

Abstract
Excessive manganese (Mn) uptake by brain cells, particularly in regions like the basal ganglia, can lead to toxicity. Mn2+ is transported into cells via a number of mechanisms, while Mn3+ is believed to be transported similarly to iron (Fe) via the transferrin (Tf) mechanism. Cellular Mn uptake is therefore determined by the activity of the mechanisms transporting Mn into each type of cell and by the amounts of Mn2+, Mn3+ and their complexes to which these cells are exposed; this complicates understanding the contributions of each transporter to Mn toxicity. While uptake of Fe3+ via the Tf mechanism is well understood, uptake of Mn3+ via this mechanism has not been systematically studied. The stability of the Mn3+Tf complex allowed us to form and purify this complex and label it with a fluorescent (Alexa green) tag. Using purified and labeled Mn3+Tf and biophysical tools, we have developed a novel approach to study Mn3+Tf transport independently of other Mn transport mechanisms. This approach was used to compare the uptake of Mn3+Tf into neuronal cell lines with published descriptions of Fe3+ uptake via the Tf mechanism, and to obtain quantitative information on Mn uptake via the Tf mechanism. Results confirm that in these cell lines significant Mn3+ is transported by the Tf mechanism similarly to Fe3+Tf transport; although Mn3+Tf transport is markedly slower than other Mn transport mechanisms. This novel approach may prove useful for studying Mn toxicity in other systems and cell types.
Keywords: manganese transport, transferrin, endosomal transport, mitochondria, manganese toxicity
1. Introduction
While manganese (Mn) is an essential biological element and a necessary cofactor in a number of important enzymatic reactions, excessive brain Mn accumulation particularly in the globus pallidus and striatum can lead to neurotoxicity with symptoms and signs resembling those of Parkinson's disease. The evidence indicates that in the 2+ oxidation state (Mn2+), Mn enters cells via a number of transport mechanisms, including the divalent metal transporter 1 (DMT1) (Au et al, 2008), a Mn citrate transporter (Crossgroveet al, 2003; Crossgrove and Yokel, 2004), a store activated Ca2+ channel (Yokel and Crossgrove, 2004), the ZIP8 mechanism (He et al, 2006; Liu et al, 2008), and the ZIP14 mechanism (Fujishiro et al, 2012; Girijashanker et al, 2008). In the 3+ oxidation state (Mn3+) the evidence suggests that Mn is transported via the transferrin (Tf) mechanism (Aschner and Aschner, 1990; Aschner and Gannon, 1994). Mn uptake into a specific cell type is thus determined by the activity of each type of uptake mechanism expressed in that cell type and the oxidation state of the Mn reaching the cell. Once inside the cell, most of the Mn is found in the mitochondrial and nuclear fractions (Maynard and Cotzias, 1955).
A study of Mn speciation in animal cells using X-ray absorption near edge structure (XANES) spectroscopy identified only Mn2+ and a trace amount of Mn3+ having the spectrum of the enzyme Mn superoxide dismutase (Gunter et al, 2006a; Gunter et al, 2005; Gunter et al, 2004). This is undoubtedly because Mn2+ is by far the more stable species of these two oxidation states (Latimer and Hildebrand, 1956). The concentration of free Mn3+ is not zero, but exists in a steady state in which its concentration is much lower than that of free Mn2+ and below the XANES detection limit. A number of factors influence this steady state, but generally free Mn3+ is more stable at low pH than at neutral pH (Latimer and Hildebrand, 1956). Mn3+ can also enter aqueous solution when stabilized by formation of stable complexes (Gunter et al, 2006b), including some formed with common organic anions usually found in cells and tissue, such as citrate or Tf.
The Tf system is known to transport iron (Fe) primarily into mitochondria for incorporation into hemes and other Fe-containing proteins (Sheftel et al, 2007). Fe3+ transport via the Tf mechanism involves the following steps: binding of the Fe3+ to extracellular Tf, binding of the Fe3+Tf complex to Tf receptors (TfRs) and movement of the Tf-bound TfR into clathrin-coated pits, invagination of these pits into endosomes bringing the Fe3+Tf inside the cell, movement of the endosomes into the region of the cytosol in which the mitochondrial network is found, release of Fe3+ from binding to the Tf complex by acidification of the endosomal interior, reduction of the Fe3+ to Fe2+ by Steap proteins in the endosomes, transport of the Fe2+ out of the endosomes by DMT1, and uptake of the Fe2+ by mitochondria where it can be incorporated into hemes and other iron-containing proteins (Hentze et al, 2004; Ohgami et al, 2005; Ohgami et al, 2006; Richardson and Ponka, 1997; Sheftel et al., 2007).
Mn and Fe are elements with atomic numbers 25 and 26, respectively, within the transition metal series and have many similar characteristics. Mn uptake via the Tf mechanism has usually been assumed to be analogous to that of Fe; nevertheless, there are differences in their chemistry, which could cause Mn3+ transport via Tf to differ from transport of Fe3+. First, the oxidation state in which Fe binds most strongly to Tf, Fe3+, is also its most stable oxidation state at physiological pH; in contrast, while Mn also binds Tf as Mn3+, it is most stable at physiological pH as Mn2+. Since the concentration of free blood Mn3+ is very low at physiological pH, even when Mn is present in excess, mass action would predict that Mn3+ would bind to Tf very slowly as Mn3+ transferrin (Mn3+Tf). This is supported by the observation that the procedure set up by Aisen et al. for preparing Mn3+Tf requires a week for the accumulation of the stable Mn3+Tf complex to approach completion (Aisen et al, 1969). We have studied this slow conversion of Mn2+ to Mn3+ more closely under somewhat more physiological conditions in a XANES experiment described below. Studies of transport of Fe3+ via the Tf mechanism show that the Fe3+ is reduced to Fe2+ by Steap proteins inside endosomes and transported out of the endosomes via DMT1 (Hentze et al., 2004; Ohgami et al., 2005; Ohgami et al., 2006; Richardson and Ponka, 1997). Mn2+, like Fe2+ is transported by DMT1 (Au et al., 2008) and hence should be released from the endosomes similarly to Fe2+. Furthermore, the Tf transport mechanism for Fe is known to deliver Fe2+ to the vicinity of the mitochondrial network, where it is sequestered into mitochondria for incorporation into hemes (Sheftel et al., 2007). If it also delivers Mn2+ to mitochondria, it could contribute to mitochondrial Mn toxicity associated with deficits in energy production (Brouillet et al, 1993; Galvani et al, 1995; Gavin et al, 1992; Malecki, 2001; Malthankar et al, 2004; Roth et al, 2000; Roth et al, 2002; Zwingmann et al, 2003)
The systems transporting Mn2+ into cells are complex and difficult to functionally isolate experimentally. Furthermore, the expression of each type of transporter varies with cell type. However, we have been able to purify the Mn3+Tf complex and also to covalently bind a fluorescent label (Alexa green) to Mn3+Tf because of the stability of this complex. This has permitted us to study the transport of Mn via the Tf mechanism independently of the other cellular Mn transport mechanisms, to follow the transport of Mn3+Tf into neuronal cells and into the region of the mitochondrial network using confocal microscopy, and to confirm that Mn transport via the Tf system functions analogously to the transport of ferric iron (Fe3+) by this mechanism. Using the purified Mn3+Tf and atomic absorption spectroscopy (AA), we have also been able to measure the accumulation of Mn via Mn3+Tf independently of the other Mn transport mechanisms using atomic absorption and to compare it with the accumulation from a similar concentration of Mn2+. The primary goal of the work reported here is to introduce this novel approach to the study of transport of Mn3+ via the Tf system to those interested in its role in Mn neurotoxicity so that it may be applied to studies of Mn3+Tf transport in additional systems and cell types and to more complete studies of its uptake kinetics.
