

Abstract
RNA viruses exist in their hosts as populations of different but related strains. The virus population, often called quasispecies, is shaped by a combination of genetic change and natural selection. Genetic change is due to both point mutations and recombination events. We present a jumping hidden Markov model that describes the generation of viral quasispecies and a method to infer its parameters from next-generation sequencing data. The model introduces position-specific probability tables over the sequence alphabet to explain the diversity that can be found in the population at each site. Recombination events are indicated by a change of state, allowing a single observed read to originate from multiple sequences. We present a specific implementation of the expectation maximization (EM) algorithm to find maximum a posteriori estimates of the model parameters and a method to estimate the distribution of viral strains in the quasispecies. The model is validated on simulated data, showing the advantage of explicitly taking the recombination process into account, and applied to reads obtained from a clinical HIV sample.
Key words: evolution, HMM, statistical models, viruses
1. Introduction
Next-generation sequencing (NGS) technologies have transformed experiments previously considered too labor-intensive into routine tasks (Metzker, 2010). One application of NGS is the sequencing of genetically heterogeneous populations to quantify their genetic diversity. The genetic diversity is of primary interest, for example, in infection by RNA viruses such as HIV and HCV. In these systems, the combination of a high mutation rate of the pathogen with recombination between pathogens gives rise to a population of different but related individuals, referred to as a viral quasispecies. Recombination can occur, as a subsequent event, when different viral particles infect a single cell. We denote the different viral strains in this population as haplotypes. Studying the features of the viral quasispecies can shed light on the mechanisms of pathogen evolution in the host and it is of direct clinical relevance. For example, the diversity of the quasispecies has been shown to affect virulence (Vignuzzi et al., 2006), immune escape (Nowak et al., 1991), and drug resistance (Johnson et al., 2008).
The quasispecies equation is a mathematical model for RNA virus populations evolving according to a mutation-selection process (Eigen, 1971). The dynamics of the model are described by a mutation term accounting for transformation of one viral haplotype (or strain) into another at the time of replication, and a selection term that accounts for varying replication rates of different strains. The mutation process is generally considered as the result of point mutations only, although recombination is known to be frequent in many clinically relevant viruses, including HIV and HCV. For example, the recombination rate of HIV is estimated to be about tenfold higher than its point mutation rate. Therefore, the quasispecies model has been extended to account for both mutation and recombination (Boerlijst et al., 1996). At equilibrium, the model predicts the viral population to be dominated by one or a few haplotypes, which are surrounded by a cloud of constantly generated, low-frequency mutants.
Recent NGS technologies allow for observing viral quasispecies at an unprecedented level of detail by producing millions of DNA reads in a single experiment. However, this high yield comes at a cost. Reads are usually short, up to 700 bp with the latest technology and much shorter than the smallest viral genomes, and they are error prone due to sample preparation and sequencing errors (Gilles et al., 2011). As a result, since the data obtained are incomplete and noisy, a meaningful characterization of viral populations by means of NGS requires careful analysis of the sequencing data (Beerenwinkel and Zagordi, 2011).
In this article, we aim at inferring viral quasispecies based on NGS data by explicitly modeling mutation and recombination. We use a hidden Markov model (HMM) to generate viral populations, i.e., haplotype distributions, and their probing by means of NGS. In our model, the haplotypes are originating from a small number of generating sequences via recombination, described as switch of state in the HMM that selects from which sequence the haplotype derives, and mutation, described by position-specific probability tables for the generating sequences. The sequencing reads are obtained from the haplotypes subject to observation error.
HMMs allowing for a switch between generating sequences, termed jumping HMMs, have been applied, for example, to sequence alignment of protein domains (Spang et al., 2002) and to detecting inter-host HIV-circulating recombinant forms (Schultz et al, 2006). A related model has been used in human genetics to infer the haplotypes of diploid genomes from genotype data (Kimmel and Shamir, 2005; Scheet and Stephens, 2006). The model presented here differs from previous approaches in several ways, including an unknown and possibly large number of haplotypes, erroneous sequence read data, and high mutation and recombination rates. In particular, our goal is to reliably identify the haplotypes shaping intra-host quasispecies, including variants of low frequencies. Since sequencing errors will confound the true variation present in the sample, methods for error correction have been proposed, including clustering of reads or flowgrams followed by removal of any remaining within-cluster variation (Eriksson et al., 2008; Quince et al., 2011; Zagordi et al., 2010a, 2010b).
