

Abstract
Gene-derived simple sequence repeats (genic SSRs), also known as functional markers, are often preferred over random genomic markers because they represent variation in gene coding and/or regulatory regions. We characterized 544 genic SSR loci derived from 138 candidate genes involved in wood formation, distributed throughout the genome of Populus tomentosa, a key ecological and cultivated wood production species. Of these SSRs, three-quarters were located in the promoter or intron regions, and dinucleotide (59.7%) and trinucleotide repeat motifs (26.5%) predominated. By screening 15 wild P. tomentosa ecotypes, we identified 188 polymorphic genic SSRs with 861 alleles, 2–7 alleles for each marker. Transferability analysis of 30 random genic SSRs, testing whether these SSRs work in 26 genotypes of five genus Populus sections (outgroup, Salix matsudana), showed that 72% of the SSRs could be amplified in Turanga and 100% could be amplified in Leuce. Based on genotyping of these 26 genotypes, a neighbour-joining analysis showed the expected six phylogenetic groupings. In silico analysis of SSR variation in 220 sequences that are homologous between P. tomentosa and Populus trichocarpa suggested that genic SSR variations between relatives were predominantly affected by repeat motif variations or flanking sequence mutations. Inheritance tests and single-marker associations demonstrated the power of genic SSRs in family-based linkage mapping and candidate gene-based association studies, as well as marker-assisted selection and comparative genomic studies of P. tomentosa and related species.
Keywords: candidate gene-derived SSRs, cross-species transferability, in silico analysis of SSR variations, Populus tomentosa, single marker–trait association mapping
1. Introduction
Poplars (Populus spp.) are widely distributed all over the world and are an important commercial tree species for timber production. In addition to their important economic value, in environmental protection, poplars also play key pioneer roles in the stability and sustainability of forest ecosystems.1,2 Many members of the genus Populus have been physiologically and genetically characterized based on their desirable biological characteristics, such as rapid growth, easy transformation, modest genome size, and ability to make interspecific crosses, propagate vegetatively.1,2 Thus, poplars have become a model species for studies of angiosperm trees, particularly because the whole genome of Populus trichocarpa has been sequenced and annotated (http://genome.jgi-psf.org/Poptr1/Poptr1.home.html).1 Additional genomics resources include databases of expressed sequence tags (ESTs) (http://www.populus.db.umu.se/index.html) and simple sequence repeats (SSRs) (http://www.ornl.gov/sci/ipgc/ssr_resource.htm), and these resources not only provide data for comparison of a long-lived perennial to short-lived model plants (e.g. Arabidopsis and rice), but also offer new opportunities to explore the genetic basis of wood formation, perenniality, and dormancy.2,3
The Chinese white poplar (Populus tomentosa) belongs to the section Leuce within the Populus genus and is native to northern China with a distribution zone of 1 million km2. P. tomentosa is of major commercial importance in timber and pulp production and also plays an important role in ecological and environmental protection.4 A vast amount of genetic variation has arisen during the evolution of the species, as is evident in the natural populations.5,6 This accumulated genetic variation provides an important resource for the exploration of the molecular mechanisms of wood formation and is also a source of alleles for the potential improvement of wood products. However, conventional breeding programmes may not be sufficient to improve this long-growing species.6 Modern molecular breeding tools, such as molecular marker-assisted selection (MAS) breeding, could enhance important Populus agronomic traits, including growth rate, wood quality, and disease resistance. Hence, development of suitable genetic marker resources is an important foundation for MAS breeding.
Among molecular genetic markers, single nucleotide polymorphisms are often used for genetic analysis. However, DNA microsatellites, or SSR markers, are excellent genetic markers because they are hypervariable, co-dominant, and therefore highly informative.7,8 Moreover, compared with SSR markers derived from random genomic locations, SSR markers derived from genes will likely provide a much greater degree of resolution in association mapping because they occur within the gene and thus may affect gene expression or function.6,8 In addition, genic SSRs exhibit relatively high transferability to closely related species and can be used as anchor markers for comparative mapping and evolutionary studies.6–9 Gene-based microsatellites have now been developed for a limited number of Populus species based on the P. trichocarpa genome sequence.10,11 However, very limited genomic information is available for P. tomentosa,4,6,12 and as the P. tomentosa linkage map was constructed using amplified fragment length polymorphisms (AFLPs),13 functional genomics studies of this economically important species are in their infancy. Furthermore, another important approach, the use of SSRs from fully characterized genes or full-length cDNA clones has not yet been utilized in Populus.
Wood quality traits are considered to be quantitative, controlled by multiple genes, with moderate-to-high heritability.14 Currently, large numbers of candidate genes involved in wood formation have been isolated from P. tomentosa using direct sequencing methods, although many have not been published.4,6,15,16 Therefore, to improve the properties of wood using a MAS approach, identification and characterization of species-specific SSR loci from wood formation-related genes is a highly promising approach. Here, we make use of the large dataset of available gene sequences to identify a large number of gene-based SSR markers for P. tomentosa. The specific aims of our study were to: (i) characterize the genic SSR loci in P. tomentosa and evaluate SSR primers and polymorphisms in different wild-type varieties, (ii) test cross-species transferability within the genus Populus and conduct in silico analysis of SSR variation between P. tomentosa and P. trichocarpa, and (iii) examine inheritance segregation in a mapping population and analyze single SSR marker–trait association in a natural population. This study provides a valuable SSR resource for comparative genomic studies of the genus Populus, and the genic microsatellites also serve as ‘framework’ markers for construction of a physical map for alignment of the ongoing sequencing of the complete P. tomentosa genome.
2. Materials and methods
2.1. Microsatellite identification, primer design, and SSR polymorphism screening
Total genomic DNA was extracted from young leaves using the DNeasy Plant Mini kit (Qiagen China, Shanghai), following the manufacturer's protocol. The reference gene models of 150 candidate genes involved in wood formation were obtained by BLASTX analyses against the NCBI database (http://www.ncbi.nlm.nih.gov/) or from the JGI database (http://genome.jgi-psf.org/Poptr1_1/Poptr1_1.home.html) (Supplementary Table S1). Subsequently, a set of specific primers was designed for polymerase chain reaction (PCR) amplification of all 150 genes, and total genomic DNA (20 ng per reaction) from the P. tomentosa LM50 clone was used for amplification. All the PCR amplification products from LM50 were sequenced (both strands) using conserved primers, the BigDye Terminator Cycle Sequencing kit, version 3.1 (Applied Biosystems, Beijing, China), and a Li-Cor 4300 genetic analyzer (Li-Cor Biosciences, Lincoln, Nebraska, USA). In all, a total of 696 792 bp of genomic DNA sequences from these 150 unique candidate genes, with an average of 4645 bp per gene, were obtained by direct sequencing, and the sequence data of the 150 candidate genes generated in this study were deposited in the GenBank Data Library under the accession numbers JX986583–JX986732 (Supplementary Table S1). These sequences were mined for genic SSR markers using the SSRIT software (http://www.gramene.org/db/searches/ssrtool).17 All microsatellite loci were located exclusively in the coding and/or regulatory regions of candidate genes, i.e. exons, promoters, 5′ untranslated regions (UTRs), 3′ UTRs, or introns. The ideal marker contained a minimum SSR repeat of five for dinucleotide repeats, four for trinucleotide repeats, and three for tetranucleotide or longer (more than pentanucleotide) repeats. A set of compound perfect repeat units was also identified. Mononucleotide repeats and complex sequence repeats were excluded.
