

Abstract
Adenoviruses are commonly used in vitro as gene transfer vectors in multiple applications. Nevertheless, issues such as low infection efficiency and toxicity effects on host cells have not been resolved yet. This work aims at developing a new versatile tool to enhance the expression of transduced genes while working at low viral doses in a sequential manner. We developed a microfluidic platform with automatically controlled sequential perfusion stages, which includes 10 independent channels. In addition, we built a stochastic mathematical model, accounting for the discrete nature of cells and viruses, to predict not only the percentage of infected cells, but also the associated infecting-virus distribution in the cell population. Microfluidic system and mathematical model were coupled to define an efficient experimental strategy. We used human foreskin fibroblasts, infected by replication-incompetent adenoviruses carrying EGFP gene, as the testing system. Cell characterization was performed through fluorescence microscopy, followed by image analysis. We explored the effect of different aspects: perfusion, multiplicity of infection, and temporal patterns of infection. We demonstrated feasibility of performing efficient viral transduction at low doses, by repeated pulses of cell-virus contact. This procedure also enhanced the exogenous gene expression in the sequential microfluidic infection system compared to a single infection at a higher, nontoxic, viral dose.
Introduction
The precise control of in vitro viral transduction is highly desirable in several applications, i.e., in the optimization of gene therapies (1–3), RNA interference experiments (4), induced pluripotent stem cell production (5), or for promoting stem cell differentiation (6). Nonreplicating adenoviral vectors (AdVs) are increasingly used for such purposes, because they introduce transgenes into cells without uncontrollable random integrations in host DNA and minimizing other perturbations of cell homeostasis (7). In addition, they are easy to manufacture and have the ability to infect also quiescent cells (8). On the other hand, transduction by AdVs suffers from low efficiency (9), and using high viral doses to achieve sufficient transduction induces cytotoxicity, as previously demonstrated not only in vivo (7,10), but also in vitro (7,11–14), using different types of primary cells and cell lines. Cytotoxicity is dose-dependent, and in vitro studies do not show significant effects on the cells at low multiplicities of infection (MOIs) (11,12). MOIs are defined as the number of viral particles per cell.
Chuck and Palsson (15) used convective flow conditions to enhance virus-cell contact and increase the probability of virus entrance into the cells. Furthermore, microfluidics can improve the control over virus-mediated gene delivery even at very low MOIs. In fact, microfluidic devices overcome some of the limitations of conventional bench-top systems, taking advantage not only of their high surface/volume ratio but also of laminar flow in the microfluidic channels (16). Microfluidics has already been used in applications involving viral particles, especially in the field of biosensors (for example, for infectious virus detection (17,18)). However, some devices were also developed for novel interesting applications: to measure virus infectivity and antiviral drug efficacy (19), for efficiently studying virus inactivation (20), and for virus-blood cell separation (21). Furthermore, a microfluidic bioreactor for virus production (22), and a device for protein production generating virus concentration gradients (23) were designed.
The highly defined environment in a microfluidic system, coupled with the possibility of easily automating its control, makes simple to explore a huge range of experimental conditions. However, a precise understanding of the physical and biological phenomena involved is needed for a rational experimental design. Multiple mathematical models describing viral infection have been developed on different scales. Efforts to quantitatively describe viral dynamics in vivo have been reviewed by Perelson (24). More detailed computational models, based on in vitro viral infection data, include works describing virus transport to the cell surface (25,26), virus-cell interaction (25,27,28), and intracellular viral trafficking (29,30).
Tayi et al. (31) developed a very exhaustive deterministic mathematical model including the steps from virus transport in the extracellular medium to viral transgene expression. Although computationally more requiring, a stochastic approach in modeling virus transduction dynamics is more appropriate, as both viruses and cells are discrete particles. Seisenberger et al. (26) experimentally showed that viruses move within the culture medium by Brownian motion and, once they get to a cell surface, they have a certain probability of penetration through the membrane. Some of the models mentioned above partly adopted a probabilistic approach, but including only the discrete nature of viruses and not of cells (25,27–29). Thus, they could not calculate how viruses distribute within the cell population without further assumptions, such as the approximation of a Poisson distribution for the fraction of cells infected by a given number of viruses (31).
In this work, we developed a useful methodology for the accurate control of time-dependent gene delivery by AdVs, by integrating experimental and theoretical analyses. We realized a microfluidic platform to perform up to 10 parallel independent experiments in a single miniaturized chip, with accurate control of perfusion stages. Given this setup, multiple operating conditions, in terms of mass transport parameters and temporal profiles of virus delivery, could be easily tested. Enhanced green fluorescent protein (EGFP) was used as a reporter of cell infection for online detection of the temporal evolution of the experiment. In parallel, we developed a stochastic simulation algorithm to theoretically study the viral transduction process. The model accounts for the discrete nature of both cells and viruses in a three-dimensional space. The computational simulations show not only the percentage of infected cells, but also the distribution of viral particles in the cell population. With these tools, we studied the feasibility of performing efficient AdV transduction experiments at low viral dose, by performing multiple sequential pulses of viral transduction.
Materials and Methods
Cell culture
Human foreskin fibroblasts were expanded in gelatin-coated 75-mL flasks with high-glucose Dulbecco’s Modified Eagle’s Medium (DMEM 41965; Invitrogen, Carlsbad, CA), supplemented with 10% fetal bovine serum (Invitrogen) and 1% penicillin/streptomycin (Invitrogen). At 90% confluency, cells were trypsinized with trypsin-EDTA 0.25% (Invitrogen), centrifugated at 1200 rpm for 5 min, and resuspended in growth medium for seeding.