In the present article, we present a novel generative probabilistic model for making inference of viral quasispecies, i.e., for estimating the intra-patient viral haplotype distribution. Specifically, we assume that the true genetic diversity is generated by a few sequences, called generators, through mutation and recombination, and that the observed diversity results from additional sequencing errors. We present the model for local haplotype inference, meaning that we aim at inferring the population structure in a genomic region of a size that can be covered by individual reads, but extending this model to global haplotype inference, i.e., to longer genomic regions, is straightforward. Local inference will generally be more reliable and sufficient for many applications. For example, the HIV protease gene, an important target of antiretroviral therapy, is 297 bp long, and it is now standard to obtain reads of 400 bp and longer with the Roche/454 GS Junior sequencer, a common pyrosequencing platform for clinical diagnostics. Local haplotype reconstructions can also be used as a starting point for global reconstruction (Astrovskaya, 2011; Eriksson et al., 2008; Prabhakaran et al., 2010; Prosperi et al., 2011; Zagordi et al., 2011). We show that our model is able to estimate the distribution of viral haplotypes with high reliability by applying it to simulated data, where we have access to the ground truth, and we present an application to reads obtained from an HIV-infected patient.
2. Methods
2.1. Hidden Markov model
During infection of a host cell, a viral strain can change either by point mutation, when a single base is copied with error, or by recombination, when a cell is infected by more than one viral particle, and viruses in subsequent generations produce a sequence that is a mosaic of those of the progenitors. The model we present here does not aim at representing these evolutionary processes mechanistically. Rather, it is a descriptive probabilistic model, in which the quasispecies is generated by switching among K different generating sequences, each of length L. We denote by πk the probability to begin with generator k at the first sequence position. The generators are defined as sequence profiles (μjkv), indicating the probability over the alphabet 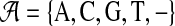 of base
of base 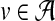 at position j of the k-th generating sequence.
at position j of the k-th generating sequence.
The set of sequences generates viral haplotypes 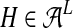 by mutation, modeled by the probability tables (μjkv), and recombination, denoted by transition matrices ρj. The transition probability ρjkl describes the recombination event in which the generating sequence k switches to l between positions j − 1 and j.
by mutation, modeled by the probability tables (μjkv), and recombination, denoted by transition matrices ρj. The transition probability ρjkl describes the recombination event in which the generating sequence k switches to l between positions j − 1 and j.
Let Zj be the hidden random variable with state space 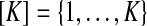 , indicating the parental sequence generating Hj, the haplotype character at position j. Each observed read R with bases Rj is obtained from a haplotype subject to noise (sequencing errors), assumed to occur independently among sites at rates ɛj. The probability of an observed read R is defined hierarchically as
, indicating the parental sequence generating Hj, the haplotype character at position j. Each observed read R with bases Rj is obtained from a haplotype subject to noise (sequencing errors), assumed to occur independently among sites at rates ɛj. The probability of an observed read R is defined hierarchically as
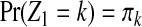 |
(1a) |
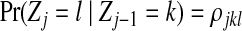 |
(1b) |
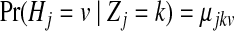 |
(1c) |
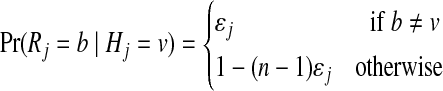 |
(1d) |
where 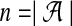 is the size of the alphabet.
is the size of the alphabet.
The full model consists, for each observation 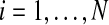 , of the hidden random variables
, of the hidden random variables  , indicating generator sequences, and
, indicating generator sequences, and 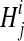 , the haplotypes of the quasispecies, and the observed reads
, the haplotypes of the quasispecies, and the observed reads 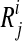 , for all sequence positions
, for all sequence positions 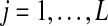 (Fig. 1). The model parameters are summarized as θ = (π, ρ, μ, ɛ).
(Fig. 1). The model parameters are summarized as θ = (π, ρ, μ, ɛ).
FIG. 1.

Graphical representation of the model. Only one observation i is depicted; for the full model, the graph is replicated for 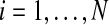 .
.
For parameter estimation, we first describe the maximum likelihood (ML) approach and develop an Expectation Maximization (EM) algorithm for ML estimation (MLE). Then, we define prior parameter distributions that enforce sparse maximum a posteriori (MAP) solutions and present a modified EM algorithm for MAP estimation of the parameters. This regularized model is used subsequently for the rest of the article.
