

Abstract
Sporadic Alzheimer's disease (AD) is an emerging chronic illness characterized by a progressive pleiotropic pathophysiological mode of actions triggered during the senescence process and affecting the elderly worldwide. The complex molecular mechanisms of AD not only are supported by cholinergic, beta-amyloid, and tau theories but also have a genetic basis that accounts for the difference in symptomatology processes activation among human population which will evolve into divergent neuropathological features underlying cognitive and behaviour alterations. Distinct immune system tolerance could also influence divergent responses among AD patients treated by immunotherapy. The complexity in nature increases when taken together the genetic/immune tolerance with the patient's brain reserve and with neuropathological evolution from early till advance AD clinical stages. The most promising diagnostic strategies in today's world would consist in performing high diagnostic accuracy of combined modality imaging technologies using beta-amyloid 42 peptide-cerebrospinal fluid (CSF) positron emission tomography (PET), Pittsburgh compound B-PET, fluorodeoxyglucose-PET, total and phosphorylated tau-CSF, and volumetric magnetic resonance imaging hippocampus biomarkers for criteria evaluation and validation. Early diagnosis is the challenge task that needs to look first at plausible mechanisms of actions behind therapies, and combining them would allow for the development of efficient AD treatment in a near future.
1. Introduction
Late-onset Alzheimer's disease (LOAD) is an aging-associated chronic neurological disease whose etiology is not well understood. Being one of the most common forms of dementia, Alzheimer's disease (AD) is recognized as a progressive neurodegenerative disorder associated with the development of dystrophic neuritic dense-core plaques, and neurofibrillary tangles (NFTs) in the atrophic cerebral cortex of dement patients [1, 2] are hallmark neuropathological features accompanied by confusion, disorientation, memory failure, and speech disturbances towards gradual loss of mental ability progressing into reduced daily living abilities as the affected individuals aged. But whether these morphological characteristics are causative of clinical symptoms is a matter of controversy.
Initial decline in cognition occurs more than 10 years before the first clinical AD symptoms are reported [3]. Once the illness is diagnosed, usually after 65 years of age or later, it can last from a few years up to 20 years depending on the condition severity. The prevalence of LOAD is rising proportionally with increasing world population and aging, contrasting with the less-prevalent early-onset Alzheimer's disease (EOAD) population that account for 2% of all AD cases [4]. According to the World Alzheimer Report 2009, the number of people living with AD is estimated at 36 million in 2010 and expected to increase to 66 million by 2030 and 115 million by 2050. The total cost was estimated at US$604 billion in 2010 which is about 1% of the world's gross domestic product. In the EU-15, the overall 2007 cost contributes for 68% informal care, 26% social care, 5% health care, and 1% productivity losses and, among them, UK had the highest health and social care costs associated with research funding [5]. Epidemiologic Western European studies indicate that women are more susceptible than men in developing AD between 60 and 100 years of age with an estimated odds ratio in the range of 1.6-fold higher [6]. The proportion of heredity risk factors transmitting the AD to their offspring are higher in men than women and could be explained by the fact that women are more susceptible to the influence of environmental risk and innate factors other than longevity difference that may account for a higher incidence of AD in women [7]. Gender-related disparity incidence could be associated with the decrease in estrogen hormone in postmenopausal women that may have a more effective neuroprotective role than testosterone against the development of AD where the incidence seems to be more accentuated in women compared to men in the oldest-old age range [8–10]. Increasing evidence shows that estrogen replacement therapy in combination with cholinergic-enhancing drugs could be considered as an effective therapeutic strategy for use in postmenopausal women during mild cognitive impairment (MCI) [6, 11].
Imaging modality technologies are increasingly employed in biomedical research. Their technology characteristics, namely, spatial resolution, temporal resolution and sensitivity of probe detection are essential to address what specific biological processes (anatomic, physiologic, metabolic, and molecular) are being conducted noninvasively [12, 13]. Nowadays scientists realized the importance of combining molecular imaging systems as one platform, for example, positron emission tomography (PET) + magnetic resonance imaging (MRI), that combine high sensitivity with spatial resolution which can be used as a more accurate diagnostic tool for evaluating molecular pathways responsible of neurologic disease progression such as AD, vascular dementia (VaD), dementia with Lewis Body (DLB), frontotemporal dementia (FTD, and Parkinson disease (PD). We anticipate that ultrasound and optical imaging are the next promising techniques that can replace high energy radioisotopes and cost effective instruments largely employed in clinical settings. As a perspective, it could be possible to combine the development of deep tissue multiphoton imaging systems with optical imaging to generate fusion image with better resolution, especially where senile plaques are produced in the cortex. Herein, the purpose of this paper is to describe current molecular imaging technologies and biomarkers employed for early clinical diagnosis as well current immunotherapies for the treatment and/or preventive approach against AD.
2. Cholinergic, Amyloid-β, Tau, and Other Hypotheses
The major culprit responsible for the initial biological event leading to behavioural and clinical AD symptoms is the subject of intensive discussion and remains unfolded. There are currently several proposed theories that explain the underlying progressive pathogenic mechanisms scope responsible for the AD neuropathological features. When all theory models are taken together, new insight of the underlying mechanisms causing AD pathogenesis, for example, neuronal cell death and beta-amyloid (Aβ) neurotoxicity, could lead to novel treatment strategies [14].
The cholinergic depletion hypothesis was the first theory proposed to explain the etiology of AD pathogenesis based on the findings that memory impairment, due to loss in cholinergic transmission, could be reversed following treatment of mild-to-moderate patients with cholinergic receptor agonist (e.g., nicotinic acetylcholine receptors and muscarinic acetylcholine receptors), acetylcholinesterase inhibitors (e.g., galantamine, donepezil, and rivastigmine), acetylcholine precursors (e.g., L-alpha glycerylphosphorylcholine), and cholinergic enzymes (e.g., choline acetyltransferase) [15–19]. Functional activity studies found that acetylcholine receptors depletion compromise in basal forebrain and hippocampal neurons occur along with accumulation of Aβ oligomers, via activation of the amyloid-precursor protein (APP) processing pathway, in the hippocampus and cortex areas underpin memory and learning process [20–23]. The cholinergic deficit model in AD indicates that nerve growth factors are the principal neurotropic agent for basal forebrain cholinergic neurons and could represent the etiology multifactorial neurodegenerative disorders. For example, an elegant study using two phenotypic lines of transgenic mice, the first where TrkA signaling is inhibited, showed that antibody-neutralizing a nerve growth factor TrkA decreases cholinergic activity and induces formation of Aβ with no apparent tau neuropathology characteristics; in contrast, the second where anti-nerve growth factor mice were crossed to p75 neurotrophin receptorexonIII(−/−) mice showed that antibody blocking the nerve growth factor abrogates p75 neurotrophin receptor signaling which recovered cholinergic activity and prevented Aβ production at all ages, but enhanced tau protein hyperphosphorylation [22, 24, 25]. It is hypothesized that AD pathophysiology could be triggered by simultaneous hypocholinergic tone and Aβ accumulation in which Aβ and apolipoprotein E epsilon 4 (ApoE4) could interact with alpha7 nicotinic receptors leading repression of glycogen synthase 3β and downstream effects towards tau protein hyperphosphorylation [26]. Past clinical trials have reported that cholinergic mechanism based drugs, such as donepezil [27], are just better than placebo-treated controls; symptomatic improvements in cognition and global functioning are temporary and offer short-term cure for AD patients carrying the ApoE4 allele. The involvement of key enzymes in lipid membrane metabolism connecting the cholinergic and glutamatergic systems such as the phospholipase A2 could play a role in cognitive alterations and neurodegenerative process in AD. Sustaining inhibition of Ca2+ dependent and independent phospholipase A2 during the early stages of AD may lead to Aβ formation through downregulation of cholinergic and glutamate receptors. When Aβ is already elevated during the AD pathophysiology, it could favor upregulation of Ca2+-dependent phospholipase A2 and secretory phospholipase A2 involved in inflammatory cytokines and oxidative stress [28].
Formulation of the amyloid cascade hypothesis proposed by Hardy and Higgins [29] was the most influential theory to explain why abnormal processing of Aβ constitutes the underlying mechanism responsible for the progressive development of AD pathogenesis. A modified amyloid cascade hypothesis claimed that both aging process of the brain and the associated risk factors act collectively rather than the initial Aβ deposition to exacerbate process of synapse dysfunction leading to neuronal cell death [30]. The Aβ metabolism involves a 2 steps selective cleavage of the APP by the α-secretase resulting in the release of the soluble short N-terminal APPα. The remaining C-terminal fragment-α is thereafter hydrolyzed by the γ-secretase to release the APP intracellular domain and the extracellular p3 fragment 17-40/42 [31]. The APP could be associated with sortilin-related receptor 1 (SORL1) whose complex can be internalized and enter the recycling endosomes before returning back to the surface membrane [32, 33]. These mechanisms do not lead to Aβ release. However, activating the amyloidogenic pathway leads to abnormalities in amyloid metabolism by generating short APPβ and APP intracellular domain-β fragments responsible for the development of AD neuropathology. The APP cleavage is initiated by β-secretase to release the N-terminal short APPβ leaving the C-terminal fragment-β in the membrane which is sequentially hydrolyzed by the γ-secretase in association with presenilin 1 and presenilin 2 enzymes complex. The γ-secretase cleaves the C-terminal fragment-β at two sites generating Aβ40 and Aβ42 which are released into the vesicle while the APP intracellular domain is released into the cytoplasm. Amyloid forms are released outside the neurons via constitutive secretory pathway whereas the C-terminal fragment-β will target the nucleus involving the signaling transcriptional activation [32, 33]. During the onset of AD, the transfer of autophagic vesicles to the lysosomes is impeded, thus promoting Aβ accumulation, and secondly microglia cells cannot destroy efficiently abundant Aβ monomers through phagocytosis [33]. In homozygotes, ApoE4 alleles increase the susceptibility or lifetime risk of developing AD by favoring the conversion of APP into Aβ40 and Aβ42, thus reducing their clearance via the degradation pathway [33]. Remodelling of the Ca2+ signaling system [34] inducing learning and memory decline involves binding extracellular Aβ42 oligomers to the cellular prion protein and alteration of ryanodine receptor expression in the nucleus following APP intracellular domain translocation. Then later, induction of apoptosis by caspase 3 could possibly involve the orphan receptor DR6 activation following its binding to the extracellular N-terminal APP (Figure 1).
Figure 1.
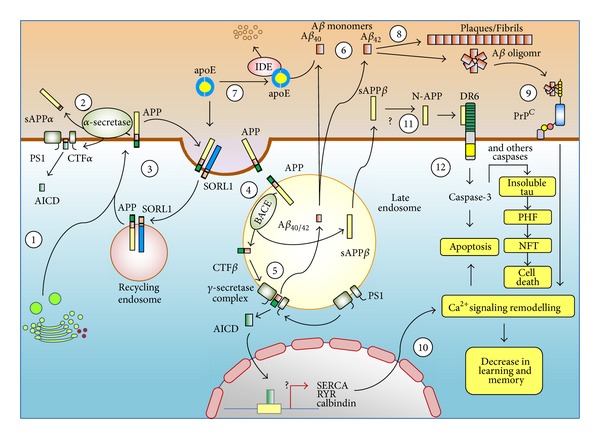
Illustration showing the implication of amyloid and tau hypotheses in the development of Alzheimer's disease. This modified illustration is reproduced with permission from [33].
Numerous neuropathological and genetic observations demonstrate that tau hypothesis consists of abnormally elevated hyperphosphorylated form (either at threonine or serine residues) of microtubule-associated tau protein which favor NFT accumulation [35–39]. Clinical correlations have shown that tau pathology is considered as a downstream pathological event, identified as an intermediate of prerequisite Aβ-induced neurotoxic effects, preceding widespread regional neurodegeneration during the AD evolution [40–43]. The mechanism of tau phosphorylation is regulated via an imbalance from kinases and phosphates. As tau becomes hyperphosphorylated, it sequesters normal tau and other proteins causing dismantlement of microtubules involved in axonal transport. Similar to Aβ, hyperphosphorylated tau proteins become prone to self-aggregates into paired helical filaments which turn into tangles formation subsequently compromising synaptic function [32, 44]. Besides the role caspase 3 has in APP cleavage for Aβ production, it is possible a set of different caspases could contribute in tau-mediated cleavage to promote its aggregation and paired helical filaments, thus linking Aβ to neurofibrillary tangles formation during the process of AD pathogenesis [45, 46] (Figure 1).
