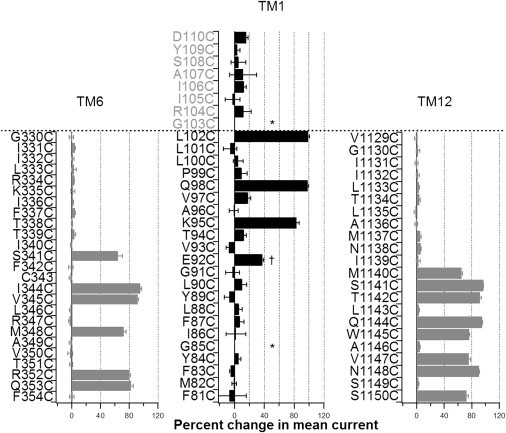
Summary of SCAM results on TM1 (middle), TM6, and TM12 (left and right). The inhibition ratio was calculated as (1-Ia/Ib), where Ia and Ib are mean currents after and before application of MTSES, respectively. Those positions colored in gray in the middle panel (TM1) represent residues on ECL1 in the traditional topological map (Fig. 1A). Stars marked above positions 85 and 103 indicate no current was detected from these two mutants, whereas the dagger (†) on position 92 specifies that the measurement was from changes of the single-channel current amplitude by MTSES due to a low expression level. The dotted line delineates the extracellular end of each segment in the traditional CFTR topology. Data were extracted from 2 to 8 patches for each mutant. Results on TM6 and TM12 were obtained from Bai et al. (12,16).
If TM1 indeed constitutes part of the pore-forming domain, the different location of the accessibility limit described previously between TM1 and TM6 raises an interesting possibility that the traditional topological map for TM1 may not be correct. In other words, some of the residues external to L102 may actually reside in the pore. To test this hypothesis, we performed whole-cell experiments in which MTS reagents could be applied from the extracellular side. Replacing each residue from 103 to 110 with cysteine results in robust cAMP-dependent whole-cell currents with the exception of position 103, where no detectable currents were observed, probably due to poor expression. Fig. 3 A shows a continuous whole-cell recording of I106C-CFTR currents. Whole-cell CFTR currents were first activated with 10 μM forskolin. Once the current reached a steady state, application of 0.5 mM MTSES nearly abolished the current completely. This inhibition is irreversible because washout of MTSES did not recover the current. Further addition of a specific CFTR inhibitor, CFTRinh-172 (39,40), caused a minor decrease of the residual current, indicating a drastic reduction of I106C-CFTR currents by external MTSES. Similar results were obtained for A107C- and Y109C-CFTR except that the magnitude of inhibition for A107C-CFTR is significantly smaller (Fig. 3 C). In contrast, minimal effects of external MTSES were seen with the other four positions (R104, I105, S108, and D110). Fig. 3 B shows an example of these negative responders. We next tested the accessibility to external MTSES on three positions identified by experiments with inside-out patches, namely K95C, Q98C, and L102C, in the same manner and all three positions turned out nonreactive (data not shown).
Figure 3.
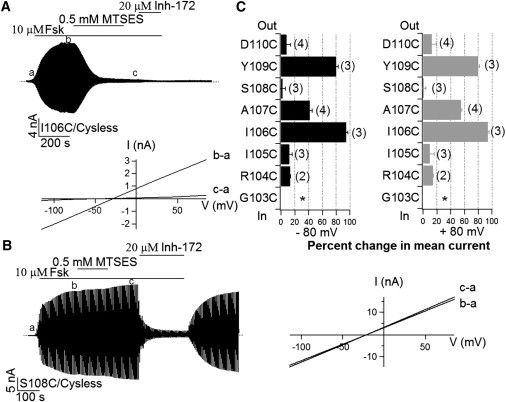
SCAM experiments with whole-cell recordings. (A) A continuous recording of whole-cell I106C/Cysless channel current in response to an external application of MTSES. The I-V relationships for net CFTR currents before (b-a) and after (c-a) MTSES modification were shown below the trace. (B) The same protocol was adopted for S108C/Cysless mutant channels, which exhibit little response to external MTSES. However, reversible inhibition by CFTRinh-172 shows that the current observed was indeed due to the activation of CFTR. (C) Summary of whole-cell SCAM results on the eight residues, G103C-D110C. Data at ±80 mV were presented to illustrate the unique voltage-dependent inhibition by MTSES at position 107. The number of cells was labeled at the end of each bar. ∗ indicates no detectable current for G103C-CFTR.
Two tentative conclusions can be made based on the results shown so far. First, when we consider all seven positive hits spanning the segment from E92 to Y109, a periodicity consistent with a secondary structure of an α-helix emerges. Second, the observation that L102 constitutes the internal accessibility limit while I106 acts as an external accessibility limit, indicates a 4-amino-acid bottleneck that prevents 6 Å MTSES from penetrating the pore from either side of the membrane. This latter conclusion bears remarkable resemblance to what has been shown for TM6 (T338–S341 (16,23)).
