

Abstract
Methyl-2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oate (CDDO-Me) is a multifunctional oleanane synthetic triterpenoid with potent anti-inflammatory and antitumorigenic properties. The mechanisms of the antisurvival and apoptosis-inducing activities of CDDO-Me and related derivatives of oleanolic acid have been defined; however, to date, no study has been carried out on the effect of CDDOs on human telomerase reverse transcriptase (hTERT) gene or telomerase activity. Here we report for the first time that inhibition of cell proliferation and induction of apoptosis by CDDO-Me in pancreatic cancer cell lines is associated with the inhibition of hTERT gene expression, hTERT telomerase activity and a number of proteins that regulate hTERT expression and activity. Furthermore, abrogation or overexpression of hTERT protein altered the susceptibility of tumor cells to CDDO-Me. These findings suggest that telomerase (hTERT) is a relevant target of CDDO-Me in pancreatic cancer cells.
Keywords: CDDO-Me, Apoptosis, hTERT, Telomerase, Pancreatic cancer
1. Introduction
Telomeres, the nucleoprotein structures present at the end of chromosomes, play a critical role in maintaining chromosome stability by preventing loss of telomeric repeats, end-to-end fusion and chromosomal rearrangement [1]. Telomeres shorten progressively during normal cell division due to a gradual loss of telomeric DNA sequence (TTAGGG) in each replication cycle [2,3]. When telomere length becomes critically short it triggers replicative senescence or apoptosis. Maintaining the length of telomere is the function of telomerase, a ribonucleoprotein polymerase that adds the hexameric DNA repeats (TTAGGG) to the 3′ flanking end of DNA strands in telomere. The human telomerase complex consists of telomerase reverse transcriptase (hTERT), telomerase RNA (hTR or TERC) and telomerase associated protein-1 (TEP-1), hsp90 and p23 [4-6]. In humans, telomerase activity is detectable only in germ line cells and certain stem cells but is repressed in somatic cells [7,8]. Deregulated telomerase activity is associated with promotion of tumorigenesis and neoplastic growth of human cancers [9-11]. In fact, approximately 90% of human cancers exhibit reactivation of telomerase activity [12,13]. Thus, cancer-specific activation of telomerase provides an attractive avenue for developing novel anticancer therapeutics that would specifically target telomere and telomerase.
Synthetic oleanane triterpenoid CDDO-Me (methyl-2-cyano-3, 12-dioxooleana-1,9(11)-dien-28-oate) is a multifunctional compound with potent anti-inflammatory and antitumorigenic properties [14]. CDDO-Me and other synthetic analogs of oleanolic acid inhibit proliferation and induce apoptosis in diverse types of tumor cell lines [15-17]. Although the anticancer mechanisms of CDDOs are not fully understood, modulation of MAPK (Erk1/2), NF-κB, TGF-β/Smad and Nrf2 signaling has been reported [18-20]. CDDOs also inhibit the growth of tumor implants and prevent development of cancer in mouse models [21-23]. We have shown that CDDOs inhibits the progression of preneoplastic lesions to adenocarcinoma in the TRAMP mouse model of prostate cancer, which was associated with the inhibition of prosurvival Akt/mTOR/NF-κB signaling [24,25]. To date, there has been no report on the effect of CDDO-Me or other CDDOs on telomerase activity in cancer cells. The purpose of this study was to investigate the effect of CDDO-Me on the expression of hTERT, its telomerase activity and proteins that regulate hTERT expression in pancreatic cancer cells. Here we show for the first time that inhibition of cell proliferation and induction of apoptosis in pancreatic cancer cells by CDDO-Me is associated with the inhibition of expression and telomerase activity of hTERT through the suppression of proteins involved in the transcriptional and post-translational regulation of hTERT.
2. Materials and methods
2.1. Reagents
CDDO-Me was obtained from the National Cancer Institute, Bethesda, MD through the Rapid Access to Intervention Development Program. Antibodies against Akt, p-Akt (ser473), NF-κB (p65), STAT3, p-STAT3 (ser736), Sp1, c-Myc and β-actin were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Anti-hTERT and p-TERT antibodies were obtained from-Abcam Inc. (Cambridge, MA). CellTiter 96® Aqueous One Solution Proliferation Assay System was from Promega (Madison, WI, USA). Annexin V-FITC apoptosis detection kit II was obtained from BD Pharmingen (San Diego, CA, USA) and TRAPeze telomerase detection kit was purchased from Millipore (Millipore, Temecula, CA).
2.2. Cell culture
Human pancreatic cancer cell lines MiaPaCa-2 and Panc-1 were obtained from the American Type Culture Collection (ATCC, Rockville, MD). Both cell lines were cultured in RPMI-1640 supplemented with 10% FBS, 1% penicillin/streptomycin, and 25 mM HEPES buffer.
2.3. MTS assay
1 × 104 cells in 100 μl of cell culture medium were seeded into each well of a 96-well plate. After incubation for 24 h, cells were treated with CDDO-Me for 48–72 h. Cell viability was then determined by the colorimetric MTS assay using CellTiter 96 Aqueous One Solution Proliferation Assay System from Promega (Madison, WI) following the instructions provided by the manufacturer.
