

Abstract
Aims
Elevated levels of the mineralocorticoid hormone aldosterone are recognized as a modifiable contributor to the pathophysiology of select cardiovascular diseases due to left heart failure. In pulmonary arterial hypertension (PAH), pulmonary vascular remodelling induces right ventricular dysfunction and heart failure in the absence of left ventricular (LV) dysfunction. Hyperaldosteronism has emerged as a promoter of pulmonary vascular disease in experimental animal models of PAH; however, the extent to which hyperaldosteronism is associated with PAH in patients is unknown. Thus, the central aim of the current study is to determine if hyperaldosteronism is an unrecognized component of the PAH clinical syndrome.
Methods and results
Plasma aldosterone levels and invasive cardiopulmonary haemodynamic measurements were obtained for 25 patients referred for evaluation of unexplained dyspnoea or pulmonary hypertension. Compared with controls (n = 5), patients with PAH (n = 18) demonstrated significantly increased plasma aldosterone levels (1200.4 ± 423.9 vs. 5959.1 ± 2817.9 pg/mL, P < 0.02), mean pulmonary artery pressure (21.4 ± 5.0 vs. 45.5 ± 10.4 mmHg, P < 0.002), and pulmonary vascular resistance (PVR) (1.41 ± 0.6 vs. 7.3 ± 3.8 Wood units, P < 0.003) without differences in LV ejection fraction or pulmonary capillary wedge pressure between groups. Among patients not prescribed PAH-specific pharmacotherapy prior to cardiac catheterization, a subgroup of the cohort with severe pulmonary hypertension, aldosterone levels correlated positively with PVR (r = 0.72, P < 0.02) and transpulmonary gradient (r = 0.69, P < 0.02), but correlated inversely with cardiac output (r = –0.79, P < 0.005).
Conclusions
These data demonstrate a novel cardiopulmonary haemodynamic profile associated with hyperaldosteronism in patients: diminished cardiac output due to pulmonary vascular disease in the absence of LV heart failure.
Keywords: Aldosterone, Pulmonary hypertension, Right ventricle, Heart failure
Introduction
Aldosterone is a mineralocorticoid hormone synthesized in the zona glomerulosa of the adrenal gland primarily in response to a decrease in circulating blood volume. Elevated levels of aldosterone (inappropriate relative to dietary sodium) are associated with a vasculopathy that is characterized by increased perivascular fibrosis, intimal hypertrophy, and abnormal vascular reactivity.1,2 When present in patients with congestive heart failure and decreased cardiac output due to left ventricular (LV) systolic dysfunction, hyperaldosteronism is associated with increased rates of morbidity and mortality.3,4
Pulmonary arterial hypertension (PAH) (referred to formerly as primary pulmonary hypertension) is a severe disease in which negative remodelling of distal pulmonary arterioles results in increased vascular tone, right ventricular (RV) pressure loading, and, ultimately, irreversible right heart failure (cor pulmonale). We have demonstrated recently that hyperaldosteronism is present in established experimental animal models of end-stage PAH characterized by decreased cardiac output in the absence of LV dysfunction.5 In these experiments, pharmacological inhibition of aldosterone attenuated perivascular fibrosis to improve cardiopulmonary haemodynamics, suggesting that aldosterone may be an unrecognized (and potentially modifiable) contributor to the pathogenesis of syndromes characterized by dysfunction of the pulmonary circulatory–RV axis. The extent to which hyperaldosteronism is present in patients with PAH, however, is unknown. Thus, in the current study, we investigated the hypothesis that hyperaldosteronism is a component of clinical PAH owing to the adverse effect of aldosterone on pulmonary vascular remodelling and the consequent changes in cardiac output in patients with severe forms of this disease.
Methods
The study was approved by the Partners Human Research Committee and complies with the Declaration of Helsinki. Informed consent for study participation was obtained for 25 patients who were referred to the Pulmonary Hypertension Center at Brigham and Women's Hospital over a 4-year period for evaluation of unexplained dyspnoea or pulmonary hypertension detected by echocardiography, or to assess response to PAH-specific pharmacotherapy. For all study patients, a right heart catheterization was performed, and blood samples were acquired with the catheter in the pulmonary artery or pulmonary capillary wedge position. Haemodynamic criteria for the diagnosis of PAH were a history of right heart catheterization demonstrating a mean pulmonary artery pressure (mPAP) >25 mmHg and pulmonary vascular resistance (PVR) >3.0 Wood units in the setting of a pulmonary capillary wedge pressure (PCWP) <16 mmHg.6 Patients for whom cardiac catheterization data did not meet these criteria were considered controls, and it was confirmed through the medical record that control patients had not been diagnosed previously with PAH.