Primer pairs specific for the SSR flanking sequences were designed using the Primer 3 program (http://frodo.wi.mit.edu/primer3/primer3.FAQ.html), according to the parameters reported by Du et al.6 All SSR primers were initially screened using genomic DNA from the P. tomentosa LM50 clone (three replications). Amplification was carried out using standard PCR conditions with annealing temperatures (Ta) set according to the primer sequences. PCR products were separated by electrophoresis in 2% agarose gels. Electrophoresis products were visualized and photographed using the Fluorchem™ 5500 (Alfa Innotech Corp., USA) gel documentation system. Finally, a subset of optimal SSR primers was identified and designated as ‘validated genic SSR markers’.
All validated genic SSR markers were scored for amplicon size polymorphisms among 15 wild P. tomentosa ecotypes that represented nearly the entire P. tomentosa geographic distribution (Supplementary Table S2).4,12 Observed product sizes and numbers of alleles per locus (NA) were calculated for each marker using POPGENE, version 1.32.18
2.2. PCR amplification and SSR genotyping
The SSR amplification reaction system and PCR amplifications were conducted following the procedure of Du et al.6 PCR product amplification was confirmed on a 2% agarose gel, and products were then separated by capillary electrophoresis using an ABI3730xl DNA Analyzer (Applied Biosystems). The polymorphic loci were analyzed using GeneMapper, version 4.0, with the LIZ 600 size standard (Applied Biosystems).
2.3. Cross-species transferability
To assess cross-species transferability and allele length polymorphisms, 30 randomly selected genic SSRs were genotyped in 26 ecotypes (24 species) belonging to 5 sections of the genus Populus, using Salix matsudana as the outgroup (Supplementary Table S3).
The summary statistical parameters reflected intra- and interpopulation genetic diversity levels, including the observed allele sizes, polymorphism information content (PIC), expected heterozygosity (HE), and number of alleles (NA), that were calculated for each marker using POPGENE, version 1.32. The discriminatory abilities of genic SSRs were estimated using cluster analyses to assess phylogenic relationships among related Populus species. A neighbour-joining (NJ) dendrogram was constructed using the proportion of shared alleles coefficient from PowerMarker, version 3.2519 and was drawn using TreeView version 1.66 (http://taxonomy.zoology.gla.ac.uk/rod/treeview.html).
2.4. In silico analysis of SSR variations
We used in silico identification of genic SSR variations between P. tomentosa and P. trichocarpa to validate the true SSR cross-species transferability within the genus Populus. All 220 available homologous sequences of P. trichocarpa were identified in the JGI Database, based on 220 specific amplicons with SSR markers selected from P. tomentosa candidate gene sequences. MEGA, version 5.1 (http://www.megasoftware.net/), was used to compare the amplified SSR alleles with the SSR-containing sequences between the two species (i.e. repeat length, repeat motif, and mutations in flanking sequences).
2.5. Testing inheritance in a linkage-mapping population
To test the power of these novel genic SSR markers for constructing a family-based linkage map, 30 random genic SSRs selected from the 188 polymorphic markers (Fig. 1 and Supplementary Table S6) were used to examine the observed segregation of markers, using 1000 F1 progeny from a controlled hybridization between a female YX01 clone (Populus alba × Populus glandulosa) and a male LM 50 clone (P. tomentosa).12 Mendelian inheritance of microsatellite variants (alleles) was determined from the observed distribution of progeny genotypes when compared with the expected segregation ratios (1:1, 1:2:1, and 1:1:1:1), based on the hypothesized genotypes of the parents by performing a chi-squared (χ2) test at the 0.01 probability level in SAS, version 8.2, (SAS Institute, Cary, NC, USA).
Figure 1.

Flow diagram of P. tomentosa genic SSR marker development and applications in this study.
2.6. SSR marker–trait association studies in a natural population
For association mapping, we used 40 genic SSRs randomly selected from the set of 188 polymorphic markers (Fig. 1 and Supplementary Table S6), to associate with several wood quality traits in a natural population of 460 unrelated wild-type ecotypes from the P. tomentosa clonal arboretum, established in the national nursery of Guan Xian County, Shandong Province, China (36°23′′N, 115°47′′E), which represent all original provenances of this species (Supplementary Table S2).4,12
In the natural population, wood property traits, including microfibre angle (MFA), fibre lengths, fibre widths, and holocellulose, α-cellulose, and lignin contents were measured according to the methods described by Du et al.20 Analysis of variance of these six phenotypic traits is shown in Supplementary Table S4. These 40 random genic SSRs were applied to test the degree of resolution of genic SSRs in association with these 6 wood property traits, using the unified mixed model method (MLM) with 104 permutations in TASSEL, version 2.0.1.21,22 In this Q + K model, the relative kinship matrix (K) was obtained using TASSEL, and the population structure matrix (Q) of the covariates was identified by Du et al.12 Corrections for multiple testing were performed using the positive false discovery rate (FDR) method with 104 permutations.23
3. Results
3.1. Frequency and distribution of genic SSR markers
By sequencing 150 candidate genes related to wood properties, we identified 544 SSR loci located in 138 (92%) unique genes from 696 792 bp total sequence, for an average of 1 SSR per 1.3 kb. The perfect SSRs were not evenly distributed, ranging from zero to seven per gene (average, 3.6), and seven loci were compound SSRs containing at least one repeat motif (Fig. 1 and Supplementary Table S1).
Analysis of all 544 gene-derived SSR loci revealed that dinucleotide (325, 59.7%) and trinucleotide repeat motifs (144, 26.5%) predominated, followed by tetranucleotide (33, 6.1%), hexanucleotide (13, 2.4%), and pentanucleotide (12, 2.2%) repeat motifs (Fig. 2A). Of the identified SSRs, slightly fewer than half (46.1%) were located in promoter regions, including dinucleotides (57.4%) and trinucleotides (28.7%) (Fig. 2A and Supplementary Table S5A). Conversely, in exons, 37.5% were dinucleotides and 62.5% were trinucleotides (Fig. 2A and Supplementary Table S5A). The dinucleotide repeat AT/TA was the most abundant motif detected in all genic SSRs (177, 32.6%), followed by ATT/TAA (53, 9.8%), AG/TC (49, 9.0%), CT/GA (48, 8.7%), and AAT/TTA (16, 3.0%) (Fig. 2B and Supplementary Table S5B). SSR length was most commonly 10–20 bp (70.5% of total SSRs), followed by 21–30 bp (20.9%) (Fig. 2C). The largest SSR found was a 68-bp dinucleotide repeat (AT/TA).
Figure 2.

(A) Distribution of repeat motifs in 544 genic SSRs identified in 138 P. tomentosa genes; (B) Distribution of dinucleotide repeat motifs detected in 138 P. tomentosa genes; (C) Distribution of repeat numbers in di- and trinucleotide SSRs. Bars of different colours show the number of genic SSRs from dinucleotide and trinucleotide.
3.2. SSR primer design, screening, and polymorphism testing
To determine whether the SSRs varied in length, and would therefore provide a useful marker, we designed flanking primers and determined the length of each SSR by capillary electrophoresis. Primer pairs specific for flanking sequences were designed for 481 of the SSR sequences (88.5%). Using the P. tomentosa LM50 clone, 413 primer pairs (85.9%) gave successful amplification, and the remaining 68 failed to generate PCR products at any annealing temperature used (Ta), and so were excluded from further analysis (Fig. 1). Of the 413 working primer pairs, 292 (70.7%) amplified PCR products of the expected sizes; however, 65 and 20 PCR products were larger or smaller, respectively, than expected, and the remaining 36 primer pairs generated multiple PCR products (Fig. 1). Details of the 292 optimal primers, including locus names, primer sequences, SSR repeat motifs, Ta, expected sizes, SSR sources (locations in genes), and GenBank accession numbers are provided in Supplementary Table S6. Distribution summaries, frequencies, and repeat motifs of these 292 genic SSR markers are provided in Supplementary Table S7A.
