

Abstract
The Ras/Raf/Mek/Erk mitogen-activated protein kinase pathway regulates fundamental processes in normal and malignant cells, including proliferation, differentiation, and cell survival. Mutations in this pathway have been associated with carcinogenesis and developmental disorders, making Mek1 and Mek2 prime therapeutic targets. In this study, we examined the requirement for Mek1 and Mek2 in skin neoplasia using the two-step 7,12-dimethylbenz(a)anthraacene/12-O-tetradecanoylphorbol-13-acetate (DMBA/TPA) skin carcinogenesis model. Mice lacking epidermal Mek1 protein develop fewer papillomas than both wild-type and Mek2-null mice following DMBA/TPA treatment. Mek1 knockout mice had smaller papillomas, delayed tumor onset, and half the tumor burden of wild-type mice. Loss of one Mek1 allele, however, did not affect tumor development, indicating that one Mek1 allele is sufficient for normal papilloma formation. No difference in TPA-induced hyperproliferation, inflammation, or Erk activation was observed between wild-type, conditional Mek1 knockout, and Mek2-null mice, indicating that Mek1 findings were not due to a general failure of these processes. These data show that Mek1 is important for skin tumor development and that Mek2 cannot compensate for the loss of Mek1 function in this setting.
Introduction
About 30% of all human tumors exhibit increased signaling through the mitogen-activated protein kinase (MAPK) pathway, which has accordingly become a target for anticancer therapy (1, 2). This finding is more pronounced in epithelial squamous cell carcinomas (SCC), the majority of which exhibit Ras GTPase activation and MAPK cascade hyperactivation (3, 4). Ras exerts its effects through downstream effector pathways that classically include the Raf/Mek/Erk MAPK cascade, type I phosphatidylinositol-3 kinases, and Ral guanine nucleotide exchange factors (RalGEF), along with a host of additional pathways (5). In mammals, the best-characterized Ras effector pathway is the MAPK cascade, which includes Raf (Raf1, B-Raf, and A-Raf), Mek (Mek1 and Mek2), and Erk (Erk1 and Erk2). This classic Erk MAPK cascade plays major roles in development, homeostasis, and cancer, with the former shown by the involvement of numerous cascade components in an array of related hereditary disorders, including Costello, Noonan, and cardio-facio-cutaneous syndromes (6). The Erk MAPK cascade signaling involves a host of scaffolding and regulatory mediators, such as KSR, MP1, RKIP, IQGAP1, and MORG1, which promote activation of specific Erk targets among the >160 known effector proteins to affect a pleiotropic array of cellular functions, including proliferation, differentiation, survival, and migration (7, 8).
In epidermis, Ras and Raf promote proliferation and oppose differentiation, although it is unclear to what extent Mek1 and Mek2 individually are responsible for these activities. The Mek isoforms share 85% sequence identity and have a similar ability to phosphorylate Erk1 and Erk2. Despite their similarity, however, the function of Mek1 and Mek2 may not be entirely redundant, as evidenced by the death of Mek1−/− mice in early gestation (9) and the phenotypic normality of Mek2−/− mice (10). Interestingly, Mek1 conditional ablation in the embryo proper results in viable mice, demonstrating that Mek1 is required solely for extraembryonic ectoderm formation (11). The only complete knockout at one level of the Erk MAPK cascade thus far, achieved by combined deletion of Mek1 and Mek2 in mouse skin, resulted in perinatal death (12). Similarly, in adult mice, inducible deletion of Mek1/2 caused apoptosis of the epidermis and lethality, indicating that Mek function is also required for survival during adulthood. Likewise, regenerated human epidermal tissue depleted of both Mek isoforms via RNA interference exhibited hypoproliferation and hypoplasia (12). In contrast, expression of constitutively active forms of H-Ras, Raf1, or Mek1, in mouse as well as human skin, promotes proliferation and hyperplasia (13, 14). Expression of constitutively active Mek2, however, has no effect, suggesting that although Mek1 and Mek2 are redundant in many settings, their spectra of action in tissues such as epidermis may not be identical.