Sample processing
All patients underwent elective cardiac catheterization. Blood was drawn from the pulmonary artery or pulmonary capillary wedge position with the patient supine. The patient's dietary status was nil per os (nothing by mouth), and thus aldosterone sample collection occurred at least 6 h following dietary salt or medication intake for all patients. In all but one patient, blood samples were collected between 8 a.m. and 12 p.m. on the day of the procedure. Whole blood samples were immediately centrifuged at 1200 r.p.m. for 10 min at 4°C. The plasma was collected and immediately stored at –80°C. Aldosterone levels were analysed by immunoassay (Cayman; detection limit (80%B/Bo) 21 pg/mL, mean intra-assay variation 10.7%, interassay variation 15.4%) by an experienced laboratory scientist who was blinded to the patients' clinical data.
Imaging
Patients underwent a two-dimensional transthoracic echocardiogram as clinically indicated using standard ultrasound equipment from three major vendors (GE Healthcare, Milwaukee, WI, USA; Phillps, Bothel, WA, USA; and Siemens, Erlanger, Germany). A cardiologist certified in echocardiography and blinded to patient plasma aldosterone levels interpreted all studies using the Syngo Cardiac Reporting (Siemens Medical Solutions USA Inc., Mountain View, CA, USA) digital system. Left atrial and LV dimensions were measured in the parasternal long axis view, and LV ejection fraction (EF) was assessed using the biplane Simpson's method.
Statistical analyses
All statistical analyses were performed using Origin 8.1 (Northampton, MA, USA). Categorical variables are reported as frequencies with percentages. Continuous data are expressed as the mean ± SD. Comparisons between two groups were performed using the unpaired t-test. Univariate logistic regression analysis was used to assess the relationship between aldosterone and various cardiopulmonary haemodynamic measurements. Statistical significance was defined as P < 0.05.
Results
Patient demographics
Of the 25 patients enrolled in the study, 20 achieved haemodynamic criteria for PAH. The patient clinical characteristics are presented in Table 1. At the time of cardiac catheterization, none of the six study patients with systemic hypertension had laboratory evidence of hypokalaemia or hypomagnesaemia, which, if present, could be indicative of primary hyperaldosteronism. There was no significant difference in age between controls and PAH patients (50.0 ± 21.0 vs. 53.5 ± 13.6 years, P = 0.7), and in each group the majority of patients were women. The most common World Health Organization (WHO) functional class in the PAH and control cohorts was category I (i.e. unlimited physical activity) (80%) and category III (i.e. comfortable at rest and symptoms with minimal activities) (70%), respectively. All PAH patients had a WHO functional class of II (i.e. comfortable at rest and symptoms with ordinary activities), III, or IV (i.e. symptoms prevent any physical activity). Among patients in the PAH cohort, 18 (90%) were diagnosed with WHO category I pulmonary hypertension (e.g. PAH) and 2 patients had WHO category IV pulmonary hypertension (e.g, chronic thrombo-embolic pulmonary hypertension). The most common aetiology for PAH was connective tissue disease, which included scleroderma and rheumatoid arthritis (Table 2).
Table 1.
Clinical characteristics of the patient cohort at the time of cardiac catheterization
| Patient characteristic | Control (n = 5) | PAH (n = 20) |
|---|---|---|
| Female | 3 (60%) | 16 (80%) |
| Age (years) | 50.0 (28–84) | 53.5 (29–76) |
| WHO functional class | ||
| 1 | 4 (80%) | 0 |
| 2 | 0 | 4 (20%) |
| 3 | 1 (20%) | 14 (70%) |
| 4 | 0 | 2 (10%) |
| Co-morbidities | ||
| Congenital heart disease | 1 | 1 |
| Asthma | 1 | 1 |
| Interstitial lung disease | 1 | 3 |
| Bronchopulmonary dysplasia | 1 | 0 |
| Coronary artery disease | 1 | 1 |
| Atrial fibrillation | 0 | 4 |
| Connective tissue disease | 0 | 5 |
| Systemic hypertension | 0 | 6 |
| Obstructive sleep apnoea | 1 | 5 |
| Liver cirrhosis | 0 | 2 |
PAH, pulmonary arterial hypertension; WHO, World Health Organization.
Table 2.
Clinical profile of pulmonary arterial hypertension patients (n = 20) at the time of cardiac catheterization
| n | |
|---|---|
| WHO category | |
| 1 | 18 |
| 2 | 0 |
| 3 | 0 |
| 4 | 2 |
| 5 | 0 |
| PAH aetiology | |
| Connective tissue disease | 9 |
| Idiopathic PAH | 6 |
| Portopulmonary hypertension | 2 |
| Chronic thrombo-embolic pulmonary hypertension | 2 |
| Congenital heart disease | 1 |
PAH, pulmonary arterial hypertension; WHO, World Health Organization.