The similarity between TM1 and TM6 was reinforced by the following results with A107C mutant channels. When recording whole-cell A107C-CFTR current with a chloride gradient (24 mM internal and 156 mM external [Cl−]), we found that instead of an expected outward rectified I-V curve due to this imposed concentration gradient, the observed I-V relationship shows significant inward rectification (Fig. 4 A). Furthermore, the degree of inhibition by MTSES under −80 mV (54.9 ± 1.4%, n = 4) was significantly higher than that under +80 mV (41.6 ± 3.7%, n = 4) for this mutant (Figs. 3 C and 4 A). These observations can be explained if the introduced cysteine at position 107 has a pKa that is lower than that of cysteine in the bulk solution (between 8.10 and 8.72 (41)). At pH 7.4, C107 can then be considered to carry a partial negative charge, a scenario analogous to placing a cysteine at position 338 in TM6 (42). We next adopted the similar strategy used in Liu et al. (42) to test if A107C-CFTR can be modulated with baths of different pH. In Fig. 4 B, A107C-CFTR channel currents were first activated with forskolin in the bath solution with a pH of 7.4; after the current level stabilized, the bath was switched to forskolin-containing solution with a pH of 6 or 8. As shown in Fig. 4 B, whole-cell currents at +80 mV increase by 182.7 ± 9% (n = 4) as the bath solution is acidified, but decrease by 44.8 ± 3.2% (n = 4) upon lowering bath [H+]. Switching among bath solutions with different pH allows us to determine the pKa value of the cysteine engineered at position 107. As shown in Fig. S1 in the Supporting Material, the pKa value of C107 is 7.25, which is nearly identical to that of T338C in TM6 (42). These results are consistent with the idea that the side chain of the introduced cysteine at position 107 is partially deprotonated resulting in a negatively charged thiol group (-S−) that impedes chloride permeation. An increase of the bath [H+] then neutralizes this -S−. As a control, neither pH 6 nor pH 8 bath alters whole-cell cysless channel currents significantly (Fig. 4 C). These results further support the idea that the side chain of residue 107 lines the pore.
Figure 4.
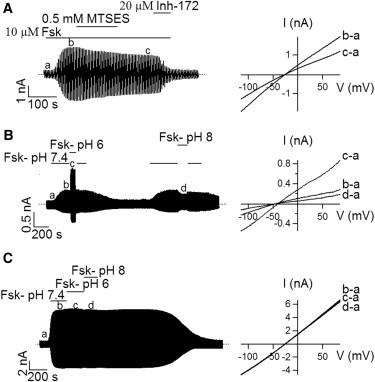
Effects of external MTSES and pH on A107C-CFTR. (A) Whole-cell A107C-CFTR currents in response to external MTSES (left) and I-V curves extracted from the whole-cell recording as marked. (B) Effects of acidic or alkaline pH on the whole-cell A107C-CFTR currents (left). Corresponding I-V relationships were shown on the right. (C) A control experiment on WT/Cysless demonstrating a lack of effect by changes of bath pH.
Modifications by MTS-ethyltrimethylammonium (MTSET) affect gating and permeation
One interesting feature about SCAM studies on TM6 is the dual effects of MTSET modification on CFTR gating and permeation (16). Similar but not identical observations were made with TM1. For instance, for L102C mutant channels, we found that macroscopic currents were increased by 108.5 ± 9.5% (n = 6) after modification by MTSET applied to the cytoplasmic side of the channel in excised inside-out patches (Fig. 5 A). Also similar to what has been reported for positions I344 and M348 in TM6, following MTSET modification of L102C-CFTR, robust activity was observed even in the complete absence of ATP. This dramatic effect on CFTR gating was readily reversed by the reducing reagent DTT. For Q98C and K95C mutant channels, the increases in the mean current amplitude following MTSET modification were ∼2- and 6-fold, respectively. Because the single-channel conductance was drastically decreased in K95C-CFTR, we were not able to assess the gating effect of MTSET modification. Fig. S2, however, shows that MTSET modification of Q98C-CFTR increases both the open probability and the single-channel amplitude.
Figure 5.
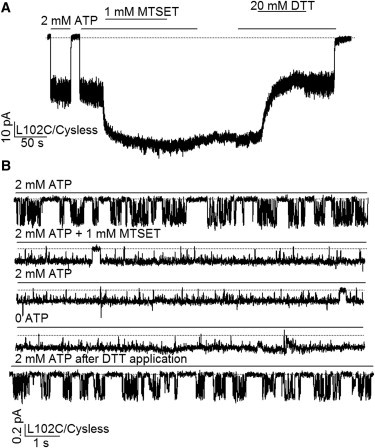
Gating of MTSET-modified L102C/Cysless channel. (A) A continuous current recording of L102C/Cysless channels showing that MTSET modification increases the macroscopic current and renders the current ATP-independent. (B) Single-channel recording of the L102C/Cysless-CFTR. Note the unusually numerous flickery closings in each opening burst before MTSET modification (compare Fig. 7A, below). The single-channel amplitude was decreased by MTSET modification, but the Po was increased to near unity. The reducing reagent, DTT, effectively reversed the effect of MTSET modification.
The remarkable effects of MTSET on L102C-CFTR currents shown in Fig. 5 A prompted us to examine this effect more closely with single-channel recordings. Fig. 5 B shows single-channel traces before and after MTSET modification of L102C-CFTR. Several intriguing findings were made. First, before MTSET modification, each opening burst was interrupted by many flickery closings (compare Fig. 7 A below). Although previous studies have provided evidence that these short-lived closures are ATP independent (43,44) and could result from voltage-dependent block of the pore by large anions from the cytoplasmic side of the channel (45), it is noted that these events are abundantly present in the double mutant L102C/E1371Q at both negative and positive membrane potentials in the absence of ATP (see Fig. S3). If we accept the premise that the NBDs of this hydrolysis-deficient channel remained in a dimeric configuration throughout the recording, these transitions could then represent autonomous opening and closing of the gate while the NBDs are dimerized. Thus, the L-to-C mutation at position 102 somehow destabilizes the gate of the channel. Second, after modification by MTSET, the single-channel current amplitude decreases by 46.4 ± 1.3% (n = 4), but the Po increases dramatically (Fig. 5 B). In fact, the modified channel remains nearly always open even after removal of ATP. Similar to what we observed for I344C- and M348C-CFTR (16), this robust ATP-independent gating was seen following modification by MTSET but not by MTS-ethylammonium (MTSEA) (Fig. S4).