To identify whether the identified SSRs varied in length in P. tomentosa, these 292 primers were subjected to further screening for polymorphisms in 15 wild P. tomentosa ecotypes. Twenty four primers amplified monomorphic products (1 band), 80 markers have identical heterozygotes among all 15 individuals (2 bands), and 188 primers (64.4%) generated clean and reproducible polymorphic bands (Fig. 1 and Supplementary Table S6). The observed product sizes and numbers of alleles per locus (NA) were calculated for each marker. A total of 861 unique alleles were identified (Supplementary Table S6). For the 188 polymorphic markers from 107 genes, the NA ranged from 2 to 7 with an average of 3.6 observed alleles per polymorphic locus; in other regions, the mean NA values varied between 2.4 for exons and 4.0 for promoter regions (Supplementary Tables S6 and S7B).
3.3. SSR cross-species transferability within the genus Populus
To determine the utility of this SSR marker set beyond P. tomentosa, we screened a subset of the markers to determine whether they could be amplified from other related species, including Populus and the closely related Salix species. The capacity of 30 representative P. tomentosa genic SSR primers to screen for polymorphisms was tested in 27 ecotypes of related Populus and Salix species (Supplementary Table 3). All tested SSR primer pairs displayed a high amplification frequency within and across sections at the subgenus level within Populus. Of the 30 examined SSR markers, 12 (40%) successfully amplified in all species. The transferability of the 30 primers tested in Populus sections was Leuce (100%), Tacamahaca (90%), Aigeros (83%), Leucoides (80%), and Turanga (72%) (Table 1). In S. matsudana, the frequency of amplification was lower, and only 50% yielded product (Table 1). Thus, the developed SSR markers could be applied across the genus Populus and provided data on polymorphisms among related species (Supplementary Fig. S1).
Table 1.
Diversity of 30 polymorphic P. tomentosa genic SSR markers for 26 genotypes within the genus Populus
| Locus | Total NA | PIC |
Tacamahaca |
Leuce |
Aigeiros |
Turanga |
Leucoides |
S. matsudana (outgroup) |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Size (bp) | NA | HE | Size (bp) | NA | HE | Size (bp) | NA | HE | Size (bp) | NA | HE | Size (bp) | NA | HE | Size (bp) | NA | HE | |||
| SSR12 | 7 | 0.663 | 230–242 | 2 | 0.802 | 230–248 | 5 | 0.363 | 230–242 | 3 | 0.607 | 246–248 | 2 | 0.667 | 0 | 0 | / | 252–256 | 2 | 1.000 |
| SSR34 | 6 | 0.601 | 98–102 | 3 | 0.675 | 96–100 | 2 | 0.539 | 0 | 0 | / | 98 | 1 | 0.000 | 96–98 | 2 | 1.000 | 0 | 0 | / |
| SSR39 | 5 | 0.457 | 119–134 | 3 | 0.607 | 119–128 | 3 | 0.750 | 119–125 | 2 | 0.508 | 0 | 0 | / | 119–125 | 2 | 1.000 | 134 | 1 | 0.000 |
| SSR43 | 7 | 0.783 | 211–217 | 4 | 0.825 | 211–227 | 5 | 0.615 | 211–217 | 4 | 0.644 | 189 | 1 | 0.000 | 211–213 | 2 | 1.000 | 195–198 | 2 | 1.000 |
| SSR47 | 4 | 0.548 | 0 | 0 | / | 192–198 | 3 | 0.458 | 0 | 0 | / | 201 | 1 | 0.000 | 195–198 | 2 | 1.000 | 0 | 0 | / |
| SSR53 | 10 | 0.820 | 117–131 | 6 | 0.864 | 115–123 | 5 | 0.750 | 111–117 | 3 | 0.714 | 0 | 0 | / | 0 | 0 | / | 0 | 0 | / |
| SSR54 | 11 | 0.841 | 146–156 | 4 | 0.661 | 136–142 | 3 | 0.538 | 134–152 | 4 | 0.607 | 138–144 | 2 | 0.667 | 132–136 | 2 | 1.000 | 136 | 1 | 0.000 |
| SSR57 | 10 | 0.818 | 245–253 | 4 | 0.778 | 227–243 | 4 | 0.561 | 221–227 | 2 | 0.533 | 253 | 1 | 0.000 | 229 | 1 | 0.000 | 261–265 | 2 | 1.000 |
| SSR58 | 6 | 0.539 | 107–119 | 4 | 0.632 | 110–121 | 4 | 0.503 | 110 | 1 | 0.000 | 110 | 1 | 0.000 | 0 | 0 | / | 0 | 0 | / |
| SSR62 | 4 | 0.529 | 0 | 0 | / | 399–403 | 3 | 0.250 | 401–403 | 2 | 0.250 | 403–405 | 2 | 0.667 | 399 | 1 | 0.000 | 0 | 0 | / |
| SSR64 | 6 | 0.632 | 377 | 1 | 0.000 | 373–379 | 4 | 0.821 | 375–377 | 2 | 0.429 | 371 | 1 | 0.000 | 375 | 1 | 0.000 | 385 | 1 | 0.000 |
| SSR65 | 6 | 0.411 | 263–273 | 4 | 0.725 | 257–263 | 3 | 0.264 | 0 | 0 | / | 265 | 1 | 0.000 | 271 | 1 | 0.000 | 269–273 | 2 | 1.000 |
| SSR66 | 5 | 0.463 | 0 | 0 | / | 215–217 | 2 | 0.440 | 199–219 | 3 | 0.644 | 221 | 1 | 0.000 | 217–221 | 2 | 1.000 | 0 | 0 | / |
| SSR67 | 7 | 0.789 | 240–242 | 2 | 0.636 | 242–246 | 3 | 0.350 | 242–250 | 3 | 0.607 | 246–250 | 2 | 0.667 | 246 | 1 | 1.000 | 260 | 1 | 0.000 |
| SSR69 | 9 | 0.834 | 213–219 | 4 | 0.928 | 221–235 | 4 | 0.333 | 215–227 | 4 | 0.786 | 219 | 1 | 0.000 | 219 | 1 | 0.000 | 209–213 | 2 | 1.000 |
| SSR70 | 8 | 0.727 | 310–322 | 4 | 0.643 | 312–318 | 4 | 0.733 | 314–324 | 3 | 0.607 | 0 | 0 | / | 314–318 | 2 | 1.000 | 0 | 0 | / |
| SSR71 | 10 | 0.771 | 192–228 | 6 | 0.849 | 201–222 | 4 | 0.636 | 228–246 | 4 | 0.821 | 234 | 1 | 0.000 | 204–234 | 2 | 1.000 | 237 | 1 | 0.000 |
| SSR73 | 10 | 0.828 | 169–183 | 4 | 0.588 | 177–191 | 5 | 0.835 | 173–191 | 6 | 0.703 | 0 | 0 | / | 0 | 0 | / | 0 | 0 | / |
| SSR74 | 7 | 0.750 | 114–120 | 3 | 0.439 | 114–124 | 4 | 0.846 | 118–126 | 4 | 0.711 | 0 | 0 | / | 0 | 0 | / | 0 | 0 | / |
| SSR75 | 8 | 0.766 | 202–220 | 4 | 0.867 | 202–210 | 4 | 0.679 | 204–210 | 4 | 0.750 | 206–208 | 2 | 0.667 | 206–208 | 2 | 1.000 | 220. | 1 | 0.000 |
| SSR77 | 8 | 0.690 | 165–179 | 5 | 0.933 | 163–169 | 3 | 0.604 | 165–175 | 4 | 0.679 | 165–169 | 2 | 0.667 | 165–169 | 2 | 1.000 | 181 | 1 | 0.000 |
| SSR91 | 5 | 0.640 | 139–151 | 4 | 0.636 | 135–147 | 3 | 0.590 | 0 | 0 | / | 147–155 | 2 | 0.667 | 0 | 0 | 0 | 0 | 0 | / |
| SSR96 | 6 | 0.739 | 111–123 | 3 | 0.725 | 108–123 | 4 | 0.632 | 114–117 | 2 | 0.596 | 0 | 0 | / | 114 | 1 | 0.000 | 0 | 0 | / |
| SSR98 | 8 | 0.798 | 224–234 | 4 | 0.642 | 224–238 | 5 | 0.846 | 226–236 | 3 | 0.607 | 226–228 | 2 | 0.667 | 0 | 0 | / | 0 | 0 | / |
| SSR113 | 6 | 0.591 | 193–199 | 3 | 0.408 | 184–196 | 4 | 0.630 | 196–202 | 3 | 0.593 | 187–193 | 2 | 0.667 | 193 | 1 | 0.000 | 184–187 | 2 | 1.000 |
| SSR165 | 8 | 0.637 | 310–322 | 4 | 0.636 | 307–322 | 4 | 0.532 | 304–316 | 5 | 0.720 | 319–325 | 3 | 0.833 | 304–310 | 2 | 1.000 | 322 | 1 | 0.000 |
| SSR169 | 7 | 0.664 | 209–219 | 4 | 0.650 | 205–217 | 4 | 0.633 | 0 | 0 | / | 205–209 | 2 | 0.667 | 211 | 1 | 0.000 | 0 | 0 | / |
| SSR170 | 6 | 0.479 | 214–220 | 4 | 0.569 | 202–220 | 4 | 0.705 | 202–214 | 2 | 0.511 | 212–220 | 3 | 0.833 | 202–208 | 2 | 1.000 | 222 | 0 | / |
