

Abstract
Staphylococcus aureus is a common human pathogen highly evolved as both a component of the commensal flora and as a major cause of invasive infection. Severe respiratory infection due to staphylococci has been increasing due to the prevalence of more virulent USA300 CA-MRSA strains in the general population. The ability of S. aureus to adapt to the milieu of the respiratory tract has facilitated its emergence as a respiratory pathogen. Its metabolic versatility, the ability to scavenge iron, coordinate gene expression, and the horizontal acquisition of useful genetic elements have all contributed to its success as a component of the respiratory flora, in hospitalized patients, as a complication of influenza and in normal hosts. The expression of surface adhesins facilitates its persistence in the airways. In addition, the highly sophisticated interactions of the multiple S. aureus virulence factors, particularly the α-hemolysin and protein A, with diverse immune effectors in the lung such as ADAM10, TNFR1, EGFR, immunoglobulin, and complement all contribute to the pathogenesis of staphylococcal pneumonia.
Keywords: Airway, Lung, Epithelial, Staphylococcus aureus, Virulence, Signaling
Introduction
Staphylococcus aureus is a ubiquitous pathogen associated with a wide range of infections affecting the respiratory tract that range from asymptomatic colonization to fulminant necrotizing pneumonia. Despite extensive epidemiological studies that include whole-genome sequencing, it remains unclear exactly which staphylococcal properties are required for invasive infection versus status as a part of the commensal flora. While in the past the analyses of S. aureus virulence mechanisms have focused almost entirely upon bacterial factors, it has become increasingly apparent that the nature of the host immune response that is evoked by these organisms is a significant determinant in the ultimate outcome of the infection. This has become especially apparent in the ongoing epidemic of MRSA infection in the USA due to the USA300 clone of S. aureus that has been associated with over 18,000 deaths in 2005 [1]. These strains elicit an intense pro-inflammatory response often associated with high morbidity and mortality [2, 3].
The success of S. aureus in general as a respiratory pathogen, and the USA300 strains specifically, may be attributed to several factors: substantial metabolic capabilities; genetic flexibility, both the ability to acquire and to mutate specific genetic elements, and the unique ability to exploit the immune responses that are evoked. In this review, we will examine a few of the important metabolic capabilities of these organisms that enable them to flourish in the airways focusing specifically on the genes involved in iron acquisition, which is a critical factor for a human pathogen. We will review the data supporting the contribution of USA300-associated virulence factors in pathogenesis and discuss the relevance of murine models in understanding the pathogenesis of staphylococcal pneumonia in humans. As the interactions of staphylococci and the immune system are likely to be the key factors in the pathogenesis of pneumonia, the many S. aureus virulence factors that interact directly with immune components (IgG, complement, T cell signaling) or activate specific immune cascades (type I IFN signaling, inflammasome activation) will be reviewed in some detail.
Epidemiology of staphylococcal pneumonia
Historically, staphylococcal pneumonia was an especially virulent infection of young infants and the elderly, particularly following influenza infection [4, 5]. A major cause of concern has been the emergence of methicillin-resistant strains of S. aureus (MRSA). Resistance to methicillin is encoded by the mec element [6]. MRSA has been typically associated with hospital-acquired strains that infect individuals with preexisting conditions. However, the early part of the twenty-first century saw the emergence of community-acquired strains of MRSA (CA-MRSA) that infect otherwise healthy individuals. The majority of cases of MRSA are caused by a single clonal group USA300. This was initially reported in athletes with close body contact to one another or other close-quartered situations such as in correctional facilities [7–11]. Pneumonias caused by USA300 have been steadily increasing over the past 10 years [12–14]. The typical pathology associated with fatal MRSA pneumonia consists of loss of alveolar architecture, hemorrhage, and consolidation of the lung parenchyma [16, 17] (Fig. 1). With the dissemination of the USA300 clones across the USA in the last decade [1, 6], MRSA pneumonia has become much more widespread [12, 13, 15], responsible for a significant number (13%) of the over 18,500 deaths attributed to MRSA in 2005 [1]. Not only did this group include the expected immunocompromised patients, but a large number were also previously healthy individuals in close contact with infected individuals [7–11].