Adenoviral vectors
Replication-incompetent adenoviral vectors (AdVs) based on the Ad5 genome and lacking the E1 and E3 regions were constructed by homologous recombination in Escherichia coli using the AdEasy vector system (Qbiogene, Carlsbad, CA). In these vectors, human cytomegalovirus promoter was used to drive expression of EGFP. Transgenic adenoviral vectors were propagated in E1-complementing HEK 293 cells, purified by cesium chloride density centrifugation, and titrated by TCID50 CPE endpoint assay according to the AdEasy production protocol (Qbiogene). Viral vector stocks were stored at 2 × 108 PFU/mL concentration in 10% glycerol at −80°C until use.
Microfluidic device
The microfluidic platform contains 10 independent channels for cell culture, each connected to a built-in medium reservoir of ∼70 μL, and to a syringe pump system for fluid handling. A schematic representation of the microfluidic system is shown in Fig. 1 A, and its dimensions are reported in Fig. 1 B.
Figure 1.
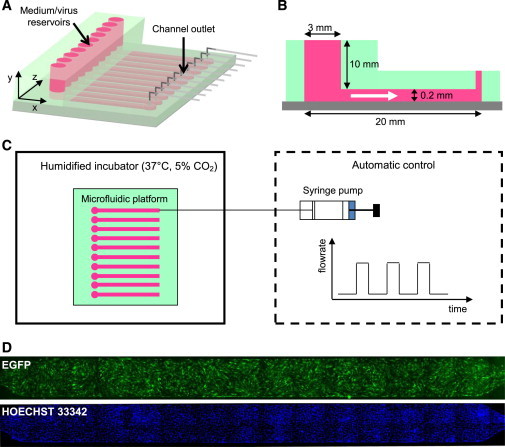
Microfluidic experimental setup. (A) Three-dimensional graphical representation of the microfluidic platform composed of 10 parallel independent channels. (B) Lateral section of one channel indicating its geometrical dimensions. (White arrow) Flow direction. (C) Overview of the whole system: the microfluidic platform (top view on the left) is placed in a biological incubator during experiments, and medium perfusion (from left to right) in every channel is provided by a set of syringe pumps, whose temporal pattern of flow rate is automatically controlled. (D) Images of the whole microfluidic channel taken with a fluorescence microscope to detect EGFP+ cells and cell nuclei.
The microfluidic chip was fabricated by common soft-lithographic techniques (32). The master was photolitographically patterned by using SU8-2100 negative photoresist (MicroChem, Newton, MA) to obtain a final thickness of 200 μm, according to manufacturer’s indications. A PDMS mold was obtained by casting a premixed 10:1 ratio prepolymer and curing agent solution (Sylgard 184 kit; Dow Corning, Midland, MI) on the silicon wafer. After curing at 70°C for 2 h, the PDMS mold was cut, peeled off, and punched with a 21G stainless steel needle (Small Part Inc., Logansport, IN) to obtain inlet/outlet holes. The PDMS mold was assembled and sealed to a 50 × 75 mm cleaned glass slide by plasma bonding. Ten independent medium reservoirs, one for each channel, were obtained by sealing an additional PDMS block to the top of the device by plasma bonding. The assembled device was cleaned with isopropanol (Sigma-Aldrich, St. Louis, MO) and sterilized in an autoclave. A syringe pump with a 10-syringe rack (Harvard Pump; Harvard Apparatus, Holliston, MA) was used to control the medium flow rate from the reservoirs into the microfluidic channels. We also used 0.5 ID Tygon tubings (Cole-Parmer, Vernon Hills, IL) and 21G stainless-steel needles with a polypropylene luer (Microtest, Taipei, Taiwan) to connect the microfluidic chip to the syringes. The entire experimental setup is shown in Fig. 1 C.
Viral transduction experiments
Before cell seeding, the microfluidic channels were coated with gelatin 0.6% and incubated at 37°C and 5% CO2 atmosphere for 24 h. The cell suspension was injected into each channel and the microfluidic chip was incubated overnight at 37°C and 5% CO2 atmosphere without perfusion. We used a proper cell seeding concentration to achieve confluent cell culture (corresponding to a final cell density of 150 cell/mm2) at 24 h after seeding. Then, discontinuous medium refill was achieved by automatically controlling the multichannel syringe pump through LabView 8.2 (National Instruments, Austin, TX), without further manual intervention. The pump was programmed to run every 3 h for 4 min with a flow rate of 3 μL/min for medium change.
Twenty-four hours after cell seeding, AdV transduction was performed. The viral stock solution was thawed and diluted with culture medium to the final concentration. Medium perfusion into the microchannels was performed with different dynamics through the automated syringe pump that aspirates medium from the channels. Each experimental condition was tested in at least two microfluidic channels in parallel. Parallel control experiments were performed in microchannels supplied with AdV-free medium during viral transduction.
The conventional bench-top system used as a reference is a 24-well tissue culture plate (Becton Dickinson, Franklin Lakes, NJ). Cells were seeded and cultured for 24 h in 1 mL medium. Then, medium was replaced with 200-μL AdV-containing medium at different concentrations. After incubation for tf = 90 min at 37°C and 5% CO2, medium was changed again with 1 mL virus-free medium that was kept until the end of the experiment. Each experimental condition was tested in at least three wells in parallel. Three wells were used as controls in every experiment and were supplied with medium without AdVs during viral transduction.