| SSR176 | 5 | 0.732 | 343–355 | 4 | 0.701 | 346–355 | 2 | 0.360 | 343–355 | 3 | 0.531 | 0 | 0 | / | 358 | 1 | 0.000 | 0 | 0 | / |
| SSR179 | 8 | 0.679 | 245–261 | 3 | 0.558 | 249–263 | 4 | 0.687 | 253–261 | 3 | 0.607 | 263–265 | 2 | 0.667 | 0 | 0 | / | 0 | 0 | / |
| Mean | 7.0 | 0.675 | / | 3.4 | 0.599 | / | 3.7 | 0.582 | / | 2.6 | 0.492 | / | 1.3 | 0.300 | / | 1.1 | 0.467 | / | 0.7 | 0.233 |
See Supplementary Table S6 for further details of the 30 genic SSR markers.
The observed number of alleles per locus (NA), PIC, expected heterozygosity (HE), and not applied (/).
We also examined the allelic variation in these ecotypes, to determine whether these SSR markers could prove useful for genetic studies such as association mapping. Of the 27 ecotypes, 213 alleles were detected using the 30 SSR primers, with an average of 7.0 alleles per locus, ranging from 4 at SSR62 to 11 at SSR54 (Table 1). The PIC mean polymorphism level of the loci was 0.675, and it ranged from 0.411 for SSR65 to 0.841 for SSR54 (Table 1). In the five Populus sections, the mean NA values were from 1.1 (Leucoides) to 3.7 (Leuce). The other diversity parameters (observed lengths and HE) are shown in Table 1.
We also tested whether these markers were informative for genetic analysis by determining whether the marker genotypes could be used to recapitulate the known phylogenetic relationship among the tested ecotypes. The NJ tree (Fig. 3) based on the shared allele coefficients from these 30 SSRs revealed the expected genetic relationships among all 27 ecotypes and clustered them into 6 main groups, in agreement with 5 putative sections, and the outgroup was derived from the basic botanical classification of these tree species.10,24,25 The relationships among species in each section are represented by the smaller branches within groups. In addition, we constructed trees based on the other functions in PowerMarker, and these were very similar to the NJ tree (Fig. 3) in that the six distinct clusters were again present, although there were slight differences in branch lengths within clusters (data not shown).
Figure 3.

Phylogenic relationship among 26 genotypes belonging to 5 sections of the genus Populus (S. matsudana as outgroup) based on 30 polymorphic genic SSR markers. The different colour branches denote the divergent clusters.
3.4. Comparison of SSR variation between P. tomentosa and P. trichocarpa
In addition to testing a subset of markers in multiple Populus and Salix species, we also examined these markers more extensively in the sequenced genome of P. trichocarpa. For all 292 genic SSR-containing sequences, only 75% (220) of homologous P. trichocarpa sequences (length, 90–593 bp) were identified (Fig. 1). Alignment and comparison of the homologous sequences between these two species for each marker revealed 10 types of mutations and variations in SSR loci and their flanking regions (Table 2 and Fig. 4). For example, the SSR53 locus, which contains an imperfect (CT) motif, showed complex mutations characterized by variation in the repeat-motif length and point mutations in both the repeat motif and the flanking sequences (data not shown). Of all 220 genic SSRs, 39.1% (86) were present in P. trichocarpa, with 57 showing only variation in repeat number in both species (conserved flanking sequences) and 29 that were monomorphic and had conserved sequences between the 2 species (Table 2). However, 41.8% (92) had polymorphisms of the SSR flanking sequences between the two species, suggesting that these SSR markers from P. tomentosa are not directly transferable to P. trichocarpa (Table 2). A comparison of the SSR markers with mutations in flanking sequences showed the highest proportion in 3′UTRs (60.0%) and the lowest in 5′UTRs (25.8%). A total of 25 new SSR markers were identified in the corresponding sequences of P. trichocarpa, of which 17 (68%) were derived from promoter regions (Table 2). Additionally, 100% of SSR markers from exon regions could be directly employed in genetic studies of P. trichocarpa (Table 2).
Table 2.
Comparison of variations and mutations of 220 genic SSR loci and their flanking sequences between P. tomentosa and P. trichocarpa
| Types of variations and mutations | Promoters | Introns | 5′UTRs | 3′UTRs | Exons | Total |
|---|---|---|---|---|---|---|
| Mutation in the flanking sequence only | 5 | 3 | 2 | 1 | 0 | 11 |
| Mutation in the flanking sequence and repeat number | 24 | 24 | 6 | 6 | 0 | 60 |
| Mutation in the flanking sequence and no SSR marker | 7 | 6 | 0 | 2 | 0 | 15 |
| Mutation in the flanking sequence and new SSR marker | 4 | 2 | 0 | 0 | 0 | 6 |
| Variation of repeat number only | 18 | 24 | 10 | 3 | 2 | 57 |
| Mutation in SSR repeat motif only | 1 | 4 | 3 | 1 | 0 | 9 |
| Mutation in SSR repeat motif and repeat number | 3 | 1 | 0 | 0 | 0 | 4 |
| No SSR marker only | 1 | 1 | 1 | 1 | 0 | 4 |
| New SSR marker only | 17 | 5 | 3 | 0 | 0 | 25 |
| Identical sequence | 9 | 7 | 6 | 1 | 6 | 29 |
| Total | 89 | 77 | 31 | 15 | 8 | 220 |
Figure 4.

Alignment and comparison of variations and mutations in genic SSRs with sequences homologous between P. tomentosa (01) and P. trichocarpa (02), based on a number of genic SSRs. Ten types of mutations and variations for genic SSR loci with their flanking regions were identified (See Table 2 for details).
We determined whether the mutations in SSR repeat motifs were transitions, transversions, insertions, or deletions related to the P. tomentosa–P. trichocarpa comparison. An insertion of even a single base in the repeat motif, e.g. (AT)5 T (AT)3, and a deletion within the repeat tract, e.g. (AGG)3 AG_ (AGG)2, disrupted the continuity of the perfect repeat units. Transversions (38/63) were the most abundant mutations in the sequences of these two species, whereas insertions (4/63) were the least abundant (Table 3). Transitions and/or transversions accounted for 86% of the total mutations in the perfect repeats, whereas insertions and/or deletions were found in only 14%. T/C transversions were the most common substitutions that disrupted repeat continuity (Table 3).