2.4. Analysis of apoptosis
Induction of apoptosis by CDDO-Me was assessed by the binding of annexin V to phosphatidylserine, which is externalized to the outer leaflet of the plasma membrane early during induction of apoptosis and from the cleavage of PARP-1 by Western blotting. For annexin V-FITC binding, MiaPaCa-2 and Panc-1 cells were treated with CDDO-Me for 48 h, harvested and resuspended in the binding buffer provided in the annexin V-FITC apoptosis detection kit. Cells were reacted with 5 μl of annexin V-FITC reagent and 5 μl of propidium iodide (PI) for 30 min at room temperature in the dark. Stained cells were analyzed by flow cytometry. For cleavage of PAR-1, cellular lysates were analyzed by Western blotting using anti-PARP-1 antibody as described below.
2.5. Western Blotting
Cell lysates were prepared using NP 40 cell lysis buffer. Lysates were clarified by centrifugation at 14,000 × g for 10 min at 4 °C and protein concentrations were determined. Samples (50 μg) were boiled in an equal volume of sample buffer (20% glycerol, 4% SDS, 0.2% Bromophenol Blue, 125 mM Tris–HCl (pH 7.5), and 640 mM 2-mercaptoethanol) and separated on pre-casted Tris–glycine polyacrylamide gels using the XCell Surelock™ Mini-Cell, in Tris–Glycine SDS running buffer, all from Novex (Invitrogen, Carlsbad, CA). Proteins resolved on the gels were transferred to PVDF membranes. Membranes were blocked with 5% milk in 10 mM Tris–HCl (pH 8.0), 150 mM NaCl with 0.05% Tween 20 (TPBS) and probed using specific antibodies against proteins of interest or β-actin (loading control) and HRP-conjugated secondary antibody. Immune complexes were visualized with enhanced chemiluminescence. Protein bands were imaged and band densities analyzed using the NIH/Scion image analysis software. The protein band densities were normalized to the corresponding β-actin band densities and percent change in signal strength was calculated.
2.6. Measurement of hTERT expression
The effect of CDDO-Me on hTERT expression was measured by analyzing hTERT mRNA and hTERT protein. For hTERT mRNA, total cellular RNA was extracted with TRI-zol reagent (GIBCO) according to the manufacturer’s recommendation. 1 μg of RNA was then reverse transcribed by oligo-dt primer and high fidelity reverse transcriptase (Boehringer Mannheim, Germany) to generate cDNAs. 1 μl of cDNA was used as the template for polymerase chain reaction (PCR) using hTERT primers: upper, 5′-TGTTTCTGGATTTGCAGGTG-3′, and lower, 5′-GTTCTTGGCTTTCAGGATGG-3′; and GAPDH primers: upper, 5′-TCC CTC AAG, ATT, GTC AGC AA-3′, and lower, 5′-AGA TCC ACA ACG GAT ACA TT-3′. The PCR conditions used were 33 cycles of denaturation (95 °C for 1 min), annealing (62 °C for 30 s), and polymerization (72 °C for 1 min). The PCR products were separated on 2% agarose gel electrophoresis and visualized by ethidium bromide staining. Gels were photographed and band densities were analyzed using the NIH/Scion image analysis software. The hTERT primers amplified a DNA fragment of 200 bp and the DNA fragment size amplified by GAPDH primers was 173 bp.
2.7. Telomerase activity assay
The telomerase activity in cell extracts was assessed by the PCR-based telomeric repeat amplification protocol (TRAP) using TRAPeze gel-based telomerase detection kit (Millipore, Temecula, CA) following the instructions provided in the kit. Briefly, cells were extracted in CHAP lysis buffer on ice for 30 min. 2 μl (100 ng) of cell extract was added to the TRAP reaction mixture containing dNTPs, TS primer, TRAP primers and Taq polymerase and incubated at 30 °C for 30 min in a thermocycler followed by 3-step PCR at 94 °C/30 s, 59 °C/30 s, and 72 °C/1 min for 33 cycles. The PCR products were fractionated on nondenaturing 12.5% polyacrilamide gel and visualized by staining with ethidium bromide. The ladder of products with 6 base pair increment indicating telomerase activity was analyzed with NIH/Scion image analysis software. The cumulative band density for each lane was normalized to the corresponding band density of internal control (36 bp) and expressed as percent of the control.
2.8. Transfections
For silencing of hTERT, cells were transfected with double stranded siRNA-hTERT or non-targeting siRNA sequence using SignalSilence siRNA kit (Cell Signaling Technology, Beverly, MA). Briefly, 2 × 106 cancer cells were plated in 60 mm Petri dish for 24 h and treated with 3 ml of transfection medium containing 20 μg LipofectAMINE and 100 nM siRNA for 48 h. hTERT expression was analyzed by immunoblotting.
For overexpression of hTERT, semi-confluent cell cultures were transfected with 10 μg of empty or hTERT expression plasmid (pCI-neo-hTERT) using LipofectAMINE Plus reagent. After transfection for 48 h, cells were analyzed for the expression of hTERT by immunoblotting.
