

Abstract
Genome-wide association studies (GWASs) have been successful at identifying single-nucleotide polymorphisms (SNPs) highly associated with common traits; however, a great deal of the heritable variation associated with common traits remains unaccounted for within the genome. Genome-wide complex trait analysis (GCTA) is a statistical method that applies a linear mixed model to estimate phenotypic variance of complex traits explained by genome-wide SNPs, including those not associated with the trait in a GWAS. We applied GCTA to 8 cohorts containing 7096 case and 19 455 control individuals of European ancestry in order to examine the missing heritability present in Parkinson's disease (PD). We meta-analyzed our initial results to produce robust heritability estimates for PD types across cohorts. Our results identify 27% (95% CI 17–38, P = 8.08E − 08) phenotypic variance associated with all types of PD, 15% (95% CI −0.2 to 33, P = 0.09) phenotypic variance associated with early-onset PD and 31% (95% CI 17–44, P = 1.34E − 05) phenotypic variance associated with late-onset PD. This is a substantial increase from the genetic variance identified by top GWAS hits alone (between 3 and 5%) and indicates there are substantially more risk loci to be identified. Our results suggest that although GWASs are a useful tool in identifying the most common variants associated with complex disease, a great deal of common variants of small effect remain to be discovered.
INTRODUCTION
Genome-wide association studies (GWASs) have been successful at identifying single-nucleotide polymorphisms (SNPs) highly associated with common traits; however, a great deal of the heritable variation associated with common traits remains unknown (1–3). Parkinson's disease (PD) is the second most common neurodegenerative disease, and is clinically characterized by rigidity, resting tremor and bradykinesia (slowed movement). The average age at onset is 68 years, however this is highly variable and can range from adolescence to old age (4–6). Generally, individuals with disease onset before age 55 are categorized as early onset, and those with disease onset after age 55 are categorized as late onset (7–9).
GWASs have identified risk variants at over two dozen loci influencing PD risk; however, these are thought to explain only a fraction of the variance in PD liability (8,10). In addition, mutations known to cause monogenic forms of PD account for only a small proportion of disease (11). These include mutations in α-synuclein (SNCA), parkin (PARK2), DJ-1 (PARK7), PTEN-induced putative kinase I (PINK1) and leucine-rich repeat kinase 2 (LRRK2) (12–14). Although additional loci that have been implicated in PD risk produce small effects, collectively they comprise a larger portion of genetic component responsible for the development of PD (15).
Twin studies are useful in differentiating the impacts of genetics and the environment as the sources of a disease. If genetic factors greatly influence the presence of a disease, it is expected that concordance in monozygotic (MZ) twins will be greater than dizygotic (DZ) twins. Twin studies of PD have shown very low pairwise concordance, estimated as ∼0.129, with no discernable difference between MZ and DZ pairs, although there is some concern over the cross-sectional nature of these studies. In addition, concordance-adjusted prevalence in twin studies has been estimated at 8.67/1000, or <0.1% (16). This is a similar prevalence value to what is seen in other population-based studies of PD prevalence (4,8,17,18).
Monogenic familial forms of PD are often early-onset, and it has been suggested that early-onset PD has a greater familial/heritable component in its etiology than later onset manifestations of the disease (19). It is likely that the etiology of early-onset PD is dissimilar to late-onset PD, which seems to occur more sporadically (15). At least one twin study has performed analyses controlled for age, with results showing significant rates of concordance for MZ pairs with early age at onset of PD, and a near lack of concordance for later onset individuals. This strongly supports the role of genetics in early-onset PD; however, the sample size for this subset of individuals was very small, and shared environmental exposure in twins cannot be overlooked (16). Furthermore, a longitudinal twin study examined both MZ and DZ pairs over an 8-year period and observed a significantly higher concordance rate among MZ pairs; over time, concordance in these individuals increased from 0 to 33% (20). These data suggest an additional environmental trigger in the etiology of PD and further demonstrate that a simple genetic model does not fit the etiology of this disease (16,20,21).
We sought a method to capture the genetic variance associated with PD. Genome-wide complex trait analysis (GCTA) estimates the components of phenotypic variance, that is, the polygenic additive variance (heritability) that can be explained by genome-wide SNPs, including those that are not significantly associated with the phenotype of interest in GWASs (22). Notably, this method provides a lower limit for trait heritability because it is improbable that all causal variants have been exactly tagged by the SNPs on the genotyping platforms used to perform initial analyses (15). GCTA works by utilizing genome-wide SNPs to quantify the phenotypic variance of all putative causal variants, as opposed to only genome-wide significant trait-associated variants. For a given disease trait, the heritability explained by all SNPs is estimated simultaneously, as opposed to testing the association of some previously identified SNPs from GWASs, which relies on shared linkage disequilibrium across common variants associated with disease status to quantify heritability (1–3).
Using GCTA, we aim to identify a larger segment of additive genetic variance not typically associated with early- and late-onset PD via GWASs in central European and Scandinavian ancestry populations, thus generating the most comprehensive estimates to date of PD heritability (23). Recent analyses have also used GCTA to examine the dichotomy of early and late-onset PD, using self-reported diagnosis data collected by the personal genetics company 23andMe (15). Do et al. (15) report estimates for early-onset PD heritability as ∼0.306 (95% CI 0.136–0.476) and for late onset as ∼0.285 (95% CI 0.2224–0.346). These estimates are supported by our meta-analysis, which provides additional support for the complex genetic structure of PD.
Our analyses offer a continuation and extension of this previous research by dichotomizing the PD phenotype based on age and subsets of SNPs; in addition, we imputed our data to test both typed and untyped variants when estimating phenotypic variance of the PD phenotype. This not only encapsulates more putatively associated variants than previous studies yielding a more informed estimate, but it is also in line with the methodologies that will be used in future GWAS studies. Critically, imputation is useful in this study because of the inclusion of multiple clinically derived studies as opposed to a single cohort study of self-reported cases. In particular, the use of imputation provided a means of facilitating more cohesive data for meta-analyses, as our genotyped data were standardized and expanded by the imputation of millions of shared SNPs per cohort. By imputing genotypes across cohorts, we are able to test the same set of SNPs regardless of the limitations of the initial platform.
RESULTS
Cohort-level heritability estimates are provided in Table 1 and accompanying forest plots are shown in Figure 1. Our heritability estimates vary from ∼16% (UK, SE = 0.027) to ∼49% (Finland, SE = 0.04). Moderate estimates were obtained for the majority of cohorts.
Table 1.
Cohort level analyses
| Heritability estimate | SE of heritability estimate | Cases (n) | Controls (n) | ||
|---|---|---|---|---|---|
| Cohort: US | |||||
| All samples | All SNPs | 0.182545 | 0.045998 | 971 | 3034 |
| PD GWAS SNPs in PD loci/regions | 0.037605 | 0.009569 | 971 | 3034 | |
| PD GWAS regions excluded | 0.143514 | 0.045902 | 971 | 3034 | |
| Early onset | All SNPs | 0.080189 | 0.214376 | 365 | 276 |
| PD GWAS SNPs in PD loci/regions | 0.013059 | 0.035854 | 365 | 276 | |
| PD GWAS regions excluded | 0.047529 | 0.214932 | 365 | 276 | |
| Late onset | All SNPs | 0.227307 | 0.080002 | 572 | 1620 |
| PD GWAS SNPs in PD loci/regions | 0.033377 | 0.014531 | 572 | 1620 | |
| PD GWAS regions excluded | 0.174017 | 0.079982 | 572 | 1620 | |
| Cohort: GER | |||||
| All samples | All SNPs | 0.259042 | 0.080298 | 742 | 944 |
| PD GWAS SNPs in PD loci/regions | 0.047032 | 0.015157 | 742 | 944 | |
| PD GWAS regions excluded | 0.228625 | 0.0807 | 742 | 944 | |
| Early onset | All SNPs | 0.22548 | 0.162678 | 302 | 670 |
| PD GWAS SNPs in PD loci/regions | 0.111 | 0.030216 | 302 | 670 | |
| PD GWAS regions excluded | 0.154317 | 0.162772 | 302 | 670 | |
| Late onset | All SNPs | 0.221245 | 0.222233 | 367 | 267 |
| PD GWAS SNPs in PD loci/regions | 0.021783 | 0.035878 | 367 | 267 | |
| PD GWAS regions excluded | 0.183247 | 0.22393 | 367 | 267 | |
| Cohort: NL | |||||
| All samples | All SNPs | 0.426036 | 0.055851 | 772 | 2024 |
| GWAS SNPs in PD loci/regions | 0.02337 | 0.010787 | 772 | 2024 | |
| PD GWAS regions excluded | 0.430828 | 0.055468 | 772 | 2024 | |
| Early onset | All SNPs | 0.133057 | 0.125479 | 366 | 871 |
| GWAS SNPs in PD loci/regions | 0.035313 | 0.022193 | 366 | 871 | |
| PD GWAS regions excluded | 0.156223 | 0.125421 | 366 | 871 | |
| Late onset | All SNPs | 0.531087 | 0.105094 | 379 | 1148 |
| GWAS SNPs in PD loci/regions | 0.026801 | 0.02065 | 379 | 1148 | |
| PD GWAS regions excluded | 0.531087 | 0.105496 | 379 | 1148 | |
| Cohort: UK | |||||
| All samples | All SNPs | 0.164206 | 0.027159 | 1705 | 5200 |
| GWAS SNPs in PD loci/regions | 0.027181 | 0.009195 | 1705 | 5200 | |
| PD GWAS regions excluded | 0.141085 | 0.027061 | 1705 | 5200 | |
| Late onset | All SNPs | 0.167639 | 0.039548 | 1258 | 2699 |
| GWAS SNPs in PD loci/regions | 0.023974 | 0.012248 | 1258 | 2699 | |
| PD GWAS regions excluded | 0.150867 | 0.039547 | 1258 | 2699 | |
| Cohort: ICE | |||||
| All samples | All SNPs | 0.491238 | 0.044370 | 604 | 4916 |
| GWAS SNPs in PD loci/regions | 0.000001 | 0.016722 | 604 | 4916 | |
| PD GWAS regions excluded | 0.517731 | 0.044039 | 604 | 4916 | |
| Late onset | All SNPs | 0.446607 | 0.060354 | 449 | 2624 |
| GWAS SNPs in PD loci/regions | 0.000001 | 0.023778 | 449 | 2624 | |
| PD GWAS regions excluded | 0.473371 | 0.059609 | 449 | 2624 | |
| Cohort: FR | |||||
| All samples | All SNPs | 0.243203 | 0.04944 | 1039 | 1984 |
| GWAS SNPs in PD loci/regions | 0.062499 | 0.011242 | 1039 | 1984 | |
| PD GWAS regions excluded | 0.198351 | 0.049936 | 1039 | 1984 | |
| Late onset | All SNPs | 0.222832 | 0.116052 | 340 | 1984 |
| GWAS SNPs in PD loci/regions | 0.047813 | 0.022223 | 340 | 1984 | |
| PD GWAS regions excluded | 0.206995 | 0.116277 | 340 | 1984 | |
| Cohort: MF | |||||
| All samples | All SNPs | 0.179096 | 0.077089 | 876 | 857 |
| GWAS SNPs in PD loci/regions | 0.018118 | 0.01266 | 876 | 857 | |
| PD GWAS regions excluded | 0.170097 | 0.076168 | 876 | 857 | |
| Cohort: FIN | |||||
| All Samples | All SNPs | 0.224504 | 0.127949 | 387 | 496 |
| GWAS SNPs in PD loci/regions | 0.058549 | 0.026548 | 387 | 496 | |
| PD GWAS regions excluded | 0.252043 | 0.126645 | 387 | 496 | |
Cohort-level heritability estimates from imputation.