The uptake of Mn3+ via the Tf mechanism in the two neuronal cell types used in the current study is slower than that of Mn2+, but isn't negligible and therefore could contribute to Mn toxicity, along with Mn transported into cells by the other transport mechanisms. However, this does not necessarily mean that transport of Mn3+ via the Tf system is so small in all cell types and so other cell types should be studied. Furthermore, Mn3+ is also a strong oxidizing agent, which might be expected to oxidize many components of the endosome in addition to the Steap proteins. Thus, transport of Mn via the Tf mechanism exposes an additional set of cell components to Mn, particularly within the endosomal system, which could be damaged by this transport process, but not by other processes of Mn transport into the cell.
2. Materials and Methods
2.1 Neuronal Cells
Mn uptake via the Tf mechanism was studied in cultured mouse hippocampal (HT22) and striatal neurons (STHdhQ7/Q7) (Trettel et al, 2000). The latter were chosen because the striatum is an area where Mn preferentially accumulates, and one of the areas most affected by Mn toxicity (Finkelstein et al, 2007). HT22 cells were grown at 37° C in an incubator with 5% CO2 in Gibco F-12 mixture (HAM) containing 5% FBS, 5% HS, 20 mM glucose, and Pen Strep. The STHdhQ7/Q7 cells were grown at 33° C with 5% CO2 in Gibco F-12 containing 10%FBS, 5% HS and Pen Strep. Both cell types were passed with 0.05% trypsin and 0.53 mM EDTA about every 3 days. For long term storage, each type of cell was frozen in 90% fetal calf serum/10% DMSO and stored in a liquid nitrogen refrigerator at 77° K. The cells were used between passages 3 and 7.
2.2 XANES Spectroscopy
XANES spectroscopy is a technique similar to extended X-ray absorption fine structure (EXAFS) in which an energy shift is observed between the absorption edges of different oxidation states of an element. Furthermore, unlike some spectroscopies, such as electron paramagnetic resonance (EPR), where some spectra may be relaxation broadened to the point where they are no longer observable, XANES spectroscopy shows the spectra of all compounds and complexes containing the element of interest weighted by their abundance, with spectra of each oxidation state found near its appropriate absorption edge. These absorption edges are separated by several electron volts, a difference which is easily measurable. This makes XANES spectroscopy the “gold standard” for determining the oxidation state or states of an unknown biological compound or complex containing the element of interest. The details of the techniques used and methods of handling the XANES sample described here are outlined in the Supplemental Material and more completely in earlier publications (Gunter et al., 2006a; Gunter et al., 2005; Gunter et al., 2006b; Gunter et al., 2004).
A sample of Mn2+ (0.2 mM) with Tf (2 mM) in HEPES (10 mM) buffered saline (160 mM Na+ and Cl− pH 7.2) was allowed to incubate at room temperature (22° C) for 24 hours. Polymerized dextran (10% w/v final concentration) was mixed into the sample, which was rapidly frozen using liquid nitrogen and maintained at −80° C until XANES spectroscopy could be carried out at beamline 9 B at the National Synchrotron Light Source at Brookhaven National Laboratory. The polymerized dextran aids in producing a glassy sample and minimizing the formation of ice crystals, which could distort the spectrum by inducing Bragg reflections. The sample's XANES spectrum was analyzed by curve fitting to a set of Mn2+ and Mn3+ standards (model compounds) as previously described and as illustrated in Figs. S1 and S2A and S2B in the Supplementary Material (Gunter et al., 2006a; Gunter et al., 2005; Gunter et al., 2006b; Gunter et al., 2004).
2.3 Construction and Characterization of Probes
2.3.1 Mn3+ Transferrin
Apotransferrin (Apo-Tf) purchased from Lee BioSolutions, St Louis, MO was shown by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) to fractionate as a single band at approximately 77 kD. Mn3+ was bound to Tf using a modification of the technique of Aisen et al (Aisen et al., 1969). Briefly, a three fold excess of a 1:1 solution of Mn2+ and citrate (pH 5.0) was mixed with a 5% solution of apotransferrin in 0.1 M KCl and incubated in a cold room (4°C) for about a week. During the incubation, the very light pink color of Mn2+ slowly assumes the dark grayish color of Mn3+. At the end of this incubation the suspension was carefully inspected visually for the presence of any precipitate. There was no evidence of a precipitate. The suspension was then spun in Vivaspin 15 tubes made by Sartorius Stedim to separate the Mn3+-bound Tf from the medium. These tubes contain a 30 kD MWCO filter which passes the small ions and molecules and retains the Tf (~ 77 kD). The retentate was collected from the filter, diluted in medium (0.1 M KCl, 0.05 M K Tris, pH 7.5), and recentrifuged up to 35 times in order to separate and remove the free Mn2+ and any other small impurities from the Tf solution. In this medium the only component which could bind the Mn was the Tf. The final solution from this purification was tested by visible/UV spectroscopy to verify that its spectrum was similar to the published Mn3+Tf spectrum (Aisen et al., 1969). It was also tested by EPR to determine the amount of Mn2+ hexahydrate (free Mn2+) remaining, by atomic absorption (AA) to determine the total Mn present, and by the Bradford assay to determine the amount of protein (i.e. Tf) present. The amount of Mn2+ found using the EPR assay represented less than 1% of the total Mn found by AA in all probes and at around 0.2 % in the probe used for quantitative Mn uptake measurements by AA in the work reported here. The results of these measurements allowed determination of the number of Mn3+ ions bound per Tf molecule. For the quantitative probes used in the current work, the Mn3+/Tf ratio was in the range 1.6 to 2.0. The purified probes were stored at −20°C until they were used in experiments.
2.3.2 Attachment of Alexa green 488 to transferrin
Alexa Fluor 488 carboxylic acid succinimidyl ester (amine binding reagent) was purchased from Invitrogen (A-20000) and attached to the Apo-Tf using the manufacturer's suggested protocol. Briefly, 16 mg of Tf was dissolved in 1 ml of 0.1 M NaHCO3 buffer (pH 8.3). The Alexa green (AG) 488 amine reactive compound was dissolved in 0.5 ml of DMSO at a concentration of 10 mg/ ml. The amine reactive solution was slowly added to the Tf solution as it was being stirred, and the resulting solution was incubated for 1 hr at 22°C. The Tf plus AG-reactive probe was dialyzed in a cold room overnight with several changes of buffer. The AG-Tf was then carried through the procedure described under section 2.3.1 “Mn3+ Transferrin” to bind Mn3+ to the labeled Tf (forming AGMn3+Tf). During the week of incubation, the beaker containing the AG-Tf was wrapped in aluminum foil and generally kept in the dark to minimize bleaching. Following the Vivaspin 15 purification centrifugations, final product was not diluted, but the concentration of Tf was measured using the Coomassie-stained SDS-PAGE technique described below. Binding of the fluorescent AG moiety to the Tf molecule is unlikely to affect the binding of Mn3+ to Tf significantly since the AG binding procedure used targets amine groups, particularly the terminal amine, and not the Mn3+ binding sites. The stability of the AG labeled probe was checked in the same way as that of the unlabeled probe, as described in the Supplementary Material, and the probe was found to be stable.