2.9. Statistical analysis
Most data are presented as means ± S.D. and outcomes for treated and untreated cells were compared by Student’s t-test. Differences were considered significant at P < 0.05.
3. Results and Discussion
3.1. CDDO-Me inhibits proliferation and induces apoptosis in pancreatic cancer cells
The effect of CDDO-Me on the proliferation of MiaPaCa-2 and Panc-1 human pancreatic cancer cell lines was measured by MTS assay. As shown in Fig. 1A, treatment with CDDO-Me for 72 h at a concentration of 0.63 μM had minimal growth inhibition (7%–16%). However, at concentrations of 1.25–5 μM the growth inhibitory effect of CDDO was significantly higher (MiaPaCa-2, 65%–84% inhibition, P < 0.01; Panc-1, 52%–80% inhibition, P < 0.01). Whether CDDO-Me also induces apoptosis in these cells was determined by the binding of annexin V-FITC to cells treated with CDDO-Me. For this, tumor cells were treated with CDDO-Me at concentrations of 0 to 5 μM for 48 h and then treated with annexin V-FITC and propidium iodide and analyzed by flow cytometry. As shown in Fig. 1B, a small percentage of untreated MiaPaCa-2 and Panc-1 cells bound annexin V-FITC (11% and 14%). However, the percentage of annexin V-FITC binding MiaPaCa-2 cells treated with CDDO-Me increased in concentration-dependent manner from 28% at 0.63 to 61% at 5 μM CDDO-Me (P < 0.01 at 1.25–5 μM compared to untreated cells). An identical increase in the percentage of annexin V-binding Panc-1 cells treated with CDDO-Me was observed (e.g., 26%, 38%, 45%, and 59% at 0.63, 1.25, 2.5 and 5 μM CDDO-Me, respectively). The induction of apoptosis by CDDO-Me was confirmed by the cleavage of native PARP-1 (110 kDa) and the appearance of the cleaved PARP-1 fragment (89 kDa) in both cell lines treated with CDDO-Me (Fig. 1C). Collectively, these data demonstrated that CDDO-Me at concentrations of 1.25–5 μM significantly inhibits proliferation and induces apoptosis in pancreatic cancer cells. We also observed that treatment with CDDO-Me at concentrations even lower than 0.63 μM when carried over a longer period of time (5 to 6 days) resulted in significant growth inhibition and apoptosis (data not shown). These findings corroborate the results of our previous studies and those of others that CDDO-Me is a potent inhibitor of cell proliferation and inducer of apoptosis in tumor cells [15-17].
Fig. 1.
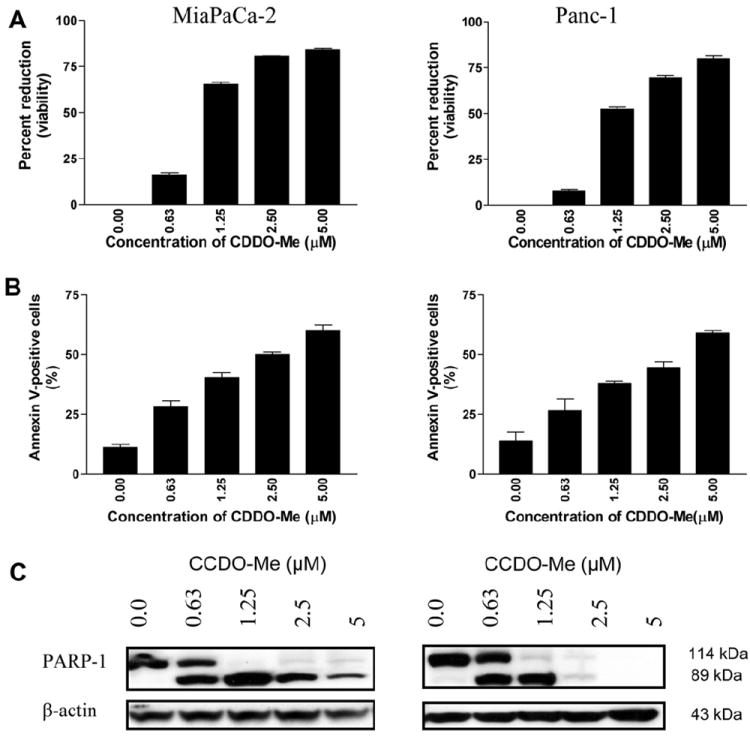
CDDO-Me inhibits proliferation and induces apoptosis in pancreatic cancer cells. (A) MiaPaCa-2 and Panc-1 pancreatic cancer cells (1 × 104/well) were treated with CDDO-Me at concentrations ranging from 0 to 5 μM for 72 h in a 96-well microtiter plate in triplicates. Cell viability was measured by MTS assay using CellTiter Aqueous Assay System. (B) Annexin V-FITC binding. Tumor cells were treated with CDDO-Me at 0–5 μM for 48 h and then reacted with 5 μl of annexin V-FITC and 5 μl PI for 30 min and the percentage of annexin V-FITC binding cells was determined by flow cytometry. (C) Cleavage of PARP-1 in cells treated with CDDO-Me was analyzed by immunoblotting. Similar results were obtained in 2 to 3 independent experiments. *P < 0.01 compared to control cells (no CDDO-Me).