In epidermal tissue, Ras has been most extensively studied in the context of murine tumorigenesis. Data generated from an array of murine genetic models, including classical 7,12-dimethylbenz(a)annthracene/12-O-tetradecanoylphorbol-13-acetate (DMBA/TPA) multistage skin carcinogenesis, indicate that Ras plays a pivotal role in initiating SCC development (15). More than 90% of the skin cancers initiated with DMBA contain the Ha-Ras activating transversion A → T at the second nucleotide of codon 61. Consistent with this, H-Ras–null mice developed ~6-fold fewer papillomas than wild-type mice in the DMBA/TPA tumor model (16). Recently, mutations in Mek1 associated with human cancers were described (17, 18). These data point to Ras and its downstream effectors as important elements in epidermal neoplasia and potential targets of clinical relevance.
Here, we investigated the effects of loss of Mek1 or Mek2 on induction of epidermal neoplasia in vivo using the DMBA/TPA two-stage skin carcinogenesis model. We show that loss of Mek1 reduced tumor formation but loss of Mek2 did not. Thus, Mek1 is selectively required during the early stages of epidermal tumorigenesis.
Materials and Methods
Mice and skin carcinogenesis protocol
Mek2−/−, Mek1fl/fl, and Mek1Δ/+ mice were maintained in a 129/SvEv background (10, 11). K14-Cre transgenic mice in a CD-1 background were obtained from Elaine Fuchs, Rockefeller University, New York (19); this line was then backcrossed to 129/SvEv mice, but animals derived from crosses with K14-Cre animals still had some CD-1 background. Littermates were used for experiments. Mice were housed and bred under standard conditions with food and water ad libitum and were maintained on a 12-h dark/light cycle. All experiments were approved by the Stanford University Animal Care and Use Committee. Mouse genotyping was performed as previously described (12). The backs of 8- to 10-wk-old mice were shaved and treated with a single application of 7,12-dimethylbenz(a)anthracene (DMBA, Sigma; 10 μg in 100 μL acetone) followed by twice weekly application of 12-O-tetradecanoylphorbol-13-acetate (TPA, Sigma: 12.5 μg in 100 μL acetone) for 20 wk. The number and sizes (mm) of papillomas were recorded once a week. The animals were sacrificed after 20 wk of TPA promotion.
Epidermal hyperplasia
TPA-induced acute hyperplasia was evaluated in wild-type, Mek1fl/flCre, and Mek2−/− animals 8 to 10 wk of age after 96-h treatment with 12.5 μg TPA in 100 μL acetone or vehicle (acetone) alone applied to the previously shaved dorsal skin.
Western blots
For immunoblotting, epidermal skin extract was obtained by incubating the dorsal skin in 1:1 dispase/PBS (Invitrogen) at 37°C for 3 h. The epidermis was lysed in lysis buffer and run on Western blots as previously described (12). For papilloma analysis, tissue was grinded and lysed in lysis buffer. Antibodies used for immunoblotting were rabbit anti–phospho-p44/42 MAPK (1:1,000, Cell Signaling Technologies), rabbit anti–p44/42 MAPK (1:1,000, Cell Signaling Technologies), rabbit anti-Mek1 (1: 1,000, Santa Cruz Biotechnologies), rabbit anti-Mek2 (1: 1,000, Santa Cruz Biotechnologies), mouse anti-actin (1:20,000, Sigma-Aldrich), donkey anti-mouse IgG conjugated to horseradish peroxidase (HRP; 1:40,000, Amersham Biosciences), and donkey anti-rabbit IgG conjugated HRP (1:40,000, Amersham Biosciences).
Histopathology and immunohistochemistry
Dorsal skin of adult mice was dissected, fixed in 10% neutral buffered formalin (accutain; Sigma-Aldrich), and embedded in paraffin, from which 5-μm sections were cut and stained with H&E or by immunohistochemistry according to standard methods. Permeabilization for antigen retrieval was achieved by microwaving samples in Antigen Unmasking Solution (Vector Laboratories), after which the sections were stained with rabbit anti–phospho-Akt (1:150) and rabbit anti–phospho-p44/p42 MAPK (1:600, Cell Signalling Technologies) as primary antibodies and biotinylated horse anti-rabbit IgG as secondary antibody (RTU Vectastain Universal Elite ABC Kit, Vector Laboratories). Staining and development were performed using the Elite ABC Reagent (Vector Laboratories) and DakoCytomation liquid DAB+ substrate chromogen system (Dako). The slides were counterstained with hematoxylin and PBS blueing. A minimum of three papillomas was analyzed per genotype.
Microscopy
H&E-stained sections were viewed using a DM LB microscope (Leica) with either a 5× Leica N Plan ∞/A, 10× Leica N Plan ∞/A, or 20× Leica HC PL Fluotar ∞/0.17 objective. Images were captured using a SPOT Insight QE camera and SPOT 4.5.9.12 software (Diagnostic Instruments, Inc.).