Following the failure of clinical trials to reduce AD progression, other factors have been proposed to be involved in this process. Among them is the emerging role of herpes simplex virus type 1 present in 90% adult brain population following childhood infection, characterized by latency and periodic reactivation causing damage over time in which apoE4 alleles carriers confer a higher risk of developing AD during aging. It has been shown that apoE4 and herpes simplex virus 1 compete with the same neuronal cell membrane receptor, called the HSPG. ApoE4 carriers, intimately binding to Aβ, influence the herpes simplex virus 1 inflammation via cytokines and iNOS and oxidative damage via lipid peroxidation processes, and as a consequence will activate the amyloidogenic pathway involving the activity of β-secretase and γ-secretase to produce more neurotoxic Aβ forms [47–49]. The homeostatic myelin repair processes represent another hypothesis underlying axonal transport interruption, axonal swellings, and neuritic plaques development, and protein deposits in Aβ and tau derivative products [50]. Some reports have recently shown that deficiency of norepinephrine in locus ceruleus projection areas is correlated with the suppression of Aβ-induced cytokine and chemokine syntheses, modulation of complement factor and impairment of microglial migration and phagocytosis, thereby reducing Aβ uptake and clearance by microglia [51, 52]. Recently, fibrinogen has been shown to interact with Aβ leading to abnormal fibrin clot formation in AD [53, 54] and could tentatively represent another hypothesis.
3. Alzheimer's Disease Risk Factors and Neuropathological Aspects
Genetic studies demonstrated that EOAD occurs with extremely high incidence in patients suffering from Down syndrome. Mutations that occur in the presenilin 1 (14q24.3), presenilin 2 (1q31-q42), and Aβ A4 precursor protein (APP; 21q21.3) [55] are responsible for autosomal-dominant early-onset familial AD. They modulate β- and γ-secretases activity that process APP cleavage for generating prematurely an age onset for the production of soluble 40 or 42 amino acid Aβ peptide. Considered as a genetic risk factor, sortilin-related receptor 1 (SORL1; 11q23.3), a low density lipoprotein receptor class A repeats, is known to regulate trafficking and processing of the APP enhancing Aβ accumulation in both EOAD and LOAD [56–58]. Although not sufficient to cause the disease by itself, inheritance of apolipoprotein E epsilon 4 (ApoE4; 19q13.2) allele constitutes a major genetic risk factor for developing EOAD as well as LOAD predisposition and hypercholesterolaemia [59, 60] and could act in synergy with other susceptible genes, for example, programmed cell death protein 4 (PDCD4) and evolutionarily conserved signaling intermediate in Toll pathway (ECSIT), in a complex interaction with environmental factors [61–65] (Table 1). The ApoE4 could be involved in cholesterol transport hindrance, diminished neuronal repair, Aβ deposition, fibrillisation, and plaque formation by acting as an Aβ interacting pathological chaperone [66, 67]. Besides APoE4, nine other candidate genes, for example, ATP-binding cassette subfamily A member 7 (ABCA7), Myc box-dependent-interacting protein 1 (BIN1), CD2-associated protein (CD2AP), CD33, clusterin (CLU), complement receptor type 1 (CR1), ephrin type-A receptor 1 (EPHA1), membrane-spanning 4A (MS4A4E/MS4A6A), and phosphatidylinositol binding clathrin assembly protein (PICALM), have been identified as AD risk loci from a three-staged meta-analysis method based on establishing the differential frequency observed between AD patients and control non-dement groups [68, 69]. Expression of ApoE4 gene with small-nucleotide polymorphism could synergistically interact with at least PICALM and, thus providing insight into their own mechanism of regulation, and serve as a diagnostic tool to predict the development of AD in nondement subjects [70, 71]. Furthermore, significant interactions of ApoE4 with SORL1 single nucleotide polymorphisms on Aβ42 cerebrospinal fluid (CSF) indicated the role that SORL1 genetic variants can have in regulating the amyloidogenic pathway [72]. Other methods have examined the relationship between small-nucleotide polymorphism loci in protein phosphatase B and calcium homeostasis modulator 1 (CALHM1), respectively, with AD quantitative biomarkers such as p-tau and Aβ42 CSF or genome-wide association study with MRI brain structure degeneration localized in temporal, parietal, and hippocampal regions [73–77].
Table 1.
Genetics and environmental risk factors associated with human AD etiology.
| GeneticsA | |
|---|---|
| AD-causative genes | |
| Presenilin 1 (PSEN1) | |
| Presenilin 2 (PSEN2) | |
| Aβ A4 precursor protein (APP) | |
| AD-susceptible genes | |
| Apolipoprotein E epsilon 4 (APOEε4) | |
| Sortilin-related receptor 1 (SORL1) | |
| Amyloid beta A4 precursor protein-binding, family B, member 2 (APBB2) | |
| Hemochromatosis (HFE) | |
| Nitric oxide synthase 3 (NOS3) | |
| PAX-interacting protein 1 (PAXIP1) | |
| Urokinase-type plasminogen activator (PLAU) | |
| Alpha-2 macroglobulin (A2M) | |
| Bleomycin hydrolase (BLMH) | |
| Myeloperoxidase (MPO) | |
| Angiotensin-converting enzyme (ACE) | |
| New AD Susceptible Loci | |
| Programmed cell death protein 4 (PDCD4) | |
| Evolutionarily conserved signaling intermediate in Toll (ECSIT) | |
| Phosphatidylinositol binding clathrin assembly protein (PICALM) | |
| Complement receptor type 1 (CR1) | |
| Myc box-dependent-interacting protein 1 (BIN1) | |
| CD2-associated protein (CD2AP) | |
| CD33 | |
| Ephrin type-A receptor 1 (EPHA1) | |
| Clusterin (CLU) | |
| ATP-binding cassette subfamily A member 7 (ABCA7) | |
| Membrane-spanning 4A4E and 6A (MS4A6A/MS4A4E) | |
| Oxidized low-density lipoprotein (LDL) receptor (OLR1) | |
| Cholesterol 24-hydroxylase (CYP46) | |
|
| |
| Environmental exposuresB | |
|
| |
| Health factors | |
| Food diets | |
| Cigarette smoking | |
| Alcohol consumption | |
| Head trauma | |
| Viral infections | |
| Systemic inflammation | |
| Metal and pesticide exposure | |
| Psychosocial factors | |
| Education | |
| Social network | |
| Leisure activities | |
| Physical activity | |
| Chronic stress | |
| Depression | |
| Somatic Factors | |
| Blood pressure | |
| Obesity | |
| Diabetes mellitus | |
| Cardiovascular diseases | |
| Cerebrovascular diseases | |
| Hyperlipidemia | |
Histopathological aromatic dyes staining using Thioflavin S but especially Congo red is the gold standard for diagnosing amyloid plaques because it only binds aggregated β-sheets [78, 79], and postmortem clinical diagnosis is still regarded as the gold standard for definitive diagnostic of AD. Other histopathological silver staining such as Bielschowsky, Bodian, or Gallyas and dyes staining such as Hematoxylin and eosin, cresyl violet, and luxol-fast blue as well as antibody specific to Aβ, phosphorylated tau, alpha-synuclein, ubiquitin, and TAR DNA-binding protein 43 (TDP-43) are routinely employed during postmortem examinations [79, 80]. Following diagnosis, neuropathological classification is assessed against Aβ, NFT, and neuritic plaque methodologies [81–83] to obtain a combined score, those Hyman et al. [84] described. The classification fall into four levels of AD neuropathological change based on the modified H. Braak and E. Braak [85] morphological staging: absence of NFT (0), low: NFT mainly in entorhinal cortex and vicinity areas (I/II), intermediate: NFT abundant in hippocampus and amygdale with some extension into the cortex (III/IV), and severe: NFT widely distributed across the neocortex (V/VI). Gradual hierarchal accumulation and distribution of NFT, neuropil threads, and dystrophic neurites take place in different brain regions during progressive AD. Anterograde Aβ expansion in brain regions has been described into 5 phases exhibiting progressive AD-related Aβ pathology [81]. In the early stages of demented elderly people, Aβ deposits are found exclusively in the frontal, parietal, temporal and occipital cortex (phase 1) which project into the hippocampal, insular, cingulate, and entorhinal cortex in addition to the amygdale where Aβ deposits occur (phase 2). Then, Aβ continues to deposit in the hypothalamus, thalamus, basal ganglia, and basal forebrain nuclei (phase 3) which are connected by multiplex afferent input regions affected from phases 1 and 2. As the disease further progress, newly senile Aβ plaques appears in the midbrain and medulla oblongata (phase 4). In the last stage (phase 5), Aβ plaques spread into the pons and cerebellum [86]. All newly affected areas receive afferent input from regions showing previously Aβ accumulation as described by Thal et al. [81].
Neuritic plaques, composed of Aβ deposits at the center of dystrophic neurites clusters containing phosphorylated tau immunoreactivity, represent another set of AD neuropathological criterion. Neuritic plaques, characterized by synapse loss and microglial activation, and NFTs are both correlated with clinical symptoms of AD. A modified method from neuritic plaque scoring system standardized protocol from postmortem assessment of dementia and normal subjects was proposed by Mirra et al. [83] is based on ranking the density of neuritic plaques in several neocortex as follows: no neuritic plaque (0), sparse (1), moderate (2), and frequent (3). Taken together, Braak NFT staging evaluation, Thal phases of Aβ deposits in neuroanatomical distribution and neuritic plaques density scoring in brain are methods used to correlate histopathological lesions during AD neuropathology changes with clinical symptoms. Molecular imaging assessment of amyloid burden in neocortex regions is a fourth method that complements neuropathological observations. A fifth method can even be utilized for discriminating soluble to aggregated peptides through biochemical assays.
4. Staging Early, Middle, and Advance AD Pathogenesis Events
Redefining the AD cascade hypothesis from a cholinergic, Aβ and tau point of view should be integrated into one refine model used to dissect out chronological distinct segments in pathogenic events leading to the development of stage-specific therapies. The presymptomatic period or prodromal stage refers to a pathophysiological process that is progressing towards developing cognitively and behavioural impairment of AD. The extent to which biomarkers in this period can predict a cognitively normal person who will subsequently develop clinical course of AD symptomatology remains to be clarified in the light of why some individuals never manifest the illness outcomes in their lifetime [87]. Therefore, it is critical to define the best factors contributing to the emergence of clinical impairment, so individual will benefit from early biomarker profile intervention [87]. Before the fifth decade of age, it is recognized that Aβ is low (about 5% of positive plaques number) among people who will develop LOAD. During the midfifties, the pathological cascade starts with Aβ accumulating in cognitively normal people. However, it is postulated that Aβ temporal lag between plaques depositions and clinical syndrome is estimated to be at least a decade interval [88–90]; therefore the earliest symptoms at midsixties of age represent the critical age onset of AD. It seems the temporal lag phase parallelism in Aβ deposits associated with clinical syndrome ends between 65- and 75-year old individuals. However, this does not mean all LOAD cases will have the same temporal lag as a shift in the temporal Aβ profile is observed among 65-year old individual between those who are ApoE epsilon4 noncarrier occurring in log phase versus those ApoE epsilon4 carrier who plateau [91]. There is little indication that shows how brain Aβ activation differs between these two groups over the lifespan [92]. A body of evidence highlight cognitive or brain reserve, genetic susceptibility (Figure 3) and/or environmental factors all could contribute to some extent as lifetime risk conditions of developing AD [93–98]. Nevertheless, the temporal Aβ profile is expected to be similar among the same group of individuals' going through the same neuropathological changes. In the hypothetical model of dynamic biomarkers set as a measurement of clinical dementia rating of the AD pathological cascade, the Aβ42-CSF PET is one of the most sensitive biomarker for earliest clinically detectable evidence for brain pathological changes (Figure 2). Measurement of Aβ42-CSF PET has been shown to be inversely correlated to increasing aggregation and plaque load in specific brain regions. During the lag phase when the individual is considered being in the so-called “normal status,” oligomeric soluble Aβ42 forms are quasi-absent at the beginning of life to very low level until the fifth decade of life. During the sixth or seventh decade, it is suggested that oligomeric soluble Aβ42-CSF PET increase logarithmically in cognitively normal individual corresponding to the presymptomatic phase. When oligomeric soluble Aβ42-CSF PET is already abnormally low during the stationary phase, then the individual has already MCI. However, fluorodeoxyglucose (FDG)-PET, tau-CSF and volumetric MRI may be more pertinent biomarkers at distinguishing early till late MCI. As abnormality in cognitive behaviour increases between early and late MCI and further progress during advance AD stage, volumetric MRI testing brain structure remains at present the sole biomarker measuring differentially those changes [99, 100].