Cell characterization
Cell count and EGFP expression were detected in situ by fluorescence microscopy (DMI 6000-B; Leica Microsystems, Milan, Italy). Cell nuclei were stained by incubating live cells with 2 μg/mL Hoechst 33342 dye (Invitrogen) in culture medium for 30 min. Bright-field and fluorescence images were taken at 10× magnification, with exposure, gain, and intensity parameters equal to the respective control in each culture system. Every channel was entirely captured for the microfluidic system (Fig. 1 D), and three randomly chosen positions in every well were acquired for the multiwell plate.
The cell density and the percentage of EGFP+ cells were quantified by image analysis, using the software MATLAB (The MathWorks, Natick, MA), as previously described in Cimetta et al. (16). We performed this image analysis using different values for the threshold of fluorescence intensity, f, that discriminates between EGFP positive and negative cells. In this way, we could quantify the number of cells expressing EGFP at different levels, and not just the number of EGFP+ cells.
Stochastic simulation algorithm
The mathematical model developed includes the extracellular virus transport in the culture medium (by diffusion and convection) and the entrance of viruses into the cells. Convective transport, present only in the microfluidic channels during filling and emptying, occurs in the x direction (Fig. 2 A) with a parabolic velocity profile, vx(y).
Figure 2.
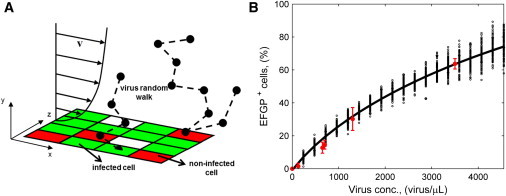
Stochastic model processes and parameter fitting. (A) Graphical representation of the phenomena included in the stochastic model: medium convection with parabolic velocity profile, virus Brownian motion in three-dimensional space, and virus entering a cell with a certain probability when it gets on its surface. Plane x-z represents the bottom surface of the channel where cells are randomly distributed within a regular square grid. (B) Results of model parameter fitting. Experimentally, transduction was performed in a 24-well plate using 200 μL of virus-containing medium for 90 min. Cell concentration was 130 cell/mm2. (Red error bars) Percentage of cells expressing EGFP 24 h after AdV transduction as a function of virus concentration, mean ± standard deviation values obtained in independent experiments, each performed in double or triple. Simulations by the stochastic model reproduce the experimental conditions. In the model, EGFP+ cells are given by cells infected by at least one virus. (Black dots) Model outcome, each dot is obtained from one simulation at the given virus concentration. One-hundred simulations were performed at each condition. (Black line) Simulation mean results.
The geometry simulated includes the whole culture channel for the microfluidic system. For the conventional multiwell plate, a representative subvolume of the well (1.73-mm2 bottom area), having the same height as the medium during viral transduction (H = 1 mm), was simulated.
Choosing a time step Δt for the simulation, the position (xi, yi, zi) of the ith virus at time t + Δt, given that at time t, was computed according to the Euler scheme of discretization for the Smoluchowski stochastic differential equation:
| (1) |
where D is the diffusion coefficient of the virus in medium, and ξx,i, ξy,i, and ξz,i are normally distributed random variables with zero mean and unit variance. The model was solved in MATLAB.
In both systems, cells were randomly distributed at the bottom within a regular square grid (Fig. 2 A), to have the same density as in the experiments. The grid size was equal to dcell = 65.7 μm, to have squares of the same cell surface area as that experimentally measured (4300 ± 560 μm2). We neglected cell division because contact inhibition prevails at the cell seeding density used, as we verified during the experiments (see the Supporting Material).
In the simulated subdomain of the multiwell plate, at time t = 0 viruses were randomly placed within the three-dimensional medium volume to match the experimental concentration. Instead, in simulating the microfluidic system, virus concentration was null at time t = 0, and then viruses progressively entered with the flowing medium, at the appropriate concentration, distributed randomly on the inlet section surface.
Virus entrance into the cells was described in the model as a partially adsorbing boundary condition at y = 0, because not all cell-virus contacts are successful for introducing the virus into the cell (26). Formally, the probability that the ith virus enters a cell in the time span Δt is given by , if the virus has yi(t +Δt) ≤ 0, where P1 is a model parameter; or
otherwise. More details on this boundary condition are provided in the Supporting Material and in Erban and Chapman (33). The other boundary conditions were set as reflection, a part for the open boundaries at medium inlet and outlet of the channel. We assumed that cells infected by at least one virus, then express EGFP.
Model parameter values
The model parameter values are summarized in Table 1. The parameters related to system geometries and operative variables were fixed to match the experimental conditions. The value of the diffusion coefficient (D) was taken from Oliver et al. (34). Parameter P1 was derived by fitting the experimental data obtained in the multiwell plate under static conditions. Specifically, we minimized the following cost function, φ,
| (2) |
where superscripts mod and exp refer to model and experimental values, respectively; and xi and σi are the mean and standard deviation of the percentage of EGFP+ cells, obtained at the ith virus concentration. Parameter fitting results are shown in Fig. 2 B in terms of both mean and standard deviation due to experimental uncertainty and model stochasticity.
Table 1.