3.2. Effect of CDDO-Me on the expression and telomerase activity of hTERT
Activated telomerase promotes tumor growth by modulating the expression of growth controlling genes and enhancing cell proliferation [26]. On the other hand, inhibition of telomerase activity with oligonucleotides against telomerase RNA or by dominant-negative mutants of hTERT in tumor cells induces telomere shortening and apoptosis [27,28]. Thus it is likely that the antiproliferative and apoptosis inducing effects of CDDO-Me may be mediated via the inhibition of telomerase. Since the expression of hTERT is a rate-limiting step for telomerase activity we first analyzed the effect of CDDO-Me on hTERT expression by measuring hTERT mRNA by RT-PCR and hTERT protein by Western blotting. Analysis of hTERT mRNA by RT-PCR showed inhibition of hTERT gene expression in response to CDDO-Me treatment for 48 h even at the lowest concentration (0.63 μM) without significantly affecting the expression of GAPDH (Fig. 2A). At higher concentrations of 1.25 and 2.5 μM CDDO-Me hTERT gene expression was almost completely inhibited. CDDO-Me also inhibited hTERT protein expression. As shown in Fig. 2B, CDDO-Me sharply reduced hTERT protein at 0.63 μM and completely inhibited it at concentrations of 1.25 μM and above in both cell lines. Since phosphorylation of catalytic subunit of hTERT is necessary for its telomerase activity we measured the effect of CDDO-Me on p-hTERT (ser826). As can be seen Fig. 2B, CDDO-Me also inhibited the levels of phosphorylated-hTERT at concentrations of 1.25 μM and above.
Fig. 2.
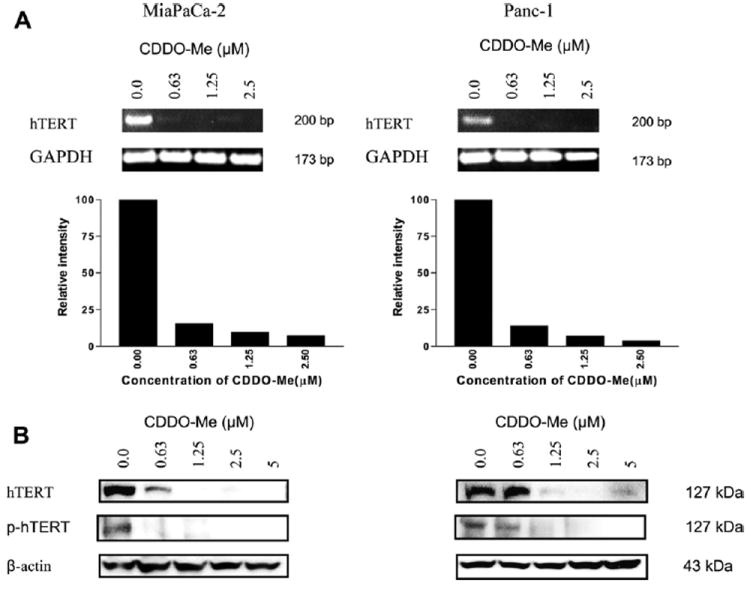
CDDO-Me inhibits hTERT gene and hTERT protein expression in pancreatic cancer cells. (A) Effect of CDDO-Me on hTERT gene expression. MiaPaCa-2 and Panc-1 cells were treated with CDDO-Me (0–5 μM) for 48 h and total cellular RNA was prepared using TRI-zole reagent. 1 μg of cellular RNA was reverse transcribed using oligo-dt primer and high fidelity reverse transcriptase. 1 μl of cDNA was amplified using hTERT or GAPDH primers. Amplified products were separated on 2% DNA agarose gel. Gels were stained with ethidium bromide and amplified DNA fragments were identified by base pair sizes. (B) Effect on hTERT protein. MiaPaCa-2 and Panc-1 cells were treated with CDDO-Me (0–5 μM) for 48 h and cell lysates were analyzed for hTERT and p-hTERT protein by Western blotting. Each experiment was repeated at least two times.
Whether CDDO-Me affects the telomerase activity of hTERT was determined next. MiaPaCa-2 and Panc-1 cells were treated with CDDO-Me (0–10 μM) for 48 h and cells were extracted in CHAP lysis buffer. The telomerase activity of extracts was measured using the PCR-based TRAP assay method. While there was ~30% reduction in the telomerase activity in cells treated with 0.63 μM CDDO-Me, telomerase activity was sharply reduced in cells treated with CDDO-Me at 1.25–10 μM CDDO-Me as identified by the loss of DNA laddering in both cell lines (Fig. 3). In fact, telomerase activity was nearly 80% inhibited at 1.25 μM CDDO-Me and almost completely inhibited at 5 and 10 μM CDDO-Me. Taken together, our data showing attenuation of hTERT gene expression and inhibition of hTERT protein and hTERT telomerase activity by CDDO-Me indicated that inhibition hTERT expression and its telomerase activity are part of the mechanism by which CDDO-Me inhibits proliferation and induce apoptosis in cancer cells. Indeed, our results are in agreement with other reports showing that inhibition of hTERT telomerase activity is necessary for the antiproliferative and apoptosis-inducing activity of compounds such as genistein, sulforaphane, amooranin and curcumin [29-32]. However, more work is required to determine whether inhibition of hTERT by CDDO-Me also leads to shortening of telomeres and if CDDO-Me directly binds and degrade the RNA template of telomerase.