Figure 3.
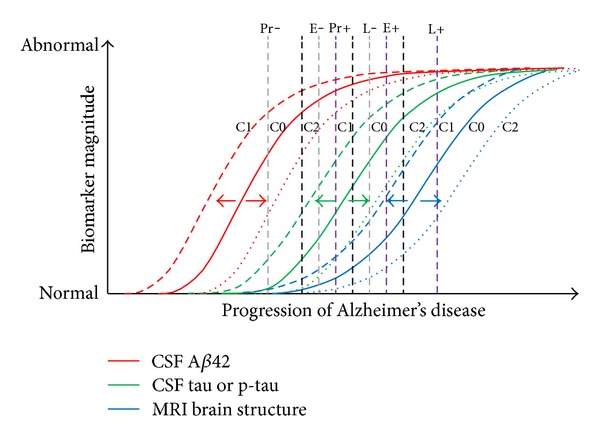
An hypothetical model showing the influence of genes, other diseases, and cognitive capacity on normal aging brain structure: C0 = CSF Aβ42, Tau or p-tau with brain structure in individuals typically observed without genetic and innate factors; C1 = CSF Aβ42, Tau or p-tau with brain structure in individuals typically observed with genetic risk loci (e.g., ApoE4) and/or together with comorbidity (e.g., cerebrovascular disease, cortical Lewy bodies, and frontal damage); C2 = CSF Aβ42, Tau, or p-tau with brain structure in individuals typically observed with high cognitive reserve or protective genetic loci (e.g., ApoE2); Pr−, Pr+, E−, E+, L−, and L+ = shifts early and late during preclinical, early, and late mild cognitive impairment stages. This modify illustration is adapted from figures in [91, 103–105].
Figure 2.
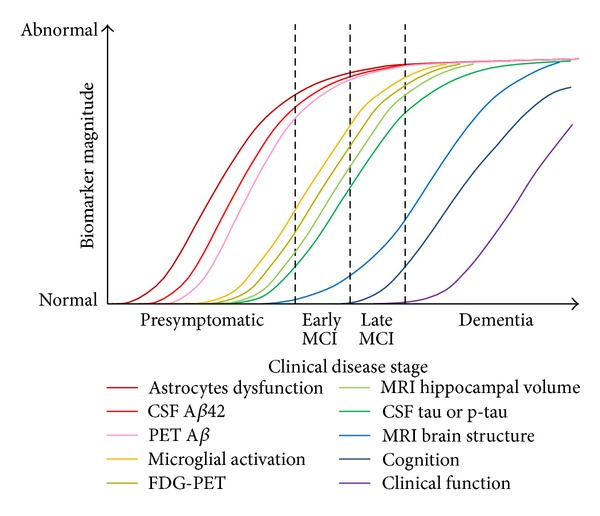
Chronobiological biomarkers to Alzheimer's disease clinical stage. This illustration is reproduced with permission from [91] and adapted from figures after [87, 101, 102].
5. Diagnostic PET and MRI Technologies Using AD Biomarkers and Criteria
Molecular imaging assessment of current clinical diagnostic criteria of MCI offered good sensitivity and specificity of AD biomarkers; however it possesses deficiencies in diagnosing early-clinical stage as gold standard postmortem analysis can detect early neuropathological observations [106, 107]. To some extent, data interpretations are regarded as the degree on which cognitive function is impaired and the effect of other causes (e.g., ApoE4) could impact as lifetime risk factors on the illness progression. To complement neuropathological observations, development of molecular imaging technologies should use reference data set as a benchmark as well as neurochemical biomarkers to detect the earliest AD pathogenic events, improve classification, track progression, and assess in prediction [108, 109]. This should facilitate the diagnosis and furthermore, supported by genetic analysis of cerebrovascular risk factors and other age-related brain disease, offer comprehensive information regarding which subtype neurodegenerative disease can potentially affect each person. Standardization in diagnostic criteria is very important because it address bias-adjusted estimates of the sensitivity and specificity [110] in which molecular imaging biomarkers are applied for better defining measurements of clinical AD stages. As well, combining molecular imaging, for example, adding magnetic resonance spectroscopy to MRI measures, could result in significantly better AD classification by improving higher sensitivity and specificity of measurements translated into better biomarkers to support the clinical diagnostic of the different AD stages [111, 112]. Standardization and/or validation in diagnostic criterion and clinical AD stages have been documented for PET and MRI biomarkers [113–119]. Diagnostic AD biomarkers are important to select population for specific selection criteria (e.g., onsets of cognitive impairment evaluated by neuropsychological testing such as the mini-mental state examination), age grouping population and increasing the statistical power of clinical trials, whereas clinical trial AD biomarkers are evaluating the type of therapeutic intervention (e.g., targeting amyloid, tau, etc.), and the clinical disease stages as well as its time-course changes during progressive pathological features [120]. To help in the AD diagnostic classification, molecular imaging should look at multimodal imaging analysis as well as pathological and functional changes associated with disease stages and brain regions in order to enrich patients in clinical trials and evaluation of treatment effects [121, 122]. For example, Zhang et al. [123] showed that combining molecular imaging (volumetric MRI, Pittsburgh Compound-B (PIB)- & FDG-PET and CSF-t-tau, -p-tau and -Aβ42 biomarkers) as reported by Fjell et al.[124] would achieve a higher diagnostic accuracy when discriminating between AD (93.2%) and MCI (76.4%) with healthy patients as well as 91.5% MCI converters and 73.4% MCI nonconverters correctly classified. The single-photon emission computed tomography (SPECT) using technetium-99m labeled hexamethylpropyleneamine oxime (99mTc-HMPAO) shows superior detection to dynamic susceptibility contrast MRI at distinguishing neuroanatomical regions between normal and AD patients and can be used together as a diagnostic tool to the medial temporal lobe of MCI patients for predicting memory dysfunction associated with AD progression [125, 126]. An initial report developed by Klunk et al. [127] demonstrated a significant higher [11C]-PIB tracer signal using PET imaging in frontal cortex, but also in parietal, temporal and occipital cortex and striatum where selective binding to Aβ deposits take place in these brain regions of living AD patients compared to healthy controls. They also show a PIB inverse correlation with the FDG tracer mainly in the parietal cortex, an observation reported across different clinical stages [121, 128–130]. It remains to address at which lowest concentration threshold of Aβ corresponding to which AD clinical stage does PIB-PET produce a positive signal. Nevertheless, Ikonomovic et al. [131] showed weakly Aβ labeled with the high fluorescent 6-CN-PIB in low Aβ brain load indicating that this AD pathology case may precede the PIB detectable level. Using PET, a compound called (E)-4-(2-(6-(2-(2-(2-([18F]-fluoroethoxy)ethoxy)ethoxy)pyridin-3-yl)vinyl)-N-methyl benzenamine, also named Florbetapir, has shown to possess a half-life of 110 min versus 20 min for PIB which allows the former to accumulate significantly more in the brain regions of AD and MCI patients associated with Aβ deposits binding compared to healthy controls [132, 133]. This observation will definitively be important as an in vivo diagnostic agent for detecting Aβ pathology during the presymptomatic stage of AD, its related hypotheses cascade and to assess the efficacy of antiamyloid therapies under clinical development [134]. Besides measuring known and new CSF biomarkers to improve the diagnostic accuracy of Aβ42 and tau [135], clinical dementia rating (CDR) scale of 0–2 indicates that CSF biomarkers are detectable at CDR 0.5, CSF biomarkers, and FDG-PET imaging at CDR 1 and all previous ones including cerebral blood flow SPECT and MRI at CDR 2 [136]. Although hypometabolism measured by FDG-PET and brain atrophy MRI show predictive patterns, their profile reveals different rates and change according to regions and disease progression [137]. For example, Villain et al. [138] assessed the relationship between hippocampal atrophy specifically related to the cingulate bundle disruption as a factor contributing to the early posterior cingulate cortex hypometabolism and peripheral connecting network regions such as the middle cingulate gyrus, thalamus, mammillary bodies, parahippocampal gyrus, and hippocampus (whole memory Papez's circuit), as well as the right tempoparietal associative cortex in AD. Shima et al. [139] cautioned that diverse atrophy patterns exist among AD subjects, but point out a significant correlation between a subset of patients affected by posterior cingulate/precuneus atrophy with greater hypometabolic manifestation. In MCI, MRI hippocampal atrophy, tempo-parietal hypo-FDG-PET, CSF Aβ42, t-tau and p-tau, and cortical amyloid deposits [11C]-PIB biomarkers were also shown to be useful for diagnostic enrichment, designed for improving clinical trials modifying drugs therapies for AD [140]. At the present time, identifying definitive surrogate markers across the wide spectrum of AD subjects using current multimodalities imaging needs further explorative evidence.
6. Methods and Approaches for the Development of Amyloid Targeted Contrast Imaging Agents
Because Aβ can be used as a gauge for measuring early AD neuropathology, developing appropriate contrast imaging agents for in vivo early detection and quantitative brain deposits can benefit antiamyloid therapies. Florbetapir F18-PET has been employed to image cortical Aβ between patients with mild-to-moderate AD as well as MCI to healthy controls [133, 141]. Amyloid PET ligand florbetapir F18 has been shown to correlate closely with the localization and density of Aβ plaques identified by silver and thioflavin S staining, and immunohistochemistry [142, 143], and their results support the notion that future studies are necessary for establishing florbetapir-PET imaging as a clinical diagnosis of AD and as a reference biomarker used for the prediction of the illness progression. Similarly, Wolk et al. [144] demonstrate a concordance between flutemetamol F18-PET imaging tested in seven patients with previous biopsy, obtained from same patients at the site of ventriculo-eritoneal, used for measuring Aβ load by immunohistochemistry and histology. Yang et al. [145] were the first to report the feasibility of combining immunohistochemistry, relaxation time T2 values in regions of interest corresponding to the cortex and hippocampal (cerebellum was set as a control), and voxel-based morphometry using statistical parametric mapping to detect amyloid plaque load in AD transgenic mice following femoral injection of ultra small superparamagnetic iron oxide contrast agent (USPIO)-Aβ42. They suggested that both are quantitative methods that demonstrate prior safe use of USPIO and could be used to distinguish patients having AD from healthy controls using MRI. The question remains if this technique is capable of detecting Aβ in MCI and if can be used for prediction of AD. Moreover, MRI can look at another approach which consists at looking at the low AD signature of regional cortical thinning identified in MCI patients associated with higher risk for developing future cognitive decline and abnormally low Aβ in CSF can be useful for predicting AD [146]. No publication is currently available regarding the utility of optical imaging platform in clinical diagnostics, but detecting Aβ plaques of AD and or MCI patients noninvasively using specific contrast agents in optical technique would certainly benefit quantitative studies for amyloid neuropathological prediction. A cursory report shows that following the cerebral injection of Alexa Fluor 750-labeled antibeta amyloid mouse monoclonal antibody BAM-10, moderately superior near-infrared optical fluorescence intensity signal was observed in transgenic mice. However, Skoch et al. [147] did not test their antibody via immunohistochemistry using the same brain scanned by optical imaging. Other molecular imaging approaches have been employed in transgenic mouse. For example, polysorbate 80(PS80)-coated poly(n-butylcyano-acrylate) nanoparticles (PBCA-NP) conjugated with 6E10-Alexa488 antibody was evaluated by in vivo optical 2-photon imaging and gadobutrol PBCA-NP accumulation in brain parenchyma over time by MRI. Koffie et al. [148] demonstrated that PBCA-NP can be used as an efficient and safe delivery biodegradable nanocarrier of the blood-brain barrier (i.e., absorbed by the ApoE onto plasma membrane and internalized via receptor-mediated transcytosis) into the brain and to transport blood-brain barrier impermeable targeted Aβ contrast agents into the brain in sufficient amount for enhancing measurement of signal-to-noise ratio during noninvasively optical imaging and MRI scans. These prove to be clinically relevant for diagnosis Aβ cellular and neuropathological changes associated with AD. Another approach used magnetic resonance imaging-guided FUS (transcranial focused ultrasound) following intravenous tail vein administration of biotinylated BAM-10 antibody. Jordão et al. [149] notice that a low dose of 40 μg BAM-10 bound to Aβ plaques in cortical regions remained associated with plaques for at least four days in brain sections and reduced plaques number density, mean size, and surface area, but did not change Aβ levels.
