

The control of auto-ignition can allow to increase the efficiency of internal combustion engines with clear potential positive effects on the problem of global warming.[1] The design of internal combustion engines,[2] as well as the improvement of safety in oxidation processes,[3] rely on a good understanding of the kinetic mechanism of the auto-ignition of organic compounds. Here we experimentally demonstrate a key assumption of this mechanism, which has been accepted for more than 20 years but never proven.[4-6] A detailed speciation of the hydroperoxides responsible for the gas-phase auto-ignition of organic compounds has been achieved for the first time, thanks to the development of a new system coupling a jet stirred reactor to a molecular-beam mass spectrometer combined with tunable synchrotron vacuum ultraviolet (SVUV) photoionization. The formation of alkylhydroperoxides (ROOH) and of carbonyl compounds including a hydroperoxide function (ketohydroperoxide) has been observed under conditions close to those actually observed before the auto-ignition. This result gives the experimental confirmation of an assumption made in all the detailed kinetic mechanisms developed to model auto-ignition phenomena.
A good understanding of the kinetic mechanism governing the oxidation and the auto ignition of organic compounds is important in two significant applications: the design of combustion engines and the safety of oxidation processes in chemical industry (e.g. petrochemistry). Auto-ignition governs many features of the combustion in internal combustion engines (e.g. knock and related phenomena, octane and cetane rating).[2] The potential explosive properties of mixtures of organic compounds with oxygen seldom to catastrophic consequences, but it very often imposes stringent constraints on the operating conditions of partial oxidation processes.[3]
In addition to their propensity to auto-ignition, mixtures of organic compounds with oxygen possess the following specific reactive features. Single or multiple small temperature pulsations (a few tenths of K) accompanied by weak blue light emission due to excited formaldehyde, so called “cool flames”,[7] are often observed under conditions preceding those of auto-ignition (usually from 550 K). Closely linked with “cool flames”, a zone of temperature (usually around 650 K) where the reactivity decreases with temperature, commonly called “negative temperature coefficient” (NTC) zone, is a second well known characteristic of these systems.[2,7] These intriguing features have made the gas phase low temperature oxidation of organic compounds a fascinating field of investigations for kineticists since the end of the 19th century. It has been proven early that the auto-ignition of hydrocarbons cannot be explained by solely thermal theory,[8] but was mainly due to free radicals chain reactions.[9] Following Semenov,[9] who proposed the concept of degenerate branched chain reactions (production of free radicals from products formed in a chain reaction) to explain explosive reactions, much work has been devoted to elucidate the species responsible for degenerate branched chain reactions during the oxidation of organic compounds.[7]
Figure 1 presents the nowadays commonly accepted mechanism which is representing the oxidation of hydrocarbons at low temperature (below 1000 K).[4] After a short initiation period, a hydrocarbon (RH) mainly reacts with a hydroxyl radical (•OH) to give an alkyl radical (R•) via reaction 1 followed by the formation of alkylperoxy radicals (ROO•) after a barrierless reaction with an oxygen molecule (reaction 2). The existence of the NTC zone is mainly due to the enhanced reversibility of this reaction as the temperature increases. From ambient temperature up to about 550 K, the only prevalent reaction of alkylperoxy radicals is with •HO2 radicals (reaction 3) or by H-abstractions from an organic molecule to yield alkylhydroperoxides (ROOH). The fragility of the RO-OH bond of hydroperoxides (the bond dissociation energy is around 43 kcal/mol), which can easily break when temperature increases, has made them very good candidates as species responsible for the degenerate branched chain reactions explaining auto-ignition and cool flame occurrence.[4,7]
Figure 1.

Simplified scheme of the primary mechanism of oxidation of alkanes at low temperature.