Figure 7.
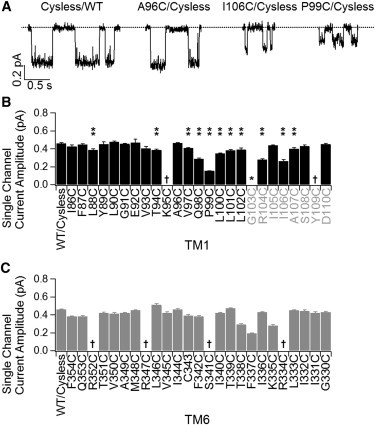
Effects of cysteine substitution on the single-channel current amplitude. (A) Representative single-channel traces for WT/Cysless, A96C/Cysless, I106C/Cysless, and P99C mutant channels. (B and C) Summary of single-channel current amplitudes for WT/Cysless and all cysteine-substituted mutant channels in TM1 (B) or TM6 (C). Single star specifies no channel activity was detected for G103C. The daggers indicate that the single channel current amplitude is too small or too variable to be measured accurately. Double stars indicate positions where cysteine substitution alters the single channel current amplitude significantly (p < 0.05, student’s t-test). Data were acquired from 3 to 4 patches for each mutant. Results on TM6 were obtained from Bai et al. (16).
State-dependent modification of E92C-, K95C-, Q98C-, and L102C-CFTR
The observation that MTSET modification of Q98C and L102C alters CFTR gating suggests that TM1 indeed participates in gating motions of CFTR. However, because only one position in each helical turn of the segment from E92 to L102 reacts with MTS reagents, it seems hard to envision that similar rotational movements proposed for TM6 and 12 can be applied to TM1. To better understand the molecular motion TM1 may undergo during gating transitions, we adopted the protocol originally designed by Yellen’s group (46) and lately applied to SCAM studies of CFTR (12). In these experiments, channels were first activated by PKA plus ATP; after ATP washout, two brief pulse applications of ATP were implemented to ensure a stationary recording condition is fulfilled. Subsequently, we applied ATP for 3 s until the current reached a steady state, followed by an 8-s washout phase to allow channel closure; MTSES was then applied for 3 s in the absence of ATP followed by a 2-s washout of MTSES. The same cycle would be repeated 12 times. As this series of experiments requires macroscopic currents, we could not test the E92C mutants, which express poorly (see Discussion for details). However, Fig. 6 A shows a representative recording of current response for K95C-CFTR mutants. Strikingly, the macroscopic current amplitude remains fairly constant over a recording time longer than 3 min. Similar results were obtained from experiments on L102C (Fig. 6 B) and Q98C mutants. These data suggest that the modification rate of cysteines on these positions is exceedingly low, if at all, when the channels are in the closed state.
Figure 6.
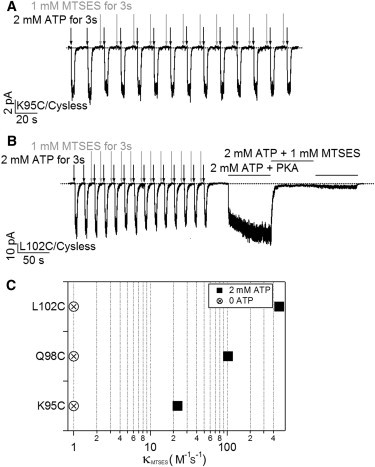
State-dependent modification by MTSES at positions 95, 98, and 102. (A) A representative recording for MTSES modification in the absence of ATP for K95C/Cysless channels. (B) A real-time recording with a similar protocol shown in (A) for L102C/Cysless channels. Although very little current was affected by application of MTSES in the absence of ATP, exposure of the same patch to MTSES in the presence of ATP readily diminished the current. (C) Summary of the modification rates for K95C, Q98C, and L102C in the presence of ATP (solid squares). The modification rates in the absence of ATP (crossed circles), were all set at 1 because the modification, if at all, was too slow to be quantified accurately. n = 5 for each construct.
On the contrary, in the presence of ATP when the channels stay in the open state for a significant amount of time, the introduced cysteine at all these positions can be readily modified by MTSES (e.g., Fig. 1 B, Fig. 6 B). We measured the modification rate in the presence of ATP by fitting the current decay with a single exponential function and the resulting time constant was converted to the second order rate constant as described previously (12). Fig. 6 C summarizes these results. Thus, these positions in TM1 are accessible to MTSES when channels are open but not when they are closed, hinting at a gating-associated structural rearrangement of TM1 relative to other parts of CFTR.