2.2. Maximum likelihood estimation
The likelihood Pr(R | θ) of the model defined in Eqs. 1a–d factorizes into the product over independent reads, and for each read, it can be computed efficiently using the Markov property,
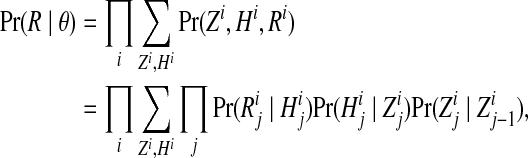 |
where 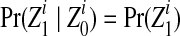 . Using the distributive law, each sum in this expression can be factored along the Markov chain, which gives rise to the forward algorithm (Rabiner, 1989). In this manner, the likelihood can be computed in O(NLK2) time.
. Using the distributive law, each sum in this expression can be factored along the Markov chain, which gives rise to the forward algorithm (Rabiner, 1989). In this manner, the likelihood can be computed in O(NLK2) time.
The EM algorithm (Dempster, 1977) is an iterative procedure to find local maxima of the likelihood as a function of θ by maximizing the auxiliary Baum's function Q(θ, θ′), defined as the expected hidden log-likelihood of the data with respect to the posterior distribution of (Z, H) given θ′,
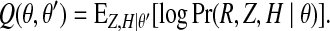 |
Here, θ′ is the previous estimate of the parameters (π, ρ, μ, ɛ). Baum's function bounds the log-likelihood from below, and repeated iterations of maximizing Q with respect to θ (M-step) alternated with estimation of the posterior Pr(Z, H | R, θ) (E-step) are guaranteed to find a local maximum of the likelihood function.
For the E-step, we compute
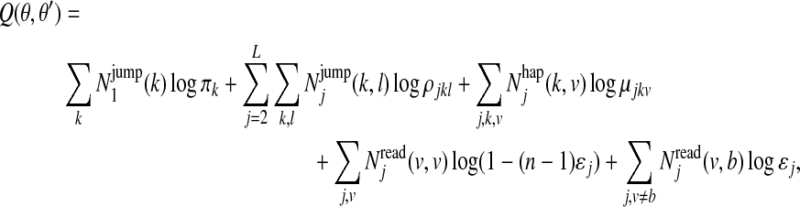 |
where 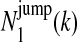 is the expected number of times a Markov chain starts in state k at position 1,
is the expected number of times a Markov chain starts in state k at position 1, 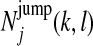 is the expected number of times that a Markov chain switches from state k to state l right before position j,
is the expected number of times that a Markov chain switches from state k to state l right before position j, 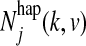 is the expected number of times the Markov chain is in state k and emits haplotype character v at position j, and
is the expected number of times the Markov chain is in state k and emits haplotype character v at position j, and 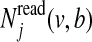 is the expected number of times the Markov chain emits character v at position j and character b is observed in the reads. These expected counts are estimated for all reads by computing posterior probabilities of the hidden variables H and Z given the data and the current estimate of θ, using the forward and backward algorithm (Rabiner, 1989).
is the expected number of times the Markov chain emits character v at position j and character b is observed in the reads. These expected counts are estimated for all reads by computing posterior probabilities of the hidden variables H and Z given the data and the current estimate of θ, using the forward and backward algorithm (Rabiner, 1989).
In the M-step, the parameters are updated by maximizing Q(θ, θ′) with respect to θ. This is achieved by setting
 |
2.3. Maximum a posteriori estimation
The HMM defined by Eqs. 1a–d is non-identifiable (Ito et al., 1992). In this case, multiple solutions of θ share the same posterior. Hence, MLEs are not uniquely defined and the EM algorithm suffers from poor convergence. To address this limitation, we define a prior distribution for the model parameters and estimate them by maximizing the posterior probability
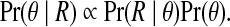 |
We assume independent priors, Pr(θ) = Pr(π)Pr(ρ)Pr(μ)Pr(ɛ), and Pr(π), and Pr(ɛ) to be flat. For the recombination probabilities ρ and the nucleotide probability tables μ, we define independent and identical Dirichlet distributions for all sequence positions j and all generators k,
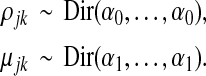 |
The hyperparameter α0 controls the sparsity of recombination events. As α0 approaches zero, the transition matrix ρj, at the MAP estimate, approaches the K × K identity matrix IK, and recombination becomes more unlikely. Similarly, α1 controls the variability of haplotype character emissions. For small values of α1, mutations become increasingly unlikely. The Dirichlet priors can enforce sparse MAP solutions and for a high degree of regularization (small α0 and α1), the model becomes identifiable.