Statistics
Post hoc analyses using Student’s t test and χ2 test were used.
Results
Disruption of Mek1, but not of Mek2, decreases tumor burden
To address the role of Mek1 and Mek2 in skin papilloma development, we generated mice deficient in either of these genes. Because Mek1 knockout mice display embryonic lethality, Mek1 conditional knockout mice were generated. To conditionally ablate Mek1 in skin, we crossed floxed Mek1 mice (Mek1fl/fl) to keratin 14-Cre transgenic mice expressing Cre recombinase specifically in stratified epithelia, including the skin. In a previous study, the expression of Mek1 in mouse skin was assessed, which confirmed efficient Cre-mediated disruption of Mek1 (12). To study Mek2, we used Mek2 knockout animals because these are viable (10). We subjected cohorts of Mek1fl/fl, Mek1fl/flCre, Mek2+/+, Mek2+/−, and Mek2−/− adult mice to two-step skin carcinogenesis. Mice were exposed to one treatment of DMBA followed by 20 weeks of twice-weekly TPA treatment, and the number and size of papillomas that developed during this period were assessed. In Mek1fl/fl mice, the onset of papillomas was at 9 weeks and these papillomas grew progressively larger throughout the treatment period. At the end of the study, these mice each had an average of 24 papillomas. In contrast, Mek1fl/flCre mice lacking Mek1 expression in the skin exhibited a 1-week delay in the onset of papillomas and developed fewer papillomas (Fig. 1A). When the animals were euthanized, the average number of papillomas in the Mek1-deficient animals was 2-fold less than that of wild-type mice. However, no significant difference was seen between Mek2+/+, Mek2+/−, and Mek2−/− mice. Papillomas began to appear in all groups at about the same time, 9 weeks after treatment initiation, and their growth rates were similar (Fig. 1B).
Figure 1.
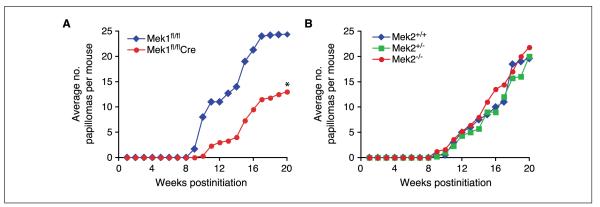
Induction of skin neoplasia in wild-type, Mek1, and Mek2 mutant mice. Skin-restricted Mek1 knockout mice (Mek1fl/flCre; A) and Mek2 knockout mice (Mek2−/−; B) were subjected to the DMBA/TPA two-stage chemical carcinogenesis protocol. A and B, average number of papillomas per mouse, n = 5. Mice were euthanized at wk 20 due to the large tumor burden. *, P < 0.02.
To further investigate the role of Mek1 in papilloma formation, both in the skin and in other tissues, we used Mek1 heterozygous mice (Mek1fl/Δ), which express only one Mek1 allele in all tissues (11), crossing them to floxed Mek1 mice and Cre transgenic mice. Further, we aimed to test whether loss of one Mek1 allele would be sufficient to lower the tumor occurrence in the DMBA/TPA tumor model, because the breeding setup of the previous experiment did not allow the investigation of Mek1 heterozygous mice. The same two-stage carcinogenesis model was applied to the following mouse groups: Mek1fl/+, Mek1fl/+Cre, Mek1fl/Δ, and Mek1fl/ΔCre. Similar to the previously described conditional Mek1 knockout mice (Mek1fl/flCre), Mek1fl/ΔCre animals had a 1-week delay in tumor onset and a 2-fold reduction in tumor burden at the end of the experiment, compared with wild-type mice (Fig. 2A). Interestingly, both groups of mice heterozygous for the Mek1 allele (Mek1fl/+Cre, Mek1fl/Δ) developed as many papillomas as did wild-type mice, indicating that one copy of Mek1 is sufficient to facilitate papilloma development. Further, the effect of loss of one Mek1 allele in all tissues, skin and non-skin, is equivalent to that of skin-specific Mek1 knockout, suggesting that the Mek1 functions involved in papilloma formation take place in the skin.
Figure 2.