Discussion
SCAM studies on CFTR’s TM1 in the current manuscript are consistent with the hypothesis that portions of this segment could line the anion-conducting pore. The reactivity of cysteines substituted in the presumed, pore-lining region of TM1 toward internally applied, polar, thiol-specific reagents is strikingly favored in the open state of the channel. This result suggests that conformational changes underlying channel closing can restrict the accessibility of cysteines substituted in this region of TM1 to reagents that enter the pore from the cytoplasmic side. Furthermore, a direct comparison of the reactivity of cysteines substituted into TM1 toward externally and internally applied, thiol-directed reagents is consistent with previous studies that identify a bottleneck in the pore that restricts the permeation of MTS reagents and may be rate-limiting for anion conduction (23).
With the completion of cysteine scanning on TM1, we now have two TMs (1,6) thoroughly investigated with channel impermeant probes applied from both the extracellular and intracellular end of the CFTR pore (15,16,22,35). Although some differences exist between these two sets of data, they do share remarkable similarities. In the following sections, we will not only elaborate the structure/function implications of the data on TM1 in its own right, but also discuss how these differences and similarities between TM1 and TM6 may shed light on the evolutionary relationship between CFTR and ABC exporters.
However, we need to first point out a few puzzling differences between current results and those published by others (35). First, the four positions identified by cytoplasmic application of MTSES are not exactly the same (E92, K95, Q98, and L102 in the current study but K95, Q98, P99, and L102 in (35)). Second, although we failed to detect any reactivity to extracellular or intracellular application of MTSES at position 104, Zhou et al. (47) reported that a cysteine placed at this position does react with external MTS reagents (see Fig. S5 for detail). Third, in the report by Wang et al. (35), K95C, but not Q98C, P99C, or L102C, can react with internal MTSES even before the channel is activated by PKA and ATP, implying a regulated barrier between positions 95 and 98. However, we found all four positions appear to be only accessible to internal MTS probes in the open state.
Notably, these discrepancies are not limited to data on TM1, numerous differences between our results on TM6 and 12 (12,16) and those reports by the same group (18,22) were observed. Though the exact reasons behind these discrepancies are unknown to us for the time being, here we discuss several technical differences in experimental design and data presentation. First, whenever possible, we presented real-time recordings of our data that offer our readers a direct view of the time course of current responses to the applied reagents. This kind of detailed, long-lasting current recording also allows our readers to more readily discern any unusual changes of the recorded current as well as the baseline leakage current. Second, a fast solution change system used in our experiments is essential for accurately assessing the reaction rate of the introduced thiols to MTS reagents as the solution change itself does not impose a rate-limiting step. Third, to determine the state-dependency of those reactive cysteines to MTSES, we adopted a method similar to that designed by Yellen’s lab (46). In contrast, Linsdell’s group routinely used the preincubation method (see (35) for details), which negated any opportunity to monitor either the time course or the extent of the reaction, or to test its reversibility, leaving some room for undesirable effects. Finally, different expression systems were used (baby hamster kidney cells in Linsdell’s group versus Chinese hamster ovary cells in our study).
Cysteine scanning on TM1 confirms a bottleneck region in the CFTR pore
Previous studies have come to a general consensus that the whole ion permeation pathway of CFTR consists of an inner vestibule and an outer vestibule flanking a rate-limiting region (42,48,49). SCAM experiments using channel impermeant MTS reagents have specified the relative locations of each of these components in TM6 (15,16). By identifying the accessibility limits of intracellularly and extracellularly applied MTS reagents, one can safely conclude that residues 338–341 in TM6 constitute the bottleneck region of the pore (15,16). Interestingly, the present studies also pinpoint a similar 4-amino-acid region (102–106) in TM1 that prevents MTS reagents from passing through the pore. Because the residues on either side of this presumed narrow region of the pore can be readily accessed by the bulky MTS reagents, it appears that the physical dimensions of both internal and external vestibules need to be at least 6 Å wide. As 4 amino acids are only sufficient to span ∼1 helical turn of an α-helix, if both TM1 and TM6 assume this type of secondary structure, the CFTR pore indeed looks like an hourglass (47) with a narrow region of ∼5–6 Å in length. Considering the diameter of 3.6 Å for a chloride ion, we reckon that this narrow region can only accommodate one chloride ion at a time.
Is this bottleneck region CFTR’s anion selectivity filter? Although alanine substitution experiments with residues in TM6 do support a role of this region in discriminating monovalent and divalent anions (50), it is questionable if a well-defined selectivity filter exists in the CFTR pore (48). Recent molecular dynamics simulations of a CFTR homology model (23) reveal potential candidates near this region that may coordinate the chloride ion in the pore (namely, S341 in TM6, T1134 in TM12, and K95 in TM1), but the current study suggests that K95 may be positioned too far to make significant contributions to the role of this presumed bottleneck for chloride permeation. It is nonetheless interesting to note that although CFTR is an anion channel, positively charged MTSET, just like negatively charged MTSES, can reach position 102 from the cytoplasmic side as well as position 106 from the external side of the channel (see Fig. S6). Thus, neither internal nor external vestibule seems to impose a strict anion-over-cation selectivity. Nevertheless, many positively charged amino acids scattered in TM1, TM5, and TM6 have been proposed to serve to concentrate chloride ions in both internal and external vestibules (47,49,51). One needs to caution that the existence of positively charged amino acids in the pore-forming segments does not guarantee that the side chains of these amino acids actually protrude into the ion permeation pathway. R347 in TM6 is a good example. Although early work suggests a pivotal role this residue plays in chloride binding in the pore (52), subsequent studies indicate otherwise (16,22,53). For TM1, R104 has been implicated in assuming a similar role in chloride selectivity (47), but this work did not find corroborating evidence.