Above 550 K (in the zone of cool flames), a second consumption pathway becomes faster for alkylperoxy radicals: their isomerisation (reaction 4) by internal transfer of a hydrogen atom to give hydroperoxyalkyl radicals (•QOOH). The latter radicals can easily decompose to yield oxygenated products, amongst which cyclic ethers are the most abundant species (reaction 5). As cyclic ethers are stable compounds easily analyzed by gas chromatography, the first steps of the oxidation of organic compounds through reactions 1-4 (in Figure 1) are relatively well known.[7]
A second important fate of •QOOH radicals is the second addition to oxygen (reaction 6). The formation and the reactions of the resulting •OOQOOH radicals have not yet been directly experimentally investigated. However, following Fish,[10] all the detailed kinetic mechanisms developed nowadays to model the low-temperature ignition of organic compounds consider decompositions of •OOQOOH radicals yielding three radicals, amongst them two •OH radicals. Cox and Cole[5] have proposed this global step to occur through the formation of alkylhydroperoxides including a carbonyl group (reaction 8), mainly ketohydroperoxides, which in turn can easily decompose to give two radicals (reaction 9) and lead to another possible chain branched reaction. Many modelling studies have suggested that the direct formation of hydroperoxides by reactions of peroxy radicals was not fast enough and that the inclusion of a chain branching step deriving from the second addition to oxygen was necessary to satisfactorily simulate the experimental observations.
While several detailed models can now reproduce most features of the oxidation and auto-ignition of alkanes representative of those present in automotive fuels,[6] experimental evidence supporting this commonly used reaction scheme is extremely scarce.[11]
The purpose of this study is to give experimental evidence to the formation of hydroperoxides under conditions which mimic those observed during the reaction period prior the auto-ignition of alkanes and using a method allowing distinguishing between alkylhydroperoxides and ketohydroperoxides. N-butane has been studied as it is the smallest alkane which has an oxidation behavior close to that of the species present in gasoline and diesel fuel.
Minimizing possible reactions of hydroperoxides between their formation and their detection can be obtained by creating a molecular beam from a high pressure reactive zone toward a detection device maintained under vacuum, which results in the “freezing” of the chemical composition of the sampled gas. Since hydroperoxides are expected to be formed in very low amounts amongst a large number of other oxidation products, a highly sensitive mass spectrometer with low photon energy to avoid the fragmentation of the products has been used, i.e. a time-of-flight mass spectrometer combined with tunable SVUV photoionization. This analytical method has already been successfully used to investigate many reactive systems,[12] such as laminar premixed flames.[13,14] Due to the need of accumulating several scans to obtain a good sensitivity, this analysis device requires coupling with a reactive system working under steady conditions. Our purpose was then not to really observe unsteady phenomena, such as cool flames or auto-ignition, but to mimic the chemistry leading to these phenomena. We have then used a heated jet-stirred reactor. This type of reactor coupled with gas chromatography analyses has already been used many times for studying the low-temperature oxidation of organic compounds,[15,16] showing the presence of a NTC zone and the formation of the same type of products as what is actually analyzed prior auto-ignition or during cool flames.[17] Figure 2 presents a simplified scheme of the jet-stirred reactor associating a molecular-beam sampling system to a reflectron time-of-flight mass spectrometer combined with SVUV photoionization.
Figure 2.

Simplified scheme of the experimental device.
The study of the oxidation of n-butane was performed at temperatures between 560 and 720 K, with a mean residence time of 6s and for an n-butane/oxygen/argon mixture composition of 4/26/70 (in mol %) corresponding to a stoichiometric mixture for complete combustion reaction.
Figure 3a presents the evolution of the mole fraction of n-butane with temperature. The mole fraction of n-butane (m/z 58) was directly derived from the normalized ion signal at mass 58 (obtained with a photon energy of 11.0 eV (1 eV = 96.5 kJ/mol)) assuming that no reaction occurred below 590 K. Raw signals of every compound were normalized by the ion signal obtained for argon (m/z 40) with a photon energy of 16.2 eV which acted as an internal standard. Many products were obtained, the major C4 ones being butenes at mass 56 and C4H8O products at mass 72. Figure 3b also displays the normalized ion signal at masses 56 and 72 obtained with a photon energy of 10.0 eV to avoid ion fragmentation. The ionization energy (IE) of butenes is between 9.11 and 9.55 eV.[18] The signal at mass 72 corresponds to the C4H8O oxygenated products which have an IE below 10 eV, e.g. butanal, butanone and all cyclic ethers (except 2 ethyl-oxirane which has an IE larger than 10 eV).