7. Methods Development for Early Diagnosis and Therapies in AD Clinical Research
Transgenic AD mouse model provides an excellent tool to support the amyloid cascade hypothesis as the foundation of AD pathogenesis. These animals have been designed for Aβ production, but more specifically for plaques and tangles formation [150, 151] to recapitulate the disease aspects. Although the use of transgenic mice in preclinical research is very useful, it has some limitations when assessment is conducted in human clinical trials applications as to which extent the transgenic mouse models are accurate representing sporadic human AD neuropathology when evaluating the safety and the potential use of therapeutics products [31]. The use of double transgenic 85Dbo/J mice harboring PSEN1dE9 and APPSWE transgenes devoid of tau expression is pertaining in the evaluation of cerebral amyloid hypothesis in AD mouse model. Indeed, Lewis et al. [152] observed high Aβ42 : 40 ratios among 7- to 12-month-old APP695SWE × PS1A246E animals compared to APP695SWE mice and suggest that Aβ40 represents a minor component of AD pathogenesis. Supportive of this, Qu et al. [153] designed an elegant study in which serum antibody Aβ42 trimer shows higher concentration than its monomer when associated with amyloid plaques in the brains of APP/PS1 TG mice. They suggest that Aβ42 trimer can be used as a potential therapeutic value during immunization in preventing brain Aβ42 plaque formation.
Since the initial study of Gilman et al. [154] showing 6% of patients developed meningoencephalitis side effect due to the QS-21 added to Aβ in adjuvant vaccine, Cao et al. [155] demonstrated that adjuvant-free vaccine utilizing different Aβ carrying diverse mutations in the T-cell epitope shows a promise as effective and safe immunotherapy against AD. Other methods have shown promising efficient and safe vaccine towards inflammatory response in human and transgenic AD models [156–160]. Still more studies are needed to ascertain vaccine validation and standardization at preventing autoimmune response, vasogenic edema and microhemorrhage although detrimental side effects have been reported in transgenic mice [161–163] as well for bapineuzumab in patients [164–167].
There are currently several immunotherapy approaches that focus on effective treatments in clinical trials but should be administrated at the earliest stages to see if therapy can prevent or delay the progression of AD as suggested by Panza et al. [168] since detrimental side effects have been reported in transgenic mice [169–171]. The Aβ immunotherapy has great potential of clearing cerebral Aβ via microglia independent pathway [172], as being the most investigated molecular target for intervention in AD which is in constant development for achieving better safer and efficacy treatments. Emerging therapies should address time-course administration to observe for any differential treatment effects associated with various AD stages as described by Breitner et al. [173]. Clearance of amyloid plaques in preclinical animal model studies provides protection and reversal of neuropathology, but failed to show significant cognitive stimulation in patients with moment symptomatic changes [174–176]. Discrepancies noted between rodents and humans should be examined whether species' and strains' or races' epitope specificity and functionality occur. Among different Aβ immunotherapy approaches, the active immunization involving synthetic Aβ42 absorbed to a carrier protein and passive immunization involving monoclonal antibodies directed against Aβ synthesis elicit plaques clearance in AD subjects and more specifically for the treatment of presymptomatic to maximize potential disease modifying drugs in clinical trials [177–180]. Gamma-secretase inhibitors are known to inhibit Aβ40 in the brain, CSF, and plasma in rat [181], but significant reduction in Aβ concentrations are noticed in human plasma and CSF without clearance change [182, 183]. Another therapy called 8-hydroxy quinoline analog (PBT2) is a metal-protein attenuating compound which reduces copper- and zinc-mediated toxic oligomerization of Aβ has shown significant reduction in CSF Aβ42 concentration compared to the placebo associated with cognitive improvement during clinical trial phase 2 [184]. Crouch et al. [185] demonstrated that PBT2 having metal chaperone activity translocated extracellular zinc and copper into the cells by promoting Aβ aggregates dissolution via matrix metalloprotease 2 could provide a mechanism by which PBT2 improves cognitive function in AD subjects. Plausible mechanisms of actions propose (1) synthetic fragment of Aβ are designed at crossing the blood-brain barrier via their epitope to opsonize Aβ whereas their Fc fragment is recognized by receptor-mediated phagocytosis by microglia, (2) administrating into the vasculature system antibody targeting Aβ whose complex will be cleared from blood circulation preventing Aβ to enter into the brain to form new plaques or modified antibody that altered the blood-brain barrier permeability used to drain out Aβ from the brain into the blood circulation to enhance clearance of soluble Aβ and (3) administration of antibody inhibitors preventing amyloid accumulate in plaque [180]. Since Aβ immunotherapy shows limited clearance of tau aggregates in dystrophic neurites and neuropil threads [186, 187] and weather clearance of tau pathology modulates Aβ [188], it proposes that tau therapy should be developed separately to directly target pathological tau in AD and related tauopathies [180]. Using a mouse model of accelerated tangle development, targeting tau immunotherapy was marked with extensive abnormal tau clearance into the brain, especially the decrease of NFTs accumulation, and prevents cognitive decline in mice [188, 189]. Evidence suggested that cognitive impairment is correlated with the degree of tau pathology, and as such clearing phosphorylated tau may be a promising therapeutic approach to slow or prevent cognitive impairment in animal tauopathy model [190]. Although few clinical studies have been reported so far, active and passive immunotherapies for tau are possible at preventing the accumulated intracellulartau pathology, neurospheroids, and associated symptoms [188, 191]. Amyloid-β immunotherapy acts by preventing phosphorylated tau accumulation and demonstrates a link between these proteins, that both therapies should be allowed as treatment to reduce or block the progression of the illness (Figure 4). In other words, targeting early Aβ and tau pathology stages may have beneficial effects as smaller Aβ and tau assemblies should be cleared faster than mature NFTs [188]. As an exemple, apomorphine treatment in 6-month old triple transgenic AD mice shows improvement of memory function as a result of Morris water maze time-course evaluation and, in this mouse model, apomorphine has been shown to promote significantly decrease in intraneuronal Aβ, phosphorylated tau, p-53, and heme oxygenase-1 proteins in cultured SH-SY5Y cells [192]. In a near future, apomorphine may be evaluated in clinical pilot AD studies to address drug efficacy in translational medicine.
Figure 4.
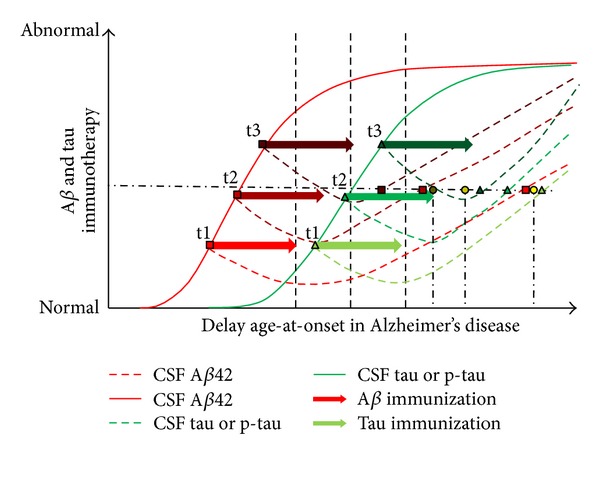
Hypothetical model illustrating the synergic effect Aβ and Tau immunotherapies combined together could have on the delay predilection of age-at-onset in progressing throughout Alzheimer's disease stages. The literature supports the notion that Aβ is not enough to treat AD symptoms, and recently tau immunotherapy may help in preventing cognitive decline in mouse AD model. This graph shows the effect each immunotherapy could have over time when giving at different pathological timelines (t1, t2, and t3). Excluding time-course, time-interval, and efficacy of Aβ and tau immunization might influence the outcome of neuropathological changes; both immunotherapies may show promise in delaying neurofibrillary tangles burden clearance into the brain, cognition decline, and clinical stages. Resulting code color glow circles are from t1 Aβ + t1 tau, t2 Aβ + t2 tau, and t3 Aβ + t3 tau, respectively.
Other therapies have shown to improve shortly cognitive performance of AD subjects. Etanercept is a fusion protein based on tumor necrosis factor alpha receptor which decreases the neuroinflammatory activity of its ligand tumor necrosis factor alpha. Clinical trial study indicates that methylthioninium chloride dissolve tau tangles during mild to moderate AD stage. Moreover, antibiotic and antiviral therapies could be used in the near future as an effective treatment. Some evidence suggests that insulin sensitizers, for example, rosiglitazone and thiazolidinedione, increase dendritic spine density of primary cortical neurons translated into cognitive recovery in a subset of AD patients [193, 194].
8. Future Prospects: Treatment and Prevention
At present the etiology that triggers progressive pleiotropic deregulated intertwined targeting pathways which contribute to synaptic lesions and thereof gradual memory loss during aging stages are based on hypothetic theory explanations, and only available treatments help on short-term to attenuate symptoms of MCI patients. Evidence point out that immunotherapy, in near future clinical trials, has the potential to slow down the progression of the disease assessed by using diagnostic PET and MRI, the two most promising molecular imaging technologies. As of December 2012, it has been found 1184 studies were conducted to treat the disease as listed in the clinicaltrial.gov website and about 20%, 25%, 15%, and 10% of these compounds are in Phase I, II, III, and IV trials, respectively. We optimistically expect that some compounds may have adequate efficacy, tolerability, and safety towards treating MCI subjects as the global economic health care burden continued to grow every day. A challenging approach, but yet feasible, would be treating patients with personalized medicine at the earliest clinical stage of dementia and follow up their cognitive evolution as compared with untreated same category patients (i.e., based on standardization of quantitative metrics and population's genetic susceptibility profile) to provide insight into the AD biomarkers' mechanisms of regulation influencing on progressive neuropathological features translated into cognitive and behaviour impairments.
Conflict of Interests
The authors do not have any conflict of interests.
Acknowledgment
This work was supported by the Molecular Imaging in Alzheimer's Disease Grant obtained from the Alzheimer's Association Research Grant.