A revised topology for TM1
Hydropathy analysis has been the method of choice for assigning membrane spanning segments of an integral membrane protein without any prior knowledge about the structure (54,55). CFTR’s TMD topology was first determined by such means more than two decades ago (1). Research based on this topology has provided ample mechanistic insights into pore architecture, ion selectivity, and gating motions. Lately, computational studies of CFTR based on crystal structures of ABC transporters (23,32–34) also shed light on the structure/function relationship of this molecule. However, although a gross picture of CFTR’s TMDs seems consistent in different CFTR models, detailed structures of each TM could differ considerably (23,32). We thus chose the original TM1 topology as our starting point.
Our experimental results show that cysteines introduced at three positions external to L102, the outermost residue in traditional TM1, can be modified by extracellularly applied MTS reagents (Fig. 3 C). These data, together with the observation that titrating the cysteine introduced at position 107 can affect ion permeation (Fig. 5 and Fig. S1), support the idea that residues 103–109 form part of the external pore. We thus propose a revised topology for TM1: two helical turns formed by residues 103 to 109 are part of the more structured ion permeation pathway. The idea that this segment constitutes part of the pore predicts that mutations of some of these residues may alter ion permeation. Fig. 7 indeed shows that cysteine substitution at several of these positions decreases the single-channel amplitude. (Previous results on TM6 are presented for comparison.) It is noted that those positions of which mutations alter the single-channel conductance include not only the aforementioned bottleneck region, but also areas flanking this segment. Interestingly, if we align TM1 and TM6 using the outermost position whose mutation affects the single-channel amplitude as the extracellular end for each TM (Y109 in TM1 and R334 in TM6), we also successfully align the intracellular accessibility limits (i.e., L102 in TM1 and S341 in TM6) as well as the extracellular accessibility limit (i.e., I106 in TM1 and T338 in TM6).
After this adjustment, L102 will be located close to the middle of the newly defined TM1 (cf. (35)). It follows that the three other positive hits (C92, C95, and C98) will reside within three helical turns down from L102. Incidentally, since S341 and M1140 are the internal accessibility limit for TM6 and TM12, respectively, the other positive hits in these two TMs are also vested within 3–4 helical turns from the referenced positions. On the basis of this revised TM1, we reason that residues internal to E92 may not be important for ion permeation, a conjecture consistent with the observations that cysteines introduced into these positions are not reactive to MTSES (Fig. 2) or reactive to MTSES but without any effects on chloride permeation, and that almost none of the cysteine substitutions at these positions significantly affects the single-channel amplitude (Fig. 7). Of note, a different alignment between TM1 and TM6 was deduced based on cross-linking experiments for paired cysteines introduced into these two TMs (35).
Possible gating motions of TM1 revealed by state-dependent modifications and crystal structures of ABC exporters
Our previous work on TM6 and TM12 provides data supporting the hypothesis that CFTR evolves from a primordial ABC exporter by degenerating its cytoplasmic gate ((12), but cf. (13)). Specifically, we showed that cysteines placed at positions 344, 348 in TM6 and 1141, 1148 in TM12 can react readily with cytoplasmic Texas Red MTSEA (∼13 Å wide for its headgroup) even when the channel is closed. We were therefore surprised to observe negligible modification of cysteines introduced at positions 95, 98, and 102 in the absence of ATP. Although we were not able to measure the modification rate for E92C because of poor expression, in five patches yielding microscopic current, MTSES appears to modify the introduced cysteine when the channel is opened (see Fig. S7). Thus, all four positive hits for cytoplasmic application of MTSES exhibit strict state dependency (cf. (35)).
Many of the data presented in our previous SCAM studies on TM6 and TM12 are consistent with a helix rotational movement during CFTR’s gating cycle (12,16). However, this scenario is unlikely applicable to TM1 because those four reactive positions are aligned on one face of the helix but none of the neighboring positions are reactive. We therefore reasoned that at least the intracellular half of TM1 may undergo conformational rearrangements between lining the pore in the open state and hiding behind other parts of the protein in the closed state.
As the crystal structure of CFTR is not yet available, we decided to resort to homology models built on the template of CFTR’s close cousin, ABC exporters, to gain some insights into this unique state-dependency of introduced cysteines in the cytoplasmic side of TM1. Several such models of CFTR have been published in recent years (23,32,34). We first examined the homology model of CFTR in the presumed open conformation based on the crystal structure of the outward-facing configuration of Sav1866. The modeled structure clearly shows that TM1, TM6, and TM12 line the open pore (Fig. 8 A) (32). On the other hand, if we accept the premise that the inward-facing structures of ABC exporters may share some resemblances with the closed state of CFTR, they may shed some light on the plausible orientation of TM1 in the closed conformation of CFTR. We then examined the homology model based on the inward-facing configuration of MsbA (34). Interestingly, TM1 now moves to a position where its contribution to pore-lining is extremely limited (Fig. S8).
Figure 8.