2.4 Protein Measurements
Protein measurements were carried out with the Bradford technique except in those cases where Alexa green 488 had been bound to Tf. The Bradford assay could not be used with AGMn3+Tf, because the intense absorbance of the fluorescent label dominated the spectrum necessary for protein measurements. For measurements of the amount of protein in AGMn3+Tf solutions, the protein was run on an SDS-PAGE gel, labeled with Coomassie blue and the total color of the band integrated and compared with that of several known concentrations of Mn3+Tf.
2.5 EPR analysis of probes
EPR was used to compare the amount of Mn2+ hexahydrate (free Mn2+) present at the beginning of the Mn3+Tf incubation, the amount present at the end of the incubation (after approximately 1 week at 4° C), and the amount present at the end of the “clean up spins”. At that time, the amount of Mn2+ hexahydrate present was compared with the total Mn present measured using atomic absorption spectroscopy (see “Mn Transferrin”). The measurements were carried out using an X-band Bruker EPR spectrometer modified as described (Black and Swarts, 2010) and utilizing an IBM Instruments Inc. microwave bridge system (ERO42MRH). During these measurements, samples were in quartz EPR flat cells in one part of a Varian dual cavity system; the other part of the dual cavity held a CuCl2 standard. The six line derivative spectrum of Mn2+ hexahydrate in the sample was compared with that of a Mn2+ hexahydrate standard of known concentration at 22 °C. While the spectra of the pre- and post- incubation samples could be easily seen because of the excess Mn2+ concentration used with respect to the concentration of transferrin, the EPR spectra of the samples carried through the Vivaspin 15 centrifugations required extensive computing of average transients in order to bring the noise down to a level where the spectrum could be interpreted. During all of these measurements, the klystron power and modulation amplitudes were kept the same for all samples varying only the signal gain. The EPR spectrum of the Mn3+Tf itself cannot be seen in these spectra because of extreme relaxation broadening commonly seen with low symmetry Mn3+ compounds at room temperature (Abragam and Bleaney, 1970; Vincent and Love, 2012). Additional information and typical EPR spectra are shown in Fig. S3B of the Supplemental Material.
2.6 UV-visible analysis of probes
UV-visible spectra were taken of the probes used in order to visualize the appearance of the UV-visible spectrum of Mn3+Tf both before the incubation and at the end of the “clean up” spins. These measurements were carried out on a Varian Cary 100Bio UV-Visible spectrometer. The spectrum of Mn3+Tf which had not been bound to Alexa green 488 looked identical to that published in Fig. 1 of Aisen et al (Aisen et al., 1969), while the spectrum of AGMn3+Tf also showed the strong absorbance of the Alexa green with a peak at 488 nm. Additional information on the UV/visible spectra of Mn3+Tf in these probes showing the known UV/visible spectrum of Mn3+Tf is shown in Fig. S3B of the Supplemental Material.
Fig. 1. XANES study of Mn3+Tf formation.

Best fit of first derivative model compound XANES spectra to the first derivative of the edge normalized XANES spectrum of Mn2+ incubated with apotransferrin. The sample was made up by mixing 2 mM Tf and 0.2 mM MnCl2 in HEPES (10 mM, pH 7.2) buffered NaCl (160 mM) and incubated for 24 hours at 22° C. Also shown are the residual (Mn transferrin spectrum - best fit spectrum) and the spectrum of 0.17 times the edge-normalized first derivative spectrum of Mn3+ porphine for comparison. Mn transferrin ( ); best fit (
); best fit ( ); residual (
); residual ( ); 0.17 times d(Mn3+ porphine)/dE (
); 0.17 times d(Mn3+ porphine)/dE ( ). The best fit is the sum of the first derivatives of the edge-normalized XANES spectra of 0.65 Mn2+ EGTA, 0.168 Mn2+ ATP, and 0.182 Mn3+ porphine. The R2 of the fit is 0.996.
). The best fit is the sum of the first derivatives of the edge-normalized XANES spectra of 0.65 Mn2+ EGTA, 0.168 Mn2+ ATP, and 0.182 Mn3+ porphine. The R2 of the fit is 0.996.
2.7 Atomic absorption analysis of the probes and of Mn uptake
Total Mn present in the probes and in cell samples was measured using a Perkin-Elmer AAnalyst 600 atomic absorption spectrometer equipped with a THGA (transversely-heated graphite furnace) and Mn Lumina™ hollow-cathode lamp (Perkin-Elmer Part No. N305-0145)(279.5 nm, slit 0.2 nm). To calibrate the spectrometer for Mn, reference solutions (containing 0, 2, 5, 10, and 20 μg/l Mn) were prepared daily in 2% HNO3 from a commercial spectroscopically pure 1000 mg/L Mn stock solution (SPC Science Cat. No. 140-051-250). Nine measurements were made for each sample. For each measurement, 20 μl of sample (or standard) was mixed with 5 μl of a 0.05% (w/v) Pd + 0.03% (w/v)Mg(NO3)2 matrix modifier and injected into the furnace with an automated sampler (Perkin-Elmer AS800). The temperature program used pyrolysis at 1300°C and atomization at 2000º C. Ultra-pure nitric acid (Seastar Chemicals Inc.; Sidney, BC, Canada) was used throughout sample preparation and standards and the matrix modifier was prepared from 10 g/L Pd and 10 g/L Mg(NO3)2 (Perkin-Elmer Part Nos. BO19-0635 and Bo 19-0634, respectively). Ultrapure water, resistivity 18 MΩ cm−1 obtained from a Barnstead/Thermolyne Diamond Life Science (Dubuque, IA)(UV/UF) ultrapure water system was used for all solutions.
2.8 Confocal Microscopy
AGMn3+Tf was used to follow the binding of Tf to the TfR on the plasma membrane of the neuronal cells and the passage of the Tf through the endosomes into the region of the mitochondrial network. Mitotracker red and Mitotracker green were used in different experiments to identify mitochondria within each neuronal cell. If Mitotracker red or green were to be used in the experiment, they were added to the medium above the cells and incubated for 20 min prior to mounting the cover slip on the stage of the confocal microscope. The neuronal cells had settled on and attached to a 15 mm cover slip, which was placed in a Warner Instruments RC-25F chamber. The imaging buffer used contained 137 mM NaCl, 0.56 mM MgCl2, 4.7 mM KCl, 1 mM Na2HPO4, 10 mM HEPES (pH 7.4), 5.5 mM glucose and 1.26 mM CaCl2. The RC-25F chamber containing the cover slip was mounted in a Warner Instruments Platform-Series 20 PH1 system on the microscope stage, which allowed fresh medium to flow into the RC-25F chamber over the cover slip. At a time designated as time zero the desired amount of AGMn3+Tf was added to the RC-25F chamber and allowed to incubate for 1 min. The unbound AGMn3+Tf was then washed away with fresh medium. The progress of the AGMn3+Tf in binding to the TfR, being incorporated into endosomes, and passing into the region of the cytosol near the mitochondrial network was then followed by periodically taking images of the cells. White light images and in some cases Z stacks were also taken to better show the cell boundaries (membranes) and the distribution of the probes within the cells.
Images were acquired simultaneously for each dye using a Nikon C1-LU3 visible light laser scanning confocal microscope with a 60× (1.4 NA) oil immersion objective. The suite of lasers and capture of images was controlled through EZ-C1 software. Alexa green 488 and mitotracker green were excited by the 488 nm argon laser line and emission was collected through a 543 nm bandpass filter and photomultiplier. Mitotracker red CMXRos was excited by the 543 nm line from a He-Ne laser, and emission was collected through a 585 ± 30 nm bandpass filter and photomultiplier.