DMBA/TPA treatment induced fewer tumors in Mek1 mutant mice. A, induction of skin tumors in Mek1 mutant mice in response to initiation with DMBA and promotion with TPA. Average number of tumors per mouse, n = 4 to 6. *, P = 0.12. B, columns represent the average numbers and sizes of papillomas ≥1 mm in mice as function of time of TPA promotion. Color scale indicates the sizes of papillomas. Mice were euthanized at wk 20 due to large tumor burden. *, P < 0.02; **, P < 0.001.
Next, we analyzed the size of DMBA/TPA–induced papillomas. Both types of conditional Mek1 knockout mice, Mek1fl/flCre and Mek1fl/ΔCre, had not only fewer tumors but also smaller tumors, compared with controls (Fig. 2B). This trend was seen from the onset of papilloma formation. At 20 weeks postinitiation, these knockout mice had 1.2- to 1.5-fold fewer 1- to 3-mm tumors, 4- to 8-fold fewer 4- to 7-mm tumors, and no tumor larger than that. All the other genotypes averaged at least one tumor ≥8 mm (Table 1). The deletion of Mek2 did not significantly affect papilloma number or size. Thus, one Mek1 allele seems to be sufficient for papilloma formation in the DMBA/TPA tumor model.
Table 1.
Progression of number and size of tumors in mice subjected to DMBA/TPA treatment
| Weeks postinitiation | Genotype | Papilloma size |
||
|---|---|---|---|---|
| ≤ 3 mm ± SE | 4-7 mm ± SE | ≥ 8 mm ± SE | ||
| 12 | Mek1fl/fl | 10.4 ± 1.8 | 0.6 ± 0.5 | 0 ± 0 |
| Mek1fl/flCre | 3.0 ± 0.7 | 0 ± 0 | 0 ± 0 | |
| Mek1fl/+ | 10.2 ± 0.8 | 0.8 ± 0.4 | 0 ± 0 | |
| Mek1fl/+Cre | 6.6 ± 3.8 | 0 ± 0 | 0 ± 0 | |
| Mek1fl/Δ | 8.4 ± 8.0 | 0 ± 0 | 0 ± 0 | |
| Mek1fl/ΔCre | 3.0 ± 2.4 | 0 ± 0 | 0 ± 0 | |
| Mek2+/+ | 5.0 ± 0.7 | 0 ± 0 | 0 ± 0 | |
| Mek1+/− | 4.2 ± 1.1 | 0 ± 0 | 0 ± 0 | |
| Mek2−/− | 5.2 ± 3.5 | 0 ± 0 | 0 ± 0 | |
| 16 | Mek1fl/fl | 18.4 ± 2.4 | 2.8 ± 1.5 | 0 ± 0 |
| Mek1fl/flCre | 8.4 ± 1.5 | 1.0 ± 1.0 | 0 ± 0 | |
| Mek1fl/+ | 15.0 ± 4.8 | 2.4 ± 2.3 | 0 ± 0 | |
| Mek1fl/+Cre | 12.6 ± 5.3 | 2.4 ± 1.1 | 0 ± 0 | |
| Mek1fl/Δ | 15.8 ± 7.3 | 2.4 ± 2.8 | 0 ± 0 | |
| Mek1fl/ΔCre | 8.0 ± 4.0 | 0.2 ± 0.4 | 0 ± 0 | |
| Mek2+/+ | 9.0 ± 2.0 | 1.0 ± 0.7 | 0 ± 0 | |
| Mek1+/− | 8.0 ± 1.4 | 1.0 ± 1.2 | 0 ± 0 | |
| Mek2−/− | 12.0 ± 4.0 | 1.4 ± 1.1 | 0 ± 0 | |
| 20 | Mek1fl/fl | 18.6 ± 4.2 | 4.6 ± 1.5 | 1.2 ± 0.4 |
| Mek1fl/flCre | 12.0 ± 3.1 | 1.0 ± 1.4 | 0 ± 0 | |
| Mek1fl/+ | 15.4 ± 4.2 | 4.2 ± 1.3 | 1.0 ± 0.7 | |
| Mek1fl/+Cre | 15.4 ± 5.2 | 4.0 ± 0.7 | 1.0 ± 0.7 | |
| Mek1fl/Δ | 15.6 ± 5.8 | 4.2 ± 1.5 | 1.0 ± 1.0 | |
| Mek1fl/ΔCre | 12.8 ± 5.8 | 0.2 ± 0.4 | 0 ± 0 | |
| Mek2+/+ | 14.2 ± 3.7 | 4.0 ± 1.6 | 1.4 ± 1.3 | |
| Mek1+/− | 15.0 ± 3.5 | 4.0 ± 1.4 | 1.0 ± 0.7 | |
| Mek2−/− | 17.0 ± 3.3 | 3.8 ± 1.3 | 1.0 ± 1.0 | |
Tumors from wild-type, conditional Mek1 knockout mice, and Mek2-null mice are benign and well differentiated
The difference in papilloma number and size between wild-type and Mek1-deficient mice throughout the 20-week course of DMBA/TPA treatment suggests that Mek1 is important not only for papilloma initiation but also for efficient development (Fig. 3A and B). The gross appearance of DMBA/TPA–treated mice at 16 and 20 weeks of TPA promotion clearly depicts the difference between tumor burden in wild-type, Mek1, and Mek2 knockout animals, with Mek1 knockout animals displaying fewer and smaller tumors. However, there was no obvious difference between genotypes in the histologic appearance of the papillomas (Fig. 3C). All papillomas were benign and well differentiated; none of them progressed to malignant carcinoma during the 20-week experiment.