Fig. 3.
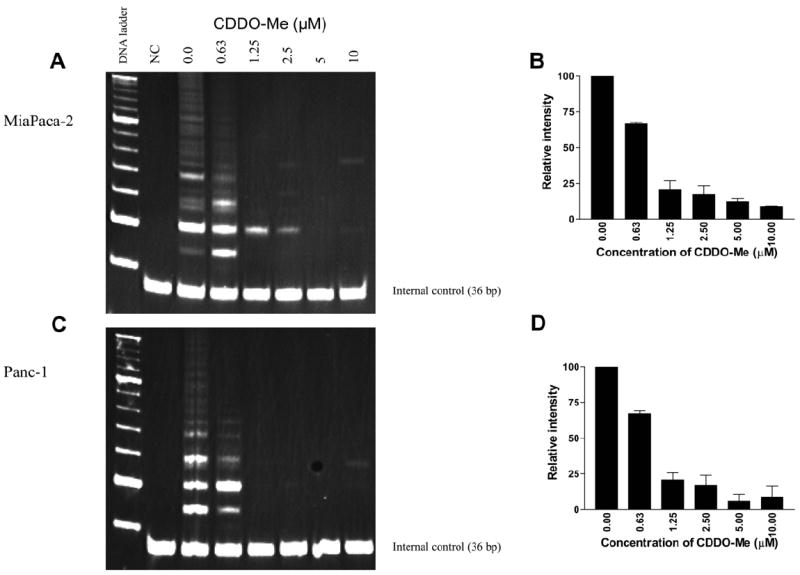
CDDO-Me inhibits telomerase activity. (A) Measurement of telomerase activity. MiaPaCa-2 and Panc-1 cells were treated with CDDO-Me (0–10 μM) for 48 h and telomerase activity of cell extracts was measured by TRAP assay as described in Section 2. (A and C) DNA laddering patterns under different treatment conditions. (B and D) Quantification of telomerase activity in (A) and (C). Experiments were repeated two times. NC, negative control (no cell extract).
3.3. CDDO-Me inhibits hTERT regulatory proteins
A number of transcription factors and molecules that regulate hTERT expression and telomerase activity have been identified. The hTERT core promoter contains several transcription factor binding sites including those of c-Myc, Sp1, NF-κB and STAT-3 [33-35]. Post-translationally, phosphorylation of hTERT on Ser826 by protein kinase B/Akt is required for activation of its telomerase activity [35,36]. Therefore, we assessed the effect of CDDO-Me on the levels of these proteins. Treatment with CDDO-Me (0–5 μM) for 48 h significantly reduced the levels of these proteins in a does-dependent manner (Fig. 4A), suggesting that inhibition of these transcription factors likely contributes to the inhibition of hTERT transcription by CDDO-Me. In addition, the inhibition of NF-κB also suggests that CDDO-Me might also impair nuclear accumulation of hTERT since hTERT associates with NF-κB for nuclear translocation. It is not surprising to find that CDDO-Me affects post-translational modification of hTERT through the inhibition of p-Akt, since we have recently shown that CDDO-Me specifically inhibits Akt kinase activity without affecting the activity of PDK1, the upstream kinase that phosphorylates and activates Akt [37]. Thus inhibition of p-Akt would be expected to negatively affect the activation of hTERT telomerase activity. Further, hTERT is also regulated epigenetically through histone acetylation and DNA methylation at the promoter sites [38]. Whether CDDO-Me impacts these epigenetic events remains to be determined.
Fig. 4.
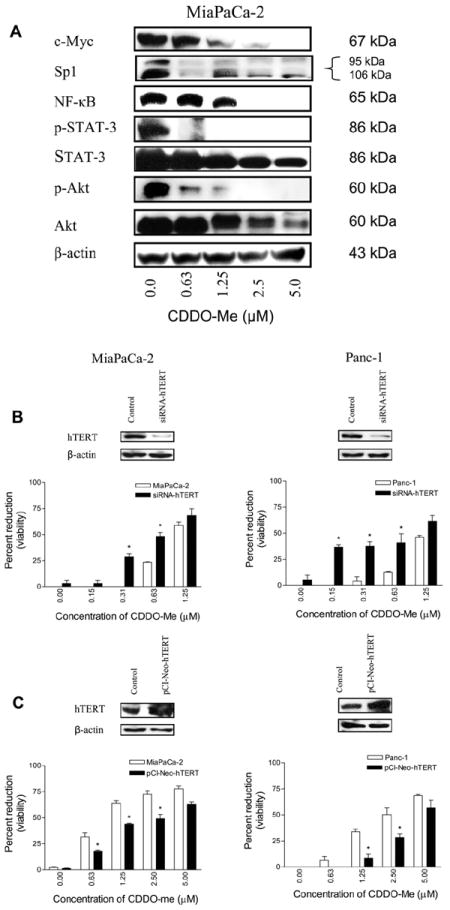
CDDO-Me inhibits hTERT regulatory proteins. (A) MiaPaCa-2 cells were treated with CDDO-Me (0–5 μM) for 48 h and cell lysates were analyzed for c-Myc, Sp1, NF-κB, STAT-3, p-STAT-3, Akt and p-Akt by western blotting (A). hTERT is a target of CDDO-Me. Tumor cells were transfected with siRNA-hTERT (A) or hTERT expression plasmid (pCI-neo-hTERT) (B) for 48 h. hTERT levels were measured by immunoblotting (insets) and response to CDDO-Me was assessed in 48 h MTS assay. Each experiment was repeated two times.