Figure 1.
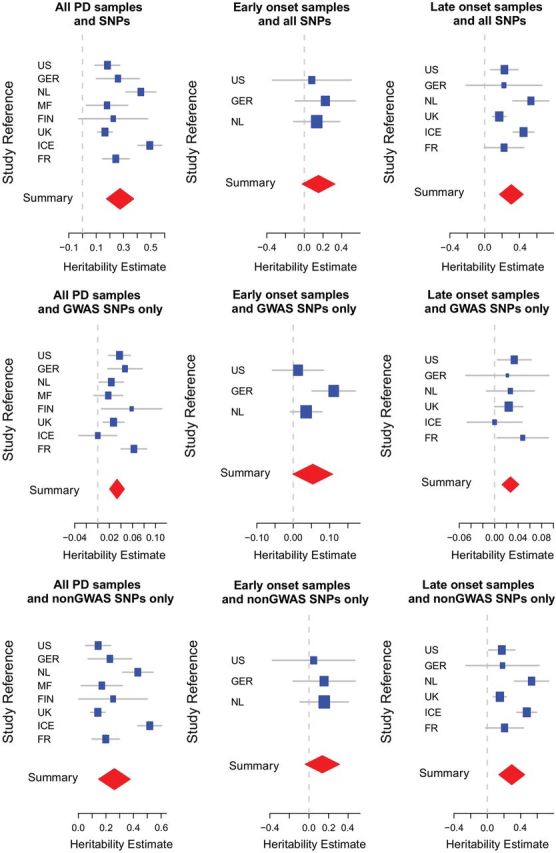
Forest plots of heritability estimates across cohorts. Cohort-specific heritability estimates are shown in blue, the size of the square is proportional to the size of the study. Confidence intervals of the summary heritability estimates are shown as red diamonds, with the centerline of each diamond representing the summary heritability estimate for that particular subset of data.
Results of our meta-analysis are presented in Table 2. We generated statistically significant heritability estimates for all PD types and late-onset PD across all SNP sets at the meta-analysis level. Our results identify 27% (95% CI 17–38, P = 8.08E − 08) phenotypic variance associated with all PD samples, 15% (95% CI −0.2 to 33, P = 0.09, non significant) phenotypic variance associated with early-onset PD and 31% (95% CI 17–44, P = 1.34E – 05) phenotypic variance associated with late-onset PD. These estimates are a substantial increase from the genetic variance identified by GWA top SNPs alone (3–5%). The estimates for all PD and late-onset PD types increased moderately using imputed data (estimate using only genotyped SNPs—all samples: 24%, P = 1.33E − 07; estimate using only genotyped SNPs—late-onset samples: 26%, P = 7.34E − 07); however, these estimates are not significantly different and are attributed to unexplained variation. The early-onset estimate decreased drastically and lost significance when imputed data were used (estimate using only genotyped SNPs—early-onset samples: 33%, P = 3.91E − 04; see Table 3 for additional detail). Despite the apparent difference between genotyped and imputed early-onset samples, the reported estimates remain within the 95% confidence interval ranges of each data type.
Table 2.
Summary of random-effects meta-analysis from imputation
| PD type | SNPs included in analysis | Heritability estimate from random effects | Lower 95% confidence interval | Upper 95% confidence interval | P-value from random effects | Heterogeneity of variance from random effects (%) | Heterogeneity P-value |
|---|---|---|---|---|---|---|---|
| All | All SNPs | 0.27 | 0.17 | 0.38 | 8.80E − 08 | 0.02 | 0.00E + 00 |
| GWAS SNPs in PD loci/regions | 0.03 | 0.02 | 0.05 | 5.23E − 07 | 0.00 | 3.20E − 02 | |
| Non-GWAS regions | 0.26 | 0.15 | 0.38 | 9.69E − 06 | 0.02 | 0.00E + 00 | |
| Early onset | All SNPs | 0.15 | −0.02 | 0.33 | 9.16E − 02 | 0.00 | 8.44E − 01 |
| GWAS SNPs in PD loci/regions | 0.05 | 0.00 | 0.11 | 5.50E − 02 | 0.00 | 6.20E − 02 | |
| Non-GWAS regions | 0.14 | −0.04 | 0.31 | 1.30E − 01 | 0.00 | 9.01E − 01 | |
| Late onset | All SNPs | 0.31 | 0.17 | 0.44 | 1.34E − 05 | 0.02 | 1.00E − 03 |
| GWAS SNPs in PD loci/regions | 0.03 | 0.01 | 0.04 | 2.59E − 04 | 0.00 | 7.85E − 01 | |
| Non-GWAS regions | 0.29 | 0.14 | 0.45 | 2.30E − 04 | 0.03 | 0.00E + 00 |
Meta-analysis of heritability estimates from imputed data. Results are significant for All and late-onset subset of PD.
Table 3.
Summary of random-effects meta-analysis from genotyping
| PD type | SNPs included in analysis | Heritability estimate from random effects | Lower 95% confidence interval | Upper 95% confidence interval | P-value from random effects | Heterogeneity of variance from random effects (%) | Heterogeneity P-value |
|---|---|---|---|---|---|---|---|
| All | All SNPs | 0.24 | 0.16 | 0.31 | 6.27E + 00 | 0.01 | 4.00E − 03 |
| GWAS SNPs in PD loci/regions | 0.02 | 0.01 | 0.03 | 1.77E − 04 | 0.00 | 3.30E − 02 | |
| Non-GWAS regions | 0.23 | 0.14 | 0.32 | 5.05E − 07 | 1.30 | <1.00E − 16 | |
| Early onset | All SNPs | 0.33 | 0.15 | 0.52 | 3.91E − 04 | 0.00 | 7.45E − 01 |
| GWAS SNPs in PD loci/regions | 0.02 | −0.01 | 0.05 | 1.38E − 01 | 0.00 | 5.94E − 01 | |
| Non-GWAS regions | 0.32 | 0.13 | 0.50 | 7.33E − 04 | 0.00 | 6.45E − 01 | |
| Late onset | All SNPs | 0.26 | 0.16 | 0.36 | 7.34E − 07 | 1.00 | 2.10E − 02 |
| GWAS SNPs in PD loci/regions | 0.01 | 0.00 | 0.02 | 1.98E − 02 | 0.00 | 5.63E − 01 | |
| Non-GWAS regions | 0.25 | 0.15 | 0.35 | 9.11E − 07 | 1.00 | 2.70E − 02 |
Meta-analysis of heritability estimates from genotyped data.
The data suggest a large portion of heritability in PD has not yet been accounted for in current GWASs. Although a small degree of heritable genetic variation is tagged by common SNPs (i.e. those included on micro-array genotyping assays), recent studies point toward the role of rare variants in disease etiology (24–26). It was expected that combined heritability estimates would show early-onset PD as having a higher portion of phenotypic variance than late-onset PD; however, this was not observed in the pooled analyses or the meta-analysis. The remaining variability in PD etiology not ascribed to heritable factors in our analyses suggests a contribution of combined effects of rare variants not tagged by current genotyping, possible environmental factors or a stochastic component. In addition, other factors including non-additive genetic effects and artifacts in the SNP data may also contribute to this discrepancy. We noted a priori 3 263 728 of rare variants were of lower quality and poorly tagged by the available microarray genotype data (57.69%).