Parameter values of the model
| Parameter | Value |
|---|---|
| Cell equivalent size (dcell) | 65.7 μm |
| Culture volume height (H) | 1000 μm (multiwell)200 μm (microfluidics) |
| Culture volume length (L) | 1314 μm (multiwell)17 × 103μm (microfluidics) |
| Culture volume width (W) | 1314 μm (multiwell)1500 μm (microfluidics) |
| Virus diffusion coefficient (D) | 3.67 μm2/s |
| AdV-cell interaction constant (P1) | 0.0079 s−0.5 |
| Cell seeding density (ρcell) | 130–150 cell/mm2 |
| AdV transduction duration (tf) | 90 min |
| Simulated time step (Δt) | 0.16 s |
Results and Discussion
Spatial heterogeneity in microfluidics
To perform transduction experiments in a microfluidic channel, virus-containing medium needs to be perfused over the cells. As medium flows from inlet to outlet, it gets depleted of the viruses that enter the cells in between; hence, cells located in the downstream part of the channel are exposed to a lower MOI than those near the inlet. We computationally investigated how relevant is spatial heterogeneity in a perfused channel.
To begin, we simulated a channel perfused with a constant flow rate of 0.1 μL/min (Peclet number equal to 300). The medium entering the system had a virus concentration corresponding to an instantaneous MOI of 20 in the system, given a cell density of 130 cell/mm2. We defined the MOI as instantaneous because it represents the number of viruses per cell within the channel at a given time. On the other hand, a global MOI can be defined by considering the total number of viruses entering the system during the whole time span of infection. The time of perfusion was set to 90 min, because it is the time (tf) required for a viral particle to travel a distance s = 200 μm (the channel height) by Brownian motion, calculated from the Einstein’s relation,
| (3) |
where D is the diffusion coefficient of the virus in medium. One-hundred stochastic simulations were performed. We verified that the whole channel was perfused by calculating that, at the flow rate simulated, 90 min correspond to 1.7 residence times.
The length of the channel was divided in 10 sectors, and in each sector the percentage of EGFP+ cells, i.e., of cells infected by at least one virus, was calculated. The results are shown in Fig. 3, A (top) and C. The percentage of positive cells in the last sector, near the outlet, is ∼20% less than that in the first sector, at the inlet. Thus, spatial heterogeneity can be a relevant phenomenon introduced by perfusion that affects the quality of the resulting cell population. We also studied this aspect at different MOIs in the Supporting Material. Although at high MOI all the cells are infected, we verified that heterogeneity is still present in terms of virus distribution within the cell population, producing a gradient of infection level along the channel (Fig. 3 A, bottom).
Figure 3.
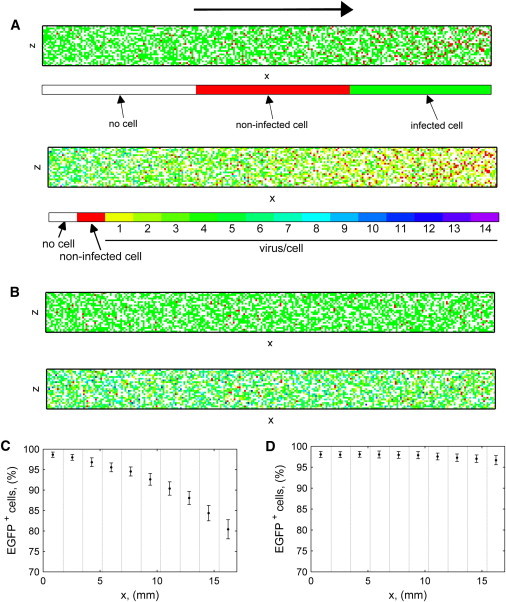
Computational study of spatial heterogeneity in the microfluidic system. (A and C) Results of stochastic simulations of continuous channel perfusion with a flow rate of 0.1 μL/min for 90 min. (B and D) Results of simulations of discontinuous perfusion: 2 min of inflow at 6 μL/min, 90 min without perfusion, and 2 min of outflow at 6 μL/min. (A and B) Bottom of a microfluidic channel: the EGFP+ cells (top) and the number of virus/cell (bottom) are shown; color meaning is explained in color bars in panel A. (Black arrow) Flow direction. (C and D) (Dotted lines) Percentage of EGFP+ cells in each of the 10 equal sectors of the channel. (Error bars) Mean ± standard deviation of 100 simulations. (A–D) Cell concentration is 130 cell/mm2, instantaneous MOI is 20.
To avoid this issue, we developed a different strategy for providing virus-containing medium to the cells, based on discontinuous perfusion. Keeping the same system geometry, cell density, and virus concentration as in the previous case, we simulated a perfusion of 2 min at a flow rate of 6 μL/min, 90 min without perfusion, and then 2 min of outflow. We verified that the shear stress occurring at 6 μL/min, ∼6.8 mPa, is well below the threshold of cell damage (35), and can be effectively used during biological experiments. As shown in Fig. 3, B and D, by performing the infection under these discontinuous conditions, spatial heterogeneity is within the stochastic noise of the system. EGFP+ cells are homogeneously distributed along the channel, because the 2 min of inflow and outflow negligibly affect the overall outcome.
The whole channel is filled in less than a minute at this flow rate and virus adsorption does not occur fast enough to produce a gradient in the infection level along the channel. On the contrary, in the previous case, channel filling took ∼50 min, a time of the same order of magnitude of the characteristic time of adsorption. Other conditions of flow were simulated confirming this hypothesis, whose results are presented in the Supporting Material. Furthermore, perfusion conditions may also affect the overall cell quality by changing the accumulation of endogenous factors in cell microenvironment, as recently reported in Moledina et al. (36). A discontinuous flow protocol better preserves cell microenvironment while ensuring a homogenous transfection along the microfluidic channel.