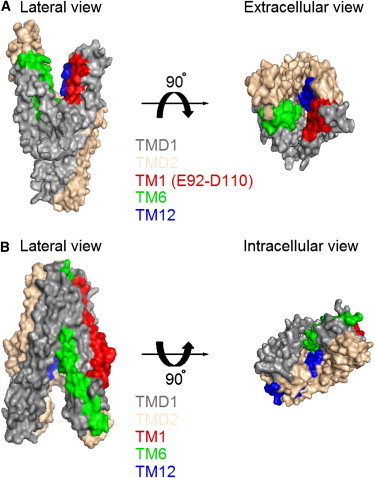
Relative orientations of TM1 in a homology model of CFTR and positions of TM1 in the crystal structure of an ABC exporter, TM287-TM288. (A) Surface views of TMDs in the homology model of CFTR based on the outward configuration of Sav1866 (32). Domains and segments were presented in different colors as indicated. (B) Surface views of the crystal structure of TM287-TM288 (31) in an inward-facing conformation. Two NBDs were removed for a better view from the intracellular side. Detailed cross-section views can be found in Fig. S9. Figures were prepared with PyMol (V0.99; Schrödinger).
As the resolution of the inward-facing structure of MsbA is not ideal, we also turned to the crystal structure of recently solved TM287-TM288 in an inward-facing conformation (31). Fig. 8 B shows that the first transmembrane segment of TM287-TM288 is located mainly in the periphery of the whole structure. Looking into the substrate translocation pathway from the cytoplasmic side, we see a very minor contribution of TM1 in the inner contour of the pore (Fig. 8 B). Similar observations were made with the crystal structure of the inward-facing configuration of another ABC exporter, P-glycoprotein (30). If, and we admit that it is a big if, CFTR and these ABC exporters share similar conformational changes during the gating/transport cycle, CFTR’s TM1 does undergo a fairly large-scale rearrangement during gating transitions.
Once we accept the possibility that TM1 undergoes major conformational changes during gating transitions, it is not surprising to see alterations in gating by the mutation itself (e.g., L102C), as well as by subsequent chemical modifications (Fig. 5). It is nevertheless interesting to note that in L102C/E1371Q-CFTR, the gate can open and close repeatedly even when the NBDs are in a dimeric configuration (Fig. S3). On the other hand, modification of L102C by MTSET literally renders the channel permanently open even long after ATP is removed and presumably NBDs have been separated. In other words, what these manipulations reveal is the potential autonomy of CFTR’s gate in TMDs irrespective of the status of its NBDs, an assertion echoing a revised gating model featuring an energetic coupling between CFTR’s NBDs and TMDs (10). In summary, the data presented in this work support the idea that contrary to TM6 and TM12, at least the cytoplasmic half of TM1 contributes to pore-lining only in the open channel conformation. Because only one face of TM1 helix lines the ion permeation pathway, suggests that even in the open state, side chains of the majority of amino acids in TM1 remain concealed by other parts of the CFTR protein or membrane lipids. We contend that these structural implications from our functional studies should be taken into consideration for future computer modeling of the CFTR protein.
Acknowledgments
We thank Shenghui Hu and Cindy Chu for their technical assistance.
This work was supported by National Institutes of Health (NIH) grant (NIHR01DK55835) and a grant (Hwang11P0) from the Cystic Fibrosis Foundation to T.-C. Hwang. This investigation was conducted in a facility constructed with support from Research Facilities Improvement Program (grant C06 RR-01648901) from the National Center for Research Resources, NIH.
Supporting Material
References
- 1.Riordan J.R., Rommens J.M., Kerem B., Alon N., Rozmahel R., Grzelczak Z., Zielenski J., Lok S., Plavsic N., Chou J.L. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 2.Bear C.E., Li C.H., Riordan J.R. Purification and functional reconstitution of the cystic fibrosis transmembrane conductance regulator (CFTR) Cell. 1992;68:809–818. doi: 10.1016/0092-8674(92)90155-6. [DOI] [PubMed] [Google Scholar]
- 3.Dean M., Annilo T. Evolution of the ATP-binding cassette (ABC) transporter superfamily in vertebrates. Annu. Rev. Genomics Hum. Genet. 2005;6:123–142. doi: 10.1146/annurev.genom.6.080604.162122. [DOI] [PubMed] [Google Scholar]
- 4.Gadsby D.C., Vergani P., Csanády L. The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature. 2006;440:477–483. doi: 10.1038/nature04712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen T.Y., Hwang T.C. CLC-0 and CFTR: chloride channels evolved from transporters. Physiol. Rev. 2008;88:351–387. doi: 10.1152/physrev.00058.2006. [DOI] [PubMed] [Google Scholar]
- 6.Hwang T.C., Sheppard D.N. Gating of the CFTR Cl- channel by ATP-driven nucleotide-binding domain dimerisation. J. Physiol. 2009;587:2151–2161. doi: 10.1113/jphysiol.2009.171595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsai M.F., Shimizu H., Hwang T.C. State-dependent modulation of CFTR gating by pyrophosphate. J. Gen. Physiol. 2009;133:405–419. doi: 10.1085/jgp.200810186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsai M.F., Li M., Hwang T.C. Stable ATP binding mediated by a partial NBD dimer of the CFTR chloride channel. J. Gen. Physiol. 2010;135:399–414. doi: 10.1085/jgp.201010399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Szollosi I., King S.J., Naughton M.T. Tachycardia in adults with cystic fibrosis is associated with normal autonomic function. Intern. Med. J. 2011;41:455–461. doi: 10.1111/j.1445-5994.2009.02039.x. [DOI] [PubMed] [Google Scholar]