3.4. hTERT is a target of CDDO-Me
To demonstrate the involvement of hTERT in CDDO-Me-induced inhibition of cell survival and proliferation, we investigated the effect of knocking-down or overexpressing hTERT on response to CDDO-Me. MiaPaCa-2 and Panc-1 cells were transfected with hTERT siRNA or hTERT expression plasmid for 48 h and response to CDDO-Me was measured in MTS assay after confirming changes in the levels of hTERT by immunoblotting. As shown in Fig. 4B, transfection with siRNA-hTERT significantly increased the susceptibility of both cell lines to concentrations of CDDO-Me which are otherwise inactive or only slightly active (i.e., 0.15–0.625 μM, P < 05). On the other hand, overexpression of hTERT significantly reduced the susceptibility to CDDO-Me at concentrations (0.625–2.5 μM, P < 0.05) that inhibit the proliferation of these cells (Fig. 4C). Transfection with a non-targeting siRNA or empty plasmid had no effect on response of cells to CDDO-Me (not shown). These data demonstrated that hTERT is a molecular target of CDDO-Me in pancreatic cancer cells.
In conclusion, we have shown for the first time that inhibition of proliferation and induction of apoptosis in pancreatic cancer cells by CDDO-Me involves the inhibition of telomerase activity. CDDO-Me inhibited hTERT gene expression, hTERT protein and hTERT telomerase activity. Further, CDDO-Me also inhibited hTERT regulatory proteins such as c-Myc, Sp1, NF-κB, p-STAT-3 and p-Akt. Thus, targeting telomere and/or telomerase with CDDO-Me is a promising strategy for the treatment of pancreatic cancer.
Acknowledgments
This work was supported by NIH grant 1R01 CA130948-01.
Abbreviations
- CDDO-Me
methyl-2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oate
- MTS
(3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium)
- PARP-1
Poly [ADP-ribose] polymerase 1
- Akt (protein kinase B)
a serine/threonine protein kinase
- hTERT
human telomerase reverse transcriptase
- NF-κB
nuclear factor kappa B
- STAT-3
signal transducer and activator of transcription 3
References
- 1.Greider CW. Chromosome first aid. Cell. 1991;67:645–647. doi: 10.1016/0092-8674(91)90058-7. [DOI] [PubMed] [Google Scholar]
- 2.Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, Shay JW. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- 3.Shay JW, Bacchetti S. A survey of telomerase activity in human cancer. Eur J Cancer. 1997;33:787–791. doi: 10.1016/S0959-8049(97)00062-2. [DOI] [PubMed] [Google Scholar]
- 4.Kilian A, Bowtell DD, Abud HE, Hime GR, Venter DJ, Keese PK, Duncan EL, Reddel RR, Jefferson RA. Isolation of a candidate human telomerase catalytic subunit gene, which reveals complex splicing patterns in different cell types. Hum Mol Genet. 1997;6:2011–2019. doi: 10.1093/hmg/6.12.2011. [DOI] [PubMed] [Google Scholar]
- 5.Feng J, Funk WD, Wang SS, Weinrich SL, Avilion AA, Chiu CP, Adams RR, Chang E, Allsopp RC, Yu J, et al. The RNA component of human telomerase. Science. 1995;269:1236–1241. doi: 10.1126/science.7544491. [DOI] [PubMed] [Google Scholar]
- 6.De Boeck G, Forsyth RG, Praet M, Hogendoorn PC. Telomere-associated proteins: cross-talk between telomere maintenance and telomere-lengthening mechanisms. J Pathol. 2009;217:327–344. doi: 10.1002/path.2500. [DOI] [PubMed] [Google Scholar]
- 7.Wright WE, Piatyszek MA, Rainey WE, Byrd W, Shay JW. Telomerase activity in human germline and embryonic tissues and cells. Dev Genet. 1996;18:173–179. doi: 10.1002/(SICI)1520-6408(1996)18:2<173::AID-DVG10>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 8.Holt SE, Wright WE, Shay JW. Regulation of telomerase activity in immortal cell lines. Mol Cell Biol. 1996;16:2932–2939. doi: 10.1128/mcb.16.6.2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blasco MA, Hahn WC. Evolving views of telomerase and cancer. Trends Cell Biol. 2003;13:289–294. doi: 10.1016/s0962-8924(03)00085-0. [DOI] [PubMed] [Google Scholar]
- 10.Newbold RF. The significance of telomerase activation and cellular immortalization in human cancer. Mutagenesis. 2002;17:539–550. doi: 10.1093/mutage/17.6.539. [DOI] [PubMed] [Google Scholar]