The regularized model is not only computationally more convenient, but sparse recombination is also a biologically plausible assumption. Indeed, despite high recombination rates, real RNA virus populations always display genomic regions that are conserved or nearly conserved, and that define the virus. In these regions, the different generating sequences cannot be distinguished because there is no or little diversity. Therefore, recombination among different sequences can only be observed in regions with higher diversity, which are a small fraction of genomic sites, and thus is expected to be a rare event.
For solving the MAP estimation problem, we use the Variational Bayes approach suggested and elaborated in Beal (2003) and Johnson (2007). With the Dirichlet priors defined above, it can be solved by a modification of the EM algorithm introduced in the previous section. Specifically, only the M-step needs to be modified to update ρ and μ as follows:
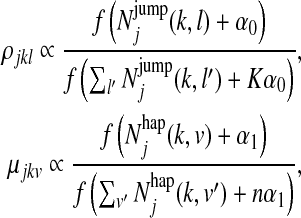 |
where the scaling function f (x) = eψ(x) is defined in terms of the digamma function ψ, the derivative of the log gamma function, and the constants of proportionality are given by the constraints 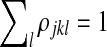 and
and 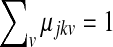 , respectively.
, respectively.
2.4. Model selection
For fixed sequence alphabet 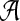 and length L, the dimension of the model is determined by the number K of generator sequences. For model selection, i.e., for choosing the optimal K, we consider the Bayesian information criterion defined as
and length L, the dimension of the model is determined by the number K of generator sequences. For model selection, i.e., for choosing the optimal K, we consider the Bayesian information criterion defined as
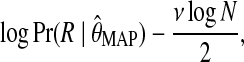 |
where ν is the dimension, or number of free parameters, of the model (Schwarz, 1978). However, the size of the model makes it infeasible to compute its dimension directly by standard methods. Instead, we resort to a heuristic inspired by the results in (Yamazaki, 2005) and approximate ν by the number of nonzero parameters in the MAP estimate 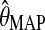 . In our empirical tests, this heuristic worked very well for values of K up to at least 5. For practical purposes, this appears sufficient, as in the quasispecies model the number of dominant haplotypes is assumed to be small and hence can be generated by a small number K of low-entropy probability tables over
. In our empirical tests, this heuristic worked very well for values of K up to at least 5. For practical purposes, this appears sufficient, as in the quasispecies model the number of dominant haplotypes is assumed to be small and hence can be generated by a small number K of low-entropy probability tables over  . For model selection, the smallest K is chosen within one standard error of the K with the maximum BIC (Tibshirani et al., 2001).
. For model selection, the smallest K is chosen within one standard error of the K with the maximum BIC (Tibshirani et al., 2001).
As an alternative model selection strategy, the goodness of fit of the model may be assessed in a cross-validation setting, but this approach is computationally much more expensive and would drastically slow down the effective runtime.
2.5. Prediction
The main object of interest that we derive from the model is the haplotype distribution Pr(H), i.e., the structure of the viral quasispecies. For given model parameters θ, estimated from read data R, we can compute the probability of each haplotype efficiently using the forward algorithm, and the distribution Pr(H) might be estimated by computing the probability of each haplotype. However, since there are 4L possible haplotypes, enumeration is infeasible already for moderate sequence lengths L. Instead, we estimate Pr(H) by sampling from the model using Eqs. 1a–c. In the applications below, we sampled 10,000 haplotypes at the MAP estimate of θ. This procedure is efficient, because the entropy of almost all model parameter probability tables μjk and ρjk is close to zero, and hence the probability mass of Pr(H) will be centered on a few haplotypes.
Although not employed in the present article, other quantities of interest can be predicted from the model. For example, read error correction can be done by replacing each read Ri with the haplotype it most probably originated from, i.e., with argmaxh Pr(Hi = h | Ri = r). If the allele frequency spectrum is sought after, for example, because the effect of specific single-nucleotide variants (SNVs) is known, then the posterior probability of each SNV given all observed reads can be computed as Pr(Hj = v | R).
2.6. Implementation
For the EM algorithm, we iterated the E-step and the M-step until convergence, which was detected by a relative change of the log-likelihood smaller than 10−8. Since the EM algorithm is only guaranteed to find a local maximum, we performed 50 random restarts and chose the solution with the largest likelihood.
The initial parameter values for μjk are constant,  , and for π and ρjk, they are drawn at random from the distributions
, and for π and ρjk, they are drawn at random from the distributions  and
and 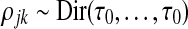 , where τ0 controls the sparsity. If necessary, the values of ρjk are reordered such that the k-th entry always has the highest value of this vector, which is achieved by switching two entries. The resulting initial transition matrix encodes rare transitions across the generators. Empirical analysis indicated good performance of MAP estimation with prior and initialization Dirichlet hyperparameters set to α0 = α1 = τ0 = 0.01.