DISCUSSION
Our estimates of phenotypic variance provide unequivocal and compelling evidence of yet-to-be-discovered additional genetic factors that contribute to the etiology of PD. While imputation can capture up to ∼50% of the genetic variation associated with PD in some cohorts, our study was limited by the sensitivity of microarray-based genotyping methods utilized in our calculation of heritability. In addition, disparate demographic histories between cohorts likely contributed to the differences shown in the cohort-level heritability estimates. In particular, the Icelandic cohort produced the highest genome-wide estimates and the lowest GWAS SNPs estimates. The population of Iceland is remarkably homogenous compared with the populations of France and the UK, for example, and their shared ancestry is reflected in their genetic structure as measured by GCTA.
Our results also provide support for the hypothesis that rare variants of potentially large effect are less likely to be accurately tagged by microarray, potentially biasing heritability estimates as appearing lower than what should be expected, particularly for early-onset PD. Large-scale genome and exome sequencing in conjunction with denser genotyping in large cohorts may help to better quantify the heritability of complex diseases. These efforts will also aid in the identification of loci that contribute to ‘missing heritability’ previously undetected in earlier generation technologies used for capturing genomic variation, as were implemented in this study. For example, the GBA locus contains approximately 17 rare mutations, not all of which were originally detected by GWASs (27,28). In addition, the use of newer technologies is likely to revise disease heritability estimates upward.
Heterogeneity in the heritability estimates reported here could result from heterogeneity in the coverage of the genome as well as from patient acquisition biases. A large proportion of the dbGaP cohort (herein referred to as MF, further described in Materials and Methods) is comprised of familial PD cases; however, the heritability estimates for this cohort are among the lowest. This may reflect differences in genotyping platform. The US-NIA (US), Dutch (NL), UK: WTCCC2/Cardiff (UK), French (FR) and German (GER) cohorts were genotyped using the 610 and 550K Illumina arrays containing 500K SNPs, whereas the MF, Finnish (FIN) and Icelandic (ICE) cohorts were genotyped using the Illumina 370K. The use of MACH (Markov Chain-based haplotyper) for imputation allows comparisons between cohorts of different genotyping platforms to be made, because genotypes are imputed based on the observed haplotype structure of each cohort, and therefore analysis occurs across the same set of SNPs.
It is important to note that the choice of prevalence value has a minor impact on our heritability estimates. Our study model dichotomizes the PD phenotype based on age, and because PD prevalence increases with age, we used a prevalence value standardized for age and gender, specified here as 0.002. To account for the larger prevalence within the case population and to control for ascertainment, we used GCTA to transform the explained variance estimate from the observed scale (V(1)/Vp) to the underlying scale (V(1)/Vp_L). Previous work has shown that assumed prevalence has only a small impact on GCTA estimates. In particular, Do et al. (15) show that prevalence values ranging 3-fold (0.005–0.015) impact on their estimates by only ∼5%. Our subsequent analyses show that using a conservative prevalence value still provides ample evidence for identifying increased phenotypic variance of PD.
An alternative method of analysis would employ a conditional analysis within GCTA, using the GWAS-significant PD SNPs as covariates to statistically correct for known signals during the restricted maximum likelihood (REML) analysis. It is expected that the known SNPs would capture the same variance as the whole set of SNPs used in the GWAS region analysis; however, three of the top SNPs (rs1491942, rs6710823 and rs76763715) were not available in our imputed data for multiple cohorts. As no proxies were available, the results are not comparable with the unconditional analysis and are not reported here.
Our analyses indicate that the estimates of heritability presented here are minimal estimates, despite the significant increase over what has been identified by GWASs. The genetic additive heritability identified by the GWAS SNPs is also likely an underestimate of the genetic variance due to common PD variants, as the associated GWAS SNPs are not necessarily causal: their effects are lower than those of the true susceptibility variants. Genotyping these variants would likely produce greater estimates of the common genetic additive variance due to the GWAS PD loci. Indel and structural variants were not considered here, although a portion of these was likely tagged by the considered SNPs. In addition, the portion of variance explained by GWAS SNPs is underestimated by GCTA, as the model places a prior centered zero as the effect size of the SNPs that are used in the calculation of the genetic relationship matrix (GRM). This has been previously shown using sparse regression techniques, which account for 6–7% of total variance in GWAS SNPs, further suggesting the majority of genetic variants that contribute to PD lay outside of GWAS-significant SNPs (15). Large-scale sequencing efforts often identify many variants that are not easily tagged using microarray genotyping. This is apparent in evaluations of the success of capturing rare variants, most commonly defined as having a minor allele frequency (MAF) of ≤0.05. In addition, rare variants are thought to harbor larger deleterious effects than more common variants (29,30). In this context, it is likely that further GWA studies that include greater numbers of typed SNPs will identify additional risk for PD.
In conclusion, our estimates do not confirm hypotheses of a greater contribution of genetic risk for early-onset PD compared with late-onset PD; however, we expect the contributions of rare variants not tagged by microarray genotyping to have a substantial impact on the genetic contributions to disease risk. Although our imputed analysis did not provide significant results for this subset of PD, we note a priori that a significant portion of rare variants are not well captured in microarrays (∼58%); therefore, our heritability estimates are lower than what might be expected. In addition, very rare variants (MAF < 0.005) require reference panels with at least 1200 subjects in order to impute, as it is necessary to observe multiple copies of an SNP in order to accurately constitute the haplotypes that will be used for imputation (31). Future analyses incorporating denser genome and exome-wide assays in conjunction with newer sequencing technologies will likely see increased heritability estimates associated with PD and other complex traits, as a significantly larger genetic contribution to disease risk is identified.
MATERIALS AND METHODS
Study populations
We utilized six GWAS data sets drawn from the International Parkinson's Disease Genomics Consortium (IPDGC), comprised of US and European participants previously described in detail elsewhere (7,32–37). The IPDGC includes data from Iceland (ICE, n = 5520), the UK: WTCCC2/Cardiff (UK, n = 6905), the Netherlands (NL, n = 2796), Germany (GER, n = 1686), France (FR, n = 3023) and the USA-NIA (US, n = 4005). Additional US participant data were obtained via dbGaP, made available by the NINDS Human Genetics Resource Center and have been described by Pankratz et al. (35) (MF, n = 1733); a Finnish case–control cohort was also incorporated into this study based on the Vantaa 85+ Study (38) (FIN = 883; cases from Oulu, n = 387; controls from mitoPARK and Vantaa 85+, n = 496). In total, 8 cohorts comprising 7096 cases and 19 455 controls were used. Study descriptives for each cohort are outlined in Table 4. Cohorts containing individuals with age data were further subset to account for early- and late-onset cases and controls. Age at onset was defined here as age at initial diagnosis in cases and matched to controls using age at study ascertainment. Early onset was quantified as ≤55 years old and late onset was quantified as >55 years old. Early-onset data sets were drawn only from individuals in the US, German and Dutch cohorts, as the remainder of the cohorts did not contain control individuals ≤55 years of age. Late-onset data sets were drawn from individuals in the US, German, Dutch, Icelandic, French and UK cohorts.
Table 4.
Study descriptives
| Cases |
Controls |
Study details |
||||||
|---|---|---|---|---|---|---|---|---|
| Cohort | Sample size | Female (%) | Mean age at onset [years (SD)] | Sample size | Female (%) | Mean age [years (SD)] | Number of SNPs used for imputation | Genomic inflation factor (λ) |
| US | 937 | 40.23 | 57.81 (13.16) | 3033 | 52.82 | 63.3 (10.06) | 545 066 | 1.035 |
| GER | 740 | 39.59 | 49.33 (22.21) | 944 | 47.99 | 47 (13.25) | 561 467 | 1.025 |
| NL | 771 | 36.45 | 53.39 (16.62) | 2024 | 55.94 | 55.56 (6.60) | 546 155 | 1.061 |
| UK | 1705 | 42.22 | 48.95 (13.84) | 5200 | 49.42 | 53 (0) | 532 616 | 1.034 |
| ICE | 604 | 47.85 | 73.26 (13.84) | 5520 | 55.87 | 85.12 (10.77) | 316 905 | 1.011 |
| FR | 1039 | 41.2 | 48.9 (12.8) | 1984 | 42.9 | 73.7 (5.4) | 492 929 | 1.03 |
| MF | 876 | 40.41 | 36.42 (11.08) | 857 | 60.21 | NA | 325 770 | 1.013 |
| FIN | 387 | 45.99 | 48.28 (6.97) | 496 | 0 | 91.98 (7.46) | 302 463 | 1.066 |
Cohort-level descriptive statistics and study details.
As per our study design, older controls were excluded from our early-onset analyses due to previous research suggesting demographic factors such as age influence genetic sub-structure. In particular, increasing homozygosity has been associated with chronological age (39). As this trend could influence the frequency of the variants used in our analyses of early-onset PD heritability and introduce stochastic change or other potential bias that is not controlled for, we have excluded older controls from early-onset analyses.
Stringent quality control measures were applied to all data sets prior to GCTA analysis, in order to control for ascertainment bias and any artifacts introduced into the data by the genotyping process (32). It was necessary to employ more stringent quality control that what is generally acceptable for GWASs because systematic differences between cases and controls could be picked up as genetic variance (3). Cases and controls in all cohorts were analyzed together to reduce bias. SNPs with different call rates in cases and controls were excluded. Additional detail regarding specific cohort-level quality control measures is described elsewhere (32,40).