Table 3.
Number and type of mutations found in perfect microsatellite motifs in P. tomentosa and P. trichocarpa
| Transition (16) |
Transversion (38) | Deletion (5) | Insertion (4) | ||||
|---|---|---|---|---|---|---|---|
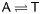 |
 |
 |
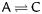 |
 |
 |
A (1), T (2), C (1), G (1) | A (2), T (2), C (0), G (0) |
| 13 (24%) | 3 (6%) | 11 (20%) | 5 (9%) | 17 (31%) | 5 (9%) | ||
3.5. Inheritance test of genic SSR markers
To further validate the usefulness of these genic SSR markers in genetic studies, we genotyped a randomly selected subset of 30 of the markers in a linkage mapping population of 1000 progeny from a controlled hybridization and tested for Mendelian segregation. Of the 30 genic SSRs, 5 loci (17%) were not used for analysis due to the presence of null alleles or unexpected lengths in the female parent. The remaining 25 loci segregated in the population (Table 4). Of the segregating loci, 2 were heterozygous in the female parent only, 4 were heterozygous in the male parent only, 17 were heterozygous in both parents, and 2 (SSR211 and SSR249) were homozygous in both parents, and thus resulted in offspring identical to the parents and with the expected heterozygote genotype (Table 4).
Table 4.
Mendelian inheritance patterns of 30 genic SSR markers based on segregation analyses of a cross between a female YX01 clone (P. alba × P. glandulosa) and a male LM 50 clone (P. tomentosa)
| Locus | The allele genotype of P1(♀) | The allele genotype of P2(♂) | The segregation genotypes of F1 progeny | The expected segregation ratio | P-value |
|---|---|---|---|---|---|
| SSR2 | 331/341 (+) | 329/335 (+) | 329/331, 329/341, 331/335, 335/341 | 1:1:1:1 | NS |
| SSR7 | 156/162 (+) | 159/165 (+) | 156/159, 156/165, 159/162, 162/165 | 1:1:1:1 | P < 0.01 |
| SSR9 | 250/256 (+) | 250/256 (+) | 250, 250/256, 256 | 1:2:1 | NS |
| SSR13 | 155/164 (+) | 155/158 (+) | 155, 155/158, 155/164, 158/164 | 1:1:1:1 | NS |
| SSR16 | 341 (−) Unexpected size | 363 (−) | / | / | / |
| SSR21 | 167/179(+) | 169/175(+) | 167/169, 167/175, 169/179, 175/179 | 1:1:1:1 | NS |
| SSR33 | 92 (−) | 92/98 (+) | 92, 92/98 | 1:1 | NS |
| SSR44 | 151/156 (+) | 156 (−) | 151/156, 156 | 1:1 | P < 0.01 |
| SSR45 | 188/192 (+) | 192/196 (+) | 188/192, 188/196, 192/196, 192 | 1:1:1:1 | NS |
| SSR47 | 186/195 (+) | 192/195 (+) | 186/192, 186/195, 192/195, 195 | 1:1:1:1 | P < 0.01 |
| SSR50 | 233/240 (+) | 233/240 (+) | 233, 233/240, 240 | 1:2:1 | NS |
| SSR52 | 145 (−) | 145/148 (+) | 145, 145/148 | 1:1 | NS |
| SSR56 | 228 (−) | 221/228 (+) | 221/228, 228 | 1:1 | NS |
| SSR61 | No amplification | 237 (−) | / | / | / |
| SSR63 | 202 (−) | 202/206 (+) | 202, 202/206 | 1:1 | NS |
| SSR71 | 204/210 (+) | 204/213 (+) | 204, 204/210, 204/213, 210/213 | 1:1:1:1 | NS |
| SSR77 | No amplification | 171/175 (+) | / | / | / |
| SSR107 | 169/172 (+) | 169 (−) | 169, 169/172 | 1:1 | NS |
| SSR108 | 177/183 (+) | 171/179 (+) | 171/177, 177/179, 171/183, 179/183 | 1:1:1:1 | NS |
| SSR114 | 102/110 (+) | 104/108(+) | 102/104, 102/108, 104/110, 108/110 | 1:1:1:1 | P < 0.01 |
| SSR118 | 191/194 (+) | 191/197 (+) | 191/197, 191/194, 194/197, 191 | 1:1:1:1 | NS |
| SSR124 | 192/196 (+) | 188/192 (+) | 188/192, 188/196, 192/196, 192 | 1:1:1:1 | NS |
| SSR142 | 227/231 (+) | 229/237 (+) | 227/229, 227/237, 229/231, 231/237 | 1:1:1:1 | NS |
| SSR150 | No amplification | 240/244 (+) | / | / | / |
| SSR188 | 207/211 (+) | 207/213 (+) | 207, 207/213, 207/213, 211/213 | 1:1:1:1 | P < 0.01 |
| SSR197 | 430 (−) Unexpected size | 417/419 (+) | / | / | / |
| SSR211 | 157 (−) | 160 (−) | 157/160 | / | / |
| SSR233 | 291/295 (+) | 289/299 (+) | 289/291, 289/295, 291/299, 295/299 | 1:1:1:1 | NS |
| SSR249 | 179 (−) | 179 (−) | 179 | / | / |
| SSR254 | 283/289 (+) | 283/289 (+) | 283, 283/289, 289 | 1:2:1 | NS |
See Supplementary Table S6 for further details of these 30 genic SSR markers.
‘P1’ represents the female clone ‘YX01’ (P. alba × P. glandulosa), ‘P2’ represents the male clone ‘LM 50’ (P. tomentosa), ‘+’ represents heterozygote, and ‘–’ represents homozygote; the χ2 significance level was P < 0.01, Ns, not significant; /, Not applied.
A chi-squared test was used to compare the segregation ratios of the 23 informative markers in these 1000 offspring. Eighteen were in accordance with Mendelian expectations (P ≥ 0.01), with a segregation ratio close to 1:2:1 for 3 SSR loci, 1:1 for 5 loci, and 1:1:1:1 for the remaining 10 markers (Table 4). Thus, these novel genic SSR markers may represent a useful resource for linkage mapping in P. tomentosa.
3.6. Genic SSR polymorphisms associated with wood property traits
Finally, to directly show that these markers could also prove useful for association mapping, we conducted a trial association mapping study using a subset of the genic SSRs to associate with traits affecting wood properties in Populus. For a random selection of 40 genic SSRs, single-marker association tests (240; 40 SSRs × 6 traits) were conducted using MLM. Twenty associations were found to be significant at a threshold of P < 0.05 (Supplementary Table S8). Multiple test corrections using the FDR method reduced this number to 12 at a significance threshold of Q < 0.10 (Table 5). These loci accounted for the phenotypic variance, with individual effects ranging from 4.0 to 8.9% (Table 5). Of these, α-cellulose and fibre length had three significant associations each, and holocellulose, MFA, and lignin had two significant associations each. However, no association with fibre width was detected (Q < 0.10, Table 5). The 12 associations represent 9 SSR loci from different regions of 9 candidate genes (Table 5). For example, SSR205 was located in the coding region (PtoC4H1 exon 1) that was associated exclusively with lignin (R2 = 8.9%, Q = 0.0211) (Table 5). For the holocellulose trait, SSR47 and SSR163, located in the non-coding regions of two candidate genes (PtoKorB and PtoCslA4), accounted for 4.3–8.7% of the phenotypic variance, and the SSR47 marker was similarly associated with α-cellulose content (Table 5).
Table 5.