, where τ0 controls the sparsity. If necessary, the values of ρjk are reordered such that the k-th entry always has the highest value of this vector, which is achieved by switching two entries. The resulting initial transition matrix encodes rare transitions across the generators. Empirical analysis indicated good performance of MAP estimation with prior and initialization Dirichlet hyperparameters set to α0 = α1 = τ0 = 0.01.
The forward and backward calculations of the E-step will underflow for long reads; therefore, these probabilities are rescaled at each step rather than computed on a logarithmic scale, which would take much more time. Reads are hashed at the beginning in order to identify identical ones and to avoid unnecessary computations. Thus, the effective runtime is O(NuLK2), where Nu is the number of unique reads. The E-step can be independently computed for each read. In addition, all random restarts of the EM can be computed separately.
We have implemented MAP estimation, model selection, and prediction of the haplotype distribution in a Java program called QuasiRecomb. It runs on any operating system supporting Java version 1.7 (Linux, OSX, Solaris, Windows). The software is open source and licensed under the GNU General Public License. It is available online at www.cbg.ethz.ch/software/quasirecomb/
3. Results
3.1. Simulation study
We assessed the performance of our model on seven different datasets, corresponding to different distributions of haplotypes of 300 bp length. Datasets 1, 2, 3, 5, and 6 have one recombination breakpoint, dataset 4 has two recombination breakpoints, and dataset C serves as a negative control without any recombinants. Generator sequences differed by between 6 and 10% of nucleotides. The haplotype distribution of each dataset is reported in Figure 2.
FIG. 2.

Frequencies in percent for the haplotypes of each dataset. The symbols for the original haplotypes and recombinants are filled dots and open squares, respectively. The numbers in parentheses on top of each plot report the number of original haplotypes plus the number of recombinants in the respective dataset.
For each distribution of haplotypes, we sampled 50 datasets of 2,000 reads each with point mutations at an error rate of 0.03% per base and evaluated the BIC score for model selection. The error rate reflects the amount of substitution errors that can be expected in a typical NGS experiment using 454/Roche after filtering low-quality reads and removing frameshift-causing indels in the alignment step. Figure 3 reports the BIC scores for the seven datasets. In all cases, the correct number of generators is chosen, applying our model selection. Except for the last dataset, the BIC score is maximum at the correct number of generators.
FIG. 3.

BIC score for the seven simulated datasets. The model correctly chooses K for up to five original haplotypes. The boxplots summarize results of 50 independent datasets. The numbers in parentheses report the number of original generators plus the number of recombinants. The maximum BIC is attained at the true number K of generators for all datasets except #6, where the maximum BIC is at K = 7, but our model selection selects the true K = 6 (dashed lines report upper and lower boundaries of the one standard error of K = 7).
In order to study the impact of the sample size, we sampled instances of the first dataset of different sizes and repeated the model selection procedure. We analyzed datasets of 300, 400, and 500 reads. The results are reported in Figure 4. For the first two samples, the BIC score erroneously selects K = 1, whereas for 500 and more reads, the procedure correctly selects K = 2 generating sequences, indicating that sufficient coverage is an important prerequisite.
FIG. 4.
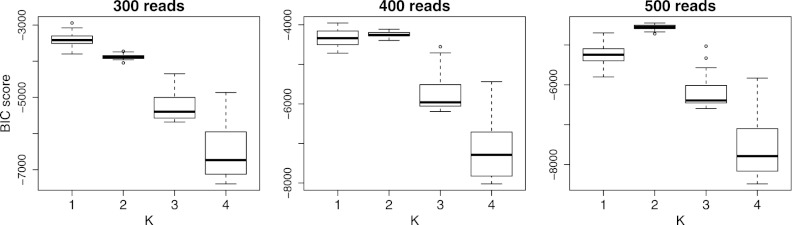
BIC score for simulated dataset 1 at different sample sizes between 300 and 500 reads. The model selection correctly selects K = 2 already with 500 reads. The boxplots summarize results on 50 independent datasets.