Figure 3.

Skin-restricted Mek1 knockout mice, but not Mek2 knockout mice, are resistant to papilloma formation. Representative images of Mek1fl/fl, Mek1fl/flCre, Mek2+/+, Mek2+/−, and Mek2−/− mice showing the dramatic difference in papilloma number and size at wk 16 (A) and wk 20 (B). C, H&E staining of a portion of a representative papilloma in wild-type (WT), Mek2 knockout (Mek2−/−), and skin-restricted Mek1 knockout mice (Mek1fl/flCre). Original magnification, 5×.
Phospho-Erk1/2 and phospho-Akt levels are not genotype dependent
To determine the effects of Mek1 and Mek2 ablation in skin papilloma development, we investigated activation of major effector pathways downstream of oncogenic Ras. Despite the loss of two Mek alleles, epidermal extract of skin-restricted Mek1 knockout mice as well as that of Mek2-null mice showed levels of Erk1/2 and phospho-Erk1/2 protein equivalent to that of wild-type epidermal extract (Fig. 4A). No compensatory changes in Mek isoform expression were seen upon loss of Mek1 or Mek2, consistent with previous findings (9, 10, 12). Further, immunohistochemical analyses of phospho-Akt and phospho-Erk1/2 in papillomas did not reveal any significant differences in expression level of either protein between wild type, Mek1, and Mek2 knockout mice (Fig. 4B). To address isoform-specific activation of Erk1/2, we analyzed the levels of phospho-Erk1 and phospho-Erk2 by Western blotting. Two papilloma extracts for each genotype were analyzed and no significant changes were seen (Fig. 4C). Thus, Mek1 knockout animals had fewer tumors, but these tumors did not display altered phospho-Erk1, phospho-Erk2, or phospho-Akt levels compared with Mek2 knockout and wild-type papillomas.
Figure 4.

Phospho-Akt and phospho-Erk1/2 levels are not genotype dependent. A, Western blot analysis of Erk1/2 and phospho-Erk1/2 levels in adult epidermal skin extracts of wild-type, Mek1fl/flCre, and Mek2−/− mice. B, immunohistochemical staining of phospho-Erk1/2 and of phospho-Akt on representative papilloma sections. Genotypes studied are noted at the top of each column and markers stained are noted at the right. Original magnification, 10×. C, Western blot analysis of Erk1/2 and phospho-Erk1/2 in papilloma skin extracts.
The epidermal hyperplasia and Erk activation induced by TPA or DMBA/TPA treatment is not affected by loss of Mek1 or Mek2
Decreased papilloma induction in Mek1 knockout mice could be due to a global proliferation defect caused by Mek1 loss, which would cause Mek1 knockout cells to undergo less DMBA/TPA–induced proliferation. To address the individual roles of Mek1 and Mek2 during stress-induced epidermal proliferation, skin was treated with TPA or vehicle (acetone) alone. The phorbol ester TPA induces a robust proliferative response leading to substantial epidermal hyperplasia. After four daily topical treatments of TPA, wild-type mouse skin exhibited marked hyperplasia and hyperkeratosis. The effect of TPA on Mek1fl/flCre and Mek2−/− mouse skin was similar, with no obvious difference in cell proliferation or cell death seen (Fig. 5A). Further, as observed in other studies of TPA treatment, epidermal inflammation was noted; however, it did not vary in extent histologically with respect to mouse genotype. These data indicate that Mek1 is not required for hyperplasia or inflammation in response to TPA treatment.
Figure 5.