Fig. 1.

Pathology of USA300 lung infections. H&E sections of C57Bl6/J mouse lung. Mice were infected with 2×107 cfu S. aureus USA300 for 24 h. In the uninfected lung, two bronchioles and a blood vessel are visible, along with clear alveolar architecture. The USA300-infected animal shows loss of alveolar architecture, necrosis, hemorrhage, infiltration of immune cells, and consolidation of the lung parenchyma. Scale bars, 100 μm
Animal models of pneumonia have shown that USA300 S. aureus is more virulent than other strains of MRSA [3, 18, 19]. There is substantial debate as to which virulence factors are responsible for the increased lethality, and it is likely that the success of the pathogen is due to the co-expression of several virulence determinants. At the genomic level, USA300 contain mobile elements with unique coding sequences, such as the arginine catabolic mobile element that appears to have been transferred from Staphylococcus epidermidis. This may confer a growth advantage in systemic infection [20–22] and effect the expression of other virulence factors [23]. A typical feature of MRSA strains acquired in the community is a large (20%) portion of the genome that contains unique genomic elements from horizontal transfer. These genomic elements include pathogenicity islands and exotoxins that cause host damage as well as counteract the immune response [24]. USA300 strains express the toxin Panton–Valentine leukocidin (PVL) which, along with the increased expression of core genome virulence factors such α-hemolysin and phenol-soluble modulins [19], appears to contribute to overall virulence. Perhaps the single most important virulence factor is the α-hemolysin which contributes significantly to the pathogenesis of pneumonia in murine models of infection through mechanisms detailed below [25–27]. However, it is important to note that the susceptibility of mice to staphylococcal infection is significantly less than that of humans due to the inability of S. aureus to release iron from murine hemoglobin [28]. Thus, while the data generated in mouse models have been very important, particularly in understanding the genetics of the host response to infection, the data that delineate the importance of specific virulence factors in the pathogenesis of pneumonia are subject to considerable debate [29].
Regulation of MRSA virulence factors in pneumonia
An important property of staphylococci is their metabolic flexibility and ability to adapt to environmental pressures. Although typically considered extracellular aerobic pathogens, they also grow and disseminate intracellularly [30–32] and tolerate anaerobic conditions [33], all of which are relevant to pulmonary infection.
A number of regulatory systems are present in the genome of S. aureus to coordinate virulence factor expression. One of the more important of these is the accessory gene regulator (Agr) system which coordinates the expression of both surface proteins and secreted toxins, divergently (such as the positive regulation of α-hemolysin and the negative regulation of protein A expression) [34]. The regulon encodes a quorum sensing peptide (AgrD) and its export protein (AgrB), a sensor histidine kinase (AgrC), and a response regulator (AgrA) [35]. AgrA autoregulates by binding its own promoter. Regulation by Agr is accomplished by an RNA molecule, RNAIII. The expression of both Agr and RNAIII has been shown to be higher in USA300 strains as well as community- versus hospital-acquired infection, which may provide some rationale for the prevalence and severity of this clonal group [3, 19, 36]. Agr mutations are also found in clinical isolates [37]. Although Agr and its regulon are dispensable for inflammatory signaling in the lung, they are necessary for invasive pulmonary infection [38], and the role of Agr in animal models of pneumonia has been well documented [25, 38, 39].
Intracellular lifestyle of an extracellular organism
One role Agr plays in pathogenesis relates to the intracellular life stage of S. aureus. S. aureus persists intracellularly within a variety of cells types, including epithelial cells, macrophages, and neutrophils (Figs. 2 and 3) [30–32]. The expression of Agr is required for these intracellular organisms to escape from endosomes. Induction of agr expression has been observed just prior to endosome release and no doubt is correlated with the expression of toxins that facilitate its escape and contribute to virulence in the lung (Fig. 2) [40, 41]. Although epithelial cells are considered non-phagocytic, small numbers of S. aureus have been observed in some epithelial cells lines [40, 42–44] and associated with apoptosis (mammary epithelial cells) [32]. In phagocytic cells following uptake, S. aureus can persist inside vacuoles for up to 4 days before the escape into the cytoplasm and the induction of cell lysis (Fig. 3) [45]. S. aureus can survive for prolonged periods of time in neutrophils and in some cases divide within dying cells [31], an effect which may contribute to a systemic dissemination of the organisms [46]. Staphylococcal isolates from the community (CA-MRSA) display greater propensity to avoid neutrophil killing and also cause more lysis of neutrophils compared with strains from hospital infections [47]. An appreciation for the potential of staphylococci to assume these diverse lifestyles is critical for the development of vaccines as expecting antibodies alone to clear these organisms, which have an intracellular niche, seems unlikely to be successful.