The medication profile for patients at the time of cardiac catheterization is provided in Figure 1. In the PAH cohort, nine patients (45%) were prescribed PAH-specific pharmacotherapy, defined as a phosphodiesterase type-5 inhibitor, dihydropyridine calcium channel antagonist, endothelin receptor antagonist, or prostacyclin analogue. In these patients, PAH-specific therapy was initiated antecedent to study enrolment, based on strong clinical evidence or prior cardiac catheterization suggesting a diagnosis of PAH. In the control group, one patient was prescribed a dihydropyridine calcium channel antagonist, although in this case the indication for treatment was essential hypertension.
Figure 1.

Patient medications at the time of cardiac catheterization. ACE-I, angiotensin-converting enzyme inhibitor; ARB, angiotensin receptor blocker; ETR, endothelin receptor antagonist; HMG-CoA reductase inhibitor (‘statin’), 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitor; PAH, pulmonary arterial hypertension; PDE-5, phosphodiesterase type-5 inhibitor; PGI2, prostacyclin I2.
Haemodynamics
Compared with controls, PAH patients demonstrated significantly increased pulmonary artery systolic pressure (PASP) (30.9 ± 5.1 vs. 69.5 ± 15.5 mmHg, P < 0.0001), mPAP (21.4 vs. 45.5 ± 10.4 mmHg, P < 0.002), and PVR (1.41 ± 0.6 vs. 7.3 ± 3.8 Wood units, P < 0.003) (Table 3). Abnormal cardiopulmonary haemodynamics in PAH patients were not secondary to LV systolic dysfunction or left heart remodelling: compared with controls, no significant differences were observed in PAH patients with respect to LVEF (61.4 ± 6.1% vs. 65.8 ± 5.9%, P = 0.15), LV end-diastolic dimension (45.0 ± 9.3 vs. 42.1 ± 8.1 mm, P = 0.56), left atrial size (38.4 ± 6.0 vs. 37.2 ± 7.6 mm, P = 0.74), or PCWP (12.4 ± 6.0 vs. 12.8 ± 6.2 mmHg, P = 0.91).
Table 3.
The haemodynamic profile of the patient cohort assessed by right heart catheterization
| Measure | Control (SD) | PAH (SD) | P-value |
|---|---|---|---|
| HR (b.p.m.) | 69.2 (23.6) | 77.6 (16.9) | 0.36 |
| MAP (mmHg) | 96.8 (8.3) | 96.2 (15.3) | 0.93 |
| RAP (mmHg) | 8.0 (4.2) | 12.7 (7.7) | 0.2 |
| PASP (mmHg) | 30.9 (5.1) | 69.5 (15.5) | <0.0001 |
| PADP (mmHg) | 13.8 (7.1) | 29.5 (9.4) | <0.002 |
| mPAP (mmHg) | 21.4 (5.0) | 45.5 (10.4) | <0.0001 |
| PCWP | 12.4 (6.0) | 12.8 (6.2) | 0.91 |
| CO (L/min) | 6.4 (0.8) | 5.1 (1.5) | 0.09 |
| CI (L/min/m2) | 3.4 (0.4) | 2.7 (0.6) | 0.06 |
| PVR (Wood units) | 1.41 (0.6) | 7.3 (3.8) | <0.003 |
| SVR (dyn/s/cm5) | 1123.9 (196.1) | 1439.3 (570.9) | 0.24 |
CI, cardiac index; CO, cardiac output; HR, heart rate; MAP, mean arterial pressure; mPAP, mean pulmonary arterial pressure; PADP, pulmonary artery diastolic pressure; PAH, pulmonary arterial hypertension; PASP, pulmonary artery systolic pressure; PCWP, mean pulmonary capillary wedge pressure; PVR, pulmonary vascular resistance; RAP, right atrial pressure; SVR, systemic vascular resistance.
Aldosterone and cardiopulmonary haemodynamics
The mean concentration of plasma aldosterone was elevated significantly in PAH patients (1200.4 ±423.9 vs. 8935.7 ±9545.8 pg/mL, P < 0.05). In two PAH patients, aldosterone levels were >30-fold above the mean value for controls, raising a suspicion of sample contamination, sample handling error, or the presence of confounding (and undiagnosed) clinical conditions in these patients with which to account for these results. Thus, additional analyses were performed with these patients excluded. Among the remaining cohort, a 4.8-fold increase in plasma aldosterone levels was observed in the PAH group compared with controls (5959.1 ±2817.9 vs. 1200.4 ±423.9 pg/mL, P < 0.02) (Figure 2A).
Figure 2.

Elevated levels of aldosterone (ALDO) in pulmonary arterial hypertension (PAH) are associated with increased mean pulmonary arterial pressure (mPAP). (A) Plasma samples acquired from the pulmonary artery or pulmonary capillary wedge position during right heart catheterization were analysed for levels of aldosterone by enzyme immunoassay. (B) The relationship between plasma aldosterone levels and mPAP among controls (n = 5) and patients with PAH (n = 18).