Mouse skin response to short-term TPA or DMBA/TPA treatment. Epidermal hyperplasia induced by four daily treatments of TPA (A) or by DMBA initiation followed by 2 wk of twice-weekly TPA promotion (B) occurs to a similar extent in control, Mek1 knockout, and Mek2 knockout skin. H&E staining of dorsal skin from mice treated with vehicle (acetone) or TPA for 4 d (n = 5). Original magnification, 20×. C, Western blot analysis of Erk1/2 and phospho-Erk1/2 levels in adult epidermal skin extracts of wild-type, Mek1fl/flCre, and Mek2−/− mice treated as in B.
To further confirm the absence of differential responses to TPA by Mek1 and Mek2 knockout skin, we assessed the effect of a TPA treatment regimen identical to that used in tumorigenesis experiments. Specifically, wild-type, Mek1fl/flCre, and Mek2−/− mice were treated with DMBA followed by 2 weeks of twice-weekly TPA treatment, at which point the skin was harvested. Similar to the short-term TPA treatment studies, no differences were observed among all three genotypes in the extent of TPA-induced hyperplasia or epidermal inflammation (Fig. 5B). Finally, immunoblotting of epidermal protein extract prepared from skin harvested at this time point confirmed no significant differences in Erk1 or Erk2 activation among the three genotypes (Fig. 5C). Thus, alterations in response to TPA do not appear to underlie the effect of Mek1 or Mek2 knockout on DMBA/TPA–induced tumorigenesis in mice.
Discussion
Here, we have used genetically engineered knockout mice to examine the roles of Mek1 and Mek2 in a two-stage skin carcinogenesis model. Whereas knockout of Mek2 or deletion of one Mek1 allele had no effect on the number or size of induced papillomas, loss of both Mek1 alleles delayed papilloma onset and resulted in smaller and fewer tumors.
The overlapping and unique functions of isoforms at each level of the Ras/Raf/Mek/Erk signaling pathway are just beginning to be elucidated. Often, no distinction is made between the functions of Mek1 and Mek2 or Erk1 and Erk2, and few studies have examined the individual contributions of each isoform to biological processes. The only identified Mek1 and Mek2 substrates are Erk1 and Erk2, which are phosphorylated by Mek1 and Mek2. It is clear from previous genetic studies that Mek1 and Mek2 are functionally redundant in some contexts (12). Mice expressing only one of their four Mek1 or Mek2 alleles in the skin are viable and fertile irrespective of whether the remaining allele is Mek1 or Mek2, demonstrating that Mek1 and Mek2 do not have distinct roles for normal skin homeostasis.
These prior studies, however, focused primarily on Mek functions in development and tissue homeostasis. In contrast, our findings here suggest that Mek1 plays a unique role in tumor development. This distinguishing role of Mek1 is in accordance with the requirement of Mek1 specifically for extraembryonic ectoderm formation (11), cancer cell line colony formation (20), and with our previous demonstration that activation of Mek1, but not of Mek2, induces skin hyperplasia (13). Interestingly, in the latter study, we found that constitutively active forms of Mek1 and Mek2 activated Erk1/2 similarly, although only active Mek1 induced hyperplasia. Similarly, we find in the present study that Mek1 and Mek2 knockout mice display equivalent levels of Erk1/2 phosphorylation upon DMBA/TPA treatment. Furthermore, in another study, we find that activation of oncogenic Ras in mouse skin containing only two of the four Mek1/2 alleles produces equivalent hyperplasia and Erk1/2 phosphorylation regardless of whether the remaining alleles are both Mek1, both Mek2, or one of each isoform (21). Together, these results suggest that differential expression of Mek- and Erk-interacting proteins, and not different levels of Erk1/2 phosphorylation, may underlie the difference in tumorigenesis in Mek1 and Mek2 knockout mice. For instance, it has been shown that the binding partner interactions of Erk1/2, including homodimerization and binding to scaffold proteins, can alter the ability of Erk1/2 to activate their substrates, independent of their own phosphorylation state (22). Furthermore, two new studies hint toward a crucial role of subcellular localization and differential use of scaffolding proteins to regulate the biological outcome of MAPK activation (23, 24). Future experiments should therefore investigate differential expression and localization of proteins that modulate activity of MAPK cascade activity in the settings of Mek1 and Mek2 loss. Alternatively, it is possible that our present experiments did not detect a difference in Erk1/2 activity between Mek1 and Mek2 knockout mice because a difference appeared only during the initial response of the skin to DMBA treatment, when Mek1 knockout cells may have been more susceptible than Mek2 knockout cells to genotoxic stress, thereby incurring higher rates of apoptosis and leading to the differential tumor burden in these knockout mice.