Significant SSR marker–trait pair associations in the natural P. tomentosa population (n = 460), after correction for multiple testing [FDR (Q) ≤ 0.10]
| Trait | Gene symbol | Locus | P | R2 (%) | Q |
|---|---|---|---|---|---|
| Lignin | PtoPAL 2 Intron1 | SSR198 | 0.0021 | 4.6 | 0.0250 |
| PtoC4H1 Exon 1 | SSR205 | 0.0016 | 8.9 | 0.0211 | |
| Holocellulose | PtoKorB Intron 3 | SSR47 | 0.0031 | 7.0 | 0.0371 |
| PtoCslA4 Promoter | SSR163 | 0.0106 | 6.4 | 0.0475 | |
| α-Cellulose | PtoKorB Intron 3 | SSR47 | 0.0095 | 4.0 | 0.0475 |
| PtoWuA Promoter | SSR53 | 0.0200 | 4.5 | 0.0702 | |
| PtoSuSy6 Promoter | SSR71 | 0.0001 | 7.4 | 0.0172 | |
| MFA | PtoPAL2 Intron 1 | SSR198 | 0.0205 | 8.0 | 0.0750 |
| PtoCCR2 5′UTR | SSR224 | 4.33E–04 | 7.5 | 0.0105 | |
| Fibre length | PtoExp10 Promoter | SSR79 | 0.0022 | 5.3 | 0.0278 |
| PtoCslA4 Promoter | SSR163 | 0.0175 | 5.5 | 0.0669 | |
| PtoCslD9 Exon 4 | SSR187 | 0.0027 | 6.4 | 0.0346 |
P = the significant level for association (the significance is P ≤ 0.05); R2 = percentage of the phenotypic variance explained; Lignin = lignin content; Holocellulose = holocellulose content; and α-cellulose = α-cellulose content.
4. Discussion
4.1. Development and characterization of genic SSR markers
Here, we used a candidate gene approach to identify a set of SSR markers in P. tomentosa genes for wood properties, showing that this approach can provide useful genomics resources for linkage or association mapping and eventually for marker-assisted breeding to improve wood quality in this important timber crop. We successfully mined for genic SSRs in the sequences of 150 candidate genes associated with wood property traits. Our analysis of these SSR markers from P. tomentosa demonstrated that this approach may be useful for characterization of other pathways in P. tomentosa and for development of molecular markers for related model species with similar genomic resources. In our study, the frequency of genic SSRs was ∼1 SSR/1.3 kb. Higher or lower frequencies of genic SSRs have been reported elsewhere.11,26,27 However, these frequency variations may be the result of the application of different SSR search criteria, methods, or the abundances of the sources of DNA sequences searched.8
Dinucleotide motifs were the most abundant genic SSR markers (Fig. 2A and Supplementary Table S5A). This result is in contrast to previous findings identifying trinucleotide motifs as the most frequent genic repeats in most plant species,8,28,29 which may be attributable to the sources of the DNA sequences used (i.e. EST, cDNA, or gene sequences). In general, previous genic SSR makers were located in EST databases, and information about promoters and introns was not considered. Genic microsatellites are located mainly in promoters, introns, and UTRs of sequenced genes and are found at a lower frequency in conserved exons.8 This agrees with the distribution pattern we report here (Fig. 2A), suggesting that genic microsatellites may have a role in regulation of gene expression.8,11,28–30 Furthermore, polymorphism tests of 15 wild P. tomentosa ecotypes indicated that exonic SSRs contained less allelic variability than did non-coding SSRs (Supplementary Table S7B), which reflects the higher selection pressure on the coding portion of the genome.31,32
Our analysis of gene-derived SSRs found that SSRs with AT/TA motifs (32.6%) were the most frequent. This is similar to the cases of P. trichocarpa and Arachis hypogaea.25,33 Previous studies have indicated that (AT)n is the most common dinucleotide motif in plant genomes.8,33 Thus, the genic SSR pattern identified in this study is likely to be a good reflection of genome-wide SSR frequencies. In trinucleotide SSRs, the polymorphism rate of the ATT/TAA SSRs is three times higher than that of AAT/TTA SSRs. This distribution suggests that AT-rich SSR loci may have a relatively high variability in P. tomentosa and also confirms the finding that AT-rich repeats (those repeats containing two or more A and/or T nucleotides per motif) are more common in non-coding regions.9,17,25,34
In this study, 91.8% of the 292 optimal genic SSR primers produced at least 2 clean amplified bands, possibly due to the naturally occurring excess of heterozygotes in the P. tomentosa genome.12 Allelic diversity estimated for these polymorphic markers was an average of 3.6 alleles per locus, (range 2–7 alleles), and the value is lower than the NA (4.3) in a population of 460 P. tomentosa and some other related species, such as Populus nigra, P. trichocarpa, Populus tremuloides, and Populus euphratica.6,35,36 The level of allelic diversity reported in this study may be due to the limited sample size and/or the relatively conserved character of genic SSR markers. For generation of genetic maps, genic SSRs can determine the relative positions of transferable markers and directly compare candidate gene-containing SSRs and quantitative trait locus (QTL) locations across a broad variety of genetic backgrounds.8,25,29,32 The result of inheritance tests for 30 genic SSR markers suggests that many genic SSRs can be used for linkage mapping in P. tomentosa, and they are also useful for QTL and marker-aided selection of important traits. In addition, segregation distortion is increasingly recognized as a potentially powerful evolutionary force that may be beneficial for QTL mapping.37,38 The actual causes of the observed segregation distortions for markers are genes subjected to gametic or zygotic selection.37 Studies have suggested that epistasis contributes to segregation distortion, and segregation distortion may also be important for the evolution of many fundamental aspects of sexual reproduction.37–39
The presence of SSRs in transcribed regions can result in changes in function, transcription, or translation. For example, SSRs in coding regions that result in amino acid changes can cause either loss or gain of function, SSRs in the 5′UTR can affect gene transcription or translation, SSRs in the 3′UTR can be responsible for gene silencing or transcription slippage, and SSRs in introns might act as transcriptional enhancers of gene expression.8,32,40 Previous study that phenylalanine ammonia-lyase (PAL) transcripts have been localized to develop xylem cells in aspen (P. tremuloides) stem, was consistent with its involvement in lignin biosynthesis.41 This report supported the identification of a single-marker non-coding association in PtoPAL2 that explained 4.6% of the phenotypic variation in lignin content (Table 5). Cinnamate 4-hydroxylase (C4H1) is proposed to be associated with G lignin deposition,42,43 and a marker with significant association was located in the coding region (PtoC4H1 exon 1) that was associated exclusively with lignin (R2 = 8.9%, Q = 0.0211) (Table 5). Physiological studies of C4H genes describe unique functions for the isoforms within the lignin biosynthetic pathway.43 Similarly, other significant associations located in different candidate genes, such as sucrose synthase (SUSY), Cellulase (KOR), and Cinnamoyl-CoA reductase (CCR) (Table 5), were also supported by studies finding that they were involved in lignocellulosic cell wall development.3,14–16,43–45 Association analysis in this study suggests that genic SSRs have considerable power in candidate gene-based association studies to identify allelic variation in genes associated with important wood quality traits,20 and these markers have the potential to be useful in genetic improvement of lignin and cellulose biosynthesis in poplar.
4.2. SSR variation among related species within the genus Populus
The genus Populus contains six subgenera: Abaso, Leuce, Leucoides, Aigeiros, Turanga, and Tacamahaca, and Turanga is the most distant from Leuce.10,24,25,46,47 We found that genic SSR markers in P. tomentosa have relatively high amplification rates among closely related taxa, including species of the Leuce, Tacamahaca, Aigeiros, and Leucoides sections, but a lower amplification rate in Turanga (Table 1). This successful amplification rate in different sections was positively correlated with mean NA and PIC values (Table 1) that are due mainly to the phylogenetic divergence from P. tomentosa and is also related to the sampling species and their genetic backgrounds in each section. Furthermore, the amplification success rates were higher for members of the other subgenera, with the exception of Turanga (Table 1), suggesting that the amplification frequency varies in tandem with evolutionary distance, and this may be because the flanking regions of the microsatellite, where the PCR primers bind to the DNA, are more similar in phylogenetically close species than phylogenetically distant species.10,17,25,32 It should, therefore, be possible to predict the utility of these primers according to the genetic distance from P. tomentosa.