Furthermore, the sensitivity has been tested using the first dataset, with fixed haplotype distances. We sampled 1,000 error-free reads from the original haplotypes at frequencies 80% and 20%, and replaced arbitrarily two reads of the sample with the two recombinants from the first dataset. QuasiRecomb was able to reconstruct these recombinants at a frequency of 0.1%. Since the scenario of a technical error-free sample is unrealistic, we again sampled as before, but with an error-rate of 0.03% per base, and again we are able to reconstruct the low-frequency recombinants. Even in the case of a tenfold higher error-rate of 0.3% per base, QuasiRecomb successfully identifies and reconstructs the recombinants. This can be explained by the fact that an accumulation of about 10 technical errors on a length of 300 bp is very unlikely. One might also investigate the sensitivity w.r.t. the distances among haplotypes and fixed frequencies.
For parameter estimation, we sampled an additional set of 2,000 reads from the haplotype distribution and ran the EM algorithm with the value of K inferred before. Then, we inspected the MAP estimates of the parameters μ, π, and ρ. Whenever the correct K had been chosen, the entropy of the estimates of the tables μjk is very close to zero. Regarding the recombination parameters, except for the negative control dataset, there is always a position j after the last variable site and before the recombination hotspot such that 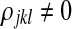 for two different generators k and l.
for two different generators k and l.
In the negative control dataset, where no recombinants are present, the three generating sequence profiles corresponded exactly to the original haplotypes, i.e., the entropy of all μjk tables was very close to zero, and no recombination was detected, i.e., ρj = IK for all j. In this case, π represents the frequency of the original haplotypes and its estimate was very close to the original distribution. The remaining discrepancy can be explained by the sampling variance of the reads alone.
We assessed the accuracy of the inferred quasispecies by comparing it to the original set of sequences using the proportion close measure, φq, defined as the fraction of reconstructed haplotypes that match an original one with at most q mismatches (Eriksson, 2008). Figure 5 reports the proportion close, as a function of the number of allowed mismatches, for the models learned from datasets 1 to 4. For comparison, we additionally learned a model in which recombination is not possible, i.e., where ρj = IK for all j.
FIG. 5.

Proportion close, φq, as a function of q. The fraction of the population reconstructed with at most q mismatches is higher than 99% already for q ≥ 1 if one allows recombination, but only for q = 3 to 15 if one does not allow recombination.
Allowing recombination, model selection chose the correct number of generators for all four datasets. Without recombination, the correct number of generators (and of haplotypes) is 4 = 2 + 2, 9 = 3 + 6, 27 = 3 + 24, and 16 = 4 + 12, respectively (Fig. 3). Identifying nine generators or more is hard, and running the EM algorithm becomes inefficient, because the runtime grows quadratically in K. For dataset 1, without recombination, BIC still selects K = 2, because the penalization for a larger K is too high. Visual inspection of the MAP estimate parameters shows that the first generator is completely concentrated on one profile, and the second generator explains the other three haplotypes by flat generator distributions μj2. Setting K = 4, all generators concentrate on the original haplotypes, but as expected, the runtime is higher and many more EM restarts are needed to find this MAP estimate, because the likelihood surface is very flat.
The advantage of modeling recombinants is evident as the fraction of the population reconstructed is always higher than in the recombinant-free case. For all four datasets, the proportion close is at least 99% for q ≥ 1 if recombination is accounted for, whereas the recombination-free model fails to reconstruct the quasispecies structure in these cases (Fig. 5). This is a consequence of the poor performance of the model selection in this case, namely K = 2, 4, 5, and 5 for datasets 1, 2, 3, and 4, respectively.
3.2. Real HIV dataset
Using QuasiRecomb, we analyzed a set of experimental NGS reads obtained by sequencing a clinical sample from an HIV-infected patient in the context of a study of viral tropism (Archer et al., 2010) (Sequence Read Archive run SRR069887). We selected 1,517 reads overlapping a 179 bp long region of the env gene (positions 6321–6499 in the HXB2 reference strain). We ran the EM algorithm on 50 datasets, generated by bootstrapping 1,517 reads each, and selected the model with K = 2 generators (Fig. 6, left). The estimated quasispecies is dominated by a single haplotype with an estimated frequency of 31%. This master sequence is surrounded by a swarm of mutants, 12 of which have relative frequencies over 1% and many others have lower frequencies (Fig. 6, right). The sequence similarities of the estimated haplotypes with relative frequencies greater than 1% are between 93 and 99%. Each generator has four positions with a positive recombination probability.
FIG. 6.

BIC scores for a clinical sample based on 50 bootstrap samples (left) and the haplotype distribution of the inferred quasispecies on a log–log scale for K = 2 (right).