Among the entire patient cohort, we observed a trend towards a modest, positive correlation between plasma aldosterone levels and mPAP (r = 0.38, P = 0.07) (Figure 2B). To explore this trend further, we next analysed the relationship between aldosterone and cardiopulmonary haemodynamics in patients undergoing cardiac catheterization without prior use of PAH-specific pharmacotherapy (treatment naïve), a subpopulation of PAH patients likely to demonstrate severe pulmonary hypertension associated with decreased cardiac output. We observed that compared with patients prescribed PAH-specific pharmacotherapy at the time of cardiac catheterization (n = 14), PVR was significantly greater among treatment-naïve PAH patients (n = 6) (6.1 ±3.1 vs. 10.6 ±4.0 Wood units, P < 0.04), confirming our hypothesis that PAH treatment-naïve patients represented a segment of the cohort with more advanced pulmonary vascular disease.
Among controls and treatment-naïve PAH patients, a positive correlation between plasma aldosterone levels and PVR (r = 0.71, P < 0.02) (Figure 3A) and between aldosterone and transpulmonary gradient (calculated by the difference between mPAP and PCWP) was observed (r = 0.68, P < 0.02) (Figure 3B). Furthermore, no significant differences were observed between controls and PAH treatment-naïve patients with respect to PCWP or LVEF, and aldosterone levels did not correlate with either of these indices. Despite these findings suggesting no effect of PCWP or LVEF on plasma aldosterone levels, however, we observed a strong, inverse correlation between plasma aldosterone levels and cardiac output (r = –0.79, P < 0.005) (Figure 4A). The interrelatedness of cardiac output and PVR as a function of plasma aldosterone levels is illustrated in Figure 4B. Collectively, these data implicate diminished cardiac output due to pulmonary vascular disease, RV failure, or both as potential mechanism(s) by which to account for hyperaldosteronism in PAH.
Figure 3.

Aldosterone (ALDO) levels correlate positively with haemodynamic measures indicative of pulmonary vascular remodelling in pulmonary arterial hypertension (PAH). The relationship between plasma aldosterone levels and (A) pulmonary vascular resistance (PVR) and (B) transpulmonary gradient is illustrated for PAH-specific therapy-naïve patients (n = 6) and controls (n = 5). WU, Wood units.
Figure 4.
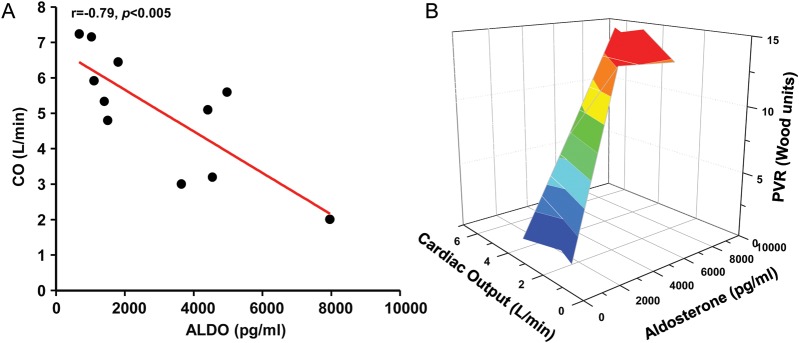
Aldosterone levels are inversely associated with cardiac output (CO) in pulmonary arterial hypertension (PAH). (A) The relationship between plasma aldosterone (ALDO) levels and CO is illustrated for pulmonary arterial hypertension (PAH)-specific therapy-naïve patients (n = 6) and controls (N = 5). (B) A three-dimensional matrix surface plot demonstrates the interrelatedness of CO (x-axis) and pulmonary vascular resistance (PVR) (y-axis) as a function of plasma aldosterone levels (z-axis) for the PAH-specific therapy-naïve controls and PAH patients. Graded changes in surface colour represent incremental increases in PVR.