Six distinct groups were identified within 27 samples, based on 30 genic SSR markers (Fig. 3). The dendrogram grouping showed the expected segregation of the different sections within the genus Populus, which agreed with the subgenus botanical classification level of Salicaceae,24,25,46,47 although a few species were not identified based on their putative relationships or as reported in other diversity studies.46,47 For example, Populus bolleana is a variety of Populus alba; however, these species did not group together in the closest branches, despite their shared ancestry (Fig. 3). Overall, this analysis roughly supports the botanical classification of the germplasm surveyed and hints at the usefulness of the genic SSR markers for genetic diversity studies and other Populus genotyping applications.
In a comparison of genic SSR variations using homologous P. tomentosa and P. trichocarpa sequences (Table 2), we identified both repeat motif variations and mutations in the flanking sequences in most markers, and this shows that microsatellite mutation patterns are often complex in cross-species amplification. However, our findings indicate that the effects of mutations accumulated over evolutionary time can readily be studied. Point mutations disrupted the repeat pattern, and in addition to a new class of repeat motifs, size variations at microsatellite loci caused by indels (insertions or deletions) in flanking sequences have also been reported.32,40,48,49 We found the highest proportion of SSRs with mutations in flanking sequences in the 3′UTR (60.0%) and the lowest in the 5′UTR (25.8%). This suggests that variations and mutations in microsatellites may be influenced by the nature and functional composition of the flanking sequences.48,49 Transitions and/or transversions accounted for 86% of the total mutations of perfect repeat motifs in the two species (Table 3), indicating that base substitution of microsatellites is more common than length variation of homologous loci between closely related species.49 It also suggests that base substitution data are vital because genotyping using SSR alleles among related species is often erroneous, if based only on microsatellites of identical allele lengths obtained in electrophoresis. In silico identification of genic SSR variations in Populus is particularly useful for evolutionary studies and for increasing our understanding of the origin, mutational processes, and structure of microsatellites. Further reciprocal studies of microsatellite allelic variation in different species would shed additional light on directional evolution and genetic toxicology.40,48
Overall, the reliability of comparison of microsatellite variation data among related species is higher than cross-species amplification success rates or polymorphisms. Comparison of microsatellite variation in these two species suggests that use of cross-species SSR primers to investigate functionally important allelic polymorphisms related to the traits of interest may be inefficient. Therefore, the direct development of species-specific primers is necessary for association mapping, due to the variation in repeat number only at each SSR locus (conserved flanking sequences) among all individuals of the same species.
Supplementary Data
Supplementary Data are available at www.dnaresearch.oxfordjournals.org.
Acknowledgements
This work was supported by the State Key Basic Research Program of China (No. 2012CB114506), and the Project of the National Natural Science Foundation of China (No. 31170622, 30872042).
Footnotes
Edited by Dr Satoshi Tabata
References
- 1.Tuskan G.A., Difazio S., Jansson S., et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray) Science. 2006;313:1596–604. doi: 10.1126/science.1128691. [DOI] [PubMed] [Google Scholar]
- 2.Brunner A.M., Busov V.B., Strauss S.H. Poplar genome sequence: functional genomics in an ecologically dominant plant species. Trends Plant Sci. 2004;9:49–56. doi: 10.1016/j.tplants.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 3.Li L., Lu S., Chiang V.L. A genomic and molecular view of wood formation. Crit. Rev. Plant Sci. 2006;25:213–33. [Google Scholar]
- 4.Zhang D.Q., Du Q.Z., Xu B.H., Zhang Z.Y., Li B.L. The actin multigene family in Populus: organization, expression and phylogenetic analysis. Mol. Genet. Genomics. 2010;284:105–19. doi: 10.1007/s00438-010-0552-5. [DOI] [PubMed] [Google Scholar]
- 5.Zhu Z., Zhang Z. The status and advances of genetic improvement of Populus tomentosa Carr. J. Beijing Forestry Univ. 1997;6:1–7. [Google Scholar]
- 6.Du Q.Z., Zhang D.Q., Li B.L. Development of 15 novel microsatellite markers from cellulose synthase genes in Populus tomentosa (Salicaceae) Am. J. Bot. 2012;99:e46–8. doi: 10.3732/ajb.1100308. [DOI] [PubMed] [Google Scholar]
- 7.Blair M.W., González L.F., Kimani P.M., Butare L. Genetic diversity, inter-gene pool introgression and nutritional quality of common beans (Phaseolus vulgaris L.) from Central Africa. Theor. Appl. Genet. 2010;121:237–48. doi: 10.1007/s00122-010-1305-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Varshney R.K., Graner A., Sorrells M.E. Genic microsatellite markers in plants: features and applications. Trends Biotechnol. 2005;23:48–55. doi: 10.1016/j.tibtech.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 9.Morgante M., Hanafey M., Powell W. Microsatellites are preferentially associated with nonrepetitive DNA in plant genomes. Nat. Genet. 2002;30:194–200. doi: 10.1038/ng822. [DOI] [PubMed] [Google Scholar]
- 10.Yin T.M., Zhang X.Y., Gunter L.E., et al. Microsatellite primers resource developed from the mapped sequence scaffolds of Nisqually-1 genome. New Phytol. 2009;181:498–503. doi: 10.1111/j.1469-8137.2008.02663.x. [DOI] [PubMed] [Google Scholar]
- 11.Li S.X., Yin T.M., Wang M.X., Tuskan G.A. Characterization of microsatellites in the coding regions of the Populus genome. Mol. Breeding. 2011;27:59–66. [Google Scholar]
- 12.Du Q.Z., Wang B.W., Wei Z.Z., Zhang D.Q., Li B.L. Genetic diversity and population structure of Chinese white poplar (Populus tomentosa) revealed by SSR markers. J. Hered. 2012 doi: 10.1093/jhered/ess061. In press. [DOI] [PubMed] [Google Scholar]
- 13.Zhang D.Q., Zhang Z.Y., Yang K., Li B.L. , Genetic mapping in (Populus tomentosa × P. bolleana) and P. tomentosa Carr. using AFLP markers. Theor. Appl. Genet. 2004;108:657–62. doi: 10.1007/s00122-003-1478-7. [DOI] [PubMed] [Google Scholar]
- 14.Thumma B.R., Nolan M.F., Evans R., Moran G.F. Polymorphisms in cinnamoyl coa reductase (CCR) are associated with variation in microfibril angle in Eucalyptus spp. Genetics. 2005;171:1257–65. doi: 10.1534/genetics.105.042028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang D.Q., Yang X.H., Zhang Z.Y., Li B.L. Expression and nucleotide diversity of the poplar COBL gene. Tree Genet. Genomes. 2010;6:331–44. [Google Scholar]
- 16.Zhang D.Q., Xu B.H., Yang X.H., Zhang Z.Y., Li B.L. The sucrose synthase gene family in Populus: structure, expression, and evolution. Tree Genet. Genomes. 2011;7:443–51. [Google Scholar]
- 17.Temnykh S., Declerck G., Lukashova A., Lipovich L., Cartinhpur S., Mccouch S. Computational and experimental analysis of microsatellites in rice (Oryza sativa L.): frequency, length variation, transposon associations, and genetic marker potential. Genome Res. 2001;11:1441–52. doi: 10.1101/gr.184001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yeh F.C., Yang R.C., Boyle T. Canada: University of Alberta; 1999. POPGENE version 1.32: Microsoft Windows-based freeware for population genetic analysis, quick user guide. Center for International Forestry Research. [Google Scholar]