Fig. 2.

Virulence factors and host signaling cascades activated by S. aureus in the airway epithelium. Depicted are some of the virulence factors mentioned in the text and their effects on the host epithelium. See text for details
Fig. 3.

S. aureus interactions with neutrophils and monocytes. S. aureus is able to persist intracellularly in neutrophils for days; their escape from vacuole compartments is reliant upon exotoxin production. Both PVL and PSMs have shown an ability to lyse neutrophils. In monocytes, interaction with S. aureus and the action of α-hemolysin lead to the activation of the inflammasome
Contribution of specific virulence factors to the pathogenesis of pneumonia
The coordinated expression of many staphylococcal virulence components is required for the establishment of a successful infection. This includes surface proteins (such as the microbial surface components recognizing adhesive matrix molecules, MSCRAMMS) to establish colonization, the expression of iron acquisition systems for proliferation in mammals where iron is tightly sequestered and secreted, and surface components to elude innate and adaptive immunity. There are several toxins whose activity appears to be primarily the lysis of target cells, but increasing data have documented that many staphylococcal virulence factors act by triggering pathological immune response as well as avoid immune clearance by thwarting complement and IgG-mediated phagocytosis.
Iron acquisition
Iron is essential for both prokaryotic and eukaryotic metabolic pathways, and as such, numerous mechanisms have evolved to assure its acquisition from the environment. Mammals normally express a number of high-affinity binding proteins such as transferrin and hemoglobin to sequester iron for storage and prevent access by invading pathogens. S. aureus produces siderophores that bind free iron for utilization by the cell [48–50]. S. aureus express two heme acquisition systems that are able to utilize hemoglobin as an iron source and a transport heme, heme transport system (Hts) and iron-regulated surface determinant (Isd) groups of proteins. Both of these systems contribute to virulence in mouse models of infection [51, 52]. One disadvantage of working with murine models of staphylococcal infection is that S. aureus has greater affinity for human as compared with murine hemoglobin. Mice that express human hemoglobin are more susceptible to systemic S. aureus infection [28]. S. aureus expresses a homologue of the iron regulatory protein ferric uptake regulator (Fur) that senses iron limitation and regulates a number of virulence factors, including α-hemolysin and PVL [53]. In a murine pneumonia model, Fur was required for full virulence, with fur mutants expressing increased levels of exotoxins and reduced immunomodulary proteins (Fig. 4). These immunomodulary proteins include factors that decrease the organisms' ability to avoid neutrophil killing as well as evade the complement cascade.
Fig. 4.

Iron-dependent regulation of virulence by Fur. In the presence of iron, Fur is able to increase the expression of iron transport systems (Hts, Isd) and the complement evasion proteins CHIPS and SCIN while decreasing exotoxin production. Other proteins known to aid in complement evasion are also shown
Microbial surface components recognizing adhesive matrix molecules
The pathogenesis of staphylococcal pneumonia is usually initiated by aspirated organisms that first colonize the nasal cavity. There are substantial epidemiological data confirming the greater likelihood of colonized patients to develop invasive infection than those who are non-colonized, and the “de-colonization” of such patients prior to surgery is common practice and cost-effective [54–56]. Thus, there is significant interest in identifying the critical staphylococcal proteins that mediate nasal colonization. Magnus Hook first coined the term MSCRAMMS to describe the numerous surface proteins that specifically recognize host components, especially those relevant to soft tissue infection, such as collagen, fibrinogen, and fibronectin (Fig. 2) [57]. These have been extensively analyzed in models of murine infection, trying to identify a “critical” factor for staphylococcal adherence. However, the obvious redundancy in the binding capabilities of the staphylococcal surface components suggests that a single entity is unlikely to emerge as “THE” adhesin required for the pathogenesis of either nasal colonization or invasive infection.