As a note, three SNPs, ACMSD, GAK, and HLA-DRB5, were initially found to be associated in a GWAS that was performed on the same data sets used in the present study (32). Known inherited Mendelian loci were not directly accounted for in our estimates.
Quality control and imputation
After samples were collected from each cohort, and standard quality control was applied, MACH (version 1.0.16) was applied to each cohort to impute genotypes for all European ancestry participants. Haplotypes were derived from 500 European ancestry samples in the 1000 Genomes Project (as of 23 September 2011), based on initial low coverage and exome sequencing. A quality threshold of a minimum 0.30 squared correlation between proximal, experimental and imputed genotypes was applied, indicated by the R2 metric from MACH (41,42). We used the default settings of MiniMac (41,42) to impute variants into each cohort, and the total number of imputed variants per cohort is shown in Table 4.
Data were imputed in a two-stage design. The first stage of imputation generated error and crossover maps on a random subset of 200 samples per study, for 100 iterations of the statistical model. These maps were used as parameter estimates for imputation, to generate maximum likelihood estimates of allele dosages per SNP, on the basis of reference haplotypes from the 1000 Genomes Project during the second stage of the imputation. SNPs were excluded if their R2 quality estimates were <0.30, as estimated by MACH, or if their MAFs were <0.01, because imputed genotypes below these values are likely poor in quality and are more susceptible to errors in imputation.
Cryptic relatedness among samples was addressed at both cohort and meta-analysis levels. Within the cohorts, samples sharing >0.15 proportion of alleles or samples identified as first or second degree relatives according to identity by descent estimates were excluded (32). As an additional quality control measure, we calculated genomic control for individual data sets. Genomic control is based on the χ2 statistic and is estimated as the deviance of the median test statistic distribution from the expected null. The product of this analysis is a λ-value; λ-values <1.05 are standard in GWASs (32). λ-Values were obtained before imputation, and indicate that population stratification is minimal. Genome-wide λ-values for each cohort are reported in Table 4.
Statistical analysis
We applied GCTA to estimate heritability within each of the stratified data sets per cohort. Heritability is defined here as the proportion of phenotypic variation in a population that is due to genetic variance between individuals. GRMs were calculated for each subset of data to determine the genetic relationship between pairs of individuals (3,22). The GRMs in this analysis were estimated using imputed dosage score SNP data; therefore, the estimation of variance explained by the SNPs relies on the R2 cutoff used to select the SNPs.
GRMs were input into an REML analysis to produce estimates of the proportion of phenotypic variance explained by the SNPs within each subset of data (22). In addition to cohort-level quality control, SNPs with MAFs <0.01 and R2 values <0.3 were excluded from the REML analysis. All analyses were adjusted for eigenvectors 1–20 from principal component analyses to account for possible confounding by population substructure within each cohort (22,43). Within the analysis, the component vectors were used as basic covariates to identify random genomic differences between genotyped data from cases and controls, in order to adjust statistical models for covariates accounting for possible population substructure. Summary statistics from these estimates were produced by every data set and were included in the meta-analyses.
The disease prevalence for PD was estimated from a general European ancestry populations identified by the literature. PD prevalence increases with age; therefore, the prevalence value was standardized for age and gender, and is specified here as 0.002 (4,8,17,18). To control for ascertainment, GCTA transformed the explained variance estimate from the observed scale (V(1)/Vp) to the underlying scale (V(1)/Vp_L) to provide more robust heritability estimates.
Using PLINK version 1.07 (44), imputed SNPs were used to subset three basic data sets for each cohort. These included:
All imputed SNPs;
All known SNPs ±1 MB located within a region identified by replicated GWASs as associated with PD (described in Table 5); and
SNPs not located within ±1 MB of a region associated with PD by GWASs. Twenty-seven highly significant and well-replicated SNPs were used to define the PD regions for the second and third data sets, as described in Table 5.
Table 5.
Loci associated with PD based on GWASs
| Gene name(s) | Primary SNP | chr | Position (bp) | Citation(s) |
|---|---|---|---|---|
| GBA | N370S/i4000416 | 1 | 153 451 576–153 472 258 | Lill et al. (48); Do et al. (15) |
| SYT11/RAB25 | chr1:154105678 | 1 | 154 105 678 | Lill et al. (48); Nalls and colleagues (32) |
| RAB7L1/PARK16 | rs708723–rs947211 | 1 | 204 019 288 | Lill et al. (48); Plagnol and colleagues (34) |
| SLC41A1 | rs823156 | 1 | 204 031 263 | Do et al. (15) |
| ACMSD | rs6710823 | 2 | 135 308 851 | Nalls and colleagues (28) |
| STK39 | rs2102808, rs2390669 | 2 | 168 800 188–168 825 271 | Lill et al. (48); Nalls and colleagues (32) |
| NMD3 | rs34016896 | 3 | 34 016 896 | Plagnol and colleagues (34) |
| MCCC1/LAMP3 | rs10513789, rs11711441 | 3 | 184 242 767–184 303 969 | Lill et al. (48); Do et al. (15) |
| GAK | chr4: 811311, rs6599389 | 4 | 911 311 | Nalls and colleagues (32) |
| DGKQ | rs11248060 | 4 | 929 113–954 359 | Lill et al. (48); Do et al. (15) |
| STBD1 | rs6812193 | 4 | 6 812 193 | Plagnol and colleagues (34) |
| BST1 | rs11724635 | 4 | 15 346 199 | Lill et al. (48); Nalls and colleagues (32) |
| SCARB2 | rs6812193 | 4 | 77 418 010 | Do et al. (15) |
| SNCA | rs356220, rs6532194 | 4 | 90 999 925–90 860 363 | Lill et al. (48); Nalls and colleagues (32); Do et al. (15) |
| HLA-DRB5 | chr6:32588205 | 6 | 32 588 205 | Nalls and colleagues (32) |
| GPNMB | rs156429 | 7 | 156 429 | Plagnol and colleagues (34) |
| FGF20 | rs591323 | 8 | 591 323 | Plagnol and colleagues (34) |
| MMP16 | chr8:89442157 | 8 | 89 442 157 | Plagnol and colleagues (34) |
| ITGA8 | rs7077361 | 10 | 15 601 549 | Lill et al. (48) |
| LRRK2 | rs1491942, rs34637584 | 12 | 38 907 075–39 020 469 | Lill et al. 2011 (48); Nalls and colleagues (32); Do et al. (15) |
| CCDC62/HIP1R | rs10847864, rs12817488 | 12 | 121 862 247–121 892 551 | Lill et al. (48); Nalls and colleagues (32) |
| STX1B | rs4889603 | 16 | 4 889 603 | Plagnol and colleagues (34) |
| SREBF1/RAI1 | rs11868035 | 17 | 17 655 826 | Do et al. (15) |
| MAPT/STH | rs12185268, rs2942168 | 17 | 41 149 582–42 131 818 | Lill et al. (48); Nalls and colleagues (32); Do et al. (15) |
| RIT2/SYT4 | rs4130047 | 18 | 38 932 233 | Do et al. (15) |
| USP25 | rs2823357 | 21 | 15 836 776 | Do et al. (15) |
Gene names and primary locations are provided for 27 highly significant and well-replicated SNPs. These SNPs were used to define the PD regions for our subsetted data sets.
Heritability estimates were compared between three data sets, when using:
All SNPs simultaneously;
SNPs identified by GWASs as located within regions associated with PD; and
SNPs not identified by GWASs (i.e. ‘missing’ or potentially hidden heritability).
We stratified our analysis to compare estimates between early and late PD onset. This allowed for a comparison of ‘heritability’ estimates for PD dependent on age at onset. REML analyses were repeated for GER, US and NL cohorts to compare early-onset (≤55 years) cases and controls, and for GER, US, NL, UK, FR and ICE cohorts to compare late-onset cases (>55 years) and controls. In addition to cohort-level analyses, a meta-analysis was conducted to combine summary statistics.
We performed a random-effects meta-analysis using R 2.15. This produced powerful phenotypic variance and heterogeneity estimates across cohorts (Table 2). To quantify heterogeneity across cohorts, Cochran's Q statistic was estimated using R. Cochran's Q is based on a χ2 distribution, and is calculated as the weighted sum of squared differences between each effect of the individual study and the pooled effect across studies (45–47):
This statistic does not inform the type or cause of heterogeneity, only of its presence or absence.
In order to quantify the impact of coverage of microarray genotyping on our heritability estimates, we determined the number of rare variants imputed into each cohort. Variants with an MAF of <1% were considered rare.
WEB RESOURCES
R Development Core Team (2011). R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria, ISBN 3-900051-07-0, http://www.R-project.org/.
1000 Genomes Project: A Deep Catalog of Human Genetic Variation (2008–2010), http://www.1000genomes.org/.
MACH: MiniMac (2011), http://genome.sph.umich.edu/wiki/Minimac.