2.9 Uptake of Mn3+Tf by Neuronal Cells
The protocol developed for washing the cells and preparing them for measurement of uptake by atomic absorption was very similar to that used to pass the cells and was designed to be both thorough and gentle. Cells of both types were grown to 70 – 80% of confluence in 6 well dishes containing 2.25 ml of medium per well (Gibco F-12 mixture (HAM) containing 5% FBS, 5% HS, 20 mM glucose, and Pen Strep) and throughout the following procedure kept at either 37° C for the HT22 cells or 33° C for the STHdhQ7/Q7 cells. During a change of medium (2.5 ml of fresh medium), 1.11 μM cyclosporin A was also added as insurance against induction of the mitochondrial permeability transition. After 15 minutes, in three separate experiments, either 100 μM purified Mn3+Tf (final concentration), 100 μM Mn2+ (chloride) (final concentration), or the amount of Mn2+ (chloride) measured by EPR at the end of the purification (0.2 μM), diluted by the same factor that the Mn3+Tf stock had been diluted to reach 100 μM (another factor of ten or 0.02 μM), was added to each well. At the appropriate time, the medium containing either Mn3+Tf or Mn2+ was removed with a pipette and the cells carefully washed twice with 2 ml of phosphate buffered saline (PBS) (137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, and 1.4 mM KH2PO4, pH 7.2). The cells were then removed from the dishes as follows: 0.25 mg/ml pronase in 1 ml of medium was added to the dish which was swirled gently and allowed to stand for 20 min. This medium was then removed and the dish was washed twice with PBS medium. At each step in this protocol the cells were inspected for damage by light microscopy. Cell medium containing 0.05% trypsin and 0.53 mM EDTA (as described above under “Neuronal Cells”) was added, and the cells were removed from the bottom of the well by the stream of medium from a transfer pipette. Next, the the trypsin-containing medium was transferred to an Eppendorf tube, and the cells were centrifuged in a Hermle centrifuge for 1 min at 10,000 rpm. The supernatant was then poured off and the pellet carefully rinsed 3 times with 1 ml of PBS. The cells were then resuspended in 1 ml of PBS. The centrifugation and rinsing steps were repeated once or twice. The resulting pellet was resuspended. Aliquots of some pellets were replated and inspected with a light microscope to ensure that the cells were intact and appeared healthy. The resuspended cells were lysed by suspension in 1 ml of double distilled water and maintained at 4° C until the end of the uptake experiment when all of the samples from the experiment were frozen. When all the samples were complete, they were thawed, carefully vortexed to insure proper mixing, and measured with AA and the Bradford assay, as described above.
2.10 Statistics
Means and standard deviations of the data and fitting lines to the data were determined using Excel© software. While it appears obvious that the points representing uptake from 100 μM Mn3+Tf and from 0.2 μM Mn2+ in Fig. 4 A and B and those representing uptake from 100 μM Mn3+Tf and 100 μM Mn2+ in Fig. 5 A and B are significantly different, this was quantified by the usual procedure of assuming the null hypothesis of a common population and showing that this hypothesis is rejected at above the 99% level. This was done by comparing the 24, 36, and 48 hour points in Fig. 4 and the 6 and 12 hour points in Fig. 5 using Willcoxon Mann-Whitney tests as described in “Statistical Methods” (Snedecor and Cochran, 1989). We used the Vassar Statistical Computation Web Site established by Prof. Richard Lowy (faculty.vassar.edu/lowy/utest.html).
Fig. 4. A comparison of the amount of Mn uptake of Mn3+Tf with that of the amount of Mn2+impurity measured in the Mn3+Tf sample in two neuronal cell lines.


A. Uptake of Mn into HT22 (hippocampal) neurons and B. STHdhQ7/Q7 (striatal) neurons using atomic absorption. Experiments showing this Mn uptake were carried out as described under “Uptake of Mn3+Tf by Neuronal Cells” in the Methods section. Uptake of Mn from 100 μM purified Mn3+Tf ( ) is compared with Mn uptake from 0.02 μM Mn2+ (chloride) (
) is compared with Mn uptake from 0.02 μM Mn2+ (chloride) ( ). 0.2 μM Mn2+ was the concentration of impurity Mn2+ estimated to be present in the Mn3+Tf stock from the EPR measurements. This was diluted by a further factor of ten in the low concentration Mn2+ experiment similar to the dilution of the Mn3+Tf stock when 100 μM of the purified Mn3+Tf was added to the neurons. The data represent averages of three AA measurements each from 2 independent experiments. Error bars represent ± 1 standard deviation. The lines fit to the data were determined by weighting each point by its reliability. Results of Willcoxon Mann-Whitney tests carried out on the 24, 36, and 48 hour points indicate that for both the HT22 data and the STHdhQ7/Q7 data, the probability that the 100 μM Mn3+Tf data is a separate population from the 0.2 μM Mn2+ data is approximately 99.9%. In other words uptake from impurity Mn2+ in the Mn3+Tf sample cannot explain the uptake observed from the Mn3+Tf sample.
). 0.2 μM Mn2+ was the concentration of impurity Mn2+ estimated to be present in the Mn3+Tf stock from the EPR measurements. This was diluted by a further factor of ten in the low concentration Mn2+ experiment similar to the dilution of the Mn3+Tf stock when 100 μM of the purified Mn3+Tf was added to the neurons. The data represent averages of three AA measurements each from 2 independent experiments. Error bars represent ± 1 standard deviation. The lines fit to the data were determined by weighting each point by its reliability. Results of Willcoxon Mann-Whitney tests carried out on the 24, 36, and 48 hour points indicate that for both the HT22 data and the STHdhQ7/Q7 data, the probability that the 100 μM Mn3+Tf data is a separate population from the 0.2 μM Mn2+ data is approximately 99.9%. In other words uptake from impurity Mn2+ in the Mn3+Tf sample cannot explain the uptake observed from the Mn3+Tf sample.
Fig. 5. A comparison of the amount of Mn uptake of Mn3+Tf with that of the same molar amount of Mn2+ in two neuronal cell lines.


A. Initial uptake measurements of Mn into HT22 (hippocampal) neurons and B. STHdhQ7/Q7 (striatal) neurons using atomic absorption comparing uptake from solutions containing 100 μM Mn3+Tf ( ) with that from 100 μM Mn2+ (chloride) (
) with that from 100 μM Mn2+ (chloride) ( ). Experiments were carried out as described under “Uptake of Mn3+Tf by Neuronal Cells” in the Methods section. The data represent averages of three AA measurements each from 2 independent experiments. Error bars represent ± 1 standard deviation. The lines fit to the data were determined by weighting each point by its reliability. Results of Willcoxon Mann-Whitney tests carried out on the 6 and 12 hour points indicate that for both the HT22 data and the STHdhQ7/Q7 data, the probability that the 100 μM Mn3+Tf data is a separate population from the 100 μM Mn2+ data is greater than 99.4%. This indicates that uptake of 100 μM Mn2+ is significantly greater than uptake from 100 μM Mn3+Tf in both these neuronal samples.