- 11.Cech TR. Beginning to understand the end of the chromosome. Cell. 2004;116:273–279. doi: 10.1016/s0092-8674(04)00038-8. [DOI] [PubMed] [Google Scholar]
- 12.Janknecht R. On the road to immortality: hTERT upregulation in cancer cells. FEBS Lett. 2004;564:9–13. doi: 10.1016/S0014-5793(04)00356-4. [DOI] [PubMed] [Google Scholar]
- 13.Meyerson M, Counter CM, Eaton EN, Ellisen LW, Steiner P, Caddle SD, Ziaugra L, Beijersbergen RL, Davidoff MJ, Liu Q, Bacchetti S, Haber DA, Weinberg RA. HEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell. 1997;90:785–795. doi: 10.1016/s0092-8674(00)80538-3. [DOI] [PubMed] [Google Scholar]
- 14.Liby KT, Yore MM, Sporn MB. Triterpenoids and rexinoids as multifunctional agents for the prevention and treatment of cancer. Nat Rev Cancer. 2007;7:357–369. doi: 10.1038/nrc2129. [DOI] [PubMed] [Google Scholar]
- 15.Ito Y, Pandey P, Sporn MB, Datta R, Kharbanda S, Kufe D. The novel triterpenoid CDDO induces apoptosis and differentiation of human osteosarcoma cells by a caspase-8 dependent mechanism. Mol Pharmacol. 2001;59:1094–1099. doi: 10.1124/mol.59.5.1094. [DOI] [PubMed] [Google Scholar]
- 16.Konopleva M, Tsao T, Estrov Z, Lee RM, Wang RY, Jackson CE, McQueen T, Monaco G, Munsell M, Belmont J, Kantarjian H, Sporn MB, Andreeff M. The synthetic triterpenoid 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid induces caspase-dependent and -independent apoptosis in acute myelogenous leukemia. Cancer Res. 2004;64:7927–7935. doi: 10.1158/0008-5472.CAN-03-2402. [DOI] [PubMed] [Google Scholar]
- 17.Gao X, Deeb D, Jiang H, Liu Y, Dulchavsky SA, Gautam SC. Synthetic triterpenoids inhibit growth and induce apoptosis in human glioblastoma and neuroblastoma cells through inhibition of prosurvival Akt, NF-kappaB and Notch1 signaling. J Neurooncol. 2007;84:147–157. doi: 10.1007/s11060-007-9364-9. [DOI] [PubMed] [Google Scholar]
- 18.Konopleva M, Contractor R, Kurinna SM, Chen W, Andreeff M, Ruvolo PP. The novel triterpenoid CDDO-Me suppresses MAPK pathways and promotes p38 activation in acute myeloid leukemia cells. Leukemia. 2005;19:1350–1354. doi: 10.1038/sj.leu.2403828. [DOI] [PubMed] [Google Scholar]
- 19.Ahmad R, Raina D, Meyer C, Kharbanda S, Kufe D. Triterpenoid CDDO-Me blocks the NF-kappaB pathway by direct inhibition of IKKbeta on Cys-179. J Biol Chem. 2006;281:35764–35769. doi: 10.1074/jbc.M607160200. [DOI] [PubMed] [Google Scholar]
- 20.Suh N, Roberts AB, Birkey Reffey S, Miyazono K, Itoh S, ten Dijke P, Heiss EH, Place AE, Risingsong R, Williams CR, Honda T, Gribble GW, Sporn MB. Synthetic triterpenoids enhance transforming growth factor beta/Smad signaling. Cancer Res. 2003;63:1371–1376. [PubMed] [Google Scholar]
- 21.Ling X, Konopleva M, Zeng Z, Ruvolo V, Stephens LC, Schober W, McQueen T, Dietrich M, Madden TL, Andreeff M. The novel triterpenoid C-28 methyl ester of 2-cyano-3, 12-dioxoolen-1, 9-dien-28-oic acid inhibits metastatic murine breast tumor growth through inactivation of STAT3 signaling. Cancer Res. 2007;67:4210–4218. doi: 10.1158/0008-5472.CAN-06-3629. [DOI] [PubMed] [Google Scholar]
- 22.Liby K, Royce DB, Williams CR, Risingsong R, Yore MM, Honda T, Gribble GW, Dmitrovsky E, Sporn TA, Sporn MB. The synthetic triterpenoids CDDO-methyl ester and CDDO-ethyl amide prevent lung cancer induced by vinyl carbamate in A/J mice. Cancer Res. 2007;67:2414–2419. doi: 10.1158/0008-5472.CAN-06-4534. [DOI] [PubMed] [Google Scholar]