The homogeneity of transfection was confirmed also by experimental observations in our microfluidic system, as shown in the Supporting Material. All the results presented in the next sections, both computational and experimental, are obtained applying a discontinuous flow.
Effect of MOI after single viral infection
We computationally investigated the effect of MOI on the percentage of EGFP+ cells and the infecting-viruses distribution. We simulated the transduction process in a microfluidic channel for a cell density of 150 cell/mm2 at four different MOIs: 20 (equal to a concentration of 15,000 PFU/μL), 10 (7500 PFU/μL), 5 (3750 PFU/μL), and 1 (750 PFU/μL). Fig. 4 shows the resulting probability and complementary cumulative distributions of the percentage of cells infected with different numbers of viruses, as calculated by the model. Despite the spatial homogeneity of performing AdV transduction in microfluidics under discontinuous flow (Fig. 3), the discrete nature of cells and viruses, and how they are randomly distributed on the bottom surface and in the medium volume, respectively can still generate a heterogeneous cell population for the number of viruses infecting each cell. At MOI 1, almost all the cells infected have only one virus, but at higher MOIs, heterogeneity is visible in the cell population (Fig. 4).
Figure 4.
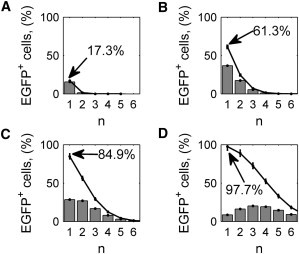
Computational study of viral transduction at different MOIs. Results of 100 stochastic simulations at MOI 1 (A), 5 (B), 10 (C), and 20 (D), in a microfluidic channel under discontinuous flow conditions, at a cell concentration of 150 cell/mm2. Bar plots represent the percentage of EGFP+ cells infected by n viruses. (Solid lines) Complementary cumulative distribution. In each plot the percentage of EGFP+ cells (i.e., cells infected by at least one virus) is also indicated. (Error bars) Mean ± standard deviation of the 100 simulations.
Furthermore, the effective mean number of viruses per cell is much lower than the theoretical one, given by the MOI, confirming previous work in Andreadis et al. (37). For example, at MOI 20 the mean number of viruses per cell is only between 3 and 4 (Fig. 4 D). Fig. 4 shows also the percentage of cells infected by at least one virus, i.e., of EGFP+ cells, according to the hypotheses described in Materials and Methods. As expected, the efficiency of the infection process increases at higher MOIs.
Experimentally, given the very great surface/volume ratio of microfluidic systems, we first verified that there were not substantial AdV losses due to binding on the surfaces of the channels. The results are reported in the Supporting Material. Then, we performed experiments of viral transduction in the microfluidic platform under the same conditions simulated above and characterized the cell population at 24 h after the beginning of infection. We assumed that this period of time is sufficient for infected cells to express EGFP protein, as reported previously in de Martin et al. (38). The very good agreement between the percentages of EGFP+ cells obtained from experimental data and predicted by the model (Fig. 5 A), shows the stochastic model fairly describes the overall infection process within the microfluidic platform. This result is particularly relevant considering that all extra- and intracellular biological phenomena of viral infection (excluding those related to extracellular viral particle mass transport) are lumped in an overall limiting step, whose parameter was obtained by experimental data collected under static conditions in a multiwell plate.
Figure 5.
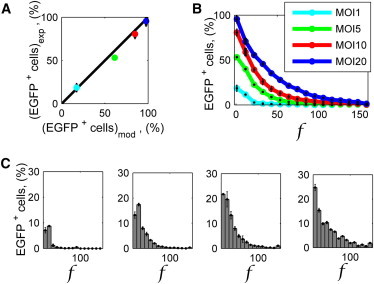
Experimental study of viral transduction in the microfluidic platform at different MOIs. (A) Comparison of experimental (subscript exp) and simulated (subscript mod) percentage of EGFP+ cells. Marker colors are defined by legend in panel B. (B) Complementary cumulative distribution of the percentage of EGFP+ cells showing a fluorescence intensity > f (see Cell Characterization in main text). (C) Probability distribution of the percentage of EGFP+ cells showing a fluorescence intensity f at MOI 1, 5, 10, and 20 (from left to right). (A–C) The percentage of cells expressing EGFP was detected 24 h after AdV transduction. Cell concentration was 150 cell/mm2. (Error bars) Mean ± standard deviation values obtained in independent experiments, each performed in double or triple.
We investigated the experimental heterogeneity of the cell population as for the EGFP produced at the single-cell level. We quantified the intensity of fluorescence, f, of each cell by image analysis, as described in Materials and Methods. The results are shown in Fig. 5, B and C, in terms of complementary cumulative distribution and of probability distribution, respectively. Besides increasing the percentage of cells expressing EGFP, AdV transduction performed at higher MOIs enhances the amount of exogenous RNA and consequently the amount of EGFP produced by single cells. This could be related to the increase in the number of viruses infecting each cell, as explored computationally (Fig. 4). However, multiple other processes overlap (such as virus intracellular transport, cell cycle, regulation of transcription and translation), and a straight correlation between the two variables, number of virus per cell, and intensity of cell fluorescence, is not possible. A simplified model that correlates these two variables is presented in the Supporting Material.