References
- 1.Blocq P, Marinesco G. Sur les lésions et la pathogénie de l’épilepsie dite essentielle. La Semaine Médicale. 1892;12:445–446. [Google Scholar]
- 2.Alzheimer A. Ober eine eigenartige Erkrankung der Hirnrinde. Zentralblatt Für Nervenheilkunde Psychiatrie. 1907;18:177–179. [Google Scholar]
- 3.Amieva H, Le Goff M, Millet X, et al. Prodromal Alzheimer’s disease: successive emergence of the clinical symptoms. Annals of Neurology. 2008;64(5):492–498. doi: 10.1002/ana.21509. [DOI] [PubMed] [Google Scholar]
- 4.Rademakers R, Cruts M, van Broeckhoven C. Genetics of early-onset Alzheimer dementia. The Scientific World Journal. 2003;3:497–519. doi: 10.1100/tsw.2003.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luengo-Fernandez R, Leal J, Gray AM. Cost of dementia in the pre-enlargement countries of the European Union. Journal of Alzheimer’s Disease. 2011;27(1):187–196. doi: 10.3233/JAD-2011-102019. [DOI] [PubMed] [Google Scholar]
- 6.Gao S, Hendrie HC, Hall KS, Hui S. The relationships between age, sex, and the incidence of dementia and Alzheimer disease: a meta-analysis. Archives of General Psychiatry. 1998;55(9):809–815. doi: 10.1001/archpsyc.55.9.809. [DOI] [PubMed] [Google Scholar]
- 7.Lautenschlager NT, Cupples LA, Rao VS, et al. Risk of dementia among relatives of Alzheimer’s disease patients in the MIRAGE study: what is in store for the oldest old? Neurology. 1996;46(3):641–650. doi: 10.1212/wnl.46.3.641. [DOI] [PubMed] [Google Scholar]
- 8.Prince MJ, editor. Alzheimer’s Disease International. World Alzheimer Report 2009. chapter 1-2. London, UK: Alzheimer’s Disease International; 2009. pp. 25–66. [Google Scholar]
- 9.Rosario ER, Chang L, Head EH, Stanczyk FZ, Pike CJ. Brain levels of sex steroid hormones in men and women during normal aging and in Alzheimer’s disease. Neurobiology of Aging. 2011;32(4):604–613. doi: 10.1016/j.neurobiolaging.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carroll JC, Rosario ER. The potential use of hormone-based therapeutics for the treatment of Alzheimer's disease. Current Alzheimer Research. 2012;9(1):18–34. doi: 10.2174/156720512799015109. [DOI] [PubMed] [Google Scholar]
- 11.Gibbs RB. Estrogen therapy and cognition: a review of the cholinergic hypothesis. Endocrine Reviews. 2010;31(2):224–253. doi: 10.1210/er.2009-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Massoud TF, Gambhir SS. Molecular imaging in living subjects: seeing fundamental biological processes in a new light. Genes and Development. 2003;17(5):545–580. doi: 10.1101/gad.1047403. [DOI] [PubMed] [Google Scholar]
- 13.Studwell AJ, Kotton DN. A shift from cell cultures to creatures: in vivo imaging of small animals in experimental regenerative medicine. Molecular Therapy. 2011;19(11):1933–1941. doi: 10.1038/mt.2011.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McDonald RJ, Craig LA, Hong NS. The etiology of age-related dementia is more complicated than we think. Behavioural Brain Research. 2010;214(1):3–11. doi: 10.1016/j.bbr.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 15.Wilson WL, Munn C, Ross RC, Harding JW, Wright JW. The role of the AT4 and cholinergic systems in the Nucleus Basalis Magnocellularis (NBM): effects on spatial memory. Brain Research. 2009;1272:25–31. doi: 10.1016/j.brainres.2009.03.025. [DOI] [PubMed] [Google Scholar]
- 16.Srivareerat M, Tran TT, Salim S, Aleisa AM, Alkadhi KA. Chronic nicotine restores normal Aβ levels and prevents short-term memory and E-LTP impairment in Aβ rat model of Alzheimer’s disease. Neurobiology of Aging. 2011;32(5):834–844. doi: 10.1016/j.neurobiolaging.2009.04.015. [DOI] [PubMed] [Google Scholar]
- 17.Prvulovic D, Hampel H, Pantel J. Galantamine for Alzheimer’s disease. Expert Opinion on Drug Metabolism and Toxicology. 2010;6(3):345–354. doi: 10.1517/17425251003592137. [DOI] [PubMed] [Google Scholar]
- 18.Moreno MDJM. Cognitive improvement in mild to moderate Alzheimer’s dementia after treatment with the acetylcholine precursor choline alfoscerate: a multicenter, double-blind, randomized, placebo-controlled trial. Clinical Therapeutics. 2003;25(1):178–193. doi: 10.1016/s0149-2918(03)90023-3. [DOI] [PubMed] [Google Scholar]
- 19.Park D, Lee HJ, Joo SS, et al. Human neural stem cells over-expressing choline acetyltransferase restore cognition in rat model of cognitive dysfunction. Experimental Neurology. 2012;234(2):521–526. doi: 10.1016/j.expneurol.2011.12.040. [DOI] [PubMed] [Google Scholar]
- 20.Roßner S, Ueberham U, Schliebs R, Perez-Polo JR, Bigl V. The regulation of amyloid precursor protein metabolism by cholinergic mechanisms and neurotrophin receptor signaling. Progress in Neurobiology. 1998;56(5):541–569. doi: 10.1016/s0301-0082(98)00044-6. [DOI] [PubMed] [Google Scholar]
- 21.Garcia-Alloza M, Zaldua N, Diez-Ariza M, et al. Effect of selective cholinergic denervation on the serotonergic system: implications for learning and memory. Journal of Neuropathology and Experimental Neurology. 2006;65(11):1074–1081. doi: 10.1097/01.jnen.0000240469.20167.89. [DOI] [PubMed] [Google Scholar]
- 22.Hernandez CM, Kayed R, Zheng H, Sweatt JD, Dineley KT. Loss of α7 nicotinic receptors enhances β-amyloid oligomer accumulation, exacerbating early-stage cognitive decline and septohippocampal pathology in a mouse model of Alzheimer’s disease. The Journal of Neuroscience. 2010;30(7):2442–2453. doi: 10.1523/JNEUROSCI.5038-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nyakas C, Granic I, Halmy LG, Banerjee P, Luiten PGM. The basal forebrain cholinergic system in aging and dementia. Rescuing cholinergic neurons from neurotoxic amyloid-β42 with memantine. Behavioural Brain Research. 2011;221(2):594–603. doi: 10.1016/j.bbr.2010.05.033. [DOI] [PubMed] [Google Scholar]
- 24.Pettit DL, Shao Z, Yakel JL. beta-Amyloid(1–42) peptide directly modulates nicotinic receptors in the rat hippocampal slice. The Journal of Neuroscience. 2001;21(1, article RC120) doi: 10.1523/JNEUROSCI.21-01-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Capsoni S, Tiveron C, Vignone D, Amato G, Cattaneo A. Dissecting the involvement of tropomyosin-related kinase A and p75 neurotrophin receptor signaling in NGF deficit-induced neurodegeneration. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(27):12299–12304. doi: 10.1073/pnas.1007181107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bencherif M, Lippiello PM. Alpha7 neuronal nicotinic receptors: the missing link to understanding Alzheimer’s etiopathology? Medical Hypotheses. 2010;74(2):281–285. doi: 10.1016/j.mehy.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 27.Doody RS, Dunn JK, Clark CM, et al. Chronic donepezil treatment is associated with slowed cognitive decline in Alzheimer’s disease. Dementia and Geriatric Cognitive Disorders. 2001;12(4):295–300. doi: 10.1159/000051272. [DOI] [PubMed] [Google Scholar]
- 28.Schaeffer EL, Gattaz WF. Cholinergic and glutamatergic alterations beginning at the early stages of Alzheimer disease: participation of the phospholipase A2 enzyme. Psychopharmacology. 2008;198(1):1–27. doi: 10.1007/s00213-008-1092-0. [DOI] [PubMed] [Google Scholar]
- 29.Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 30.Armstrong RA. The pathogenesis of alzheimer’s disease: a reevaluation of the ‘amyloid cascade hypothesis’. International Journal of Alzheimer’s Disease. 2011;2011:6 pages. doi: 10.4061/2011/630865.630865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blennow K, de Leon MJ, Zetterberg H. Alzheimer’s disease. The Lancet. 2006;368(9533):387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- 32.Querfurth HW, LaFerla FM. Alzheimer’s disease. The New England Journal of Medicine. 2010;362(4):329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 33.Berridge MJ. Calcium hypothesis of Alzheimer’s disease. Pflügers Archiv. 2010;459(3):441–449. doi: 10.1007/s00424-009-0736-1. [DOI] [PubMed] [Google Scholar]
- 34.Green KN, LaFerla FM. Linking calcium to Aβ and Alzheimer’s disease. Neuron. 2008;59(2):190–194. doi: 10.1016/j.neuron.2008.07.013. [DOI] [PubMed] [Google Scholar]
- 35.Lu PJ, Wulf G, Zhou XZ, Davies P, Lu KP. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature. 1999;399(6738):784–788. doi: 10.1038/21650. [DOI] [PubMed] [Google Scholar]
- 36.Buerger K, Ewers M, Pirttilä T, et al. CSF phosphorylated tau protein correlates with neocortical neurofibrillary pathology in Alzheimer’s disease. Brain. 2006;129(part 11):3035–3041. doi: 10.1093/brain/awl269. [DOI] [PubMed] [Google Scholar]
- 37.Babu JR, Seibenhener ML, Peng J, et al. Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. Journal of Neurochemistry. 2008;106(1):107–120. doi: 10.1111/j.1471-4159.2008.05340.x. [DOI] [PubMed] [Google Scholar]
- 38.Mateo I, Sánchez-Juan P, Rodríguez-Rodríguez E, et al. Synergistic effect of heme oxygenase-1 and tau genetic variants on Alzheimer's disease risk. Dementia and Geriatric Cognitive Disorders. 2008;26(4):339–342. doi: 10.1159/000161059. [DOI] [PubMed] [Google Scholar]
- 39.Eroglu B, Moskophidis D, Mivechi NF. Loss of Hsp110 leads to age-dependent tau hyperphosphorylation and early accumulation of insoluble amyloid β . Molecular and Cellular Biology. 2010;30(19):4626–4643. doi: 10.1128/MCB.01493-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Solé-Padullés C, Lladó A, Bartrés-Faz D, et al. Association between cerebrospinal fluid tau and brain atrophy is not related to clinical severity in the Alzheimer's disease continuum. Psychiatry Research. 2011;192(3):140–146. doi: 10.1016/j.pscychresns.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 41.Attems J, Thomas A, Jellinger K. Correlations between cortical and subcortical tau pathology. Neuropathology and Applied Neurobiology. 2012;38(6):582–590. doi: 10.1111/j.1365-2990.2011.01244.x. [DOI] [PubMed] [Google Scholar]
- 42.Hyman BT. Amyloid-dependent and amyloid-independent stages of Alzheimer disease. Archives of Neurology. 2011;68(8):1062–1064. doi: 10.1001/archneurol.2011.70. [DOI] [PubMed] [Google Scholar]
- 43.Crespo-Biel N, Theunis C, van Leuven F. Protein tau: prime cause of synaptic and neuronal degeneration in Alzheimer's disease. International Journal of Alzheimer’s Disease. 2012;2012:13 pages. doi: 10.1155/2012/251426.251426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maccioni RB, Farías G, Morales I, Navarrete L. The revitalized tau hypothesis on Alzheimer’s disease. Archives of Medical Research. 2010;41(3):226–231. doi: 10.1016/j.arcmed.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 45.Dickson DW. Apoptotic mechanisms in Alzheimer neurofibrillary degeneration: cause or effect? Journal of Clinical Investigation. 2004;114(1):23–27. doi: 10.1172/JCI22317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rohn TT, Head E. Caspases as therapeutic targets in Alzheimer's disease: is it time to “cut” to the chase? International Journal of Clinical and Experimental Pathology. 2009;2(2):108–118. [PMC free article] [PubMed] [Google Scholar]
- 47.Itzhaki RF, Wozniak MA. Herpes simplex virus type 1, apolipoprotein E, and cholesterol: a dangerous liaison in Alzheimer’s disease and other disorders. Progress in Lipid Research. 2006;45(1):73–90. doi: 10.1016/j.plipres.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 48.Itzhaki RF, Cosby SL, Wozniak MA. Herpes simplex virus type 1 and Alzheimer’s disease: the autophagy connection. Journal of NeuroVirology. 2008;14(1):1–4. doi: 10.1080/13550280701802543. [DOI] [PubMed] [Google Scholar]
- 49.Santana S, Recuero M, Bullido MJ, et al. Herpes simplex virus type I induces the accumulation of intracellular β-amyloid in autophagic compartments and the inhibition of the non-amyloidogenic pathway in human neuroblastoma cells. Neurobiology of Aging. 2012;33(2):430.e19–430.e33. doi: 10.1016/j.neurobiolaging.2010.12.010. [DOI] [PubMed] [Google Scholar]