Figure 3.

Experimental and simulated mole fractions or signals of the oxidation of n-butane, the main products and hydroperoxides with temperature: (a) the experimental (points) and simulated (line) mole fractions of n-butane, (b) the experimental ion signals (in arbitrary units) at mass 48 (green circles), 56 (white squares, the signal is divided by 7.5), 62 (purple circles), 72 (black squares), 90 (blue circles, the signal is divided by 4) and 104 (red circles) and (c) the computed mole fractions (in ppm) of CH3OOH (green line, mole fraction divided by 5), C2H5OOH (purple line), C4H9OOH (blue line), the C4 ketohydroperoxides (red line), butenes (dash line, mole fraction divided by 5) and C4H8O products (black line).
Note the NTC zone obtained above 630 K (see Figure 3a), which makes the evolution with temperature of the normalized signal of all the products have the same shape as those at mass 72 with a kind of “plateau” between 620 and 680 K. This “plateau” is not observed in the case of species corresponding to four specific masses: 48, 62, 90, and 104 (Figure 3b). For these species, a sharp peak is obtained around 590 K meaning that they are very reactive molecules which decompose very quickly when the temperature increases.
In a system containing only carbon, hydrogen and oxygen atoms, mass 48 can only be related to methylhydroperoxide (CH3OOH). For masses 62, 90 and 104, several possibilities exist. However the mass of ethylhydroperoxide (C2H5OOH) is 62, that of butylhydroperoxides (C4H9OOH) is 90 and that of C4 ketohydroperoxides (C4H8O3) is 104. Figure 4 shows a mass spectrum with the presence of masses 48, 62, 90 and 104. A substantiation of this identification is given because these four compounds are the only ones having the same particular evolution with temperature, with a sharp peak, characteristic of compounds rapidly decomposing when temperature increases.
Figure 4.

Typical mass spectrum obtained for the oxidation of n-butane. The temperature in the reactor was 590 K and the photon energy was 10.0 eV.
Additional supporting evidence for this identification relies on IEs from measurements of photoionization efficiency spectra. When such values were not available in the literature,[18] zero-point energy corrected adiabatic IEs have been calculated from the CBS-QB3 method[19] using Gaussian03.[20] An experimental sweeping of photon energies from 8.5 to 11.5 eV has been made. The obtained experimental IEs were 9.91, 9.69, 9.37 and 9.29 ± 0.05 eV for mass 48, 62, 90 and 104, respectively (see the supplementary information). The calculated values for methylhydroperoxide, ethylhydroperoxide, butylhydroperoxides and C4 ketohydroperoxides were 9.83, 9.61, 9.33-9.36 and 9.34-9.39, respectively. For the last two compounds, a range of energy is given because the calculations were made for all the expected isomers (see Table 1). Note that 1,2 dihydroxyethane, another possibility for mass 62, has an ionization energy of 10.16 eV.[18]
Table 1.
Ionization energies of most expected isomers of C4 hydroperoxides deriving from the oxidation of n-butane.
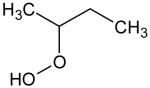
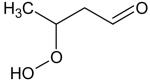
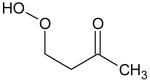
When no reference is given the value has been calculated. The mean absolute error of CBS-QB3 for the G2 test is less than 0.05 eV. For hydroperoxides which can involve hydrogen bonds, lowest energy conformers were searched for a systematic manner.
Due to kinetic reasons, the probability of having two neighbouring atoms of carbon each bonded to an oxygen atom is lower.
Finally, even further substantiation of the proposed assignments is given by simulations performed with a detailed kinetic mechanism of the oxidation of n-butane previously proposed[21] (see the supplementary information). Figures 3a and 3c show that, while small differences are encountered for the start of the reactivity and the extent of the NTC zone, the model reproduces well the variations with temperature of the mole fraction of n-butane, as well as the “plateau” obtained below 680 K for the major products, such butenes and C4H8O compounds. As shown in Figure 3c, simulations reproduce well the particular mole fraction profiles (with a sharp peak) obtained for the four hydroperoxides detected in the experiments compared to those of the major products.