Multiple pulses of viral transduction
Thanks to the developed technology, the same efficiency of infection of cell culture could be obtained by infecting cells several times at low MOI, thus preventing any cytotoxic effect due to external viral particles. For this reason, we explored the possibility of performing multiple sequential pulses of infection at low viral doses to increase protein expression over time and avoid the cell toxicity of high MOIs. We compared the outcome of a single viral transduction at MOI 20 (still low enough not to be toxic) with that of two infections at MOI 10, and of four infections at MOI 5. The total number of viruses entering the system was thus the same, but three different regular temporal sequences were used (Fig. 6). We simulated these three scenarios with the stochastic model, and found that theoretically the same outcome is achievable with a multipulse strategy, in terms of percentage of EGFP+ cells and of the distribution of the number of viruses per cell (Fig. 6).
Figure 6.
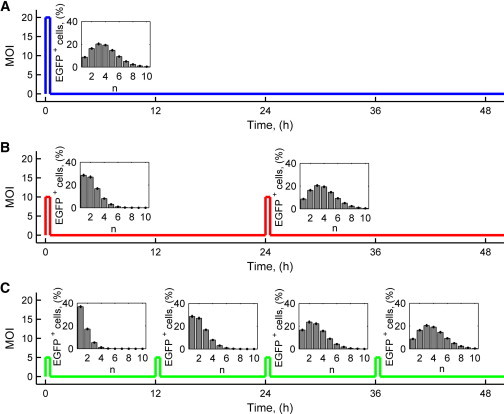
Multiple viral transductions strategy and computational results. Repeated 90-min AdV infections with MOI 20 (A), 10 (B), and 5 (C), at the time-points indicated. (Insets) Model predictions of the percentage of EGFP+ cells infected by n viruses at the end of each pulse of infection. Cell concentration was 150 cell/mm2. (Error bars) Mean ± standard deviation of 100 simulations at each condition.
Taking advantage of the precise automation of flow rate control in the microfluidic platform, we performed the experiment corresponding to the theoretical simulations described above. We infected the cells at time 0 and the other time-points given in the three protocols, using a discontinuous flow strategy, i.e., 2 min of virus-containing medium flow (or virus-free medium in the channels not to be infected), 90 min of infection under static conditions, and 2 min of virus-free medium flow. We monitored the level of cell EGFP expression 24 h after each infection, i.e., at the following time points: 24, 36, 48, and 60 h after the first infection. The results are presented in Fig. 7, in terms of probability and complementary cumulative distributions. Fig. 7 D shows a comparable EGFP gene expression with the three protocols after 48 h, while at 60 h channels treated with multipulse viral transduction at lower MOIs show a higher percentage of cells being more fluorescent (Fig. 7 D).
Figure 7.
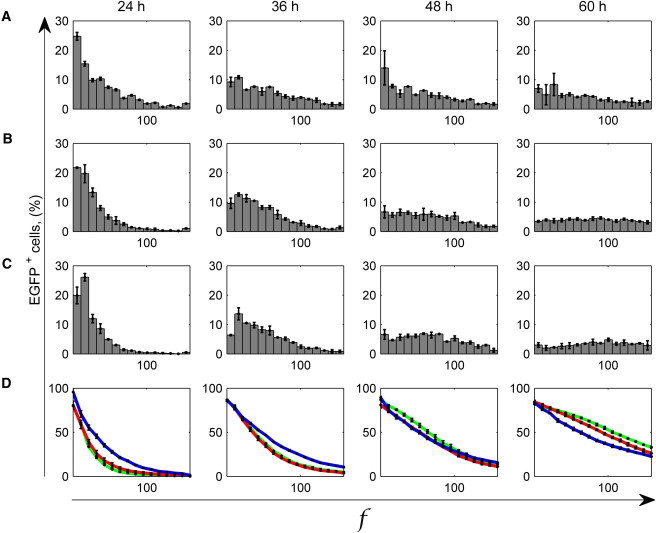
Results from experiments of multiple viral transductions within the microfluidic platform. AdV transduction timing followed the strategy shown in Fig. 6: one infection at MOI 20 (A and blue line in D), two infections at MOI 10 (B and red line in D), and 4 at MOI 5 (C and green line in D). EGFP+ cell fluorescence intensity, f, was measured at 24, 36, 48, and 60 h after the first infection. Cell concentration was 150 cell/mm2. (Error bars) Mean ± standard deviation of experiments repeated twice. (A–C) Probability distribution of the percentage of EGFP+ cells showing a fluorescence intensity f, and (D) associated complementary cumulative distribution.
In this experiment, the maximum MOI used was 20. As previously verified, this is below the threshold of cytotoxicity, identified approximately at MOI 50 (11,12). These conditions, coupled with a constant cell density due to contact inhibition, are useful to detect the effect of a sequential infection strategy at net of cell division and death phenomena, which are also not included in the stochastic model. Overall, a comparison between the percentages of EGFP+ cells (Fig. 7 D) shows the feasibility of performing sequential viral transduction experiments without loss of efficiency, and highlights the importance of the multipulse infection strategy in cases when cytotoxicity is a concern. We performed sequential transduction experiments also at very high MOIs, as reported in the Supporting Material, and the results strongly support the reduction in cytotoxicity achievable.
Furthermore, the higher EGFP expression achieved after repeated infections at MOI 5 than after a single one at MOI 20, confirms the complexity of gene expression regulation, which is not linearly dependent on the amount of exogenous DNA inserted. Thus, the sequential infection strategy may be a useful means also to study complex dynamics of gene expression in a highly controlled system.