Discussion
Under physiological conditions, aldosterone is synthesized by zona glomerulosa cells in the adrenal gland to maintain intravascular sodium, potassium, and water homeostasis.7 In turn, pathophysiological concentrations of circulating aldosterone levels may be observed in the presence of hormone-producing tumours (i.e. primary hyperaldosteronism) or due to diminished cardiac output that up-regulates the renin–angiotensin–aldosterone signalling pathway (i.e. secondary hyperaldosteronism). Hyperaldosteronism inappropriate relative to dietary sodium intake has been reported previously in association with selected cardiovascular diseases, including in patients with hypertension due to an adrenal adenoma8,9 (i.e. renin-independent hyperaldosteronism), or congestive heart failure due to impaired LV systolic/diastolic function10,11 (i.e. renin-dependent hyperaldosteronism), in which plasma aldosterone levels may be increased by ∼60-fold compared with normal.3 Likewise, the adverse consequences of hyperaldosteronism on clinical outcome are well established: plasma aldosterone levels associate positively with mortality in patients presenting with acute ST-segment elevation myocardial infarction,12 whereas prospective, placebo-controlled, randomized clinical trials have demonstrated that in the setting of LV systolic dysfunction, pharmacological inhibition of the mineralocorticoid receptor with eplerenone or spironolactone offers a significant survival benefit in patients with mild or severe congestive heart failure, respectively.4,13 As a result of these studies, mineralocorticoid receptor antagonists were given a Class IA recommendation by the European Society of Cardiology (ESC) for use in heart failure patients with New York Heart Association (NYHA) functional class III or IV and LVEF <35%.14 Furthermore, the Aldosterone receptor blockade in Diastolic Heart Failure (Aldo-DHF)15 trial and Treatment of Preserved Cardiac Function Heart Failure With an Aldosterone Antagonist Trial (TOPCAT) (NCT00094302, clinicaltrials.gov) are ongoing randomized, placebo-controlled clinical trials designed to assess the clinical efficacy of spironolactone in patients with symptomatic heart failure with preserved LVEF. Indeed, early results reported from the Aldo-DHF study suggest that independent of blood pressure-lowering effects, spironolactone attenuates LV remodelling and improves echocardiographically assessed measures of LV diastology in this patient population.16
In the current study, we performed a single-institution retrospective patient cohort analysis to demonstrate that plasma levels of aldosterone are markedly elevated in the pulmonary arteriole circulation in patients with PAH compared with controls. Our findings further indicate an inverse association between hyperaldosteronism and diminished cardiac output in PAH that was not a consequence of LV systolic dysfunction or negative remodelling of the left heart. These observations are in agreement with recently published data demonstrating increased levels of angiotensin II in the peripheral venous circulation in patients with idiopathic PAH;17 however, investigations assessing the relationship between aldosterone and PAH per se are limited to case reports18 and other studies in which patient haemodynamic data were not systematically reported.19 Thus, in these earlier studies, the possibility that pulmonary hypertension was due to cardiovascular diseases other than PAH could not be excluded. In the current study, PAH was confirmed in patients by haemodynamic assessment, and, in most cases, the presence of a clinical entity known to be associated with the development of PAH.
Our findings that demonstrate a positive correlation between plasma aldosterone levels and PVR and transpulmonary gradient provide evidence to support a novel link between aldosterone and pulmonary vascular dysfunction in patients with PAH. We also observed that aldosterone levels were inversely correlated with cardiac output in the absence of left heart failure. Taken together, these data support previous observations in various large animal models demonstrating that mechanical restriction to RV cardiac output (e.g. pulmonary artery band ligation) results in renin-dependent secondary hyperaldosteronism20 and supports our assertion that hyperaldosteronism is an unrecognized pathophysiological sequelae of pulmonary circulatory–RV axis dysfunction in patients with pulmonary vascular disease.
Nevertheless, alternative mechanisms by which to account for hyperaldosteronism in this patient cohort cannot be fully excluded. For example, in our study, some patients with PAH also had obstructive sleep apnoea,21 systemic hypertension,22 and atrial fibrillation,23 which have been linked previously to clinically evident hyperaldosteronism in the absence of significantly decreased cardiac output. Additionally, differences in patients' relative salt balance and/or circadian factors that regulate adrenal aldosterone synthesis, which could influence plasma aldosterone levels, were not explicitly measured in this study.24,25
In patients with impaired LV systolic function and moderate to severe congestive heart failure, aldosterone is a bona fide treatment target to improve quality of life and decrease mortality.13,26 The contribution of aldosterone to the clinical expression of pulmonary vascular disease in the setting of normal LV systolic function, however, is unknown. We have demonstrated recently that in inflammatory and angioproliferative experimental rodent models of PAH, increases in plasma aldosterone levels akin to those observed in PAH patients in the current study were sufficient to promote perivascular fibrosis of distal pulmonary arterioles, pulmonary vascular dysfunction, and pulmonary hypertension that was associated with RV remodelling and decreased cardiac output.5 Moreover, renin-dependent hyperaldosteronism is observed in patients with normal LV systolic function and decreased cardiac output due to constrictive pericarditis,27 heart failure due to right-sided congenital heart disease,28 and cor pulmonale due to primary lung disease.29 In these and other similar clinical scenarios, including PAH, the functional consequences of hyperaldosteronism to disease pathophysiology in patients is unresolved. Nevertheless, our current findings support these prior observations and suggest that in humans with PAH, RV dysfunction vis-à-vis pulmonary vascular remodelling is an important pathophysiological mechanism promoting hyperaldosteronism. Importantly, our findings do not establish a direct cause and effect relationship between hyperaldosteronism and PAH, or a temporal pattern by which aldosterone levels are increased in this disease. Thus, results from this study do not elucidate further whether hyperaldosteronism promotes PAH disease progression or occurs as a secondary consequence of congestive right heart failure.