This work gives the first speciation of the hydroperoxides formed during the low temperature oxidation of an organic compound. Further investigations are necessary to connect the structure of the reactant to the type of hydroperoxides obtained. This should help to increase the accuracy of detailed kinetic combustion models in order to make them really predictive[22] and able to reproduce the formation of minor pollutants, which have a deleterious impact on environment and human health.[23]
Experimental Section
The utilized spherical quartz jet-stirred reactor (diameter around 5 cm,) in which a gas mixture is continuously flowing was operated at a constant temperature and pressure.[16,24] This type of reactor, which can be heated up to 1200 K, is well adapted for kinetic studies: the gas phase inside the reactor is well stirred, meaning the concentrations and temperature are homogenous.[25] Quartz wall effects can usually be considered as negligible. This study has been performed under quasi-atmospheric pressure (1.05 atm). As the type of diluent gas is of negligible reactive importance, argon has been used instead of nitrogen, for the ease of the mass spectrometric analysis. The coupling between the quartz reactor and the mass spectrometer was made through a quartz cone-like nozzle with a 75 μm orifice (the tip of which has been inserted inside the reactor) generating a molecular beam which was skimmed before the entrance in the spectrometer chamber. In-situ detection of intermediates was made possible by combining tuneable VUV photoionization and the molecular-beam sampling mass spectrometer. The produced ions are detected by a reflectron time-of-flight mass spectrometer which can distinguish between different masses.[26]
Supplementary Material
Footnotes
This work was supported by the European Commission through the “Clean ICE” Advanced Research Grant of the European Research Council, by a grant of Région Lorraine (Soutien Jeune chercheur, Olivier Herbinet). FQ thanks the funding supports from Chinese Academy of Sciences, Natural Science Foundation of China (50925623 and 20533040), and Ministry of Science and Technology of China (2007CB815204 and 2007DFA61310). We thank Prof. J. F. Pauwels and D. Leray for their help in designing the coupling with the quartz reactor, S. Bax, H. Legall and P. Aury for technical assistance in France.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Contributor Information
Dr. Frédérique Battin-Leclerc, Département de Chimie Physique des Réactions, Nancy Université, CNRS, ENSIC, BP 20451, 1 rue Grandville, 54001 Nancy, France.
Dr. Olivier Herbinet, Département de Chimie Physique des Réactions, Nancy Université, CNRS, ENSIC, BP 20451, 1 rue Grandville, 54001 Nancy, France
Dr. Pierre-Alexandre Glaude, Département de Chimie Physique des Réactions, Nancy Université, CNRS, ENSIC, BP 20451, 1 rue Grandville, 54001 Nancy, France
Prof. René Fournet, Département de Chimie Physique des Réactions, Nancy Université, CNRS, ENSIC, BP 20451, 1 rue Grandville, 54001 Nancy, France
Dr. Zhongyue Zhou, National Synchrotron Radiation Laboratory, University of Science and Technology of China, Hefei, Anhui 230029, China
Dr. Liulin Deng, National Synchrotron Radiation Laboratory, University of Science and Technology of China, Hefei, Anhui 230029, China
Dr. Huijun Guo, National Synchrotron Radiation Laboratory, University of Science and Technology of China, Hefei, Anhui 230029, China
Dr. Mingfeng Xie, National Synchrotron Radiation Laboratory, University of Science and Technology of China, Hefei, Anhui 230029, China
Prof. Fei Qi, National Synchrotron Radiation Laboratory, University of Science and Technology of China, Hefei, Anhui 230029, China.