Conclusions
Viral vectors are increasingly used to insert exogenous genetic material in cells. Low efficiency of infection at low MOIs and cytotoxicity at high MOIs are two critical limitations affecting the process. In this work, we showed the feasibility of performing sequential infections at low viral dose with high efficiency, taking advantage of microfluidic technology to provide a highly defined temporal control over the cell culture environment.
We coupled the experiments with a theoretical analysis through a stochastic model to follow the dynamics of virus entrance into the cells under continuous and discontinuous flow conditions. In particular, being discrete in its description of viruses and cells, the model can recapitulate also the heterogeneity in the cell population without further assumptions.
Under the simplifying hypotheses of the model, one infection at high viral dose and multiple at low doses, using the same overall amount of viruses, should give the same outcome in terms of distribution of viruses in the infected cell population. However, we also found experimentally that, compared to a single infection at high MOI, performing multiple low-dose infections affects the gene expression profile of the resulting cell population. Thus, the strategy developed to perform time-controlled multipulse infections, in addition to reducing viral toxicity, could be a powerful tool to investigate the dynamic complexity of the expression of exogenous genes using a highly controlled system.
Acknowledgments
This research was supported by Progetti di Eccellenza CARIPARO and Progetti di Eccellenza Giovani Ricercatori of Ministero della Salute.
Supporting Material
References
- 1.Wu Z.J., Asokan A., Samulski R.J. Adeno-associated virus serotypes: vector toolkit for human gene therapy. Mol. Ther. 2006;14:316–327. doi: 10.1016/j.ymthe.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 2.Carty M., Bowie A.G. Recent insights into the role of Toll-like receptors in viral infection. Clin. Exp. Immunol. 2010;161:397–406. doi: 10.1111/j.1365-2249.2010.04196.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ames R.S., Lu Q. Viral-mediated gene delivery for cell-based assays in drug discovery. Expert. Opin. Drug Discov. 2009;4:243–256. doi: 10.1517/17460440902751599. [DOI] [PubMed] [Google Scholar]
- 4.Arts G.J., Langemeijer E., van Es H. Adenoviral vectors expressing siRNAs for discovery and validation of gene function. Genome Res. 2003;13:2325–2332. doi: 10.1101/gr.1332603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takahashi K., Tanabe K., Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 6.Takayama K., Inamura M., Mizuguchi H. Efficient generation of functional hepatocytes from human embryonic stem cells and induced pluripotent stem cells by HNF4α transduction. Mol. Ther. 2012;20:127–137. doi: 10.1038/mt.2011.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu Q., Muruve D.A. Molecular basis of the inflammatory response to adenovirus vectors. Gene Ther. 2003;10:935–940. doi: 10.1038/sj.gt.3302036. [DOI] [PubMed] [Google Scholar]
- 8.Ghosh S.S., Gopinath P., Ramesh A. Adenoviral vectors: a promising tool for gene therapy. Appl. Biochem. Biotechnol. 2006;133:9–29. doi: 10.1385/abab:133:1:9. [DOI] [PubMed] [Google Scholar]
- 9.Stadtfeld M., Nagaya M., Hochedlinger K. Induced pluripotent stem cells generated without viral integration. Science. 2008;322:945–949. doi: 10.1126/science.1162494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bangari D.S., Mittal S.K. Current strategies and future directions for eluding adenoviral vector immunity. Curr. Gene Ther. 2006;6:215–226. doi: 10.2174/156652306776359478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brand K., Klocke R., Strauss M. Induction of apoptosis and G2/M arrest by infection with replication-deficient adenovirus at high multiplicity of infection. Gene Ther. 1999;6:1054–1063. doi: 10.1038/sj.gt.3300914. [DOI] [PubMed] [Google Scholar]
- 12.Matkovic U., Pacenti M., Barzon L. Investigation on human adrenocortical cell response to adenovirus and adenoviral vector infection. J. Cell. Physiol. 2009;220:45–57. doi: 10.1002/jcp.21727. [DOI] [PubMed] [Google Scholar]
- 13.Hartman Z.C., Black E.P., Amalfitano A. Adenoviral infection induces a multi-faceted innate cellular immune response that is mediated by the toll-like receptor pathway in A549 cells. Virology. 2007;358:357–372. doi: 10.1016/j.virol.2006.08.041. [DOI] [PubMed] [Google Scholar]
- 14.Müller J., Thirion C., Pfaffl M.W. Electric cell-substrate impedance sensing (ECIS) based real-time measurement of titer dependent cytotoxicity induced by adenoviral vectors in an IPI-2I cell culture model. Biosens. Bioelectron. 2011;26:2000–2005. doi: 10.1016/j.bios.2010.08.075. [DOI] [PubMed] [Google Scholar]
- 15.Chuck A.S., Palsson B.O. Consistent and high rates of gene transfer can be obtained using flow-through transduction over a wide range of retroviral titers. Hum. Gene Ther. 1996;7:743–750. doi: 10.1089/hum.1996.7.6-743. [DOI] [PubMed] [Google Scholar]