Important limitations to the current pilot study include low patient enrolment and the absence of a healthy control population. The former may be explained, in part, by the low prevalence of PAH in the general population.6 In terms of the latter, important ethical considerations prohibit recruitment of individuals for invasive cardiac haemodynamic assessment in the absence of a clinical indication. Nevertheless, in this study, low patient enrolment may have contributed to a wide distribution of plasma aldosterone values in patients with PAH, thereby potentially introducing bias to our results. Moreover, data from laboratory and imaging tests that are necessary to diagnose primary hyperaldosteronism in our patients were unavailable. Thus, we are unable to establish definitively that patients in this study were free from competing causes of hyperaldosteronism independent of cardiovascular and/or pulmonary vascular disease.
As an additional limitation, we did not measure plasma levels of angiotensin II or endothelin-1, both of which are stimulators of adrenal aldosterone synthesis5,30 and are linked to pulmonary hypertension and RV heart failure.31,32 Thus, the precise biological mechanism resulting in hyperaldosteronism in the study cohort was not defined.
Collectively, our findings demonstrate a novel cardiopulmonary haemodynamic profile associated with hyperaldosteronism in patients: diminished cardiac output due to pulmonary vascular disease in the absence of LV dysfunction. Additional clinical studies are needed to define further the consequences of aldosterone to PAH pathophysiology and to assess the extent by which aldosterone modulates PAH progression and/or clinical outcome in patients afflicted with this and other clinical syndromes of pulmonary vascular dysfunction and right heart failure.
Funding
The US National Institutes of Health (grants HL105301 to J.A.L.; HL61795, HL48743, HL107192, HL070819, and HL108630 to J.L.); the American Heart Association (11POST6720000 to B.A.M.); the Lerner Foundation at Brigham and Women's Hospital (to B.A.M.).
Conflict of interest: none declared.
References
- 1.Maron BA, Zhang YY, Handy DE, Beuve A, Tang SS, Loscalzo J, Leopold JA. Aldosterone increases oxidant stress to impair guanylyl cyclase activity by cysteinyl thiol oxidation in vascular smooth muscle cells. J Biol Chem. 2009;284:7665–7672. doi: 10.1074/jbc.M809460200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leopold JA, Dam A, Maron BA, Scribner AW, Liao R, Handy DE, Stanton RC, Pitt B, Loscalzo J. Aldosterone impairs vascular reactivity by decreasing glucose-6-phosphate dehydrogenase activity. Nat Med. 2007;13:189–197. doi: 10.1038/nm1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weber KT. Aldosterone in congestive heart failure. N Engl J Med. 2001;345:1689–1697. doi: 10.1056/NEJMra000050. [DOI] [PubMed] [Google Scholar]
- 4.Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, Palensky J, Wittes J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure: Randomized Aldactone Evaluation Study Investigators. N Eng J Med. 1999;341:709–717. doi: 10.1056/NEJM199909023411001. [DOI] [PubMed] [Google Scholar]
- 5.Maron BA, Zhang Y-Y, White K, Chan SY, Handy DE, Mahoney CE, Loscalzo J, Leopold J. Aldosterone inactivates the endothelin-B receptor via a cysteinyl thiol redox switch to decrease pulmonary endothelial nitric oxide level and modulate pulmonary arterial hypertension. Circulation. 2012;126:963–974. doi: 10.1161/CIRCULATIONAHA.112.094722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McLaughlin, Archer SL, Badesch DB, Barst RJ, Farber HW, Lindner JR, Mathier MA, McGoon MD, Park MH, Rosenson RS, Rubin RJ, Tapson VF, Varga J. ACCF/AHA 2009 Expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association. J Am Coll Cardiol. 2009;53:1573–1619. doi: 10.1016/j.jacc.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 7.Maron BA, Leopold JA. Aldosterone receptor antagonists: effective but often forgotten. Circulation. 2010;121:934–939. doi: 10.1161/CIRCULATIONAHA.109.895235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wanta SM, Basina M, Chang SD, Chang DT, Ford JM, Greco R, Kingham K, Merritt RE, Kunz PL. A rare case of an aldosterone secreting metastatic adrenocortical carcinoma and papillary thyroid carcinoma in a 31-year-old male. Rare Tumors. 2011;3:e45. doi: 10.4081/rt.2011.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blacher J, Amah G, Girerd X, Kheder A, Ben Mais H, London GM, Safar ME. Association between increased plasma levels of aldosterone and decreased systemic arterial compliance in subject with essential hypertension. Am J Hypertens. 1997;10:1326–1334. doi: 10.1016/s0895-7061(97)00301-4. [DOI] [PubMed] [Google Scholar]