References
- [1].Yao M, Zheng Z, Liu H. Prog. Energ. Combust. Sci. 2009;35:398–437. [Google Scholar]
- [2].Westbrook CK, Mizobuchi Y, Poinsot TJ, Smith PJ, Warnatz J. Proc. Combust. Inst. 2005;30:125–157. [Google Scholar]
- [3].Pekalski AA, Terli E, Zevenbergen JF, Lemkowitz SM, Pasman HJ. Proc. Combust. Inst. 2005;30:1933–1939. [Google Scholar]
- [4].Walker RW, Morley C. Basic chemistry of combustion. In: Pilling MJ, editor. Comprehensive Chemical Kinetics: Low-Temperature Combustion and Autoignition. Elsevier; Amsterdam: 1997. p. 35. [Google Scholar]
- [5].Cox RA, Cole JA. Combust Flame. 1985;60:109–123. [Google Scholar]
- [6].Battin-Leclerc F. Prog. Energ. Combust. Sci. 2008;34:440–498. [Google Scholar]
- [7].Pollard RT. Hydrocarbons. In: Bamford CH, Tipper CFH, editors. Comprehensive Chemical Kinetics: Gas-Phase Combustion. Elsevier; Amsterdam: 1977. p. 17. [Google Scholar]
- [8].Frank-Kamenetskii DA. Diffusion and Heat Exchange in Chemical Kinetics. Princeton Univ. Press; Princeton: 1955. [Google Scholar]
- [9].Semenov NN. Some Problems in Chemical Kinetics and Reactivity. Vol. 2. Pergamon Press; London: 1958. [Google Scholar]
- [10].Fish A. Angewandte Chem. 1968;7:45–60. [Google Scholar]
- [11].Blin-Simiand N, Jorand F, Sahetchian K, Brun M, Kerhoas L, Malosse C, Einhorn J. Combust. Flame. 2001;126:1524–1532. [Google Scholar]
- [12].Li Y, Qi F. Acc. Chem. Research. 2010;43:68–78. doi: 10.1021/ar900130b. [DOI] [PubMed] [Google Scholar]
- [13].Hansen N, Cool TA, Westmoreland PR, Kohse-Höinghaus K. Prog. Energ. Combust. Sci. 2009;35:168–191. [Google Scholar]
- [14].Taatjes CA, Hansen N, McIlroy A, Miller JA, Senosiain JP, Klippenstein SJ, Qi F, Sheng LS, Zhang YW, Cool TA, Wang J, Westmoreland PR, Law ME, Kasper T, Kohse-Höinghaus K. Science. 2005;308:1887–1889. doi: 10.1126/science.1112532. [DOI] [PubMed] [Google Scholar]
- [15].Dagaut P, Reuillon M, Cathonnet M. Combust. Flame. 1995;101:132–140. [Google Scholar]
- [16].Hakka MH, Glaude PA, Herbinet O, Battin-Leclerc F. Combust. Flame. 2009;156:2129–2144. doi: 10.1016/j.combustflame.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Minetti R, Carlier M, Ribaucour M, Therssen E, Sochet LR. Combust. Flame. 1995;102:298–309. [Google Scholar]
- [18].NIST Chemistry Webbook . NIST Standard Reference Database 69; National Institute of Standards and Technology. Gaithersburg, MD: 2005. http://webbook.nist.gov/chemistry/ [Google Scholar]
- [19].Montgomery JA, Frisch MJ, Ochterski JW, Petersson GA. J. Chem. Phys. 1999;110:2822–2827. [Google Scholar]
- [20].Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, et al. Gaussian03, revision B05. Gaussian, Inc.; Wallingford, CT: 2004. [Google Scholar]
- [21].Buda F, Bounaceur R, Warth V, Glaude PA, Fournet R, Battin-Leclerc F. Combust. Flame. 2005;142:170–186. [Google Scholar]
- [22].Green WH. Adv. Chem. Eng. 2007;32:1–50. [Google Scholar]
- [23].Nelson PF, Tibbett AR, Day SJ. Atmos. Environ. 2008;42:5291–5303. [Google Scholar]
- [24].Biet J, Hakka MH, Warth V, Glaude PA, Battin-Leclerc F. Energy &Fuels. 2008;22:2258–2269. [Google Scholar]
- [25].Matras D, Villermaux J. Chem. Eng. Sci. 1973;28:129. [Google Scholar]
- [26].Li YY, Zhang LD, Tian ZY, Yuan T, Yang B, Yang JZ, Qi F. Energy & Fuels. 2009;23:1473–1485. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.