). Experiments were carried out as described under “Uptake of Mn3+Tf by Neuronal Cells” in the Methods section. The data represent averages of three AA measurements each from 2 independent experiments. Error bars represent ± 1 standard deviation. The lines fit to the data were determined by weighting each point by its reliability. Results of Willcoxon Mann-Whitney tests carried out on the 6 and 12 hour points indicate that for both the HT22 data and the STHdhQ7/Q7 data, the probability that the 100 μM Mn3+Tf data is a separate population from the 100 μM Mn2+ data is greater than 99.4%. This indicates that uptake of 100 μM Mn2+ is significantly greater than uptake from 100 μM Mn3+Tf in both these neuronal samples.
3. Results
3.1 Conversion of Mn2+ to Mn3+ through stabilization in a Tf complex
An experimental sample containing separately added Mn2+ (0.2 mM) and Tf (2.0 mM) was incubated at 22° C for 24 hr in HEPES (10 mM) (pH 7.2) buffered saline (160 mM NaCl). The sample was then frozen and maintained at −80° C until a XANES spectrum could be obtained (Fig. 1). The sample was analyzed with curve-fitting techniques in which its spectrum was compared with the spectra of the Mn2+ and Mn3+ model compounds shown in Fig. S2 A & S2 B in the Supplementary Material. These techniques are described in detail in four earlier papers (Gunter et al., 2006a; Gunter et al., 2005; Gunter et al., 2006b; Gunter et al., 2004). The results showed that after the 24 hr incubation, the sample had acquired approximately 18% Mn3+. This shows that Mn3+Tf does form, albeit slowly, from Mn2+ at neutral pH. It also shows that the Mn3+Tf complex is capable of holding Mn3+Tf stably over time.
3,2 Binding of Alexa green labeled Mn3+Tf to cell membranes, formation of endosomes and transport of the Alexa green labeled Mn3+Tf via the endosomes into the region of the mitochondrial network
As can be seen in Figs. 2A and 2B, AGMn3+Tf binds rapidly within a 1 min incubation period to the membranes of both HT22 neurons (Fig. 2A) and STHdhQ7/Q7 neurons (Fig. 2B). The green color shows the location of the Mn3+Tf, and the red color (Mitotracker red) shows the location of the mitochondrial network in these cells. These sets of images were taken 2 min (to account for the time required to wash away unbound AGMn3+Tf) and 16 min after addition of AGMn3+Tf. The 16 min images show that the green labeled vesicles have moved farther into the cell than they had at 2 min. Over the initial 16 min of these experiments, the movement of the AGMn3+Tf can be followed as the green fluorescence emission moves from the membrane towards the region of the mitochondrial network. Similar experiments, carried out without mitotracker dyes showed no essential difference in the behavior and movement of the green color (AG label). The mitotracker dyes were only used to indicate the area of the mitochondrial network and no attempt was made to determine whether mitochondria take up the Mn per se. As with Fe3+Tf, Mn3+Tf uptake likely takes place within endosomes (Hentze et al., 2004; Richardson and Ponka, 1997), although these organelles do not appear in the images Fig. 3A & B because they are smaller than the resolution of the optical system. The Z-plane of the images shown in Fig. 2A and 2B was about 0.5 μm above the cover slip. Apparent inclusion of a few green spots and even mitochondria in the nuclei in some of the cells indicates that a few green endosomes and mitochondria are trapped between the nuclear membrane and plasma membrane in these images.
Fig. 2. Following AGMn3+Tf uptake into cells using confocal microscopy.
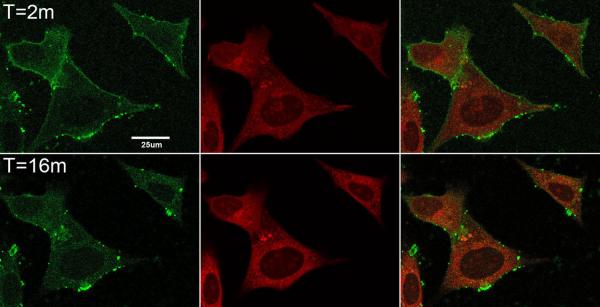
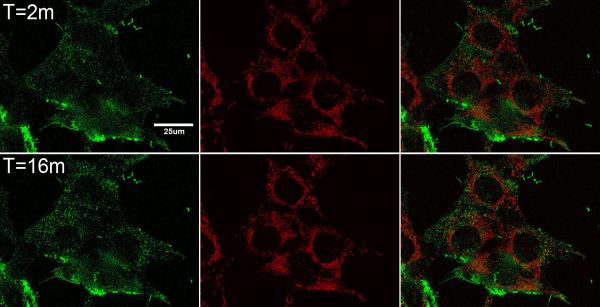
Confocal microscope images showing AGMn3+Tf and mitotracker red in A. HT22 (hippocampal neurons) and B. STHdhQ7/Q7 (striatal neurons). The images in each set from left to right are AGMn3+Tf (green) emission, mitotracker red (red) emission, and merged images. The top row in each set was taken 2 min after addition of 0.2 μM AGMn3+Tf while the bottom row was taken 16 min after addition of the AGMn3+Tf. Flow of medium through the cell holder was begun 1 min after addition of the AGMn3+Tf in order to remove unbound fluorescent probe from the system. Initially the AGMn3+Tf is found on the outside of the cell membranes but with time moves into the cell cytoplasm toward the mitochondrial network. The thickness of the Z planes in these images is between 0.15 and 0.3 μm.
Fig. 3. Competition between uptake of AGMn3+Tf and Fe3+Tf and inhibition of uptake by chlorpromazine and dynasore using confocal microscopy.


Uptake of AGMn3+Tf into the cell interior as measured by green emission ocurring within regions of interest drawn just inside the plasma membrane of A. HT22 (hippocampal) neurons and B. STHdhQ7/Q7 (striatal) neurons measured using “Image J” software obtained from NIH. 0.2 μM AGMn3+Tf was added at time zero in each of these experiments and excess AGMn3+Tf washed away with a flow of fresh medium at time 1 min with the flow of medium continuing for 1 min. Movement of the AGMn3+Tf into the neurons was determined by measuring the average emission intensity per unit area using Image J software inside the regions of interest drawn around the inside of the cell membrane as shown in supplementary figure S5. In these experiments, the regions of interest were drawn around the inside of the plasma membrane of each cell within the field of view and the average emission intensity per unit area taken and averaged over all of the cells. Control: 0.2 μM Mn3+Tf no other additions (●): 80 μM Fe3+Tf (∎) was added and washed away before time zero; 30 μM chlorpromazine (◆) was added 10 min prior to time zero; 80 μM dynasore (▴) was added 10 min prior to zero time. The error bars represent ± 1 standard deviation of three independent measurements. The curves through the data points represent fits to the equation y = m1 + m2(1-e−m3x) with each point weighted by its reliability and the values of m1, m2, and m3 determined by the fit.
The AG probe which had not reacted with the Tf molecule during synthesis of AGMn3+Tf was dialyzed away from the AGMn3+Tf using fresh medium (see “Attachment of AG488 to transferrin”), made non reactive, saved separately from the AGMn3+Tf and subsequently used as a control in experiments similar to those described in Fig. 2A and 2B above. This unattached AG could easily be seen before being washed away after 1 min of incubation with the cells; however, almost all of it was washed away when the cells were washed. There was no indication of extensive binding to the exterior of the cells and the formation of AG labeled endosomes as there was with the AGMn3+Tf treated samples.