- 19.Liu K., Muse S.V. PowerMarker: an integrated analysis environment for genetic markers analysis. Bioinformatics. 2005;21:22128–9. doi: 10.1093/bioinformatics/bti282. [DOI] [PubMed] [Google Scholar]
- 20.Du Q.Z., Pan W., Xu B.H., Li B.L., Zhang D.Q. Polymorphic SSR loci within PtoCesA genes are associated with growth and wood properties in Populus tomentosa New Phytol In press. [DOI] [PubMed]
- 21.Yu J., Pressoir G., Briggs W.H., et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006;38:203–8. doi: 10.1038/ng1702. [DOI] [PubMed] [Google Scholar]
- 22.Bradbury P.J., Zhang Z., Kroon D.E., Casstevens T.M., Ramdoss Y., Buckler E.S. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics. 2007;23:2633–5. doi: 10.1093/bioinformatics/btm308. [DOI] [PubMed] [Google Scholar]
- 23.Storey J.D., Tibshirani R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA. 2003;100:9440–5. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eckenwalder J.E. Systematics and evolution of Populus. In: Stettler R.F., Bradshaw H.D., Heilman P.E., Hinckley T.M., editors. Biology of Populus and its implications for management and conservation. Ottawa, Canada: NRC Research Press; 1996. pp. 7–32. [Google Scholar]
- 25.Tuskan G.A., Gunter L.E., Yang Z.M., Yin T.M., Sewell M.M., DiFazio S.P. Characterization of microsatellites revealed by genomic sequencing of Populus trichocarpa. Can. J. Forest Res. 2004;34:5–93. [Google Scholar]
- 26.Singh R., Zaki N.M., Ting N.-C., et al. Exploiting an oil palm EST database for the development of gene-derived SSR markers and their exploitation for assessment of genetic diversity. Biologia. 2008;63:227–35. [Google Scholar]
- 27.Feng S.P., Li W.G., Huang H.S., Wang J.Y., Wu Y.T. Development, characterization and cross-species/genera transferability of EST-SSR markers for rubber tree (Hevea brasiliensis) Mol. Breed. 2009;23:85–97. [Google Scholar]
- 28.Chagné D., Chaumeil P., Ramboer A., et al. Cross-species transferability and mapping of genomic and cDNA SSRs in pines. Theor. Appl. Genet. 2004;109:1204–14. doi: 10.1007/s00122-004-1683-z. [DOI] [PubMed] [Google Scholar]
- 29.Hisano H., Sato S., Isobe S., et al. Characterization of the soybean genome using EST-derived microsatellite markers. DNA Res. 2007;14:271–81. doi: 10.1093/dnares/dsm025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cardle L., Ramsay L., Milbourne D., Macaulay M., Marshall D., Waugh R. Computational and experimental characterization of physically clustered simple sequence repeats in plants. Genetics. 2000;156:847–54. doi: 10.1093/genetics/156.2.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kutter C., Watt S., Stefflova K., et al. Rapid turnover of long noncoding RNAs and the evolution of gene expression. PLoS Genet. 2012;8:e1002841. doi: 10.1371/journal.pgen.1002841. doi:10.1371/journal.pgen.1002841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y.C., Korol A.B., Fahima T., Nevo E. Microsatellites within genes: structure, function, and evolution. Mol. Biol. Evol. 2004;21:991–1007. doi: 10.1093/molbev/msh073. [DOI] [PubMed] [Google Scholar]
- 33.Wang H., Penmetsa R.V., Yuan M., et al. Development and characterization of BAC-end sequence derived SSRs, and their incorporation into a new higher density genetic map for cultivated peanut (Arachis hypogaea L.) BMC Plant Biol. 2012;12:10. doi: 10.1186/1471-2229-12-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Toth G., Gaspari Z., Jurke J. Microsatellites in different eukaryotic genomes: survey and analysis. Genome Res. 2000;10:967–81. doi: 10.1101/gr.10.7.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Slavov G.T., Zhelev P. Salient biological features, systematics, and genetic variation of Populus. In: Jansson S., Bhalerao R., Groover A.T., editors. Genetics and genomics of Populus. New York: Springer; 2010. pp. 15–38. [Google Scholar]
- 36.Wang J., Li Z., Guo Q., Ren G., Wu Y. Genetic variation within and between populations of a desert poplar (Populus euphratica) revealed by SSR markers. Ann. Forest Sci. 2011;68:1143–9. [Google Scholar]
- 37.Xu S.Z. Quantitative trait locus mapping can benefit from segregation distortion. Genetics. 2008;180:2201–8. doi: 10.1534/genetics.108.090688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taylor D.R., Ingvarsson P.K. Common features of segregation distortion in plants and animals. Genetica. 2003;117:27–35. doi: 10.1023/a:1022308414864. [DOI] [PubMed] [Google Scholar]
- 39.Tao Y., Hartl D.L., Laurie C.C. Sex-ratio segregation distortion associated with reproductive isolation in Drosophila. Proc. Natl. Acad. Sci. USA. 2001;98:13183–8. doi: 10.1073/pnas.231478798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang L., Zuo K., Zhang F., et al. Conservation of noncoding microsatellites in plants: implication for gene regulation. BMC Genomics. 2006;7:323. doi: 10.1186/1471-2164-7-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kao Y.Y., Harding S.A., Tsai C.J. Differential expression of two distinct phenylalanine ammonia-lyase genes in condensed tannin-accumulating and lignifying cells of quaking aspen. Plant Physiol. 2002;130:796–807. doi: 10.1104/pp.006262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu S.F., Zhou Y.H., Li L.G., Chiang V.L. Distinct roles of cinnamate 4-hydroxylase genes in Populus. Plant Cell Physiol. 2006;47:905–14. doi: 10.1093/pcp/pcj063. [DOI] [PubMed] [Google Scholar]
- 43.Wegrzyn J.L., Eckert A.J., Choi M., et al. Association genetics of traits controlling lignin and cellulose biosynthesis in black cottonwood (Populus trichocarpa, Salicaceae) secondary xylem. New Phytol. 2010;188:515–32. doi: 10.1111/j.1469-8137.2010.03415.x. [DOI] [PubMed] [Google Scholar]
- 44.Boerjan W., Ralph J., Baucher M. Lignin biosynthesis. Annu. Rev. Plant Biol. 2003;54:519–46. doi: 10.1146/annurev.arplant.54.031902.134938. [DOI] [PubMed] [Google Scholar]
- 45.Coleman H.D., Yan J., Mansfield S.D. Sucrose synthase affects carbon partitioning to increase cellulose production and altered cell wall ultrastructure. Proc. Natl. Acad. Sci. USA. 2009;106:13118–23. doi: 10.1073/pnas.0900188106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sterck L., Rombauts S., Jansson S., Sterky F., Rouzé P., Vande Peer Y. EST data suggest that poplar is an ancient polyploidy. New Phytol. 2005;167:165–70. doi: 10.1111/j.1469-8137.2005.01378.x. [DOI] [PubMed] [Google Scholar]
- 47.Leskinen E., Alström-Rapaport C. Molecular phylogeny of Salicaceae and closely related Flacourtiaceae: evidence from 5.8 S, ITS 1 and ITS 2 of the rDNA. Plant Syst. Evol. 1999;215:209–27. [Google Scholar]
- 48.Prasad M.D., Muthulakshmi M., Madhu M., Archak S., Mita K., Nagaraju J. Survey and analysis of microsatellites in the silkworm, Bombyx mori: frequency, distribution, mutations, marker potential and their conservation in heterologous species. Genetics. 2004;169:197–214. doi: 10.1534/genetics.104.031005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ellegren H. Microsatellites: simple sequences with complex evolution. Nature Rev. Genet. 2004;5:435–45. doi: 10.1038/nrg1348. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.