The collagen-binding protein of S. aureus (encoded by the cna gene) binds collagen substrates and tissues. While the involvement of collagen binding in virulence has been shown in models of endocarditis and keratinitis [58, 59], with respect to the airway, only an association with PVL-positive isolates that cause increased pulmonary pathology has been observed [60]. Fibronectin-binding proteins are involved in the adherence of S. aureus to undifferentiated human airway epithelial cells. This does not extend to differentiated cells, where binding is reduced [61]. The ability of S. aureus to bind fibronectin does not facilitate nasal colonization, but does mediate internalization into epithelial cells [62]. Lung injury, as well as bacterial load, was increased in a rat model of pneumonia with a fibronectin null strain [63]. Adherence to fibrinogen by S. aureus is accomplished by clumping factors A and B. Clumping factor A (ClfA) plays an antiphagocytic role in both neutrophils and macrophages [64, 65]. ClfA is important in animal models of arthritis, sepsis, and endocarditis, but not in models of respiratory tract infection [66–69].
Clumping factor B
Clumping factor B (ClfB) is involved in respiratory infection, mediating the attachment to cytokeratins on nasal epithelial cells. Mutants lacking ClfB reduce the ability of S. aureus to adhere [70]. Similarly, a sortase mutant lacking the gene that links surface proteins during cell wall assembly via cleavage of an LPXTG motif [71] was also reduced in nasal colonization. Immunization with ClfB or treatment with monoclonal ClfB antibody both decreased the ability of S. aureus to persist in the nares [62]. The role of ClfB in nasal colonization was further confirmed in human subjects as those infected with a clfB strain cleared S. aureus faster (median clearance, 3 days) than the wild-type organism (7 days) [72]. Following nasal colonization, via ClfB or other staphylococcal ligands, the participation of several conserved gene products, both surface components and secreted toxins, all contribute to invasive infection.
α-Hemolysin
Following aspiration into the lower respiratory tract, the secretion of multiple cytolytic toxins has been thought to contribute to pathology, as reviewed below. It is important to note that many studies have relied upon recombinant or purified toxins delivered at concentrations unlikely to be achieved in vivo. Studies comparing mutant versus wild-type strains seem more likely to reflect the impact of specific virulence factors expressed by S. aureus.
The α-toxin or α-hemolysin (encoded by hla) of S. aureus is a major pore-forming toxin that assembles into heptamers at the cell membrane to create small pores sufficient for the movement of ions [73]. Hla expression is Agr-dependent and increased upon interaction with epithelial cells, infection in vivo, and is higher in USA300 strains [3, 19, 32, 74]. In airway epithelial cells α-hemolysin has been associated with calcium fluxes, pro-inflammatory signaling [75], and alteration of ciliary beat frequency of cells (Fig. 2) [76]. α-Hemolysin binds to the metal-loproteinase ADAM10, which is necessary for the toxin to cause cytotoxicity at low concentrations [77]. The interaction of ADAM10 with α-hemolysin activates a number of intracellular signaling events. The application of α-hemolysin to perfused lungs results in increased vascular leakage and damage, and increased permeability of airway epithelial cells [74, 78–80]. Infection with α-hemolysin null strains is associated with significantly lower mortality compared with wild-type strains in pneumonia models [25–27], associated with reduced pulmonary inflammation, neutrophil influx, and bacterial counts. The mechanism of α-hemolysin toxicity is associated with the activation of pyroptosis, induction of the inflammasome via caspase-1 activation, its targeting pro-IL-1β, and the generation of IL-1β and IL-18, highly pro-inflammatory cytokines (Fig. 3). This has been well described in immune cells and is likely to be important in other cells in the infected lung [81–86]. The lack of pathology associated with murine infection with hla mutants is consistent with studies demonstrating a correlation between α-hemolysin expression levels and virulence in the lung [87]. The use of α-hemolysin as a vaccine target for pneumonia has shown promise. Immunization of mice with mutant forms of α-hemolysin that cannot form pores protect mice from lethal pulmonary challenge [87]. Mice also show a decreased bacterial burden when immunized with an inactive α-hemolysin variant. Antibodies raised against α-hemolysin can be transferred and subsequently protect naive mice, while the treatment of epithelial cells in vitro with α-hemolysin antibody reduces epithelial injury [87]. Monoclonal antibodies raised against α-hemolysin as well as β-cyclodextrin compounds (that have symmetry to α-hemolysin) are also effective in preventing mortality in mice and in decreasing epithelial cell injury [88, 89].