- 10.Jih K.Y., Sohma Y., Hwang T.C. Identification of a novel post-hydrolytic state in CFTR gating. J. Gen. Physiol. 2012;139:359–370. doi: 10.1085/jgp.201210789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rees D.C., Johnson E., Lewinson O. ABC transporters: the power to change. Nat. Rev. Mol. Cell Biol. 2009;10:218–227. doi: 10.1038/nrm2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bai Y., Li M., Hwang T.C. Structural basis for the channel function of a degraded ABC transporter, CFTR (ABCC7) J. Gen. Physiol. 2011;138:495–507. doi: 10.1085/jgp.201110705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang W., Linsdell P. Alternating access to the transmembrane domain of the ATP-binding cassette protein cystic fibrosis transmembrane conductance regulator (ABCC7) J. Biol. Chem. 2012;287:10156–10165. doi: 10.1074/jbc.M112.342972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jih K.Y., Hwang T.C. Nonequilibrium gating of CFTR on an equilibrium theme. Physiology (Bethesda) 2012;27:351–361. doi: 10.1152/physiol.00026.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alexander C., Ivetac A., Dawson D.C. Cystic fibrosis transmembrane conductance regulator: using differential reactivity toward channel-permeant and channel-impermeant thiol-reactive probes to test a molecular model for the pore. Biochemistry. 2009;48:10078–10088. doi: 10.1021/bi901314c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bai Y., Li M., Hwang T.C. Dual roles of the sixth transmembrane segment of the CFTR chloride channel in gating and permeation. J. Gen. Physiol. 2010;136:293–309. doi: 10.1085/jgp.201010480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou J.J., Li M.S., Linsdell P. Regulation of conductance by the number of fixed positive charges in the intracellular vestibule of the CFTR chloride channel pore. J. Gen. Physiol. 2010;135:229–245. doi: 10.1085/jgp.200910327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qian F., El Hiani Y., Linsdell P. Functional arrangement of the 12th transmembrane region in the CFTR chloride channel pore based on functional investigation of a cysteine-less CFTR variant. Pflugers Arch. 2011;462:559–571. doi: 10.1007/s00424-011-0998-2. [DOI] [PubMed] [Google Scholar]
- 19.Wilson G.G., Karlin A. The location of the gate in the acetylcholine receptor channel. Neuron. 1998;20:1269–1281. doi: 10.1016/s0896-6273(00)80506-1. [DOI] [PubMed] [Google Scholar]
- 20.Engh A.M., Maduke M. Cysteine accessibility in ClC-0 supports conservation of the ClC intracellular vestibule. J. Gen. Physiol. 2005;125:601–617. doi: 10.1085/jgp.200509258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pascual J.M., Wang D., De Vivo D.C. Structural signatures and membrane helix 4 in GLUT1: inferences from human blood-brain glucose transport mutants. J. Biol. Chem. 2008;283:16732–16742. doi: 10.1074/jbc.M801403200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.El Hiani Y., Linsdell P. Changes in accessibility of cytoplasmic substances to the pore associated with activation of the cystic fibrosis transmembrane conductance regulator chloride channel. J. Biol. Chem. 2010;285:32126–32140. doi: 10.1074/jbc.M110.113332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Norimatsu Y., Ivetac A., Sansom M.S. Cystic fibrosis transmembrane conductance regulator: a molecular model defines the architecture of the anion conduction path and locates a “bottleneck” in the pore. Biochemistry. 2012;51:2199–2212. doi: 10.1021/bi201888a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beck E.J., Yang Y., Raghuram V. Conformational changes in a pore-lining helix coupled to cystic fibrosis transmembrane conductance regulator channel gating. J. Biol. Chem. 2008;283:4957–4966. doi: 10.1074/jbc.M702235200. [DOI] [PubMed] [Google Scholar]
- 25.Akabas M.H., Kaufmann C., Archdeacon P. Amino acid residues lining the chloride channel of the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 1994;269:14865–14868. [PubMed] [Google Scholar]
- 26.Wang W., Linsdell P. Relative movements of transmembrane regions at the outer mouth of the cystic fibrosis transmembrane conductance regulator channel pore during channel gating. J. Biol. Chem. 2012;287:32136–32146. doi: 10.1074/jbc.M112.385096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dawson R.J., Locher K.P. Structure of a bacterial multidrug ABC transporter. Nature. 2006;443:180–185. doi: 10.1038/nature05155. [DOI] [PubMed] [Google Scholar]
- 28.Dawson R.J., Locher K.P. Structure of the multidrug ABC transporter Sav1866 from Staphylococcus aureus in complex with AMP-PNP. FEBS Lett. 2007;581:935–938. doi: 10.1016/j.febslet.2007.01.073. [DOI] [PubMed] [Google Scholar]
- 29.Ward A., Reyes C.L., Chang G. Flexibility in the ABC transporter MsbA: alternating access with a twist. Proc. Natl. Acad. Sci. USA. 2007;104:19005–19010. doi: 10.1073/pnas.0709388104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aller S.G., Yu J., Chang G. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009;323:1718–1722. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hohl M., Briand C., Seeger M.A. Crystal structure of a heterodimeric ABC transporter in its inward-facing conformation. Nat. Struct. Mol. Biol. 2012;19:395–402. doi: 10.1038/nsmb.2267. [DOI] [PubMed] [Google Scholar]