In order to appreciate the compactness of the model inferred with the jumping HMM, we compared its solutions with those of another tool to reconstruct haplotypes, implemented in the software ShoRAH (Zagordi, 2011). This method, which does not take recombination into account, identified 15 haplotypes in the same dataset, which can be further reduced to 10 if one excludes those with frequencies lower than 1% and those which harbor a frameshift due to a deletion.
4. Discussion
We have presented a probabilistic model based on an HMM that infers the distribution of haplotypes in a viral quasispecies from NGS data. The model describes these different viral strains present in the population as originating from different generating sequences by means of two processes: point mutation and recombination. Point mutation is captured by the fact that the sequences are modeled as probability tables over the sequence alphabet. Recombination is modeled via a change of the sequence from which the haplotype is drawn, as indicated by a change of state, or a jump, in the hidden Markov chain. Due to the possibility of switching between sequences, the number of tables necessary to describe the population structure remains small, while offering an excellent fit to the data. This results in a more compact and structured description of the viral population.
We have introduced regularization to achieve sparse MAP estimates accounting for the fact that mutation and recombination are rare events. Using the EM algorithm, MAP estimates can be computed efficiently. Our results on simulated data demonstrate the usefulness of enforcing this sparsity when inferring recombinant haplotypes from read data.
There are several ways to extend the methodology presented here. Estimating the haplotype distribution is currently done by sampling from the model. Another approach would be to compute the top suboptimal haplotypes from the recombinant sequence generators. In previous work on the analysis of NGS data to estimate genetic diversity, model selection has been approached in a non-parametric way by using the Dirichlet process mixture (Prabhakaran et al., 2010; Zagordi, 2010a). Extension of the HMM in this direction has been proposed and might be explored in this context as well (Beal et al., 2002).
We have presented our results in a local reconstruction setting, but our implementation QuasiRecomb is already adapted to accept global read alignments in BAM format. In this scenario, the population structure inferred locally is extended to genomic regions that are longer than the typical read length. This is achieved by allowing for longer generating sequences, along with two additional silent states, to describe the unobserved regions before and after each read in the same fashion as the pair-HMM can be used for semi-global sequence alignment.
The quasispecies inference approach we propose here is designed for sequencing technologies with long reads. In general, we expect the accuracy of our method to decrease for shorter reads. We note, however, that the read length of most sequencing technologies is constantly improving and that some NGS platforms can produce reads over 1,000 bp. With such long reads, the probability to observe recombinations on a single read will be higher, and the necessity to keep the number of generators small will be even more compelling.
Acknowledgment
Eran Halperin is a Faculty Fellow of the Edmond J. Safra Center for Bioinformatics at Tel-Aviv University. He was supported by the Israeli Science Foundation (grant 04514831).
Disclosure Statement
The authors declare that no competing financial interests exist.
References
- Archer J. Rambaut A. Taillon B.E., et al. The evolutionary analysis of emerging low frequency hiv-1 cxcr4 using variants through time—an ultra-deep approach. PLoS Comput. Biol. 2010;6:e1001022. doi: 10.1371/journal.pcbi.1001022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astrovskaya I. Tork B. Mangul S., et al. Inferring viral quasispecies spectra from 454 pyrosequencing reads. BMC Bioinformatics. 2011:12. doi: 10.1186/1471-2105-12-S6-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal M. Ghahramani Z. Rasmussen C. The infinite hidden Markov model. Advances in neural information. 2002;14:577–584. [Google Scholar]
- Beal M.J. University College London; 2003. Variational algorithms for approximate bayesian inference [Tech. rep.,] [Google Scholar]
- Beerenwinkel N. Zagordi O. Ultra-deep sequencing for the analysis of viral populations. Current Opinion in Virology. 2011;1:413–418. doi: 10.1016/j.coviro.2011.07.008. [DOI] [PubMed] [Google Scholar]
- Boerlijst M. Bonhoeffer S. Nowak M. Viral quasi-species and recombination. Proceedings: Biological Sciences. 1996;263:1577–1584. [Google Scholar]
- Dempster A. Laird N. Rubin D. Maximum likelihood from incomplete data via the EM algorithm (with discussions) J. R. Statist. Soc. B. 1977;39:1–38. [Google Scholar]
- Eigen M. Self-organization of matter and the evolution of biological macromolecules. Naturwissenschaften. 1971. www.springerlink.com/index/q47866457218x543.pdf. www.springerlink.com/index/q47866457218x543.pdf [DOI] [PubMed]
- Eriksson N. Pachter L. Mitsuya Y., et al. Viral population estimation using pyrosequencing. PLoS Computational Biology. 2008;4:e1000074. doi: 10.1371/journal.pcbi.1000074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilles A. Meglécz E. Pech N., et al. Accuracy and quality assessment of 454 GS-FLX Titanium pyrosequencing. BMC Genomics. 2011;12:245. doi: 10.1186/1471-2164-12-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H. Amari S.I. Kobayashi K. Identifiability of hidden Markov information sources and their minimum degrees of freedom. IEEE Transactions on Information Theory. 1992;38:324–333. [Google Scholar]
- Johnson J.A. Li J.F. Wei X., et al. Minority HIV-1 drug resistance mutations are present in antiretroviral treatment-naïve populations and associate with reduced treatment efficacy. Plos Med. 2008:e158. doi: 10.1371/journal.pmed.0050158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M. Why doesn't EM find good HMM POS-taggers? 2007. www.aclweb.org/anthology/D/D07/D07-1031 pp. 296–305.www.aclweb.org/anthology/D/D07/D07-1031 EMNLP-CoNLL.