- 50.Bartzokis G. Alzheimer’s disease as homeostatic responses to age-related myelin breakdown. Neurobiology of Aging. 2011;32(8):1341–1371. doi: 10.1016/j.neurobiolaging.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Heneka MT, Nadrigny F, Regen T, et al. Locus ceruleus controls Alzheimer’s disease pathology by modulating microglial functions through norepinephrine. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(13):6058–6063. doi: 10.1073/pnas.0909586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kong Y, Ruan L, Qian L, Liu X, Le Y. Norepinephrine promotes microglia to uptake and degrade amyloid β peptide through upregulation of mouse formyl peptide receptor 2 and induction of insulin-degrading enzyme. The Journal of Neuroscience. 2010;30(35):11848–11857. doi: 10.1523/JNEUROSCI.2985-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cortes-Canteli M, Paul J, Norris EH, et al. Fibrinogen and beta-amyloid association alters thrombosis and fibrinolysis: a possible contributing factor to Alzheimer's disease. Neuron. 2010;66(5):695–709. doi: 10.1016/j.neuron.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ahn HJ, Zamolodchikov D, Cortes-Canteli M, Norris EH, Glickman JF, Strickland S. Alzheimer’s disease peptide β-amyloid interacts with fibrinogen and induces its oligomerization. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(50):21812–21817. doi: 10.1073/pnas.1010373107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sorbi S, Forleo P, Tedde A, et al. Genetic risk factors in familial Alzheimer’s disease. Mechanisms of Ageing and Development. 2001;122(16):1951–1960. doi: 10.1016/s0047-6374(01)00308-6. [DOI] [PubMed] [Google Scholar]
- 56.Rogaeva E, Meng Y, Lee JH, et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nature Genetics. 2007;39(2):168–177. doi: 10.1038/ng1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schmidt V, Sporbert A, Rohe M, et al. SorLA/LR11 regulates processing of amyloid precursor protein via interaction with adaptors GGA and PACS-1. The Journal of Biological Chemistry. 2007;282(45):32956–32964. doi: 10.1074/jbc.M705073200. [DOI] [PubMed] [Google Scholar]
- 58.Reitz C, Mayeux R. Endophenotypes in normal brain morphology and Alzheimer’s disease: a review. Neuroscience. 2009;164(1):174–190. doi: 10.1016/j.neuroscience.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reiman EM, Chen K, Langbaum JBS, et al. Higher serum total cholesterol levels in late middle age are associated with glucose hypometabolism in brain regions affected by Alzheimer’s disease and normal aging. NeuroImage. 2010;49(1):169–176. doi: 10.1016/j.neuroimage.2009.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Genin E, Hannequin D, Wallon D, et al. APOE and Alzheimer disease: a major gene with semi-dominant inheritance. Molecular Psychiatry. 2011;16(9):903–907. doi: 10.1038/mp.2011.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Soler-López M, Zanzoni A, Lluís R, et al. Interactome mapping suggests new mechanistic details underlying Alzheimer's disease. Genome Research. 2011;21(3):364–376. doi: 10.1101/gr.114280.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ghebranious N, Mukesh B, Giampietro PF, et al. A pilot study of gene/gene and gene/environment interactions in Alzheimer disease. Clinical Medicine and Research. 2011;9(1):17–25. doi: 10.3121/cmr.2010.894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ganguli M, Kukull WA. Lost in translation: epidemiology, risk, and alzheimer disease. Archives of Neurology. 2010;67(1):107–111. doi: 10.1001/archneurol.2009.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Migliore L, Coppedè F. Genetics, environmental factors and the emerging role of epigenetics in neurodegenerative diseases. Mutation Research. 2009;667(1-2):82–97. doi: 10.1016/j.mrfmmm.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 65.Qiu C, de Ronchi D, Fratiglioni L. The epidemiology of the dementias: an update. Current Opinion in Psychiatry. 2007;20(4):380–385. doi: 10.1097/YCO.0b013e32816ebc7b. [DOI] [PubMed] [Google Scholar]
- 66.Sadowski M, Pankiewicz J, Scholtzova H, et al. A synthetic peptide blocking the apolipoprotein E/β-amyloid binding mitigates β-amyloid toxicity and fibril formation in vitro and reduces β-amyloid plaques in transgenic mice. The American Journal of Pathology. 2004;165(3):937–948. doi: 10.1016/s0002-9440(10)63355-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhong N, Scearce-Levie K, Ramaswamy G, Weisgraber KH. Apolipoprotein E4 domain interaction: synaptic and cognitive deficits in mice. Alzheimer’s & Dementia. 2008;4(3):179–192. doi: 10.1016/j.jalz.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Naj AC, Jun G, Beecham GW, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nature Genetics. 2011;43(5):436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hollingworth P, Harold D, Sims R, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nature Genetics. 2011;43(5):429–435. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jun G, Naj AC, Beecham GW, et al. Meta-analysis confirms CR1, CLU, and PICALM as Alzheimer disease risk loci and reveals interactions with APOE genotypes. Archives of Neurology. 2010;67(12):1473–1484. doi: 10.1001/archneurol.2010.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Logue MW, Schu M, Vardarajan BN, et al. A comprehensive genetic association study of Alzheimer disease in African Americans. Archives of Neurology. 2011;68(12):1569–1579. doi: 10.1001/archneurol.2011.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Alexopoulos P, Guo LH, Kratzer M, et al. Impact of SORL1 single nucleotide polymorphisms on Alzheimer's disease cerebrospinal fluid markers. Dementia and Geriatric Cognitive Disorders. 2011;32(3):164–170. doi: 10.1159/000332017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cruchaga C, Kauwe JSK, Mayo K, et al. SNPs associated with cerebrospinal fluid Phospho-tau levels influence rate of decline in alzheimer’s disease. PLoS Genetics. 2010;6(9) doi: 10.1371/journal.pgen.1001101.e1001101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kauwe JSK, Cruchaga C, Bertelsen S, et al. Validating predicted biological effects of Alzheimer’s disease associated SNPs using CSF biomarker levels. Journal of Alzheimer’s Disease. 2010;21(3):833–842. doi: 10.3233/JAD-2010-091711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stein JL, Hua X, Morra JH, et al. Genome-wide analysis reveals novel genes influencing temporal lobe structure with relevance to neurodegeneration in Alzheimer’s disease. NeuroImage. 2010;51(2):542–554. doi: 10.1016/j.neuroimage.2010.02.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Furney SJ, Simmons A, Breen G, et al. Genome-wide association with MRI atrophy measures as a quantitative trait locus for Alzheimer's disease. Molecular Psychiatry. 2011;16(11):1130–1138. doi: 10.1038/mp.2010.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ge T, Feng J, Hibar DP, et al. Increasing power for voxel-wise genome-wide association studies: the random field theory, least square kernel machines and fast permutation procedures. Neuroimage. 2012;63(2):858–873. doi: 10.1016/j.neuroimage.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wu C, Pike VW, Wang Y. Amyloid imaging: from benchtop to bedside. Current Topics in Developmental Biology. 2005;70:171–213. doi: 10.1016/S0070-2153(05)70008-9. [DOI] [PubMed] [Google Scholar]
- 79.Perl DP. Neuropathology of Alzheimer’s disease. Mount Sinai Journal of Medicine. 2010;77(1):32–42. doi: 10.1002/msj.20157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Castellani RJ, Alexiev BA, Philips D, et al. Microscopic investigations in neurodegenerative diseases. In: Méndez-Vilas A, Díaz J, editors. Modern Research and Educational Topics on Microscopy. Vol. 1. Formatex; 2007. pp. 171–182. [Google Scholar]
- 81.Thal DR, Rüb U, Orantes M, Braak H. Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58(12):1791–1800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- 82.Nagy Z, Yilmazer-Hanke DM, Braak H, Braak E, Schultz C, Hanke J. Assessment of the pathological stages of Alzheimer’s disease in thin paraffin sections: a comparative study. Dementia and Geriatric Cognitive Disorders. 1998;9(3):140–144. doi: 10.1159/000017038. [DOI] [PubMed] [Google Scholar]
- 83.Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41(4):479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 84.Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimer’s & Dementia. 2012;8(1):1–13. doi: 10.1016/j.jalz.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathologica. 1991;82(4):239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 86.Thal DR, Griffin WST, Braak H. Parenchymal and vascular Aβ-deposition and its effects on the degeneration of neurons and cognition in Alzheimer’s disease. Journal of Cellular and Molecular Medicine. 2008;12(5B):1848–1862. doi: 10.1111/j.1582-4934.2008.00411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s & Dementia. 2011;7(3):280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hebert LE, Scherr PA, Beckett LA, et al. Age-specific incidence of Alzheimer’s disease in a community population. Journal of the American Medical Association. 1995;273(17):1354–1359. [PubMed] [Google Scholar]
- 89.Ganguli M, Dodge HH, Chen P, Belle S, DeKosky ST. Ten-year incidence of dementia in a rural elderly US community population. The MoVIES project. Neurology. 2000;54(5):1109–1116. doi: 10.1212/wnl.54.5.1109. [DOI] [PubMed] [Google Scholar]
- 90.Kukull WA, Higdon R, Bowen JD, et al. Dementia and Alzheimer disease incidence: a prospective cohort study. Archives of Neurology. 2002;59(11):1737–1746. doi: 10.1001/archneur.59.11.1737. [DOI] [PubMed] [Google Scholar]
- 91.Jack CR, Jr., Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. The Lancet Neurology. 2010;9(1):119–128. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jagust WJ, Mormino EC. Lifespan brain activity, β-amyloid, and Alzheimer's disease. Trends in Cognitive Sciences. 2011;15(11):520–526. doi: 10.1016/j.tics.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Arenaza-Urquijo EM, Bosch B, Sala-Llonch R, et al. Specific anatomic associations between white matter integrity and cognitive reserve in normal and cognitively impaired elders. American Journal of Geriatric Psychiatry. 2011;19(1):33–42. doi: 10.1097/JGP.0b013e3181e448e1. [DOI] [PubMed] [Google Scholar]
- 94.Lee JJ, Jo SA, Park JH, et al. Choline acetyltransferase 2384 G>a polymorphism and the risk of Alzheimer disease. Alzheimer Disease & Associated Disorders. 2011 doi: 10.1097/WAD.0b013e31821cbcaf. [DOI] [PubMed] [Google Scholar]
- 95.Elias-Sonnenschein LS, Viechtbauer W, Ramakers IH, et al. Predictive value of APOE-ε4 allele for progression from MCI to AD-type dementia: a meta-analysis. Journal of Neurology, Neurosurgery and Psychiatry. 2011;82(10):1149–1156. doi: 10.1136/jnnp.2010.231555. [DOI] [PubMed] [Google Scholar]
- 96.Steenland K, Karnes C, Seals R, et al. Late-life depression as a risk factor for mild cognitive impairment or Alzheimer's disease in 30 US Alzheimer's disease centers. Journal of Alzheimer’s Disease. 2012;31(2):265–275. doi: 10.3233/JAD-2012-111922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schmidt C, Wolff M, von Ahsen N, et al. Alzheimer's disease: genetic polymorphisms and rate of decline. Dementia and Geriatric Cognitive Disorders. 2012;33:84–89. doi: 10.1159/000336790. [DOI] [PubMed] [Google Scholar]
- 98.Bégin ME, Plourde M, Pifferi F, et al. What Is the link between docosahexaenoic acid, cognitive impairment, and Alzheimer’s disease in the elderly? In: Montmayeur JP, le Coutre J, editors. Fat Detection: Taste, Texture, and Post Ingestive Effects. chapter 19. Boca Raton, Fla, USA: CRC Press; 2010. [Google Scholar]
- 99.Vemuri P, Wiste HJ, Weigand SD, et al. Serial MRI and CSF biomarkers in normal aging, MCI, and AD. Neurology. 2010;75(2):143–151. doi: 10.1212/WNL.0b013e3181e7ca82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fjell AM, Walhovd KB. New tools for the study of Alzheimer's disease: what are biomarkers and morphometric markers teaching us? Neuroscientist. 2011;17(5):592–605. doi: 10.1177/1073858410392586. [DOI] [PubMed] [Google Scholar]