- 32.Mornon J.P., Lehn P., Callebaut I. Atomic model of human cystic fibrosis transmembrane conductance regulator: membrane-spanning domains and coupling interfaces. Cell. Mol. Life Sci. 2008;65:2594–2612. doi: 10.1007/s00018-008-8249-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Serohijos A.W., Hegedus T., Riordan J.R. Phenylalanine-508 mediates a cytoplasmic-membrane domain contact in the CFTR 3D structure crucial to assembly and channel function. Proc. Natl. Acad. Sci. USA. 2008;105:3256–3261. doi: 10.1073/pnas.0800254105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mornon J.P., Lehn P., Callebaut I. Molecular models of the open and closed states of the whole human CFTR protein. Cell. Mol. Life Sci. 2009;66:3469–3486. doi: 10.1007/s00018-009-0133-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang W., El Hiani Y., Linsdell P. Alignment of transmembrane regions in the cystic fibrosis transmembrane conductance regulator chloride channel pore. J. Gen. Physiol. 2011;138:165–178. doi: 10.1085/jgp.201110605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li Y., Yu W.P., Chen T.Y. Oxidation and reduction control of the inactivation gating of Torpedo ClC-0 chloride channels. Biophys. J. 2005;88:3936–3945. doi: 10.1529/biophysj.104.055012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu X., Alexander C., Dawson D.C. Variable reactivity of an engineered cysteine at position 338 in cystic fibrosis transmembrane conductance regulator reflects different chemical states of the thiol. J. Biol. Chem. 2006;281:8275–8285. doi: 10.1074/jbc.M512458200. [DOI] [PubMed] [Google Scholar]
- 38.Csanady L. Rapid kinetic analysis of multichannel records by a simultaneous fit to all dwell-time histograms. Biophys. J. 2000;78:785–799. doi: 10.1016/S0006-3495(00)76636-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ma T., Thiagarajah J.R., Verkman A.S. Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J. Clin. Invest. 2002;110:1651–1658. doi: 10.1172/JCI16112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kopeikin Z., Sohma Y., Hwang T.C. On the mechanism of CFTR inhibition by a thiazolidinone derivative. J. Gen. Physiol. 2010;136:659–671. doi: 10.1085/jgp.201010518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lindley H. A study of the kinetics of the reaction between thiol compounds and choloracetamide. Biochem. J. 1960;74:577–584. doi: 10.1042/bj0740577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu X., Zhang Z.R., Dawson D.C. CFTR: a cysteine at position 338 in TM6 senses a positive electrostatic potential in the pore. Biophys. J. 2004;87:3826–3841. doi: 10.1529/biophysj.104.050534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zeltwanger S., Wang F., Hwang T.C. Gating of cystic fibrosis transmembrane conductance regulator chloride channels by adenosine triphosphate hydrolysis. Quantitative analysis of a cyclic gating scheme. J. Gen. Physiol. 1999;113:541–554. doi: 10.1085/jgp.113.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vergani P., Nairn A.C., Gadsby D.C. On the mechanism of MgATP-dependent gating of CFTR Cl-channels. J. Gen. Physiol. 2003;121:17–36. doi: 10.1085/jgp.20028673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou Z., Hu S., Hwang T.C. Voltage-dependent flickery block of an open cystic fibrosis transmembrane conductance regulator (CFTR) channel pore. J. Physiol. 2001;532:435–448. doi: 10.1111/j.1469-7793.2001.0435f.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu Y., Holmgren M., Yellen G. Gated access to the pore of a voltage-dependent K+ channel. Neuron. 1997;19:175–184. doi: 10.1016/s0896-6273(00)80357-8. [DOI] [PubMed] [Google Scholar]
- 47.Zhou J.J., Fatehi M., Linsdell P. Identification of positive charges situated at the outer mouth of the CFTR chloride channel pore. Pflugers Arch. 2008;457:351–360. doi: 10.1007/s00424-008-0521-6. [DOI] [PubMed] [Google Scholar]
- 48.Smith S.S., Steinle E.D., Dawson D.C. Cystic fibrosis transmembrane conductance regulator. Physical basis for lyotropic anion selectivity patterns. J. Gen. Physiol. 1999;114:799–818. doi: 10.1085/jgp.114.6.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Linsdell P. Mechanism of chloride permeation in the cystic fibrosis transmembrane conductance regulator chloride channel. Exp. Physiol. 2006;91:123–129. doi: 10.1113/expphysiol.2005.031757. [DOI] [PubMed] [Google Scholar]
- 50.McCarty N.A., Zhang Z.R. Identification of a region of strong discrimination in the pore of CFTR. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001;281:L852–L867. doi: 10.1152/ajplung.2001.281.4.L852. [DOI] [PubMed] [Google Scholar]
- 51.Aubin C.N., Linsdell P. Positive charges at the intracellular mouth of the pore regulate anion conduction in the CFTR chloride channel. J. Gen. Physiol. 2006;128:535–545. doi: 10.1085/jgp.200609516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tabcharani J.A., Rommens J.M., Hanrahan J.W. Multi-ion pore behaviour in the CFTR chloride channel. Nature. 1993;366:79–82. doi: 10.1038/366079a0. [DOI] [PubMed] [Google Scholar]
- 53.Cotten J.F., Welsh M.J. Cystic fibrosis-associated mutations at arginine 347 alter the pore architecture of CFTR. Evidence for disruption of a salt bridge. J. Biol. Chem. 1999;274:5429–5435. doi: 10.1074/jbc.274.9.5429. [DOI] [PubMed] [Google Scholar]
- 54.Kyte J., Doolittle R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 55.White S.H., Wimley W.C. Membrane protein folding and stability: physical principles. Annu. Rev. Biophys. Biomol. Struct. 1999;28:319–365. doi: 10.1146/annurev.biophys.28.1.319. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.