β-toxin
The contribution of the beta and delta toxins of S. aureus to pulmonary infection is less well studied. As both target erythrocytes, they may contribute to the iron-scavenging mechanisms for the organism, but that has not been demonstrated experimentally. The β-toxin of S. aureus (expressed in 96% versus 56%) bovine versus human carrier isolates [90] is a sphingomyelinase that targets the membranes of host cells to generate ceremide (Fig. 2) [91]. β-toxin lyses a variety of cells types, including red blood cells, monocytes, lymphocytes, and neutrophils [92–94]. In airway epithelial cells, purified beta toxin has been shown to inhibit ciliary beat frequency [95]. Hayashida et al. [96] observed a β-toxin-associated pathology of the lung with increased airway permeability as well as a neutrophilic response, which was decreased in infections with β-toxin null strains. Instillation of a recombinant toxin caused comparable loss of alveolar architecture as in S. aureus-infected mice. The effect of beta toxin in the lung may not be direct as injury was ameliorated in neutropenic mice, indicating that the immune response played a significant role in injury.
Phenol-soluble modulins
Phenol-soluble modulins (PSMs) are small peptides originally isolated in the phenol-soluble lysate during extraction from S. epidermidis [97]. They are referred to as modulins due to their ability to induce cytokine release. The PSMs identified in USA300 MRSA [98] include four short (alpha, ~20 amino acids) and two longer (beta, ~40 amino acids) types that are produced in greater amount than in other S. aureus strains [19]. The PSMs are sensed by the formyl peptide 2 receptor and lead to the recruitment, activation, and lysis of neutrophils and play a role in virulence in skin models of infection (Fig. 3)[99, 100]. PSMs also possess antimicrobial activity against other species such as Streptococcus pyogenes [101]. This antimicrobial function may be a factor that has influenced the prevalence of USA300 within the community. It has also been proposed that PSMs impact upon biofilm development, as seen in S. epidermidis. Due to their surfactant-like properties, they may be involved in biofilm structure as well as detachment and dissemination [102]. Since the initial identification of PSMs in S. aureus, another PSM has been identified in the methicillin resistance locus (mec). This mec-PSM also plays a role in skin models when the expression of the other PSMs is low [100]. The role PSMs play in pneumonia models of infection have yet to be determined, but an increased PSM expression has been detected in epidemic (USA300) isolates. The δ-toxin of S. aureus is a 26-amino acid cytolysin encoded by RNAIII [103] and is similar to the α-PSMs. δ-toxin is lytic to a range of cell types and structures such as erythrocytes, bacterial protoplasts, and lysosomes [104]. δ-toxin acts synergistically along with β-toxin to facilitate staphylococcal escape from endosomes in airway epithelial cells (Fig. 2); however, its role in vivo is yet to be determined [42].
Panton–Valentine leukocidin
PVL is a toxin encoded by two co-transcribed genes, lukF-PV and lukS-PV, carried on a bacteriophage [105]. PVL forms octomeric protein pores at the cell membranes of neutrophils and macrophages and is specific to humans and rabbits, but not mice or non-human primates [106–112]. PVL causes the apoptosis of neutrophils via caspases 3 and 9 (Fig. 3) [113], and a role for TLR2 in causing inflammation in the lung by PVL has been observed [114]. TLR2 null mice displayed reduced production of IL-1β, TNF, and KC in the lung after the administration of PVL [114], and the toxin alone has dermonecrotic capabilities [115].