Figs 3A and 3B show a time sequence of AGMn3+Tf uptake in typical samples of HT22 neurons (Fig. 3A) and STHdhQ7/Q7 neurons (Fig. 3B). Uptake was quantified by drawing regions of interest just inside the plasma membrane of these cells as shown in the Supplementary Material (Fig. S4) and using “Image J” software (available from the NIH) to show the relative amount of green emission from inside the cell as a function of time. The average intensity of this green fluorescence is shown quantitatively in Fig. 3A for HT22 neurons and in Fig. 3B for STHdhQ7/Q7 neurons. Because it takes at least 1 min to wash off the external AGMn3+Tf from the cover slip in the field of the confocal microscope, the first uptake data that can be analyzed in this way is 2 to 3 min after addition of the AGMn3+Tf to the medium above the neurons. By this time, the amount of AGMn3+Tf found inside the cells is already significant. It continues to increase for approximately 5 to 6 more min before complete saturation. Even though the intensity of the average green fluorescence inside the cells saturated at about 5 or 6 min after addition of the AGMn3+Tf probe, movement of the green fluorescence away from the cell membranes continued for another 10 or 11 min. Therefore, 16 min was chosen in the second row of panels in Fig. 2A and 2B to provide some information on this additional movement of the AGMn3+Tf.
3.3 Competition between Fe3+Tf and Mn3+Tf for transport via the Tf mechanism and inhibition of this transport by chlorpromazine and dynasore
Figs. 3A (HT22 hippocampal neurons) and 3B (STHdhQ7/Q7 striatal neurons) show strong competitive inhibition of AGMn3+Tf transport by Fe3+Tf added to the RC25F chamber prior to addition of the same amount of AGMn3+Tf used in acquiring the images shown in Fig. 2A and 2B and the quantitative data in Figs. 3A and 3B. However, the inhibition seems stronger for the STHdhQ7/Q7 striatal neurons. Chlorpromazine and dynasore are classical inhibitors of Fe3+Tf binding to the TfR and its subsequent incorporation into clathrin-coated pits and endosomes (Ivanov, 2008). Addition of these inhibitors (both from Sigma Chem. Co.; St. Louis, MO) to the RC25F chamber prior to addition of AGMn3+Tf greatly reduces uptake of AGMn3+Tf in both HT22 (Fig. 3A) and STHdhQ7/Q7 neurons (Fig. 3B), with stronger inhibition in the STHdhQ7/Q7 neurons. This finding provides further evidence that Mn3+ uptake occurs similarly to that of Fe3+ by the Tf mechanism.
3.4 A comparison of uptake of purified Mn3+Tf with uptake of Mn2+
3.4.1. Uptake of residual Mn2+ present as an impurity
Fig. 4A compares the uptake of purified Mn3+Tf into HT22 hippocampal neurons from a 100 μM solution of purified Mn3+Tf with that of Mn2+ from a 0.02 μM solution of Mn2+ using the AA technique described in the Methods section. 0.2 μM was the concentration of free Mn2+ measured as an impurity by EPR in the purified Mn3+Tf stock samples. This 0.2 μM was then diluted by an additional factor of ten to match the dilution of the Mn3+Tf stock in setting up the 100 μM Mn3+Tf samples. Fig. 4B shows results of the same experiment using STHdhQ7/Q7 striatal neurons. In both cases, uptake from the 100 μM Mn3+Tf solution was much greater than that from 0.02 μM Mn2+. Willcoxon Mann-Whitney tests in each case indicate significance at the 99.9% level. Thus the uptake from the 0.02 μM Mn2+ impurity cannot account for the uptake observed from the purified Mn3+Tf solutions. Therefore, the uptake from Mn3+Tf is real and significant. The actual uptake of Mn from the 100 μM Mn3+Tf samples should be thought of as the difference between these two curves.
3.4.2. Uptake rates of equal amounts of Mn3+Tf and Mn2+
Figs. 5A and 5B compare initial rates of uptake of Mn from 100 μM purified Mn3+Tf with that from the same molar concentration (100 μM) of Mn2+ in hippocampal and striatal neurons, respectively. The uptake of Mn2+ was markedly faster in both cases than that of transport via the Tf mechanism. That these data represent separate populations with uptake from 100 μM Mn2+ significantly greater than that from 100 μM Mn3+Tf was established using Willcoxon Mann-Whitney tests at the 99.4% level.
4. Discussion
While Fe uptake via the Tf mechanism has been thoroughly studied and described in a large number of publications over the last 30 years, the transport of Mn via this mechanism has not been systematically studied. Initial evidence that Mn could be transported into cells via the Tf mechanism was published over 20 years ago (Aschner and Aschner, 1990); however, many of the characteristics of this transport have remained speculative and significant differences may exist between Fe and Mn transport via this mechanism. For example, Mn2+ is the most stable state of free Mn at the pH found in extracellular fluid while Fe3+ is the most stable state of Fe under the same conditions (Latimer and Hildebrand, 1956). In addition, since Mn3+ (Standard Reduction Potential E = 1.51) is a much stronger oxidizing agent than Fe3+ (E= 0.77), there could be significant differences in the transport behavior of these two transition metals via this Tf mechanism (West, 1971). Steap proteins II through IV have evolved to reduce Fe3+ to Fe2+ within the endosomes so that the Fe2+ can be transported out of the endosomes via the DMT1 mechanism (Ohgami et al., 2005; Ohgami et al., 2006; Sheftel et al., 2007); however, Mn3+, is a strong enough oxidizing agent that it could oxidize endosomal components other than the Steap proteins, and in the process it could damage the endosomes and the cell. This type of damage would not be directly visible using confocal microscopy because of the small size of the endosomes, but it could constitute an additional mechanism of Mn toxicity. Mn transport via the Tf mechanism could also induce toxicity through the same mechanisms as transport of Mn2+ into the cell.
The XANES data shown in Fig. 1 clearly establish that Mn3+Tf can be slowly formed from Mn2+ and Tf at neutral pH in saline, thus strongly suggesting that this will also occur in extracellular fluid. The speed of formation of Mn3+Tf in extracellular fluid will undoubtedly vary with Mn and Tf concentrations, pH, and the concentration of other stabilizing compounds such as citrate.
The small green sources of emission seen moving into the cytosol in the AGMn3+Tf data shown in Fig. 2A and 2B appear similar to what would be expected if Mn3+ transport via the Tf mechanism involves endosomes and is a close analog of what Fe3+ transport via this mechanism is believed to be (Hentze et al., 2004; Ohgami et al., 2005; Ohgami et al., 2006; Richardson and Ponka, 1997; Sheftel et al., 2007).
The uptake rate of AGMn3+Tf shown in Fig. 3 A and B appears to be more rapid than the accumulation of Mn in the cells shown in Fig. 4 and 5; however, these experiments employed two different types of measurements and measured different things. Of the two types of measurement, fluorescence is much more sensitive than AA. The fluorescence data in Fig. 3 show the AG accumulation in the cell interior, which saturates when the cycling of the TfR between the endosomes and the plasma membrane reaches steady state. The AA data in Fig. 4 and 5 show the accumulation of Mn itself in the cells. It takes numerous cycles of transport via the Tf mechanism (as seen in the fluorescence experiments) to build up the amounts of Mn seen inside the cells by AA. Even this relatively slow Mn accumulation via the Tf mechanism inside the cells (Fig. 4 and 5) is undoubtedly faster than the Mn accumulation which would occur via the Tf mechanism in vivo, because the concentration of free Mn3+ prior to its binding to Tf in vivo is very low. Furthermore, because the iron concentration of the extracellular fluid is high, much of the Tf would be expected to be bound by Fe3+. Nevertheless, because the Mn3+ concentration is not zero, mass action would allow very small amounts of Mn3+ to bind to Tf and to be transported via the Tf mechanism (Vincent and Love, 2012). In the experiments shown in Fig. 4 and 5, we have avoided these concentration-induced bottlenecks by binding Mn3+ to Tf and purifying the complex prior to the experiments.