- 101.Petersen RC. Alzheimer’s disease: progress in prediction. The Lancet Neurology. 2010;9(1):4–5. doi: 10.1016/S1474-4422(09)70330-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lichtenstein MP, Carriba P, Masgrau R, et al. Staging anti-inflammatory therapy in Alzheimer's disease. Frontiers in Aging Neuroscience. 2010;2, article 142 doi: 10.3389/fnagi.2010.00142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Vemuri P, Weigand SD, Przybelski SA, et al. Cognitive reserve and Alzheimer’s disease biomarkers are independent determinants of cognition. Brain. 2011;134(5):1479–1492. doi: 10.1093/brain/awr049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Jack CR, Jr., Wiste HJ, Vemuri P, et al. Brain beta-amyloid measures and magnetic resonance imaging atrophy both predict time-to-progression from mild cognitive impairment to Alzheimer’s disease. Brain. 2010;133(11):3336–3348. doi: 10.1093/brain/awq277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Verghese PB, Castellano JM, Holtzman DM. Apolipoprotein E in Alzheimer’s disease and other neurological disorders. The Lancet Neurology. 2011;10(3):241–252. doi: 10.1016/S1474-4422(10)70325-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.de Jager CA, Honey TE, Birks J, Wilcock GK. Retrospective evaluation of revised criteria for the diagnosis of Alzheimer’s disease using a cohort with post-mortem diagnosis. International Journal of Geriatric Psychiatry. 2010;25(10):988–997. doi: 10.1002/gps.2448. [DOI] [PubMed] [Google Scholar]
- 107.DeKosky ST, Carrillo MC, Phelps C, et al. Revision of the criteria for Alzheimer’s disease: a symposium. Alzheimer’s & Dementia. 2011;7(1):e1–e12. doi: 10.1016/j.jalz.2010.12.007. [DOI] [PubMed] [Google Scholar]
- 108.Hampel H, Broich K. Enrichment of MCI and early Alzheimer’s disease treatment trials using neurochemical & imaging candidate biomarkers. The Journal of Nutrition, Health and Aging. 2009;13(4):373–375. doi: 10.1007/s12603-009-0048-3. [DOI] [PubMed] [Google Scholar]
- 109.Jack CR., Jr. Alliance for aging research AD biomarkers work group: structural MRI. Neurobiology of Aging. 2011;32(Supplement 1):S48–S57. doi: 10.1016/j.neurobiolaging.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lu Y, Dendukuri N, Schiller I, Joseph L. A Bayesian approach to simultaneously adjusting for verification and reference standard bias in diagnostic test studies. Statistics in Medicine. 2010;29(24):2532–2543. doi: 10.1002/sim.4018. [DOI] [PubMed] [Google Scholar]
- 111.Westman E, Wahlund LO, Foy C, et al. Combining MRI and MRS to distinguish between Alzheimer’s disease and healthy controls. Journal of Alzheimer’s Disease. 2010;22(1):171–181. doi: 10.3233/JAD-2010-100168. [DOI] [PubMed] [Google Scholar]
- 112.Lopez OL, McDade E, Riverol M, et al. Evolution of the diagnostic criteria for degenerative and cognitive disorders. Current Opinion in Neurology. 2011;24(6):532–541. doi: 10.1097/WCO.0b013e32834cd45b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jack CR, Jr., Shiung MM, Gunter JL, et al. Comparison of different MRI brain athrophy rate measures with clinical disease progression in AD. Neurology. 2004;62(4):591–600. doi: 10.1212/01.wnl.0000110315.26026.ef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nestor SM, Rupsingh R, Borrie M, et al. Ventricular enlargement as a possible measure of Alzheimer’s disease progression validated using the Alzheimer’s disease neuroimaging initiative database. Brain. 2008;131(part 9):2443–2454. doi: 10.1093/brain/awn146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mosconi L, Tsui WH, Herholz K, et al. Multicenter standardized 18F-FDG PET diagnosis of mild cognitive impairment, Alzheimer’s disease, and other dementias. Journal of Nuclear Medicine. 2008;49(3):390–398. doi: 10.2967/jnumed.107.045385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Vandenberghe R, van Laere K, Ivanoiu A, et al. 18F-flutemetamol amyloid imaging in Alzheimer disease and mild cognitive impairment a phase 2 trial. Annals of Neurology. 2010;68(3):319–329. doi: 10.1002/ana.22068. [DOI] [PubMed] [Google Scholar]
- 117.Schmand B, Eikelenboom P, van Gool WA. Value of neuropsychological tests, neuroimaging, and biomarkers for diagnosing Alzheimer's disease in younger and older age cohorts. Journal of the American Geriatrics Society. 2011;59(9):1705–1710. doi: 10.1111/j.1532-5415.2011.03539.x. [DOI] [PubMed] [Google Scholar]
- 118.Lo RY, Hubbard AE, Shaw LM, et al. Longitudinal change of biomarkers in cognitive decline. Archives of Neurology. 2011;68(10):1257–1266. doi: 10.1001/archneurol.2011.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chen K, Ayutyanont N, Langbaum JBS, et al. Characterizing Alzheimer’s disease using a hypometabolic convergence index. NeuroImage. 2011;56(1):52–60. doi: 10.1016/j.neuroimage.2011.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wu L, Rosa-Neto P, Gauthier S. Use of biomarkers in clinical trials of Alzheimer disease: from concept to application. Molecular Diagnosis & Therapy. 2011;15(6):313–325. doi: 10.1007/BF03256467. [DOI] [PubMed] [Google Scholar]
- 121.Forsberg A, Almkvist O, Engler H, Wall A, Långström B, Nordberg A. High PIB retention in Alzheimer’s disease is an early event with complex relationship with CSF biomarkers and functional parameters. Current Alzheimer Research. 2010;7(1):56–66. doi: 10.2174/156720510790274446. [DOI] [PubMed] [Google Scholar]
- 122.Forsberg A, Engler H, Blomquist G, et al. The use of PIB-PET as a dual pathological and functional biomarker in AD. Biochimica et Biophysica Acta. 2012;1822(3):380–385. doi: 10.1016/j.bbadis.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 123.Zhang D, Wang Y, Zhou L, Yuan H, Shen D. Multimodal classification of Alzheimer’s disease and mild cognitive impairment. NeuroImage. 2011;55(3):856–867. doi: 10.1016/j.neuroimage.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Fjell AM, Walhovd KB, Fennema-Notestine C, et al. CSF biomarkers in prediction of cerebral and clinical change in mild cognitive impairment and Alzheimer’s disease. The Journal of Neuroscience. 2010;30(6):2088–2101. doi: 10.1523/JNEUROSCI.3785-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Cavallin L, Axelsson R, Wahlund LO, et al. Voxel-based correlation between coregistered single-photon emission computed tomography and dynamic susceptibility contrast magnetic resonance imaging in subjects with suspected alzheimer disease. Acta Radiologica. 2008;49(10):1154–1161. doi: 10.1080/02841850802438512. [DOI] [PubMed] [Google Scholar]
- 126.Wang H, Golob E, Bert A, et al. Alterations in regional brain volume and individual MRI-guided perfusion in normal control, stable mild cognitive impairment, and MCI-AD converter. Journal of Geriatric Psychiatry and Neurology. 2009;22(1):35–45. doi: 10.1177/0891988708328212. [DOI] [PubMed] [Google Scholar]
- 127.Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound-B. Annals of Neurology. 2004;55(3):306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 128.Engler H, Forsberg A, Almkvist O, et al. Two-year follow-up of amyloid deposition in patients with Alzheimer’s disease. Brain. 2006;129(part 11):2856–2866. doi: 10.1093/brain/awl178. [DOI] [PubMed] [Google Scholar]
- 129.Pike KE, Savage G, Villemagne VL, et al. β-amyloid imaging and memory in non-demented individuals: evidence for preclinical Alzheimer’s disease. Brain. 2007;130(part 11):2837–2844. doi: 10.1093/brain/awm238. [DOI] [PubMed] [Google Scholar]
- 130.Kadir A, Almkvist O, Forsberg A, et al. Dynamic changes in PET amyloid and FDG imaging at different stages of Alzheimer's disease. Neurobiology of Aging. 2012;33(1):198.e1–198.14. doi: 10.1016/j.neurobiolaging.2010.06.015. [DOI] [PubMed] [Google Scholar]
- 131.Ikonomovic MD, Abrahamson EE, Price JC, et al. Early AD pathology in a [C-11]PIB-negative case: a PIB-amyloid imaging, biochemical, and immunohistochemical study. Acta Neuropathologica. 2012;123(3):433–447. doi: 10.1007/s00401-012-0943-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wong DF, Rosenberg PB, Zhou Y, et al. In vivo imaging of amyloid deposition in Alzheimer disease using the radioligand 18F-AV-45 (flobetapir F 18) Journal of Nuclear Medicine. 2010;51(6):913–920. doi: 10.2967/jnumed.109.069088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Fleisher AS, Chen K, Liu X, et al. Using positron emission tomography and florbetapir F18 to image cortical amyloid in patients with mild cognitive impairment or dementia due to Alzheimer disease. Archives of Neurology. 2011;68(11):1404–1411. doi: 10.1001/archneurol.2011.150. [DOI] [PubMed] [Google Scholar]
- 134.Vallabhajosula S. Positron emission tomography radiopharmaceuticals for imaging brain beta-amyloid. Seminars in Nuclear Medicine. 2011;41(4):283–299. doi: 10.1053/j.semnuclmed.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 135.Perrin RJ, Craig-Schapiro R, Malone JP, et al. Identification and validation of novel cerebrospinal fluid biomarkers for staging early Alzheimer's disease. PLoS ONE. 2011;6(1) doi: 10.1371/journal.pone.0016032.e16032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Morinaga A, Ono K, Ikeda T, et al. A comparison of the diagnostic sensitivity of MRI, CBF-SPECT, FDG-PET and cerebrospinal fluid biomarkers for detecting Alzheimer’s disease in a memory clinic. Dementia and Geriatric Cognitive Disorders. 2010;30(4):285–292. doi: 10.1159/000320265. [DOI] [PubMed] [Google Scholar]
- 137.Weiner MW, Veitch DP, Aisen PS, et al. The Alzheimer's disease neuroimaging initiative: a review of papers published since its inception. Alzheimer’s & Dementia. 2012;8(Supplement 1):S1–S68. doi: 10.1016/j.jalz.2011.09.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Villain N, Desgranges B, Viader F, et al. Relationships between hippocampal atrophy, white matter disruption, and gray matter hypometabolism in Alzheimer’s disease. The Journal of Neuroscience. 2008;28(24):6174–6181. doi: 10.1523/JNEUROSCI.1392-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Shima K, Matsunari I, Samuraki M, et al. Posterior cingulate atrophy and metabolic decline in early stage Alzheimer's disease. Neurobiology of Aging. 2012;33(9):2006–2017. doi: 10.1016/j.neurobiolaging.2011.07.009. [DOI] [PubMed] [Google Scholar]
- 140.Lorenzi M, Donohue M, Paternicò D, et al. Enrichment through biomarkers in clinical trials of Alzheimer’s drugs in patients with mild cognitive impairment. Neurobiology of Aging. 2010;31(8):1443.e1–1451.e1. doi: 10.1016/j.neurobiolaging.2010.04.036. [DOI] [PubMed] [Google Scholar]
- 141.Camus V, Payoux P, Barré L, et al. Using PET with 18F-AV-45 (florbetapir) to quantify brain amyloid load in a clinical environment. European Journal of Nuclear Medicine and Molecular Imaging. 2012;39(4):621–631. doi: 10.1007/s00259-011-2021-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Clark DG. Residual vectors for Alzheimer disease diagnosis and prognostication. Brain and Behavior. 2011;1(2):142–152. doi: 10.1002/brb3.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Choi SR, Schneider JA, Bennett DA, et al. Correlation of amyloid PET ligand florbetapir F18 binding with Aβ aggregation and neuritic plaque deposition in postmortem brain tissue. Alzheimer Disease and Associated Disorders. 2012;26(1):8–16. doi: 10.1097/WAD.0b013e31821300bc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wolk DA, Grachev ID, Buckley C, et al. Association between in vivo fluorine 18-labeled flutemetamol amyloid positron emission tomography imaging and in vivo cerebral cortical histopathology. Archives of Neurology. 2011;68(11):1398–1403. doi: 10.1001/archneurol.2011.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Yang J, Wadghiri YZ, Hoang DM, et al. Detection of amyloid plaques targeted by USPIO-Aβ1–42 in Alzheimer’s disease transgenic mice using magnetic resonance microimaging. NeuroImage. 2011;55(4):1600–1609. doi: 10.1016/j.neuroimage.2011.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Dickerson BC, Wolk DA. MRI cortical thickness biomarker predicts AD-like CSF and cognitive decline in normal adults. Neurology. 2012;78(2):84–90. doi: 10.1212/WNL.0b013e31823efc6c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Skoch J, Dunn A, Hyman BT, Bacskai BJ. Development of an optical approach for noninvasive imaging of Alzheimer’s disease pathology. Journal of Biomedical Optics. 2005;10(1) doi: 10.1117/1.1846075.11007 [DOI] [PubMed] [Google Scholar]