While PVL is epidemiologically linked to invasive pneumonias caused by USA300 strains, there has been significant controversy over its role in pathogenesis [5, 21, 29, 116–118]. It is clearly not required for severe CA-MRSA infections in humans [119]. PVL has shown variable roles of involvement in murine models of pneumonia [26, 120–123]. Since PVL has specificity for neutrophils from humans and rabbits [106–112], rabbit models of USA300 do show a necrotizing pneumonia that suggests the contribution of PVL to pathology [112] with lung necrosis, pulmonary edema, and other outcomes consistent with the necrotizing pneumonia seen in humans. Rabbits infected with pvl strains showed decreased mortality, and complementation was able to restore virulence to the pvl strain. However, the extremely high inocula used in these studies and the length of infection have led many to remain skeptical of PVL being important in the pathogenesis of acute MRSA pneumonia in humans [29].
Staphylococcal gene products that activate immune signaling
In addition to the immunological effects of the hemolytic toxins, other S. aureus proteins activate T and B cells and numerous other immune effectors. These include T cell activation via superantigens (TSST and SEA/SEB) [124–126], complement evasion [127], polyclonal B cell and platelet activation via protein A [128, 129], immunoglobulin sequestration by S. aureus IgG-binding protein (Sbi) and protein A [130–132], and activation of the inflammasome by α-hemolysin [83, 84]. The production of the staphylococcal superantigens that are able to activate clones of T cells through V beta recognition alone has been well characterized for decades (Fig. 5). It is unclear whether this is a direct cause of pulmonary pathology, but more likely, the massive immunoactivation caused by S .aureus superantigen production is likely to contribute to pulmonary pathology. Superantigens evoke a massive immune response, stimulating T cells by cross-linking T cell receptors with MHC class II molecules on target T cells [124–126], resulting in T cell proliferation, cytokine production, and apoptosis [133]. Macrophages are accessory cells in this process [134], although a direct interaction with superantigens can induce cytokine production [135]. Two groups of superantigens produced by S. aureus are the toxic shock syndrome (TSS) toxin (TSST-1) and the enterotoxins.
Fig. 5.
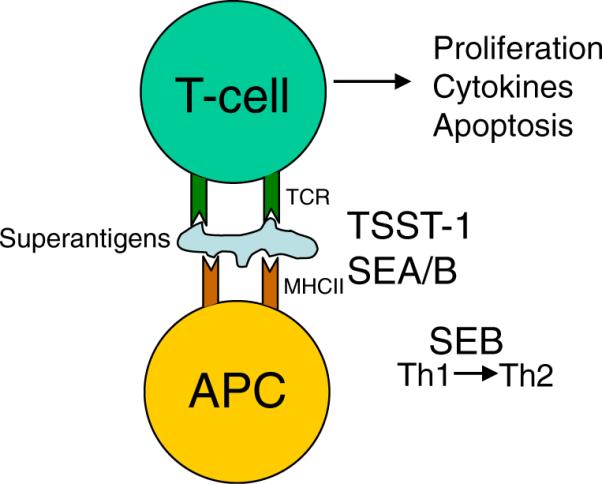
S. aureus superantigens. S. aureus superantigens cause T cell proliferation, cytokine production, and apoptosis by simultaneously cross-linking MHCII molecules on antigen presenting cells with T cell receptors
TSST-1 is the toxin responsible for TSS, a syndrome defined by fever, rash, hypotension, and multisystem involvement, initially associated with the use of highly absorbent tampons during menstruation [136–138] but is now well recognized in many patient populations [139]. Cases that involve the respiratory tract have been reported, including pneumonia, post-influenza infection, pharyngitis, and laryngotracheitis [140–144], although it is unclear what direct role TSST-1 has in pathogenesis. The enterotoxins (SE—A and B) of S. aureus are a group of structurally related toxins [145–147] suggested to play a role in the allergic response to infection in model systems [148]. A correlation exists between the levels of IgE antibodies to enterotoxin and asthma [149]. A similar observation is seen between enterotoxin IgE antibodies and higher eosinophil cationic protein, a marker for asthma and rhinitis as well as IgE levels for dust mites [150], suggesting that the presence of enterotoxin can mediate or potentiate the inflammation associated with asthma. Cytokine release by epithelial cells in response to SEA has also been observed [151].