- 10.Mizuno Y, Yoshimura M, Yasue H, Sakamoto T, Ogawa H, Kugiyama K, Harada E, Nakayama M, Nakamura S, Ito T, Shimasaki Y, Saito Y, Nakao K. Aldosterone production is activated in failing ventricles in humans. Circulation. 2001;103:72–77. doi: 10.1161/01.cir.103.1.72. [DOI] [PubMed] [Google Scholar]
- 11.Rouleau JL, Packer M, Moyé L, de Champlain J, Bichet D, Klein M, Rouleau JR, Sussex B, Arnold JM, Sestier F, Parker JO, McEwan P, Bernstein V, Cuddy TE, Lamas G, Gottlieb SS, McCans J, Nadeau C, Delage F, Chuan-Chuan CW, Pfeffer MA. Prognostic value of neurohormonal activation in patients with acute myocardial infarction: effect of captopril. J Am Coll Cardiol. 1994;24:583–591. doi: 10.1016/0735-1097(94)90001-9. [DOI] [PubMed] [Google Scholar]
- 12.Beygui F, Collet JP, Benoliel JJ, Vignolles N, Dumaine R, Barthélémy O, Montalescot G. High plasma aldosterone levels on admission are associated with death in patients presenting with acute ST-segment myocardial infarction. Circulation. 2006;114:2604–2610. doi: 10.1161/CIRCULATIONAHA.106.634626. [DOI] [PubMed] [Google Scholar]
- 13.Zannad F, McMurray JJ, Krum H, van Veldhuisen DJ, Swedberg K, Shi H, Vincent J, Pocock SJ, Pitt B EMPHASIS-HF Study Group. Eplerenone in patients with systolic heart failure and mild symptoms. N Eng J Med. 2011;364:11–21. [Google Scholar]
- 14.McMurray JJ, Adamopoulos S, Anker SD, Auricchio A, Böhm M, Dickstein K, Falk V, Filippatos G, Fonseca C, Sanchez MA, Jaarsma T, Køber L, Lip GY, Maggioni AP, Parkhomenko A, Pieske BM, Popescu BA, Rønnevik PK, Rutten FH, Schwitter J, Seferovic P, Stepinska J, Trindade PT, Voors AA, Zannad F, Zeiher A, Bax JJ, Baumgartner H, Ceconi C, Dean V, Deaton C, Fagard R, Funck-Brentano C, Hasdai D, Hoes A, Kirchhof P, Knuuti J, Kolh P, McDonagh T, Moulin C, Popescu BA, Reiner Z, Sechtem U, Sirnes PA, Tendera M, Torbicki A, Vahanian A, Windecker S, McDonagh T, Sechtem U, Bonet LA, Avraamides P, Ben Lamin HA, Brignole M, Coca A, Cowburn P, Dargie H, Elliott P, Flachskampf FA, Guida GF, Hardman S, Iung B, Merkely B, Mueller C, Nanas JN, Nielsen OW, Orn S, Parissis JT, Ponikowski P Authors/Task Force Members. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: the Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail. 2012;14:803–869. doi: 10.1093/eurjhf/hfs105. ESC Committee for Practice Guidelines (CPG).Document Reviewers. [DOI] [PubMed] [Google Scholar]
- 15.Edelmann F, Schmidt AG, Gelbrich G, Binder L, Hermann-Lingen C, Halle M, Hasenfuss G, Wachter R, Pieske B. Rationale and design of the ‘aldosterone receptor blockade in diastolic heart failure’ trial: a double-blind, randomized, placebo-controlled, parallel group study to determine the effects of spironolactone on exercise capacity and diastolic function in patients with symptomatic diastolic heart failure (Aldo-DHF) Eur J Heart Fail. 2010;12:874–882. doi: 10.1093/eurjhf/hfq087. [DOI] [PubMed] [Google Scholar]
- 16.Edelmann F, Wachter R, Schmidt A, Kraigher-Krainer E, Colantonio C, Kamke W, Duvinage A, Stahrenberg R, Dustewitz K, Löffler M, Düngen H-D, Tschöpe, Herrmann-Lingen C, Halle M, Hasenfuss G, Gelbrich G, Pieske B. Aldosterone receptor blockade I diastolic heart failure: the Aldo-DHF trial (Abstract) European Heart Society Meeting. August 2012; Munich, Germany. [Google Scholar]
- 17.de Man FS, Tu L, Handoko ML, Rain S, Ruiter G, François C, Schalij I, Dorfmüller P, Simonneau G, Fadel E, Perros F, Boonstra A, Postmus PE, van der Velden J, Vonk-Noordegraaf A, Humbert M, Eddahibi S, Guignabert C. Dysregulated renin–angiotensin–aldosterone system contributes to pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012 doi: 10.1164/rccm.201203-0411OC. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kokubu T, Kazatani Y, Hamada M, Matsuzaki K, Ito T, Nishimura K, Ochi T, Daimon F, Joh T. Is captopril effective in primary pulmonary hypertension? Jpn Circ J. 1982;46:1095–1097. doi: 10.1253/jcj.46.1095. [DOI] [PubMed] [Google Scholar]