Interestingly, we found that Mek1 and Mek2 knockout animals responded similarly to TPA treatment, which induced both inflammation and hyperplasia. This result contrasts with recent studies of Erk1 knockout animals, which exhibit reduced hyperproliferation in response to TPA alone, but are, similar to conditional Mek1 knockout animals, resistant to DMBA/TPA–induced tumorigenesis, having half the tumor burden and tumors of smaller size than those of wild-type littermates (25). Like Mek1 knockout animals, Erk1 knockout mice are viable, fertile, and of normal size. In contrast to Mek1 knockout mice, however, Erk1 knockout mice exhibit disrupted thymocyte maturation and proliferation, as well as local cutaneous lesions and hyperplastic skin. Future efforts to assess the effects of Erk2 knockout on DMBA/TPA–induced tumorigenesis will be important to determine whether the role of Erk1 in tumorigenesis, like the role of Mek1, is isoform specific.
In summary, we have shown that Mek1 function is important for the early stages of skin tumor formation and that Mek2 cannot compensate for its absence. Additional studies will be required to understand how these two isoforms differ on a mechanistic level. Understanding the individual contributions of Mek1 and Mek2, as well as those of their downstream targets Erk1 and Erk2, to normal biological processes and pathogenesis will be important in further evaluating these genes as potential therapeutic targets.
Acknowledgments
Grant support: U.S. Veterans Affairs Office of Research and Development and by AR49737 from NIAMS/NIH to P.A. Khavari, the Swiss cancer foundation grant BIL SKL-01236-02-2002 to F.A.S, and MOP-67208 from CIHR to J. Charron.
We thank A. Truong for critical reading of the manuscript, E. Fuchs for K14-Cre mice, P. Bernstein, H. Bernstein, and N. Griffiths for support.
Footnotes
Disclosure of Potential Conflicts of Interest No potential conflicts of interest were disclosed.
References
- 1.Sebolt-Leopold JS, Herrera R. Targeting the mitogen-activated protein kinase cascade to treat cancer. Nat Rev Cancer. 2004;4:937–47. doi: 10.1038/nrc1503. [DOI] [PubMed] [Google Scholar]
- 2.Wallace EM, Lyssikatos JP, Yeh T, Winkler JD, Koch K. Progress towards therapeutic small molecule MEK inhibitors for use in cancer therapy. Curr Top Med Chem. 2005;5:215–29. doi: 10.2174/1568026053507723. [DOI] [PubMed] [Google Scholar]
- 3.Dajee M, Lazarov M, Zhang JY, et al. NF-κB blockade and oncogenic Ras trigger invasive human epidermal neoplasia. Nature. 2003;421:639–43. doi: 10.1038/nature01283. [DOI] [PubMed] [Google Scholar]
- 4.Pierceall WE, Goldberg LH, Tainsky MA, Mukhopadhyay T, Ananthaswamy HN. Ras gene mutation and amplification in human nonmelanoma skin cancers. Mol Carcinog. 1991;4:196–202. doi: 10.1002/mc.2940040306. [DOI] [PubMed] [Google Scholar]
- 5.Rajalingam K, Schreck R, Rapp UR, Albert S. Ras oncogenes and their downstream targets. Biochim Biophys Acta. 2007;1773:1177–95. doi: 10.1016/j.bbamcr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 6.Schubbert S, Bollag G, Shannon K. Deregulated Ras signaling in developmental disorders: new tricks for an old dog. Curr Opin Genet Dev. 2007;17:15–22. doi: 10.1016/j.gde.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 7.Kolch W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat Rev Mol Cell Biol. 2005;6:827–37. doi: 10.1038/nrm1743. [DOI] [PubMed] [Google Scholar]
- 8.McKay MM, Morrison DK. Integrating signals from RTKs to ERK/MAPK. Oncogene. 2007;26:3113–21. doi: 10.1038/sj.onc.1210394. [DOI] [PubMed] [Google Scholar]
- 9.Giroux S, Tremblay M, Bernard D, et al. Embryonic death of Mek1-deficient mice reveals a role for this kinase in angiogenesis in the labyrinthine region of the placenta. Curr Biol. 1999;9:369–72. doi: 10.1016/s0960-9822(99)80164-x. [DOI] [PubMed] [Google Scholar]