Analogous to SEA, SEB also induces an inflammatory response in the lungs (Fig. 2). Consistent with SEA and SEB potentiating an allergic response, SEB shifts the T cell response from Th1 to Th2 and suppresses Treg activity to cause a state of persistent inflammation [152]. At low doses, SEB induces cytokine production in the lungs and leads to increases in eosinophil counts, while higher doses lead to increases in neutrophils and monocytes and are capable of producing an interstitial pneumonia [133, 153–156]. SEB also enhances the inflammation caused by allergens [157]. The interaction of human epithelial cells with SEB results in the production of IL-5 and GM-CSF [158].
Evasion of complement
The complement system performs a variety of functions to clear invading pathogens. S. aureus expresses a number of proteins to circumvent this response [127]. S. aureus expresses IgG-binding proteins that are also able to bind complement. Protein A (discussed below) is an IgG-binding protein that can bind to C1q, which is highly expressed on platelets [159]. A second IgG-binding protein, Sbi, binds to the Fc region of IgG and aids in avoiding neutrophil-mediated opsonophagocytosis (Fig. 4) [130, 160]. Sbi is also able to bind complement, interacting with the C3 portion of the cascade [161]. In vivo models have shown that Sbi promotes survival in blood [130].
Two additional complement evasion proteins are staphylococcal complement inhibitor (SCIN) and chemotaxis inhibitory protein of staphylococci (CHIPS; Fig. 4). Both SCIN and CHIPS are present on an immune evasion cluster located on a bacteriophage, which also contains staphylococcal enterotoxin B (described below) and staphylokinase. Staphylokinase activates human plasminogen to plasmin at the bacterial cell surface, creating a surface-bound protease capable of digesting IgG and complement [162]. Staphylokinase also releases α-defensins from neutrophils and interacts with α-defensins directly to inhibit bactericidal activity [163]. This immune evasion cluster is highly prevalent in carrier strains (90%), although it is not essential for nasal colonization [164]. SCIN is produced by the majority of strains. SCIN prevents the formation of the C3 convertases, resulting in a reduced phagocytosis and killing by neutrophils [165]. CHIPS is also capable of complement evasion via binding of the C5a receptors. CHIPS inhibits neutrophil and monocyte chemotaxis by interacting with the formylated peptide receptor [166, 167]. There are a number of other complement evasion proteins in the genome of S. aureus, including homologues of SCIN and CHIPS [168]. As with all of the complement evasion proteins, a direct role in pneumonia is yet to be established.
Protein A
Protein A (SpA) is perhaps the most complex of the staphylococcal components in terms of its multiple interactions with host immune signaling. Protein A (SpA) is an abundant surface protein of S. aureus expressed by virtually all strains that is shed during growth [169]. SpA contains an N-terminal signal sequence followed by five repeated domains (E, D, A, B, C) that bind IgG [131, 132], a property which is commercially utilized in biochemistry. SpA is anchored to the surface of the bacterial cell through a carboxy terminal LPXTG motif and the action of a sortase enzyme [71, 170]. The carboxy terminus of SpA contains variable numbers of a 24-bp repeat sequence known as the Xr region. The Xr region of SpA is successfully used in the typing of S. aureus isolates and has high concordance with genomic microarray data [171, 172]. This region is associated with a high mutational rate in S. aureus isolates from the airways of patients with cystic fibrosis [173]. The Xr region likely participates in immune signaling [174], although the mechanism remains to be defined.
S. aureus also induces the expansion of B cells through the binding of protein A. Protein A interacts with the Fab portion of V(H)3-type B cell receptors, reducing the adaptive immune response [175]. This induces polyclonal nonspecific B cell expansion and is thought to interfere with the use of staphylococcal mutants in vaccine development. Mutant forms of protein A unable to interact with B cells have shown promise as vaccine candidates in murine models of infection [176].