FUNDING
This work was supported by the Intramural Research Programs of the National Institute on Aging, National Institute of Neurological Disorders and Stroke, NIEHS and NHGRI of the National Institutes of Health, Department of Health and Human Services (project numbers Z01-AG000949-02 and Z01-ES101986), human subjects protocol 2003-077. This work was also supported by the US Department of Defense (award number W81XWH-09-2-0128); National Institutes of Health (grants NS057105 and RR024992); American Parkinson Disease Association (APDA); Barnes Jewish Hospital Foundation; Greater St Louis Chapter of the APDA; Hersenstichting Nederland; Neuroscience Campus Amsterdam; and the section of medical genomics, the Prinses Beatrix Fonds. The KORA (Cooperative Research in the Region of Augsburg) research platform was started and financed by the Forschungszentrum für Umwelt und Gesundheit, which is funded by the German Federal Ministry of Education, Science, Research, and Technology and by the State of Bavaria. This study was also funded by the German National Genome Network (NGFNplus number 01GS08134, German Ministry for Education and Research); by the German Federal Ministry of Education and Research (NGFN 01GR0468, PopGen); and 01EW0908 in the frame of ERA-NET NEURON and Helmholtz Alliance Mental Health in an Ageing Society (HA-215), which was funded by the Initiative and Networking Fund of the Helmholtz Association. The French GWAS work was supported by the French National Agency of Research (ANR-08-MNP-012). This study was also funded by the Michael J. Fox Foundation (MS), and sponsored by the Landspitali University Hospital Research Fund (grant to S.Sv.); Icelandic Research Council (grant to S.Sv.); and European Community Framework Programme 7, People Programme and IAPP on novel genetic and phenotypic markers of Parkinson's disease and Essential Tremor (MarkMD), contract number PIAP-GA-2008-230596 MarkMD (to H.P. and J.Hu.). Genotyping of UK replication cases on ImmunoChip was part of the WTCCC2 project, which was funded by the Wellcome Trust (083948/Z/07/Z). This study was supported by the Medical Research Council and Wellcome Trust disease centre (grant WT089698/Z/09/Z to N.W.W., J.Ha. and A.Sc.). This study was also supported by Parkinson's UK (grants 8047 and J-0804) and the Medical Research Council (G0700943). DNA extraction work that was done in the UK was undertaken at University College London Hospitals, University College London, which received a proportion of funding from the Department of Health's National Institute for Health Research Biomedical Research Centres funding. This study was supported in part by the Wellcome Trust/Medical Research Council Joint Call in Neurodegeneration award (WT089698) to the Parkinson's Disease Consortium (UKPDC), whose members are from the UCL Institute of Neurology, University of Sheffield and the Medical Research Council Protein Phosphorylation Unit at the University of Dundee. Mohamad Saad was funded by the France Parkinson Association.
ACKNOWLEDGEMENTS
We used the Biowulf Linux cluster at the National Institutes of Health, Bethesda, MD, USA, and DNA panels, samples and clinical data from the National Institute of Neurological Disorders and Stroke Human Genetics Resource Center DNA and Cell Line Repository. People who contributed samples are acknowledged in descriptions of every panel on the repository website. We thank the French Parkinson's Disease Genetics Study Group: Y. Agid, M. Anheim, A.-M. Bonnet, M. Borg, A. Brice, E. Broussolle, J.-C. Corvol, P. Damier, A. Destée, A. Dürr, F. Durif, S. Klebe, E. Lohmann, M. Martinez, P. Pollak, O. Rascol, F. Tison, C. Tranchant, M. Vérin, F. Viallet and M. Vidailhet. We also thank the members of the French 3C Consortium: A. Alpérovitch, C. Berr, C. Tzourio and P. Amouyel for allowing us to use part of the 3C cohort, and D. Zelenika for support in generating the genome-wide molecular data. We thank P. Tienari (Molecular Neurology Programme, Biomedicum, University of Helsinki), T. Peuralinna (Department of Neurology, Helsinki University Central Hospital), L. Myllykangas (Folkhalsan Institute of Genetics and Department of Pathology, University of Helsinki) and R. Sulkava (Department of Public Health and General Practice Division of Geriatrics, University of Eastern Finland) for the Finnish controls (Vantaa85+ GWAS data). We used genome-wide association data generated by the Wellcome Trust Case Control Consortium 2 (WTCCC2) from UK patients with Parkinson's disease and UK control individuals from the 1958 Birth Cohort and National Blood Service. UK population control data were made available through WTCCC1. We thank Jeffrey Barrett for assistance with the design of the ImmunoChip.
Conflict of Interest statement. None declared.
APPENDIX
INTERNATIONAL PARKINSON DISEASE GENOMICS CONSORTIUM MEMBERS
Margaux F. Keller (Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, MD, USA), Michael A. Nalls (Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, MD, USA), Vincent Plagnol (UCL Genetics Institute, London, UK), Dena G. Hernandez (Laboratory of Neurogenetics, National Institute on Aging; and Department of Molecular Neuroscience, UCL Institute of Neurology, London, UK), Manu Sharma (Department for Neurodegenerative Diseases, Hertie Institute for Clinical Brain Research, University of Tübingen, and DZNE, German Center for Neurodegenerative Diseases, Tübingen, Germany), Una-Marie Sheerin (Department of Molecular Neuroscience, UCL Institute of Neurology), Mohamad Saad (INSERM U563, CPTP, Toulouse, France; and Paul Sabatier University, Toulouse, France), Javier Simón-Sánchez (Department of Clinical Genetics, Section of Medical Genomics, VU University Medical Centre, Amsterdam, The Netherlands), Claudia Schulte (Department for Neurodegenerative Diseases, Hertie Institute for Clinical Brain Research), Suzanne Lesage [INSERM, UMR-S975 (formerly UMR-S679), Paris, France; Université Pierre et Marie Curie-Paris, Centre de Recherche de l'Institut du Cerveau et de la Moelle épinière, Paris, France; and CNRS, Paris, France], Sigurlaug Sveinbjörnsdóttir (Department of Neurology, Landspítali University Hospital, Reykjavík, Iceland; Department of Neurology, MEHT Broomfield Hospital, Chelmsford, Essex, UK; and Queen Mary College, University of London, London, UK), Sampath Arepalli (Laboratory of Neurogenetics, National Institute on Aging), Roger Barker (Department of Neurology, Addenbrooke's Hospital, University of Cambridge, Cambridge, UK), Yoav Ben-Shlomo (School of Social and Community Medicine, University of Bristol), Henk W. Berendse (Department of Neurology and Alzheimer Center, VU University Medical Center), Daniela Berg (Department for Neurodegenerative Diseases, Hertie Institute for Clinical Brain Research), Kailash Bhatia (Department of Motor Neuroscience, UCL Institute of Neurology), Rob M.A. de Bie (Department of Neurology, Academic Medical Center, University of Amsterdam, Amsterdam, The Netherlands), Alessandro Biffi (Center for Human Genetic Research and Department of Neurology, Massachusetts General Hospital, Boston, MA, USA; and Program in Medical and Population Genetics, Broad Institute, Cambridge, MA, USA), Bas Bloem (Department of Neurology, Radboud University Nijmegen Medical Centre, Nijmegen, The Netherlands), Zoltan Bochdanovits (Department of Clinical Genetics, Section of Medical Genomics, VU University Medical Centre), Michael Bonin (Department of Medical Genetics, Institute of Human Genetics, University of Tübingen, Tübingen, Germany), Jose Bras (Department of Molecular Neuroscience, UCL Institute of Neurology), Kathrin Brockmann (Department for Neurodegenerative Diseases, Hertie Institute for Clinical Brain Research), Janet Brooks (Laboratory of Neurogenetics, National Institute on Aging), David J. Burn (Newcastle University Clinical Ageing Research Unit, Campus for Ageing and Vitality, Newcastle upon Tyne, UK), Gavin Charlesworth (Department of Molecular Neuroscience, UCL Institute of Neurology), Honglei Chen (Epidemiology Branch, National Institute of Environmental Health Sciences, National Institutes of Health, NC, USA), Patrick F. Chinnery (Neurology M4104, The Medical School, Framlington Place, Newcastle upon Tyne, UK), Sean Chong (Laboratory of Neurogenetics, National Institute on Aging), Carl E. Clarke (School of Clinical and Experimental Medicine, University of Birmingham, Birmingham, UK; and Department of Neurology, City Hospital, Sandwell and West Birmingham Hospitals NHS Trust, Birmingham, UK), Mark R. Cookson (Laboratory of Neurogenetics, National Institute on Aging), J. Mark Cooper (Department of Clinical Neurosciences, UCL Institute of Neurology), Jean Christophe Corvol (INSERM, UMR_S975; Université Pierre et Marie Curie-Paris; CNRS; and INSERM CIC-9503, Hôpital Pitié-Salpêtrière, Paris, France), Carl Counsell (University of Aberdeen, Division of Applied Health Sciences, Population Health Section, Aberdeen, UK), Philippe Damier (CHU Nantes, CIC0004, Service de Neurologie, Nantes, France), Jean-François Dartigues (INSERM U897, Université Victor Segalen, Bordeaux, France), Panos Deloukas (Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Cambridge, UK), Günther Deuschl (Klinik für Neurologie, Universitätsklinikum Schleswig-Holstein, Campus Kiel, Christian-Albrechts-Universität Kiel, Kiel, Germany), David T. Dexter (Parkinson's Disease Research Group, Faculty of Medicine, Imperial College London, London, UK), Karin D. van Dijk (Department of Neurology and Alzheimer Center, VU University Medical Center), Allissa Dillman (Laboratory of Neurogenetics, National Institute on Aging), Frank Durif (Service de Neurologie, Hôpital Gabriel Montpied, Clermont-Ferrand, France), Alexandra Dürr (INSERM, UMR-S975; Université Pierre et Marie Curie-Paris; CNRS; and AP-HP, Pitié-Salpêtrière Hospital), Sarah Edkins (Wellcome Trust Sanger Institute), Jonathan R. Evans (Cambridge Centre for Brain Repair, Cambridge, UK), Thomas Foltynie (UCL Institute of Neurology), Jianjun Gao (Epidemiology Branch, National Institute of Environmental Health Sciences), Michelle Gardner (Department of Molecular Neuroscience, UCL Institute of Neurology), J. Raphael Gibbs (Laboratory of Neurogenetics, National Institute on Aging; and Department of Molecular Neuroscience, UCL Institute of Neurology), Alison Goate (Department of Psychiatry, Department of Neurology, Washington University School of Medicine, MI, USA), Emma Gray (Wellcome Trust Sanger Institute), Rita Guerreiro (Department of Molecular Neuroscience, UCL Institute of Neurology), Ómar Gústafsson (deCODE genetics and Department of Psychiatry, Oslo University Hospital, N-0407 Oslo, Norway), Clare Harris (University of Aberdeen), Jacobus J. van Hilten (Department of Neurology, Leiden University Medical Center, Leiden, The Netherlands), Albert Hofman (Department of Epidemiology, Erasmus University Medical Center, Rotterdam, The Netherlands), Albert Hollenbeck (AARP, Washington, DC, USA), Janice Holton (Queen Square Brain Bank for Neurological Disorders, UCL Institute of Neurology), Michele Hu (Department of Clinical Neurology, John Radcliffe Hospital, Oxford, UK), Xuemei Huang (Departments of Neurology, Radiology, Neurosurgery, Pharmacology, Kinesiology, and Bioengineering, Pennsylvania State University—Milton S. Hershey Medical Center, Hershey, PA, USA), Heiko Huber (Department for Neurodegenerative Diseases, Hertie Institute for Clinical Brain Research), Gavin Hudson (Neurology M4104, The Medical School, Newcastle upon Tyne, UK), Sarah E. Hunt (Wellcome Trust Sanger Institute), Johanna Huttenlocher (deCODE genetics), Thomas Illig (Institute of Epidemiology, Helmholtz Zentrum München, German Research Centre for Environmental Health, Neuherberg, Germany), Pálmi V. Jónsson (Department of Geriatrics, Landspítali University Hospital, Reykjavík, Iceland), Jean-Charles Lambert (INSERM U744, Lille, France; and Institut Pasteur de Lille, Université de Lille Nord, Lille, France), Cordelia Langford (Cambridge Centre for Brain Repair), Andrew Lees (Queen Square Brain Bank for Neurological Disorders), Peter Lichtner (Institute of Human Genetics, Helmholtz Zentrum München, German Research Centre for Environmental Health, Neuherberg, Germany), Patricia Limousin (Institute of Neurology, Sobell Department, Unit of Functional Neurosurgery, London, UK), Grisel Lopez (Section on Molecular Neurogenetics, Medical Genetics Branch, NHGRI, National Institutes of Health), Delia Lorenz (Klinik für Neurologie, Universitätsklinikum Schleswig-Holstein), Alisdair McNeill (Department of Clinical Neurosciences, UCL Institute of Neurology), Catriona Moorby (School of Clinical and Experimental Medicine, University of Birmingham), Matthew Moore (Laboratory of Neurogenetics, National Institute on Aging), Huw R. Morris (MRC Centre for Neuropsychiatric Genetics and Genomics, Cardiff University School of Medicine, Cardiff, UK), Karen E. Morrison (School of Clinical and Experimental Medicine, University of Birmingham; and Neurosciences Department, Queen Elizabeth Hospital, University Hospitals Birmingham NHS Foundation Trust, Birmingham, UK), Ese Mudanohwo (Neurogenetics Unit, UCL Institute of Neurology and National Hospital for Neurology and Neurosurgery), Sean S. O'Sullivan (Queen Square Brain Bank for Neurological Disorders), Justin Pearson (MRC Centre for Neuropsychiatric Genetics and Genomics), Joel S. Perlmutter (Department of Neurology, Radiology, and Neurobiology at Washington University, St Louis, MO, USA), Hjörvar Pétursson (deCODE genetics; and Department of Medical Genetics, Institute of Human Genetics, University of Tübingen), Pierre Pollak (Service de Neurologie, CHU de Grenoble, Grenoble, France), Bart Post (Department of Neurology, Radboud University Nijmegen Medical Centre), Simon Potter (Wellcome Trust Sanger Institute), Bernard Ravina (Translational Neurology, Biogen Idec, MA, USA), Tamas Revesz (Queen Square Brain Bank for Neurological Disorders), Olaf Riess (Department of Medical Genetics, Institute of Human Genetics, University of Tübingen), Fernando Rivadeneira (Departments of Epidemiology and Internal Medicine, Erasmus University Medical Center), Patrizia Rizzu (Department of Clinical Genetics, Section of Medical Genomics, VU University Medical Centre), Mina Ryten (Department of Molecular Neuroscience, UCL Institute of Neurology), Stephen Sawcer (University of Cambridge, Department of Clinical Neurosciences, Addenbrooke's Hospital, Cambridge, UK), Anthony Schapira (Department of Clinical Neurosciences, UCL Institute of Neurology), Hans Scheffer (Department of Human Genetics, Radboud University Nijmegen Medical Centre, Nijmegen, The Netherlands), Karen Shaw (Queen Square Brain Bank for Neurological Disorders), Ira Shoulson (Department of Neurology, University of Rochester, Rochester, NY, USA), Ellen Sidransky (Section on Molecular Neurogenetics, Medical Genetics Branch, NHGRI), Colin Smith (Department of Pathology, University of Edinburgh, Edinburgh, UK), Chris C.A. Spencer (Wellcome Trust Centre for Human Genetics, Oxford, UK), Hreinn Stefánsson (deCODE genetics), Stacy Steinberg (deCODE genetics), Joanna D. Stockton (School of Clinical and Experimental Medicine), Amy Strange (Wellcome Trust Centre for Human Genetics), Kevin Talbot (University of Oxford, Department of Clinical Neurology, John Radcliffe Hospital, Oxford, UK), Carlie M. Tanner (Clinical Research Department, The Parkinson's Institute and Clinical Center, Sunnyvale, CA, USA), Avazeh Tashakkori-Ghanbaria (Wellcome Trust Sanger Institute), François Tison (Service de Neurologie, Hôpital Haut-Lévêque, Pessac, France), Daniah Trabzuni (Department of Molecular Neuroscience, UCL Institute of Neurology), Bryan J. Traynor (Laboratory of Neurogenetics, National Institute on Aging), André G. Uitterlinden (Departments of Epidemiology and Internal Medicine, Erasmus University Medical Center), Daan Velseboer (Department of Neurology, Academic Medical Center), Marie Vidailhet (INSERM, UMR-S975, Université Pierre et Marie Curie-Paris, CNRS, UMR-7225), Robert Walker (Department of Pathology, University of Edinburgh), Bart van de Warrenburg (Department of Neurology, Radboud University Nijmegen Medical Centre), Mirdhu Wickremaratchi (Department of Neurology, Cardiff University, Cardiff, UK), Nigel Williams (MRC Centre for Neuropsychiatric Genetics and Genomics), Caroline H. Williams-Gray (Department of Neurology, Addenbrooke's Hospital), Sophie Winder-Rhodes (Department of Psychiatry and Medical Research Council and Wellcome Trust Behavioural and Clinical Neurosciences Institute, University of Cambridge), Kári Stefánsson (deCODE genetics), Maria Martinez (INSERM U563; and Paul Sabatier University), John Hardy (Department of Molecular Neuroscience, UCL Institute of Neurology), Peter Heutink (Department of Clinical Genetics, Section of Medical Genomics, VU University Medical Centre), Alexis Brice (INSERM, UMR-S975, Université Pierre et Marie Curie-Paris, CNRS, UMR-7225, AP-HP, Pitié-Salpêtrière Hospital), Wellcome Trust Case Control Consortium 2 (webappendix, p. 13), Thomas Gasser (Department for Neurodegenerative Diseases, Hertie Institute for Clinical Brain Research, and DZNE, German Center for Neurodegenerative Diseases), Andrew B. Singleton (Laboratory of Neurogenetics, National Institute on Aging), Nicholas W. Wood (UCL Genetics Institute; and Department of Molecular Neuroscience, UCL Institute of Neurology).