- 16.Cimetta E., Franzoso M., Elvassore N. Microfluidic-driven viral infection on cell cultures: theoretical and experimental study. Biomicrofluidics. 2012;6:024127. doi: 10.1063/1.4723853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng X., Chen G., Rodriguez W.R. Micro- and nanotechnology for viral detection. Anal. Bioanal. Chem. 2009;393:487–501. doi: 10.1007/s00216-008-2514-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kremser L., Blaas D., Kenndler E. Virus analysis using electromigration techniques. Electrophoresis. 2009;30:133–140. doi: 10.1002/elps.200800590. [DOI] [PubMed] [Google Scholar]
- 19.Zhu Y., Warrick J.W., Yin J. Infection on a chip: a microscale platform for simple and sensitive cell-based virus assays. Biomed. Microdevices. 2009;11:565–570. doi: 10.1007/s10544-008-9263-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bailey M.R., Chen D., Shamlou P.A. Evaluation of microfluidics reactor technology on the kinetics of virus inactivation. Biotechnol. Bioeng. 2008;99:1384–1391. doi: 10.1002/bit.21714. [DOI] [PubMed] [Google Scholar]
- 21.Zhao C., Cheng X. Microfluidic separation of viruses from blood cells based on intrinsic transport processes. Biomicrofluidics. 2011;5:32004–3200410. doi: 10.1063/1.3609262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vu H.N., Li Y., Yarmush M.L. A microfluidic bioreactor for increased active retrovirus output. Lab Chip. 2008;8:75–80. doi: 10.1039/b711577f. [DOI] [PubMed] [Google Scholar]
- 23.Walker G.M., Ozers M.S., Beebe D.J. Cell infection within a microfluidic device using virus gradients. Sens. Actuators B Chem. 2004;98:347–355. [Google Scholar]
- 24.Perelson A.S. Modeling viral and immune system dynamics. Nat. Rev. Immunol. 2002;2:28–36. doi: 10.1038/nri700. [DOI] [PubMed] [Google Scholar]
- 25.Anekal S.G., Zhu Y., Yin J. Dynamics of virus spread in the presence of fluid flow. Integr. Biol. (Camb) 2009;1:664–671. doi: 10.1039/b908197f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seisenberger G., Ried M.U., Bräuchle C. Real-time single-molecule imaging of the infection pathway of an adeno-associated virus. Science. 2001;294:1929–1932. doi: 10.1126/science.1064103. [DOI] [PubMed] [Google Scholar]
- 27.Gibbons M.M., Chou T., D’Orsogna M.R. Diffusion-dependent mechanisms of receptor engagement and viral entry. J. Phys. Chem. B. 2010;114:15403–15412. doi: 10.1021/jp1080725. [DOI] [PubMed] [Google Scholar]
- 28.Nowak S.A., Chou T. Mechanisms of receptor/coreceptor-mediated entry of enveloped viruses. Biophys. J. 2009;96:2624–2636. doi: 10.1016/j.bpj.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lagache T., Dauty E., Holcman D. Quantitative analysis of virus and plasmid trafficking in cells. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2009;79:011921. doi: 10.1103/PhysRevE.79.011921. [DOI] [PubMed] [Google Scholar]
- 30.Sidorenko Y., Reichl U. Structured model of influenza virus replication in MDCK cells. Biotechnol. Bioeng. 2004;88:1–14. doi: 10.1002/bit.20096. [DOI] [PubMed] [Google Scholar]
- 31.Tayi V.S., Bowen B.D., Piret J.M. Mathematical model of the rate-limiting steps for retrovirus-mediated gene transfer into mammalian cells. Biotechnol. Bioeng. 2010;105:195–209. doi: 10.1002/bit.22515. [DOI] [PubMed] [Google Scholar]
- 32.Gómez-Sjöberg R., Leyrat A.A., Quake S.R. Versatile, fully automated, microfluidic cell culture system. Anal. Chem. 2007;79:8557–8563. doi: 10.1021/ac071311w. [DOI] [PubMed] [Google Scholar]
- 33.Erban R., Chapman S.J. Reactive boundary conditions for stochastic simulations of reaction-diffusion processes. Phys. Biol. 2007;4:16–28. doi: 10.1088/1478-3975/4/1/003. [DOI] [PubMed] [Google Scholar]
- 34.Oliver C.J., Shortridge K.F., Belyavin G. Diffusion coefficient and molecular weight of type 5 adenovirus by photon-correlation spectroscopy. Biochim. Biophys. Acta. 1976;437:589–598. doi: 10.1016/0304-4165(76)90026-x. [DOI] [PubMed] [Google Scholar]
- 35.Hua J.M., Erickson L.E., Glasgow L.A. A review of the effects of shear and interfacial phenomena on cell viability. Crit. Rev. Biotechnol. 1993;13:305–328. doi: 10.3109/07388559309075700. [DOI] [PubMed] [Google Scholar]
- 36.Moledina F., Clarke G., Zandstra P.W. Predictive microfluidic control of regulatory ligand trajectories in individual pluripotent cells. Proc. Natl. Acad. Sci. USA. 2012;109:3264–3269. doi: 10.1073/pnas.1111478109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Andreadis S., Lavery T., Morgan J.R. Toward a more accurate quantitation of the activity of recombinant retroviruses: alternatives to titer and multiplicity of infection. J. Virol. 2000;74:1258–1266. doi: 10.1128/jvi.74.3.1258-1266.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Martin R., Raidl M., Binder B.R. Adenovirus-mediated expression of green fluorescent protein. Gene Ther. 1997;4:493–495. doi: 10.1038/sj.gt.3300408. [DOI] [PubMed] [Google Scholar]
- 39.Dinh A.T., Theofanous T., Mitragotri S. A model for intracellular trafficking of adenoviral vectors. Biophys. J. 2005;89:1574–1588. doi: 10.1529/biophysj.105.059477. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.