- 19.Martyniuk TV, Chazova IE, Masenko VP, Volkov VN, Belenkov Iu N. [Activity of renin–angiotensin–aldosterone system (RAAS) and vasopressin level in patients with primary pulmonary hypertension] Ter Arkh. 1998;70:33–36. [PubMed] [Google Scholar]
- 20.Davis JO, Olichney MJ, Brown TC, Binnion PF. Metabolism of aldosterone in several experimental situations with altered aldosterone secretion. J Clin Invest. 1965;44:1433–1441. doi: 10.1172/JCI105249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Calhoun DA, Nishizaka MK, Zaman MA, Harding SM. Aldosterone excretion among subjects with resistant hypertension and symptoms of sleep apnea. Chest. 2004;125:112–117. doi: 10.1378/chest.125.1.112. [DOI] [PubMed] [Google Scholar]
- 22.Calhoun DA, White WB, Krum H, Guo W, Bermann G, Trapani A, Lefkowitz MP, Ménard J. Effects of a novel aldosterone synthase inhibitor for treatment of primary hypertension: results of a randomized, double-blind, placebo- and active-controlled phase 2 trial. Circulation. 2011;124:1945–1955. doi: 10.1161/CIRCULATIONAHA.111.029892. [DOI] [PubMed] [Google Scholar]
- 23.Khatib R, Joseph P, Briel M, Yusuf S, Healey J. Blockade of renin–angiotensin–aldosterone system (RAAS) for primary prevention of non-valvular atrial fibrillation: a systematic review and meta analysis of randomized controlled trials. Int J Cardiol. 2012 doi: 10.1016/j.ijcard.2012.02.009. doi:10.1016/j.ijcard.2012.02.009. Published online ahead of print 13 March 2012. [DOI] [PubMed] [Google Scholar]
- 24.Nikolaeva S, Pradervand S, Centeno G, Zavadova V, Tokonami N, Maillard M, Bonny O, Firsov D. The circadian clock modulates renal sodium handling. J Am Soc Nephrol. 2012;23:1019–1026. doi: 10.1681/ASN.2011080842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bolbrinker J, Beige J, Huber M, Sharma AM, Thomas A, Deter HC, Kreutz R. Role of CYP2C9 genetic variants for salt sensitivity and the regulation of the renin–angiotensin–aldosterone system in normotensive men. J Hypertens. 2011;29:56–61. doi: 10.1097/HJH.0b013e32833f5de5. [DOI] [PubMed] [Google Scholar]
- 26.Farquharson CA, Struthers AD. Aldosterone induces acute endothelial dysfunction in vivo in humans: evidence for an aldosterone-induced vasculopathy. Clin Sci (Lond) 2002;10:425–431. doi: 10.1042/cs1030425. [DOI] [PubMed] [Google Scholar]
- 27.Anand IS, Ferrari R, Kalra GS, Wahi PL, Poole-Wilson PA, Harris PC. Pathogenesis of edema in constrictive pericarditis. Studies of body water and sodium, renal function, hemodynamics, and plasma hormones before and after pericardiectomy. Circulation. 1991;83:1880–1887. doi: 10.1161/01.cir.83.6.1880. [DOI] [PubMed] [Google Scholar]
- 28.Anand IS, Chandrashekhar Y, Ferrari R, Sarma R, Guleria R, Jindal SK, Wahi PL, Poole-Wilson PA, Harris P Pathogenesis of congestive state in chronic obstructive pulmonary disease. Studies of body water and sodium, renal function, hemodynamics, and plasma hormones during edema and after recovery. Circulation. 1992;86:12–21. doi: 10.1161/01.cir.86.1.12. [DOI] [PubMed] [Google Scholar]
- 29.Inai K, Nakanishi T, Nakawaza M. Clinical correlation and prognostic predictive value of neurohumoral factors in patients late after the Fontan operation. Am Heart J. 2005;150:588–94. doi: 10.1016/j.ahj.2004.10.030. [DOI] [PubMed] [Google Scholar]
- 30.Rossi GP, Albertin G, Neri G, Andreis PG, Hofmann S, Pessina AC, Nussdorfer GG. Endothelin-1 stimulates steroid secretion of human adrenocortical cells ex vivo via both ETA and ETB receptor subtypes. J Clin Endocrinol Metab. 1997;82:3445–3449. doi: 10.1210/jcem.82.10.4279. [DOI] [PubMed] [Google Scholar]
- 31.Rubin LJ, Badesch DB, Barst RJ, Galie N, Black CM, Keogh A, Pulido T, Frost A, Roux S, Leconte I, Landzberg M, Simmoneau G. Bosentan therapy for pulmonary arterial hypertension. N Eng J Med. 2002;346:896–903. doi: 10.1056/NEJMoa012212. [DOI] [PubMed] [Google Scholar]
- 32.Johnson JA, Hemnes AR, Perrien DS, Schuster M, Robinson LJ, Gladson S, Loibner H, Bai S, Blackwell TR, Tada Y, Harral JW, Talati M, Lane KB, Fagan KA, West J. Cytoskeletal defects in Bmpr2-associated pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2012;302:L474–l484. doi: 10.1152/ajplung.00202.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]