WELLCOME TRUST CASE CONTROL CONSORTIUM 2 MEMBERS
Management committee
Peter Donnelly (Chair)1,2, Ines Barroso (Deputy Chair)3, Jenefer M. Blackwell4,5, Elvira Bramon6, Matthew A. Brown7, Juan P. Casas8, Aiden Corvin9, Panos Deloukas3, Audrey Duncanson10, Janusz Jankowski11, Hugh S. Markus12, Christopher G. Mathew13, Colin N.A. Palmer14, Robert Plomin15, Anna Rautanen1, Stephen J. Sawcer16, Richard C. Trembath13, Ananth C. Viswanathan17 and Nicholas W. Wood18
Data and analysis group
Chris C.A. Spencer1, Gavin Band1, Céline Bellenguez1, Colin Freeman1, Garrett Hellenthal1, Eleni Giannoulatou1, Matti Pirinen1, Richard Pearson1, Amy Strange1, Zhan Su1, Damjan Vukcevic1 and Peter Donnelly1,2
DNA, genotyping, data QC and informatics group
Cordelia Langford3, Sarah E. Hunt3, Sarah Edkins3, Rhian Gwilliam3, Hannah Blackburn3, Suzannah J. Bumpstead3, Serge Dronov3, Matthew Gillman3, Emma Gray3, Naomi Hammond3, Alagurevathi Jayakumar3, Owen T. McCann3, Jennifer Liddle3, Simon C. Potter3, Radhi Ravindrarajah3, Michelle Ricketts3, Matthew Waller3, Paul Weston3, Sara Widaa3, Pamela Whittaker3, Ines Barroso3 and Panos Deloukas3
Publications committee
Christopher G. Mathew (Chair)13, Jenefer M Blackwell4,5, Matthew A. Brown7, Aiden Corvin9, Mark I. McCarthy19 and Chris C.A. Spencer1
1Wellcome Trust Centre for Human Genetics, Roosevelt Drive, Oxford OX3 7LJ, UK; 2Department of Statistics, University of Oxford, Oxford OX1 3TG, UK; 3Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge CB10 1SA, UK; 4Telethon Institute for Child Health Research, Centre for Child Health Research, University of Western Australia, 100 Roberts Road, Subiaco, Western Australia 6008, Australia; 5Cambridge Institute for Medical Research, University of Cambridge School of Clinical Medicine, Cambridge CB2 0XY, UK; 6Department of Psychosis Studies, NIHR Biomedical Research Centre for Mental Health at the Institute of Psychiatry, King's College London and The South London and Maudsley NHS Foundation Trust, Denmark Hill, London SE5 8AF, UK; 7Diamantina Institute of Cancer, Immunology and Metabolic Medicine, Princess Alexandra Hospital, University of Queensland, Brisbane, Queensland, Australia; 8Department of Epidemiology and Population Health, London School of Hygiene and Tropical Medicine, London WC1E 7HT, UK, and Department of Epidemiology and Public Health, University College London, London WC1E 6BT, UK; 9Neuropsychiatric Genetics Research Group, Institute of Molecular Medicine, Trinity College Dublin, Dublin 2, Ireland; 10Molecular and Physiological Sciences, The Wellcome Trust, London NW1 2BE, UK; 11Centre for Digestive Diseases, Queen Mary University of London, London E1 2AD, UK, and Digestive Diseases Centre, Leicester Royal Infirmary, Leicester LE7 7HH, UK, and Department of Clinical Pharmacology, Old Road Campus, University of Oxford, Oxford OX3 7DQ, UK; 12Clinical Neurosciences, St George's University of London, London SW17 0RE, UK; 13King's College London, Department of Medical and Molecular Genetics, School of Medicine, Guy's Hospital, London SE1 9RT, UK; 14Biomedical Research Centre, Ninewells Hospital and Medical School, Dundee DD1 9SY, UK; 15King's College London Social, Genetic and Developmental Psychiatry Centre, Institute of Psychiatry, Denmark Hill, London SE5 8AF, UK; 16University of Cambridge Department of Clinical Neurosciences, Addenbrooke's Hospital, Cambridge CB2 0QQ, UK; 17NIHR Biomedical Research Centre for Ophthalmology, Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology, London EC1V 2PD, UK; 18Department Molecular Neuroscience, Institute of Neurology, Queen Square, London WC1N 3BG, UK; 19Oxford Centre for Diabetes, Endocrinology and Metabolism (ICDEM), Churchill Hospital, Oxford OX3 7LJ, UK.
REFERENCES
- 1.Yang J., Benyamin B., McEvoy B.P., Gordon S., Henders A.K., Nyhold D.R., Madden P.A., Heath A.C., Martin N.G., Montgomery G.W., et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 2010;42:565–569. doi: 10.1038/ng.608. doi:10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang J., Manolio T.A., Pasquale L.R., Boerwinkle E., Caporaso N., Cunningham J.M., de Andrade M., Feenstra B., Feingold E., Hayes M.G., et al. Genome partitioning of genetic variation for complex traits using common SNPs. Nat. Genet. 2011;43:519–525. doi: 10.1038/ng.823. doi:10.1038/ng.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee S.H., Wray N.R., Goddard M.E., Visscher P.M. Estimating missing heritability for disease from genome-wide association studies. Am. J. Hum. Genet. 2011;88:294–305. doi: 10.1016/j.ajhg.2011.02.002. doi:10.1016/j.ajhg.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wickremaratchi M.M., Perera D., O'Loghlen C., Sastry D., Morgan E., Jones A., Edwards P., Robertson N.P., Butler C., Morris H.R., et al. Prevalence and age of onset of Parkinson's disease in Cardiff: a community based cross sectional study and meta-analysis. J. Neurol. Neurosurg. Psychiatry. 2009;80:805–807. doi: 10.1136/jnnp.2008.162222. [DOI] [PubMed] [Google Scholar]
- 5.Foltynie T., Brayne C.E.G., Robbins T.W., Barker R.A. The cognitive ability of an incident cohort of Parkinson's patients in the UK. The CamPaIGN study. Brain. 2004;127:550–560. doi: 10.1093/brain/awh067. doi:10.1093/brain/awh067. [DOI] [PubMed] [Google Scholar]
- 6.VanDenEeden S.K., Tanner C.M., Bernstein A.L., Fross R.D., Leimpeter A., Bloch D.A., Nelson L.M. Incidence of Parkinson's disease: variation by age, gender, and race/ethnicity. Am. J. Epidemiol. 2003;157:1015–1022. doi: 10.1093/aje/kwg068. doi:10.1093/aje/kwg068. [DOI] [PubMed] [Google Scholar]
- 7.Saad M., Lesage S., Saint-Pierre A., Corvol J.C., Zelenika D., Lambert J.C., Vidailhet M., Mellick G.D., Lohmann E., Durif F., et al. Genome-wide association study confirms BST1 and suggests a locus on 12q24 as risk loci for Parkinson's disease in the European population. Hum. Mol. Genet. 2011;20:615–627. doi: 10.1093/hmg/ddq497. doi:10.1093/hmg/ddq497. [DOI] [PubMed] [Google Scholar]
- 8.Gasser T. Genetics of Parkinson's disease. Curr. Opin. Neurol. 2005;18:363–369. doi: 10.1097/01.wco.0000170951.08924.3d. doi:10.1097/01.wco.0000170951.08924.3d. [DOI] [PubMed] [Google Scholar]
- 9.Pankratz N., Foroud T. Genetics of Parkinson disease. Genet. Med. 2007;9:801–811. doi: 10.1097/gim.0b013e31815bf97c. doi:10.1097/GIM.0b013e31815bf97c. [DOI] [PubMed] [Google Scholar]
- 10.Payami H., Zareparsi S., James D., Nutt J. Familial aggregation of Parkinson's disease: a comparative study of early-onset and late-onset disease. Arch. Neurol. 2002;59:848–850. doi: 10.1001/archneur.59.5.848. doi:10.1001/archneur.59.5.848. [DOI] [PubMed] [Google Scholar]
- 11.Alcalay R.N., Caccappolo E., Mejia-Santana H., Tang M.X., Rosado L., Ross B.M., Verbitsky M., Kisselev S., Louis E.D., Comella C., et al. Frequency of known mutations in early-onset Parkinson's disease: implication for genetic counseling: the consortium on risk for early onset Parkinson disease study. Arch. Neurol. 2010;67:1116–1122. doi: 10.1001/archneurol.2010.194. doi:10.1001/archneurol.2010.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singleton A.B., Farrer M., Johnson J., Singleton A., Hague S., Kachergus J., Hulihan M., Peuralinna T., Dutra A., Nussbaum R., et al. Alpha-synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. doi:10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 13.Pankratz N., Dumitriu A., Hetrick K.N., Sun M., Latourelle J.C., Wilk J.B., Halter C., Doheny K.F., Gusella J.F., Nichols W.C., et al. Copy number variation in familial Parkinson disease. PLoS One. 2011;6:e20988. doi: 10.1371/journal.pone.0020988. doi:10.1371/journal.pone.0020988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pirkevi C., Lesage S., Brice A., Başak A.N. From genes to proteins in Mendelian Parkinson's disease: an overview. Anat. Rec. 2009;292:1893–1901. doi: 10.1002/ar.20968. doi:10.1002/ar.20968. [DOI] [PubMed] [Google Scholar]
- 15.Do C.B., Tung J.Y., Dorfman E., Kiefer A.K., Drabant E.M., Francke U., Moutain J.L., Goldman S.M., Tanner C.M., Langston J.W., et al. Web-based genome-wide association study identifies two novel Loci and a substantial genetic component for Parkinson's disease. PLoS Genet. 2011;7:e1002141. doi: 10.1371/journal.pgen.1002141. doi:10.1371/journal.pgen.1002141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tanner C.M., Ottman R., Goldman S.M., Ellenberg J., Chan P., Mayeux R., Langston J.W. Parkinson disease in twins: an etiologic study. JAMA. 1999;281:341–346. doi: 10.1001/jama.281.4.341. doi:10.1001/jama.281.4.341. [DOI] [PubMed] [Google Scholar]
- 17.Porter B., Macfarlane R., Unwin N., Walker R. The prevalence of Parkinson's disease in an area of North Tyneside in the north-east of England. Neuroepidemiology. 2006;26:156–161. doi: 10.1159/000091657. doi:10.1159/000091657. [DOI] [PubMed] [Google Scholar]
- 18.Wirdefeldt K., Adami H.O., Cole P., Trichopoulos D., Mandel J. Epidemiology and etiology of Parkinson's disease: a review of the evidence. Eur. J. Epidemiol. 2011;26:1–58. doi: 10.1007/s10654-011-9581-6. doi:10.1007/s10654-011-9581-6. [DOI] [PubMed] [Google Scholar]