The data shown above (Fig. 3A and 3B) also show that Fe3+ competes with Mn3+ for uptake via the Tf mechanism and that this Mn3+Tf transport is inhibited by classical inhibitors of the clathrin-coated pit mechanism, chlorpromazine and dynasore, confirming that Mn transport is similar to Fe transport via the Tf mechanism (Ivanov, 2008).
In addition, the experiments comparing the rate of uptake of purified Mn3+Tf with that of Mn2+ at equal concentrations (Fig. 5 A & B) show that Mn3+Tf is transported via this classically described, Fe-specific mechanism, although it is taken up more slowly than a similar concentration of Mn2+ by one or more of the Mn2+-transporting mechanisms in both types of neurons. 100 μM Mn2+ is sequestered about 8.5 fold faster than the Mn3+Tf in HT22 hippocampal neurons and about 8.6 fold faster in STHdqQ7Q7 striatal neurons in these data. This is true even though the protocol used Mn3+ which had already been stabilized by binding to Tf, eliminating the step in which Mn3+ associates with the Tf, which must be a bottleneck to the process in vivo. Therefore, the uptake velocities measured here should not be taken as what would be seen in vivo, but should be taken to represent uptake into these two types of neurons via the Tf mechanism when the probable rate-limiting step is bypassed. Since the concentration of Mn2+ used in the uptake studies (100 μM) was in the upper end of the physiological range or lower end of the pathological range found in cells and tissue (Gunter et al., 2005; Gunter et al., 2004), it might be argued that there could be saturation effects influencing the uptake rate in the Mn2+ data. Even if this is true, the uptake of Mn2+ is so much faster than that of the Mn3+Tf at the same concentration that these two sets of data can be shown to represent two statistically distinct populations reinforcing the conclusion that uptake of Mn2+ into these cells is significantly greater than that of Mn3+Tf. These results and arguments suggest that Mn3+ uptake via the Tf mechanism into these types of neurons is small and much smaller than that of Mn2+ via other mechanisms.
The results of the experiments described here suggest that: 1) A significant amount of Mn3+ can be transported into neuronal cells via the Tf mechanism. 2) This transport shows characteristics similar to those of Fe3+ transport via this same mechanism. 3) The amount of this transport in the two lines of neuronal cells used was small and much less than transport of Mn2+ via other transport mechanisms. 4) In addition to contributing to the same mechanisms of toxicity as other mechanisms of Mn uptake into cells, because Mn3+ is a strong oxidizing agent, Mn3+ transport via the Tf mechanism, could also cause oxidative damage at the endosomal level. More complete answers to issues raised here such as transport kinetics, endosomal damage, and amounts of Mn3+Tf transport in other cell types (e.g. astrocytes) will require further study.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. William Bernhard for use of his EPR and UV-visible spectrometers and Mr. Paul Black for instructions on the use and help in operating these spectrometers. They would like to thank Dr. Marcy MacDonald for developing and permitting us to use the STHdhQ7/Q7 striatal neurons and Dr. Gail Johnson-Voll for providing us with these cells. They would like to thank Dr. Pamela Meher for providing us with the HT22 hippocampal neurons. They thank Dr. Beth Van Winkle for help with cells. They thank Dr. Lisa Miller and Dr. Nebojsa Marinkovic and other personnel of beamline 9B of the National Synchrotron Light Source (NSLS) at Brookhaven National Laboratory for use of their facilities and assistance with the XANES experiments. They thank Dr. Harold Smith for important discussions on techniques used in this work and Dr. Andrei Ivanov for helpful discussions about the clathrin-coated pit mechanism of formation of endosomes. They thank Dr. Claire Gavin for editing and suggestions for clarity in the wording of the manuscript and both Dr. William Simon and Ms. Kelly Gunter for consultations on statistics. Research described in this article was supported by NIH RO1 ES10041. The NSLS is supported by the United States Department of Energy under contract DE-AC02-98CH10886.
Abbreviations
- Tf
transferrin
- Mn3+Tf
Mn3+ bound to transferrin
- DMT1
divalent metal transporter 1
- XANES
X-ray absorption near edge structure spectroscopy
- TfRs
transferrin receptors
- EXAFS
extended X-ray absorption fine structure
- EPR
electron paramagnetic resonance
- Apo-Tf
Apotransferrin
- AA
atomic absorption
- AG
Alexa green
- AGMn3+Tf
Alexa green manganese transferrin
References
- Abragam A, Bleaney B. Electron Paramagnetic Resonance of Transition Ions. Clarendon Press; Oxford: 1970. [Google Scholar]
- Aisen P, Aasa R, Redfield AG. The chromium, manganese, and cobalt complexes of transferrin. J Biol Chem. 1969;244:4628–4633. [PubMed] [Google Scholar]
- Aschner M, Aschner JL. Manganese transport across the blood brain barrier: Relationship to iron homeostasis. Brain Res Bull. 1990;24:857–860. doi: 10.1016/0361-9230(90)90152-p. [DOI] [PubMed] [Google Scholar]
- Aschner M, Gannon M. Manganese (Mn) transport across the rat blood-brain barrier: Saturable and transferrin-independent mechanisms. Brain Res Bull. 1994;33:345–349. doi: 10.1016/0361-9230(94)90204-6. [DOI] [PubMed] [Google Scholar]
- Au C, Benedetto A, Aschner M. Manganese transport in eukaryotes: the role of DMT1. Neurotoxicology. 2008;29:569–576. doi: 10.1016/j.neuro.2008.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black PJ, Swarts SG. Ex vivo analysis of irradiated fingernails: chemical yields and properties of radiation-induced and mechanically-induced radicals. Health Physics. 2010;98:301–308. doi: 10.1097/HP.0b013e3181b0c045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouillet EP, Shinobu L, McGarvey U, Hochberg F, Beal MF. Manganese injection into the rat striatum produces excitotoxic lesions by impairing energy metabolism. Exp Neurol. 1993;120:89–94. doi: 10.1006/exnr.1993.1042. [DOI] [PubMed] [Google Scholar]
- Crossgrove JS, Allen DD, Bukaveckas BL, Rhineheimer SS, Yokel RA. Manganese distribution across the blood-brain barrier: I. Evidence for carrier-mediated influx of manganese citrate as well as manganese and manganese transferrin. Neurotoxicology. 2003;24:3–13. doi: 10.1016/s0161-813x(02)00089-x. [DOI] [PubMed] [Google Scholar]
- Crossgrove JS, Yokel RA. Manganese distribution across the blood-brain barrier: III. The divalent metal transporter-1 is not the major mechanism mediating brain manganese uptake. Neurotoxicology. 2004;25:451–460. doi: 10.1016/j.neuro.2003.10.005. [DOI] [PubMed] [Google Scholar]