- Kimmel G. Shamir R. GERBIL: Genotype resolution and block identification using likelihood. Proc. Natl. Acad. Sci. U. S. A. 2005;102:158–62. doi: 10.1073/pnas.0404730102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzker M.L. Sequencing technologies—the next generation. Nat. Rev. Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- Nowak M.A. Anderson R.M. McLean A.R., et al. Antigenic diversity thresholds and the development of AIDS. Science. 1991;254:963–9. doi: 10.1126/science.1683006. [DOI] [PubMed] [Google Scholar]
- Prabhakaran S. Rey M. Zagordi O., et al. HIV-haplotype inference using a constraint-based dirichlet process mixture model. Machine Learning in Computational Biology (MLCB) NIPS Workshop. 20102010:1–4. doi: 10.1109/TCBB.2013.145. [DOI] [PubMed] [Google Scholar]
- Prosperi M.C. Prosperi L. Bruselles A., et al. Combinatorial analysis and algorithms for quasispecies reconstruction using next-generation sequencing. BMC Bioinformatics. 2011;12:5. doi: 10.1186/1471-2105-12-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quince C. Lanzen A. Davenport R.J. Turnbaugh P.J. Removing noise from pyrosequenced amplicons. BMC Bioinformatics. 2011;12:38. doi: 10.1186/1471-2105-12-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabiner L. A tutorial on hidden Markov models and selected applications in speech recognition (with erratum) Proceedings of the IEEE. 1989;77:257–286. [Google Scholar]
- Scheet P. Stephens M. A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. The American Journal of Human Genetics. 2006;78:629–644. doi: 10.1086/502802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz A.K. Zhang M. Leitner T., et al. A jumping profile hidden Markov model and applications to recombination sites in HIV and HCV genomes. BMC Bioinformatics. 2006;7:265. doi: 10.1186/1471-2105-7-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz G. Estimating the dimension of a model. Ann. Satist. 1978;6:461–464. [Google Scholar]
- Spang R. Rehmsmeier M. Stoye J. A novel approach to remote homology detection: jumping alignments. J. Comput. Biol. 2002;9:747–60. doi: 10.1089/106652702761034172. [DOI] [PubMed] [Google Scholar]
- Tibshirani R. Walther G. Hastie T. Estimating the number of clusters in a data set via the gap statistic. J. R. Statist. Soc. B. 2001;63:411–423. [Google Scholar]
- Vignuzzi M. Stone J. Arnold J., et al. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature. 2006;439:344–348. doi: 10.1038/nature04388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki K. Watanabe S. Algebraic geometry and stochastic complexity of hidden Markov models. Neurocomputing. 2005;69:62–84. [Google Scholar]
- Zagordi O. Bhattacharya A. Eriksson N. Beerenwinkel N. Shorah: estimating the genetic diversity of a mixed sample from next-generation sequencing data. BMC Bioinformatics. 12:119. doi: 10.1186/1471-2105-12-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagordi O. Geyrhofer L. Roth V. Beerenwinkel N. Deep sequencing of a genetically heterogeneous sample: local haplotype reconstruction and read error correction. J. Comput. Biol. 2010;17:417–28. doi: 10.1089/cmb.2009.0164. [DOI] [PubMed] [Google Scholar]
- Zagordi O. Klein R. Däumer M. Beerenwinkel N. Error correction of next-generation sequencing data and reliable estimation of HIV quasispecies. Nucleic Acids Res. 38:7400–9. doi: 10.1093/nar/gkq655. [DOI] [PMC free article] [PubMed] [Google Scholar]