- 19.Thacker E.L., Ascherio A. Familial aggregation of Parkinson's disease: a meta-analysis. Mov. Disord. 2008;23:1174–1183. doi: 10.1002/mds.22067. doi:10.1002/mds.22067. [DOI] [PubMed] [Google Scholar]
- 20.Piccini P., Burn D.J., Ceravolo R., Maraganore D., Brooks D.J. The role of inheritance in sporadic Parkinson's disease: evidence from a longitudinal study of dopaminergic function in twins. Ann. Neurol. 1999;45:577–582. doi: 10.1002/1531-8249(199905)45:5<577::aid-ana5>3.0.co;2-o. doi:10.1002/1531-8249(199905)45:5<577::AID-ANA5>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 21.Wirdefeldt K., Gatz M., Schalling M., Pedersen N.L. No evidence for heritability of Parkinson disease in Swedish twins. Neurology. 2004;63:305–311. doi: 10.1212/01.wnl.0000129841.30587.9d. doi:10.1212/01.WNL.0000129841.30587.9D. [DOI] [PubMed] [Google Scholar]
- 22.Yang J., Lee S.H., Goddard M.E., Visscher P.M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. doi:10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sveinbjörnsdóttir S., Hicks A.A., Jónsson T., Pétursson H., Guðmundsson G., Frigge M.L., Kong A., Gulcher J.R., Stefánsson K. Familial aggregation of Parkinson's disease in Iceland. N. Engl. J. Med. 2000;343:1765–1770. doi: 10.1056/NEJM200012143432404. doi:10.1056/NEJM200012143432404. [DOI] [PubMed] [Google Scholar]
- 24.Pritchard J.K. Are rare variants responsible for susceptibility to complex disease? Am. J. Hum. Genet. 2001;69:124–137. doi: 10.1086/321272. doi:10.1086/321272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cirulli E.T., Goldstein D.B. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat. Rev. Genet. 2010;11:415–425. doi: 10.1038/nrg2779. doi:10.1038/nrg2779. [DOI] [PubMed] [Google Scholar]
- 26.Manolio T.A., Collins F.S., Cox N.J., Goldstein D.B., Hindorff L.A., Hunter D.J., McCarthy M.I., Ramos E.M., Cardon L.R., Chakravarti A., et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. doi:10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neumann J., Bras J., Deas E., O'Sullivan S.S., Parkkinen L., Lachmann R.H., Li A., Holton J., Guerreiro R., Paudel R., et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson's disease. Brain. 2009;132:1783–1794. doi: 10.1093/brain/awp044. doi:10.1093/brain/awp044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sidranksy E., Nalls M.A., Aasly J.O., Aharon-Peretz J., Annesi G., Barbosa E.R., Bar-Shira A., Berg D., Bras J., Brice A., et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N. Engl. J. Med. 2009;361:1651–1661. doi: 10.1056/NEJMoa0901281. doi:10.1056/NEJMoa0901281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manolio T.A. Genome-wide association studies and disease risk assessment. N. Engl. J. Med. 2010;363:166–176. doi: 10.1056/NEJMra0905980. doi:10.1056/NEJMra0905980. [DOI] [PubMed] [Google Scholar]
- 30.Singleton A.B., Hardy J., Traynor B.J., Houlden H. Towards a complete resolution of the genetic architecture of disease. Trends Genet. 2010;26:438–442. doi: 10.1016/j.tig.2010.07.004. doi:10.1016/j.tig.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li L., Li Y., Browning S.R., Browning B.L., Slater A.J., Kong Z., Aponte J.L., Mooser V.E., Chissoe S.L., Whittaker J.C., et al. Performance of genotype imputation for rare variants identified in exons and flanking regions of genes. PLoS One. 2011;6:e24945. doi: 10.1371/journal.pone.0024945. doi:10.1371/journal.pone.0024945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nalls M.A., Plagnol V., Hernandez D.G., Sharma M., Sheerin U.M., Saad M., Simón-Sànchez J., Schulte C., Lesage S., et al. International Parkinson Disease Genomics Consortium. Imputation of sequence variants for identification of genetic risks for Parkinson's disease: a meta-analysis of genome-wide association studies. Lancet. 2011;377:641–649. doi: 10.1016/S0140-6736(10)62345-8. doi:10.1016/S0140-6736(10)62345-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Simón-Sánchez J., Schulte C., Bras J.M., Sharma M., Gibbs J.R., Berg D., Paisan-Ruiz C., Lichtner P., Scholz S.W., Hernandez H.G., et al. Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat. Genet. 2009;41:1308–1312. doi: 10.1038/ng.487. doi:10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Plagnol V., Nalls M.A., Bras J.M., Hernandez D.G., Sharma M., Sheerin U.M., Saad M., Simón-Sánchez J., et al. International Parkinson's Disease Genomics, Wellcome Trust Case Control Consortium 2. A two-stage meta-analysis identifies several new loci for Parkinson's disease. PLoS Genet. 2011;7:e1002142. doi: 10.1371/journal.pgen.1002142. doi:10.1371/journal.pgen.1002142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pankratz N., Wilk J.B., Latourelle J.C., DeStefano A.L., Halter C., Pugh E.W., Doheny K.F., Gusella J.F., Nichols W.C., Foroud T., et al. Genome-wide association study for susceptibility genes contributing to familial Parkinson disease. Hum. Genet. 2009;124:593–605. doi: 10.1007/s00439-008-0582-9. doi:10.1007/s00439-008-0582-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Simón-Sánchez J., van Hilten J.J., van deWarrenburg B., Post B., Berendse H.W., Arepalli S., Hernandez D.G., de Bie R.M., Velseboer D., Scheffer H., et al. Genome-wide association study confirms extant PD risk loci among the Dutch. Eur. J. Hum. Genet. 2011;19:655–661. doi: 10.1038/ejhg.2010.254. doi:10.1038/ejhg.2010.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fung H.C., Scholz S., Matarin M., Simón-Sánchez J., Hernandez D., Britton A., Gibbs J.R., Langefeld C., Stiegert M.L., Schymick J., et al. Genome-wide genotyping in Parkinson's disease and neurologically normal controls: first stage analysis and public release of data. Lancet Neurol. 2006;5:911–916. doi: 10.1016/S1474-4422(06)70578-6. doi:10.1016/S1474-4422(06)70578-6. [DOI] [PubMed] [Google Scholar]
- 38.Laaksovirta H., Peuralinna T., Schymick J.C., Scholz S.W., Lai S.L., Myllykangas L., Sulkava R., Jansson L., Hernandez D.G., Gibbs J.R., et al. Chromosome 9p21 in amyotrophic lateral sclerosis in Finland: a genome-wide association study. Lancet Neurol. 2010;9:978–985. doi: 10.1016/S1474-4422(10)70184-8. doi:10.1016/S1474-4422(10)70184-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nalls M.A., Simon-Sanchez J., Gibbs J.R., Paisan-Ruiz C., Bras J.T., Tanaka T., Matarin M., Scholz S., Weitz C., Harris T.B., et al. Measures of autozygosity in decline: globalization, urbanization, and its implications for medical genetics. PLoS Genet. 2009;5:e1000415. doi: 10.1371/journal.pgen.1000415. doi:10.1371/journal.pgen.1000415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spencer C.C., Plagnol V., Strange A., Gardner M., Paisan-Ruiz C., Band G., Barker R.A., Bellenguez C., et al. The United Kingdom Parkinson's Disease Consortium, The Wellcome Trust Case Control Consortium 2. Dissection of the genetics of Parkinson's disease identifies an additional association 5′ of SNCA and multiple associated haplotypes at 17q21. Hum. Mol. Genet. 2011;20:345–353. doi: 10.1093/hmg/ddq469. doi:10.1093/hmg/ddq469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li Y., Willer C.J., Sanna S., Abecasis G.R. Genotype imputation. Annu. Rev. Genomics Hum. Genet. 2009;10:387–406. doi: 10.1146/annurev.genom.9.081307.164242. doi:10.1146/annurev.genom.9.081307.164242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Y., Willer C.J., Ding J., Scheet P., Abecasis G.R. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet. Epidemiol. 2010;34:816–834. doi: 10.1002/gepi.20533. doi:10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Price A.L., Patterson N.J., Plenge R.M., Weinblatt M.E., Shadick N.A., Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006;38:904–909. doi: 10.1038/ng1847. doi:10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 44.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A.R., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., et al. PLINK: a toolset for whole-genome association and population-based linkage analysis. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. doi:10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ioannidis J., Patsopoulos N., Evangelou E. Uncertainty in heterogeneity estimates in meta-analyses. BMJ. 2007;335:914–918. doi: 10.1136/bmj.39343.408449.80. doi:10.1136/bmj.39343.408449.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Higgins J.P.T., Thompson S.G., Deeks J.J., Altman D.G. Measuring inconsistency in meta-analyses. BMJ. 2003;327:557–560. doi: 10.1136/bmj.327.7414.557. doi:10.1136/bmj.327.7414.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cochran W.G. The combination of estimates from different experiments. Biometrics. 1954;10:101–129. doi:10.2307/3001666. [Google Scholar]
- 48.Lill C.M., Roehr J.T., McQueen M.B., Kavvoura F.K., Bagade S., Schjeide B.M.M., Schjeide L.M., Meissner E., Zauft U., Allen N.C., et al. Comprehensive research synopsis and systematic meta-analyses in Parkinson's disease genetics: the PDGene database. PLoS Genet. 2011;8:e1002548. doi: 10.1371/journal.pgen.1002548. doi:10.1371/journal.pgen.1002548. [DOI] [PMC free article] [PubMed] [Google Scholar]