- 10.Belanger LF, Roy S, Tremblay M, et al. Mek2 is dispensable for mouse growth and development. Mol Cell Biol. 2003;23:4778–87. doi: 10.1128/MCB.23.14.4778-4787.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bissonauth V, Roy S, Gravel M, Guillemette S, Charron J. Requirement for Mapk2k1 (Mek1) in extraembryonic ectoderm during placentogenesis. Development. 2006;133:3429–40. doi: 10.1242/dev.02526. [DOI] [PubMed] [Google Scholar]
- 12.Scholl FA, Dumesic PA, Barragan DI, et al. Mek1/2 MAPK kinases are essential for Mammalian development, homeostasis, and Raf-induced hyperplasia. Dev Cell. 2007;12:615–29. doi: 10.1016/j.devcel.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 13.Scholl FA, Dumesic PA, Khavari PA. Mek1 alters epidermal growth and differentiation. Cancer Res. 2004;64:6035–40. doi: 10.1158/0008-5472.CAN-04-0017. [DOI] [PubMed] [Google Scholar]
- 14.Tarutani M, Cai T, Dajee M, Khavari PA. Inducible activation of Ras and Raf in adult epidermis. Cancer Res. 2003;63:319–23. [PubMed] [Google Scholar]
- 15.Woodworth CD, Michael E, Smith L, et al. Strain-dependent differences in malignant conversion of mouse skin tumors is an inherent property of the epidermal keratinocyte. Carcinogenesis. 2004;25:1771–8. doi: 10.1093/carcin/bgh170. [DOI] [PubMed] [Google Scholar]
- 16.Ise K, Nakamura K, Nakao K, et al. Targeted deletion of the H-ras gene decreases tumor formation in mouse skin carcinogenesis. Oncogene. 2000;19:2951–6. doi: 10.1038/sj.onc.1203600. [DOI] [PubMed] [Google Scholar]
- 17.Estep AL, Palmer C, McCormick F, Rauen KA. Mutation analysis of BRAF, MEK1 and MEK2 in 15 ovarian cancer cell lines: implications for therapy. PLoS ONE. 2007;2:e1279. doi: 10.1371/journal.pone.0001279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marks JL, Gong Y, Chitale D, et al. Novel MEK1 mutation identified by mutational analysis of epidermal growth factor receptor signaling pathway genes in lung adenocarcinoma. Cancer Res. 2008;68:5524–8. doi: 10.1158/0008-5472.CAN-08-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vasioukhin V, Degenstein L, Wise B, Fuchs E. The magical touch: genome targeting in epidermal stem cells induced by tamoxifen application to mouse skin. Proc Natl Acad Sci U S A. 1999;96:8551–6. doi: 10.1073/pnas.96.15.8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ussar S, Voss T. MEK1 and MEK2, different regulators of the G1/S transition. J Biol Chem. 2004;279:43861–9. doi: 10.1074/jbc.M406240200. [DOI] [PubMed] [Google Scholar]
- 21.Scholl FA, Dumesic PA, Barragan DI, Charron J, Khavari PA. Mek1/2 gene dosage determines tissue response to oncogenic Ras signaling in the skin. Oncogene. 2009;28:1485–95. doi: 10.1038/onc.2008.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Casar B, Pinto A, Crespo P. Essential role of ERK dimers in the activation of cytoplasmic but not nuclear substrates by ERK-scaffold complexes. Mol Cell. 2008;31:708–21. doi: 10.1016/j.molcel.2008.07.024. [DOI] [PubMed] [Google Scholar]
- 23.Casar B, Arozarena I, Sanz-Moreno V, et al. Ras subcellular localization defines ERK1/2 substrate specificity through distinct utilization of scaffold proteins. Mol Cell Biol. 2008;29:1338–53. doi: 10.1128/MCB.01359-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Inder K, Harding A, Plowman SJ, Philips MR, Parton RG, Hancock JF. Activation of the MAPK module from different spatial locations generates distinct system outputs. Mol Biol Cell. 2008;19:4776–84. doi: 10.1091/mbc.E08-04-0407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bourcier C, Jacquel A, Hess J, et al. p44 mitogen-activated protein kinase (extracellular signal-regulated kinase 1)-dependent signaling contributes to epithelial skin carcinogenesis. Cancer Res. 2006;66:2700–7. doi: 10.1158/0008-5472.CAN-05-3129. [DOI] [PubMed] [Google Scholar]