- 23.Liby KT, Royce DB, Risingsong R, Williams CR, Maitra A, Hruban RH, Sporn MB. Synthetic triterpenoids prolong survival in a transgenic mouse model of pancreatic cancer. Cancer Prev Res (Phila) 2010;3:1427–1434. doi: 10.1158/1940-6207.CAPR-10-0197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deeb D, Gao X, Liu Y, Jiang D, Divine GW, Arbab AS, Dulchavsky SA, Gautam SC. Synthetic triterpenoid CDDO prevents the progression and metastasis of prostate cancer in TRAMP mice by inhibiting survival signaling. Carcinogenesis. 2011;32:757–764. doi: 10.1093/carcin/bgr030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao X, Deeb D, Liu Y, Arbab AS, Divine GW, Dulchavsky SA, Gautam SC. Prevention of prostate cancer with oleanane synthetic triterpenoid CDDO-Me in the tramp mouse model of prostate cancer. Cancers (Basel) 2011;3:3353–3369. doi: 10.3390/cancers3033353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith LL, Coller HA, Roberts JM. Telomerase modulates expression of growth-controlling genes and enhances cell proliferation. Nat Cell Biol. 2003;5:474–479. doi: 10.1038/ncb985. [DOI] [PubMed] [Google Scholar]
- 27.Herbert B, Pitts AE, Baker SI, Hamilton SE, Wright WE, Shay JW, Corey DR. Inhibition of human telomerase in immortal human cells leads to progressive telomere shortening and cell death. Proc Natl Acad Sci USA. 1999;96:14276–14281. doi: 10.1073/pnas.96.25.14276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang X, Mar V, Zhou W, Harrington L, Robinson MO. Telomere shortening and apoptosis in telomerase-inhibited human tumor cells. Genes Dev. 1999;13:2388–2399. doi: 10.1101/gad.13.18.2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park TW, Riethdorf S, Riethdorf L, Loning T, Janicke F. Differential telomerase activity, expression of the telomerase catalytic sub-unit and telomerase-RNA in ovarian tumors. Int J Cancer. 1999;84:426–431. doi: 10.1002/(sici)1097-0215(19990820)84:4<426::aid-ijc17>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 30.Meeran SM, Patel SN, Tollefsbol TO. Sulforaphane causes epigenetic repression of hTERT expression in human breast cancer cell lines. PLoS One. 2010;5:e11457. doi: 10.1371/journal.pone.0011457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jagadeesh S, Kyo S, Banerjee PP. Genistein represses telomerase activity via both transcriptional and posttranslational mechanisms in human prostate cancer cells. Cancer Res. 2006;66:2107–2115. doi: 10.1158/0008-5472.CAN-05-2494. [DOI] [PubMed] [Google Scholar]
- 32.Rabi T, Banerjee S. Novel semisynthetic triterpenoid AMR-Me inhibits telomerase activity in human leukemic CEM cells and exhibits in vivo antitumor activity against Dalton’s lymphoma ascites tumor. Cancer Lett. 2009;278:156–163. doi: 10.1016/j.canlet.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 33.Kyo S, Takakura M, Taira T, Kanaya T, Itoh H, Yutsudo M, Ariga H, Inoue M. Inoue Sp1 cooperates with c-Myc to activate transcription of the human telomerase reverse transcriptase gene (hTERT) Nucleic Acids Res. 2000;28:669–677. doi: 10.1093/nar/28.3.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Konnikova L, Simeone MC, Kruger MM, Kotecki M, Cochran BH. Signal transducer and activator of transcription 3 (STAT3) regulates human telomerase reverse transcriptase (hTERT) expression in human cancer and primary cells. Cancer Res. 2005;65:6516–6520. doi: 10.1158/0008-5472.CAN-05-0924. [DOI] [PubMed] [Google Scholar]
- 35.Kimura A, Ohmichi M, Kawagoe J, Kyo S, Mabuchi S, Takahashi T, Ohshima C, Arimoto-Ishida E, Nishio Y, Inoue M, Kurachi H, Tasaka K, Murata Y. Induction of hTERT expression and phosphorylation by estrogen via Akt cascade in human ovarian cancer cell lines. Oncogene. 2004;23:4505–4515. doi: 10.1038/sj.onc.1207582. [DOI] [PubMed] [Google Scholar]
- 36.Kang SS, Kwon T, Kwon DY, Do SI. Akt protein kinase enhances human telomerase activity through phosphorylation of telomerase reverse transcriptase subunit. J Biol Chem. 1999;274:13085–13090. doi: 10.1074/jbc.274.19.13085. [DOI] [PubMed] [Google Scholar]
- 37.Liu Y, Gao X, Deeb D, Gautam SC. Oleanane triterpenoid CDDO-Me inhibits Akt activity without affecting PDK1 kinase or PP2A phosphatase activity in cancer cells. Biochem Biophys Res Commun. 2012;417:570–575. doi: 10.1016/j.bbrc.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li Y, Liu L, Andrews LG, Tollefsbol TO. Genistein depletes telomerase activity through cross-talk between genetic and epigenetic mechanisms. Int J Cancer. 2009;125:286–296. doi: 10.1002/ijc.24398. [DOI] [PMC free article] [PubMed] [Google Scholar]