- 148.Koffie RM, Farrar CT, Saidi LJ, et al. Nanoparticles enhance brain delivery of blood-brain barrier-impermeable probes for in vivo optical and magnetic resonance imaging. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(46):18837–18842. doi: 10.1073/pnas.1111405108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Jordão JF, Ayala-Grosso CA, Markham K, et al. Antibodies targeted to the brain with image-guided focused ultrasound reduces amyloid-β plaque load in the TgCRND8 mouse model of Alzheimer’s disease. PLoS ONE. 2010;5(5) doi: 10.1371/journal.pone.0010549.e10549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Games D, Adams D, Alessandrini R, et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature. 1995;373(6514):523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 151.Moran PM, Higgins LS, Cordell B, Moser PC. Age-related learning deficits in transgenic mice expressing the 751-amino acid isoform of human β-amyloid precursor protein. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(12):5341–5345. doi: 10.1073/pnas.92.12.5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lewis HD, Beher D, Smith D, et al. Novel aspects of accumulation dynamics and Aβ composition in transgenic models of AD. Neurobiology of Aging. 2004;25(9):1175–1185. doi: 10.1016/j.neurobiolaging.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 153.Qu BX, Lambracht-Washington D, Fu M, Eagar TN, Stüve O, Rosenberg RN. Analysis of three plasmid systems for use in DNA Aβ42 immunization as therapy for Alzheimer’s disease. Vaccine. 2010;28(32):5280–5287. doi: 10.1016/j.vaccine.2010.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Gilman S, Koller M, Black RS, et al. Clinical effects of Aβ immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64(9):1553–1562. doi: 10.1212/01.WNL.0000159740.16984.3C. [DOI] [PubMed] [Google Scholar]
- 155.Cao C, Lin X, Wahi MM, Jackson EA, Potter H. Successful adjuvant-free vaccination of BALB/c mice with mutated amyloid β peptides. BMC Neuroscience. 2008;9, article 25 doi: 10.1186/1471-2202-9-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Okura Y, Miyakoshi A, Kohyama K, Park IK, Staufenbiel M, Matsumoto Y. Nonviral Aβ DNA vaccine therapy against Alzheimer’s disease: long-term effects and safety. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(25):9619–9624. doi: 10.1073/pnas.0600966103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Zamora E, Handisurya A, Shafti-Keramat S, et al. Papillomavirus-like particles are an effective platform for amyloid-β immunization in rabbits and transgenic mice. Journal of Immunology. 2006;177(4):2662–2670. doi: 10.4049/jimmunol.177.4.2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Qu BX, Xiang Q, Li L, et al. Abeta42 gene vaccine prevents Abeta42 deposition in brain of double transgenic mice. Journal of the Neurological Sciences. 2007;260(1-2):204–213. doi: 10.1016/j.jns.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Wang YJ, Gao CY, Yang M, et al. Intramuscular delivery of a single chain antibody gene prevents brain Aβ deposition and cognitive impairment in a mouse model of Alzheimer’s disease. Brain, Behavior, and Immunity. 2010;24(8):1281–1293. doi: 10.1016/j.bbi.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 160.Xing X, Sha S, Li Y, et al. Immunization with a new DNA vaccine for Alzheimer's disease elicited Th2 immune response in BALB/c mice by in vivo electroporation. Journal of the Neurological Sciences. 2012;313(1-2):17–21. doi: 10.1016/j.jns.2011.09.040. [DOI] [PubMed] [Google Scholar]
- 161.Kim HD, Tahara K, Maxwell JA, et al. Nasal inoculation of an adenovirus vector encoding 11 tandem repeats of Aβ1-6 upregulates IL-10 expression and reduces amyloid load in a Mo/ Hu APPswe PS1dE9 mouse model of Alzheimer’s disease. Journal of Gene Medicine. 2007;9(2):88–98. doi: 10.1002/jgm.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Wilcock DM, Gharkholonarehe N, van Nostrand WE, Davis J, Vitek MP, Colton CA. Amyloid reduction by amyloid-β vaccination also reduces mouse tau pathology and protects from neuron loss in two mouse models of Alzheimer’s disease. The Journal of Neuroscience. 2009;29(25):7957–7965. doi: 10.1523/JNEUROSCI.1339-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Minami SS, Sidahmed E, Aid S, et al. Therapeutic versus neuroinflammatory effects of passive immunization is dependent on Aβ/amyloid burden in a transgenic mouse model of Alzheimer’s disease. Journal of Neuroinflammation. 2010;7, article 57 doi: 10.1186/1742-2094-7-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Salloway S, Sperling R, Gilman S, et al. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology. 2009;73(24):2061–2070. doi: 10.1212/WNL.0b013e3181c67808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Rinne JO, Brooks DJ, Rossor MN, et al. 11C-PiB PET assessment of change in fibrillar amyloid-β load in patients with Alzheimer’s disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. The Lancet Neurology. 2010;9(4):363–372. doi: 10.1016/S1474-4422(10)70043-0. [DOI] [PubMed] [Google Scholar]
- 166.Black RS, Sperling RA, Safirstein B, et al. A single ascending dose study of bapineuzumab in patients with alzheimer disease. Alzheimer Disease and Associated Disorders. 2010;24(2):198–203. doi: 10.1097/WAD.0b013e3181c53b00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Roher AE, Maarouf CL, Daugs ID, et al. Neuropathology and amyloid-β spectrum in a bapineuzumab immunotherapy recipient. Journal of Alzheimer’s Disease. 2011;24(2):315–325. doi: 10.3233/JAD-2011-101809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Panza F, Frisardi V, Solfrizzi V, et al. Immunotherapy for Alzheimer's disease: from anti-β-amyloid to tau-based immunization strategies. Immunotherapy. 2012;4(2):213–238. doi: 10.2217/imt.11.170. [DOI] [PubMed] [Google Scholar]
- 169.Racke MM, Boone LI, Hepburn DL, et al. Exacerbation of cerebral amyloid angiopathy-associated microhemorrhage in amyloid precursor protein transgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyloid β . The Journal of Neuroscience. 2005;25(3):629–636. doi: 10.1523/JNEUROSCI.4337-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Wilcock DM, Colton CA. Immunotherapy, vascular pathology, and microhemorrhages in transgenic mice. CNS and Neurological Disorders. 2009;8(1):50–64. doi: 10.2174/187152709787601858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Luo F, Rustay NR, Seifert T, et al. Magnetic resonance imaging detection and time course of cerebral microhemorrhages during passive immunotherapy in living amyloid precursor protein transgenic mice. Journal of Pharmacology and Experimental Therapeutics. 2010;335(3):580–588. doi: 10.1124/jpet.110.172932. [DOI] [PubMed] [Google Scholar]
- 172.Garcia-Alloza M, Ferrara BJ, Dodwell SA, Hickey GA, Hyman BT, Bacskai BJ. A limited role for microglia in antibody mediated plaque clearance in APP mice. Neurobiology of Disease. 2007;28(3):286–292. doi: 10.1016/j.nbd.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Breitner JC, Baker LD, Montine TJ, et al. Extended results of the Alzheimer's disease anti-inflammatory prevention trial. Alzheimer’s & Dementia. 2011;7(4):402–411. doi: 10.1016/j.jalz.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Petrushina I, Ghochikyan A, Mktrichyan M, et al. Alzheimer’s disease peptide epitope vaccine reduces insoluble but not soluble/oligomeric Aβ species in amyloid precursor protein transgenic mice. The Journal of Neuroscience. 2007;27(46):12721–12731. doi: 10.1523/JNEUROSCI.3201-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Cribbs DH. Abeta DNA vaccination for Alzheimer’s disease: focus on disease prevention. CNS and Neurological Disorders. 2010;9(2):207–216. doi: 10.2174/187152710791012080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Zago W, Buttini M, Comery TA, et al. Neutralization of soluble, synaptotoxic amyloid β species by antibodies is epitope specific. The Journal of Neuroscience. 2012;32(8):2696–2702. doi: 10.1523/JNEUROSCI.1676-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Lemere CA, Spooner ET, LaFrancois J, et al. Evidence for peripheral clearance of cerebral Aβ protein following chronic, active Aβ immunization in PSAPP mice. Neurobiology of Disease. 2003;14(1):10–18. doi: 10.1016/s0969-9961(03)00044-5. [DOI] [PubMed] [Google Scholar]
- 178.Bard F, Barbour R, Cannon C, et al. Epitope and isotype specificities of antibodies to β-amyloid peptide for protection against Alzheimer’s disease-like neuropathology. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(4):2023–2028. doi: 10.1073/pnas.0436286100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Fisher Y, Nemirovsky A, Baron R, Monsonego A. T cells specifically targeted to amyloid plaques enhance plaque clearance in a mouse model of Alzheimer’s disease. PloS ONE. 2010;5(5):p. e10830. doi: 10.1371/journal.pone.0010830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Delrieu J, Ousset PJ, Caillaud C, et al. Clinical trials in Alzheimer's disease: immunotherapy approaches. Journal of Neurochemistry. 2012;120(Supplement 1):186–193. doi: 10.1111/j.1471-4159.2011.07458.x. [DOI] [PubMed] [Google Scholar]
- 181.Shapiro JS, Stiteler M, Wu G, et al. Cisterna magna cannulated repeated CSF sampling rat model—effects of a gamma-secretase inhibitor on Aβ levels. The Journal of Neuroscience Methods. 2012;205(1):36–44. doi: 10.1016/j.jneumeth.2011.12.010. [DOI] [PubMed] [Google Scholar]
- 182.Fleisher AS, Raman R, Siemers ER, et al. Phase 2 safety trial targeting amyloid β production with a γ-secretase inhibitor in Alzheimer disease. Archives of Neurology. 2008;65(8):1031–1038. doi: 10.1001/archneur.65.8.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Bateman RJ, Siemers ER, Mawuenyega KG, et al. A γ-secretase inhibitor decreases amyloid-β production in the central nervous system. Annals of Neurology. 2009;66(1):48–54. doi: 10.1002/ana.21623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Lannfelt L, Blennow K, Zetterberg H, et al. Safety, efficacy, and biomarker findings of PBT2 in targeting Aβ as a modifying therapy for Alzheimer’s disease: a phase IIa, double-blind, randomised, placebo-controlled trial. The Lancet Neurology. 2008;7(9):779–786. doi: 10.1016/S1474-4422(08)70167-4. [DOI] [PubMed] [Google Scholar]
- 185.Crouch PJ, Savva MS, Hung LW, et al. The Alzheimer's therapeutic PBT2 promotes amyloid-β degradation and GSK3 phosphorylation via a metal chaperone activity. Journal of Neurochemistry. 2011;119(1):220–230. doi: 10.1111/j.1471-4159.2011.07402.x. [DOI] [PubMed] [Google Scholar]
- 186.Serrano-Pozo A, William CM, Ferrer I, et al. Beneficial effect of human anti-amyloid-β active immunization on neurite morphology and tau pathology. Brain. 2010;133(part 5):1312–1327. doi: 10.1093/brain/awq056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Boche D, Denham N, Holmes C, Nicoll JAR. Neuropathology after active Aβ42 immunotherapy: implications for Alzheimer’s disease pathogenesis. Acta Neuropathologica. 2010;120(3):369–384. doi: 10.1007/s00401-010-0719-5. [DOI] [PubMed] [Google Scholar]
- 188.Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau prevents cognitive decline in a new tangle mouse model. The Journal of Neuroscience. 2010;30(49):16559–16566. doi: 10.1523/JNEUROSCI.4363-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Boimel M, Grigoriadis N, Lourbopoulos A, Haber E, Abramsky O, Rosenmann H. Efficacy and safety of immunization with phosphorylated tau against neurofibrillary tangles in mice. Experimental Neurology. 2010;224(2):472–485. doi: 10.1016/j.expneurol.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 190.Gu J, Sigurdsson EM. Immunotherapy for tauopathies. Journal of Molecular Neuroscience. 2011;45(3):690–695. doi: 10.1007/s12031-011-9576-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Chai X, Wu S, Murray TK, et al. Passive immunization with anti-tau antibodies in two transgenic models: reduction of tau pathology and delay of disease progression. The Journal of Biological Chemistry. 2011;286(39):34457–34467. doi: 10.1074/jbc.M111.229633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Himeno E, Ohyagi Y, Ma L, et al. Apomorphine treatment in Alzheimer mice promoting amyloid-β degradation. Annals of Neurology. 2011;69(2):248–256. doi: 10.1002/ana.22319. [DOI] [PubMed] [Google Scholar]
- 193.Brodbeck J, Balestra ME, Saunders AM, Roses AD, Mahley RW, Huang Y. Rosiglitazone increases dendritic spine density and rescues spine loss caused by apolipoprotein E4 in primary cortical neurons. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(4):1343–1346. doi: 10.1073/pnas.0709906104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Cocks G, Wilde JI, Graham SJ, et al. The thiazolidinedione pioglitazone increases cholesterol biosynthetic gene expression in primary cortical neurons by a PPARγ-independent mechanism. Journal of Alzheimer’s Disease. 2010;19(2):631–646. doi: 10.3233/JAD-2010-1266. [DOI] [PubMed] [Google Scholar]