- Finkelstein Y, Milatovic D, Aschner M. Modulation of cholinergic systems by manganese. Neurotoxicology. 2007;28:1003–1014. doi: 10.1016/j.neuro.2007.08.006. [DOI] [PubMed] [Google Scholar]
- Fujishiro H, Yano Y, Takada Y, Tanihara M, Himeno S. Roles of ZIP8, ZIP14, and DMT1 in transport of cadmium and manganese in mouse kidney proximal tubule cells. Metallomics. 2012;4:700–708. doi: 10.1039/c2mt20024d. [DOI] [PubMed] [Google Scholar]
- Galvani P, Fumagalli P, Santagostino A. Vulnerability of mitochondrial complex I in PC12 cells exposed to manganese. Eur J Pharmacol. 1995;293:377–383. doi: 10.1016/0926-6917(95)90058-6. [DOI] [PubMed] [Google Scholar]
- Gavin CE, Gunter KK, Gunter TE. Mn2+ sequestration by mitochondria and inhibition of oxidative phosphorylation. Toxicol Appl Pharmacol. 1992;115:1–5. doi: 10.1016/0041-008x(92)90360-5. [DOI] [PubMed] [Google Scholar]
- Girijashanker K, He L, Soleimani M, Reed JM, Li H, Liu Z, Wang B, Dalton TP, Nebert DW. Slc39a14 gene encodes ZIP14, a metal/bicarbonate symporter: similarities to the ZIP8 transporter. Mol Pharmacol. 2008;73:1413–1423. doi: 10.1124/mol.107.043588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunter KK, Aschner M, Miller LM, Eliseev R, Salter J, Anderson K, Gunter TE. Determining the oxidation states of manganese in NT2 cells and cultured astrocytes. Neurobiol Aging. 2006a;27:1816–1826. doi: 10.1016/j.neurobiolaging.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Gunter KK, Aschner MA, Miller LM, Eliseev R, Salter J, Anderson K, Hammond S, Gunter TE. Determining the oxidation states of manganese in PC12 and nerve growth factor-induced PC12 cells. Free Radic Biol Med. 2005;39:164–181. doi: 10.1016/j.freeradbiomed.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Gunter TE, Gavin CE, Aschner M, Gunter KK. Speciation of manganese in cells and mitochondria: A search for the proximal cause of manganese neurotoxicity. Neurotoxicology. 2006b;27:765–776. doi: 10.1016/j.neuro.2006.05.002. [DOI] [PubMed] [Google Scholar]
- Gunter TE, Miller LM, Gavin CE, Eliseev R, Salter J, Buntinas L, Alexandrov A, Hammond S, Gunter KK. Determination of the oxidation states of manganese in brain, liver, and heart mitochondria. J Neurochem. 2004;88:266–280. doi: 10.1046/j.1471-4159.2003.02122.x. [DOI] [PubMed] [Google Scholar]
- He L, Girijashanker K, Dalton TP, Reed J, Li H, Soleimani M, Nebert DW. ZIP8, member of the solute-carrier-39 (SLC39) metal-transporter family: characterization of transporter properties. Molecular Pharmacology. 2006;70:171–180. doi: 10.1124/mol.106.024521. [DOI] [PubMed] [Google Scholar]
- Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell. 2004;117:285–297. doi: 10.1016/s0092-8674(04)00343-5. [DOI] [PubMed] [Google Scholar]
- Ivanov AI. Pharmacological inhibition of endocytic pathways: Is it specific enough to be useful? Methods in Molecular Biology. 2008;440:15–33. doi: 10.1007/978-1-59745-178-9_2. [DOI] [PubMed] [Google Scholar]
- Latimer WM, Hildebrand JH. Reference Book of Inorganic Chemistry. The McMillan Company; New York: 1956. [Google Scholar]
- Liu Z, Li H, Soleimani M, Girijashanker K, Reed JM, He L, Dalton TP, Nebert DW. Cd2+ versus Zn2+ uptake by the ZIP8 HCO3-dependent symporter: kinetics, electrogenicity and trafficking. Biochemical & Biophysical Research Communications. 2008;365:814–820. doi: 10.1016/j.bbrc.2007.11.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malecki EA. Manganese toxicity is associated with mitochondrial dysfunction and DNA fragmentation in rat primary striatal neurons. Brain Res Bull. 2001;55:225–228. doi: 10.1016/s0361-9230(01)00456-7. [DOI] [PubMed] [Google Scholar]
- Malthankar GV, White BK, Bhushan A, Daniels CK, Rodnick KJ, Lai JC. Differential lowering by manganese treatment of activities of glycolytic and tricarboxylic acid (TCA) cycle enzymes investigated in neuroblastoma and astrocytoma cells is associated with manganese-induced cell death. Neurochem Res. 2004;29:709–717. doi: 10.1023/b:nere.0000018841.98399.ce. [DOI] [PubMed] [Google Scholar]
- Maynard LS, Cotzias GC. The partition of manganese among organs and intracellular organelles of the rat. J Biol Chem. 1955;214:489–495. [PubMed] [Google Scholar]
- Ohgami RS, Campagna DR, Greer EL, Antiochos B, McDonald A, Chen J, Sharp JJ, Fujiwara Y, Barker JE, Fleming MD. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nature Genet. 2005;37:1264–1269. doi: 10.1038/ng1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohgami RS, Campagna DR, McDonald A, Fleming MD. The Steap proteins are metalloreductases. Blood. 2006;108:1388–1394. doi: 10.1182/blood-2006-02-003681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson DS, Ponka P. The molecular mechanisms of the metabolism and transport of iron in normal and neoplastic cells. Biochim Biophys Acta. 1997;1331:1–40. doi: 10.1016/s0304-4157(96)00014-7. [DOI] [PubMed] [Google Scholar]
- Roth JA, Feng L, Walowitz J, Browne RW. Manganese-induced rat pheochromocytoma (PC12) cell death is independent of caspase activation. J Neurosci Res. 2000;61:162–171. doi: 10.1002/1097-4547(20000715)61:2<162::AID-JNR7>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Roth JA, Horbinski C, Higgins D, Lein P, Garrick MD. Mechanisms of manganese-induced rat pheochromocytoma (PC12) cell death and cell differentiation. Neurotoxicology. 2002;23:147–157. doi: 10.1016/s0161-813x(01)00077-8. [DOI] [PubMed] [Google Scholar]
- Sheftel AD, Zhang A-S, Brown C, Shirihai OS, Ponka P. Direct interorganellar transfer of iron from endosome to mitochondrion. Blood. 2007;110:125–132. doi: 10.1182/blood-2007-01-068148. [DOI] [PubMed] [Google Scholar]
- Snedecor GW, Cochran WG. Statistical Methods. Iowa State University Press; Ames, IA: 1989. [Google Scholar]
- Trettel F, Rigamonti D, Hilditch-Maguire P, Wheeler VC, Sharp AH, Persichetti F, Cattaneo E, MacDonald ME. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Human Mol Genet. 2000;9:2799–2809. doi: 10.1093/hmg/9.19.2799. [DOI] [PubMed] [Google Scholar]
- Vincent JB, Love S. The binding and transport of alternative metals by transferrin. Biochim Biophys Acta. 2012;1820:362–378. doi: 10.1016/j.bbagen.2011.07.003. [DOI] [PubMed] [Google Scholar]
- West RC, editor. CRC Handbook of Chemistry and Physics. the Chemical Rubber Co.; Cleveland, OH: 1971. [Google Scholar]
- Yokel RA, Crossgrove JS. Manganese toxicokinetics at the blood-brain barrier. Research Report - Health Effects Institute. 2004;119:7–73. [PubMed] [Google Scholar]
- Zwingmann C, Leibfritz D, Hazell AS. Energy metabolism in astrocytes and neurons treated with manganese: Relation among cell-specific energy failure, glucose metabolism, and intercellular trafficking using multinuclear NMR-spectroscopic analysis. J Cereb Blood Flow Metab. 2003;23:756–771. doi: 10.1097/01.WCB.0000056062.25434.4D. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.