The role of protein A in the virulence of S. aureus has been demonstrated in models of sepsis and pneumonia [25, 176–178]. Neonatal mice infected with a spa mutant show reduced numbers of bacteria in both the lung and spleen, in addition to a significant reduction in pro-inflammatory signaling [177]. The spa strain was also significantly attenuated in its ability to activate signaling (CXCL8) in airway epithelial cells. In an adult model of murine staphylococcal pneumonia, mortality was significantly less with a spa mutant compared with the wild-type strain [25]. Several mechanisms have been proposed for the role of protein A in virulence, including its interaction with TNFR1 and epidermal growth factor receptor (EGFR; Fig. 2).
TNFR1 signaling by SpA
The TNF receptor (TNFR1) is expressed on many different cell types and initiates the activation of the pro-inflammatory and anti-apoptotic TNF cascade [179]. In the lung, epithelial TNFR1 is localized on the cell surface before being shed into the extracellular milieu after exposure to S. aureus through the action of ADAM17 (which is also activated by protein A) [177]. Protein A directly binds to TNFR1 via its IgG-binding domains, mimicking the TNF–TNFR1 interaction [177, 180]. This TNFR1–protein A interaction induces CXCL8 via TRAF2/p38 mitogen-activated protein kinase (MAPK) and NF-κB [181].
Protein A–TNFR1 responses play an important role in pathogenesis. Tnfr1−/− mice display significantly reduced levels of pneumonia and bacteremia when infected with S. aureus compared with wild-type mice. The levels of pneumonia and bacteria observed in TNFR1 null mice with the wild-type strain are comparable to those observed with the spa strain [177]. Wild-type mice infected with the spa strain as well as Tnfr1−/− mice infected with the wild-type organism both display reduced neutrophil numbers in the lung. TNFR1 signaling in response to S. aureus is a primary mechanism for inflammatory signaling since the TLR adaptor molecule MyD88 is dispensable for S. aureus pneumonia models in vivo [182].
EGFR signaling by SpA
A slightly different domain of the IgG-binding region of SpA initiates signaling events from EGFR. The apically displayed EGFR mediates a number of signaling events in the airway epithelium and responds to several different ligands.
SpA stimulates EGFR and ERK phosphorylation and stimulates ADAM17 (or TACE-TNF alpha-converting enzyme), the TNFR1 sheddase [180]. ADAM17 activity is also responsible for shedding IL-6Rα, trans-signaling which cleaves off the cytokine receptors that would perpetuate the pro-inflammatory response. Thus, S. aureus activates not only an epithelial pro-inflammatory response via both TNFR1 and EGFR but also ADAM17 that regulates this response.
Additional consequences of EGFR activation include the stimulation of epithelial wound repair and cytoskeletal contraction. Exposure of damaged epithelial cells to heat-killed S. aureus results in an increased wound closure and transepithelial resistance. This is in addition to the increased proliferation and survival of uninjured epithelial cells [183]. The initiation of epithelial wound repair is the result of TLR-EGFR signaling. A further consequence of EGFR activation is the induction of mucin production, an important physical component of the innate defenses of the lung [184].
Host responses to S. aureus in the respiratory tract
Perhaps the most important factor in the pathogenesis of staphylococcal pneumonia is the intensity of the innate immune response to the aspirated organisms. This is a response to the intact organisms as well as to shed components from the bacteria enmeshed in airway secretions. The initial immune signaling is accomplished by airway epithelial cells that immediately recruit several types of immune cells including dendritic cells (DCs), macrophages, and T cells. The net result of this immune signaling is the recruitment of polymorphonuclear neutrophils (PMNs) which are critical in staphylococcal clearance. It has long been thought that PMNs themselves are the single most important component in the host response to S. aureus. This observation is borne out by the increased susceptibility to staphylococcal infection of humans with deficiencies in neutrophil chemotaxis, oxidative burst, and granule production [185]. However, the cells that actively recruit PMNs and activate them may also be potential targets to modulate the intensity of the inflammatory response to the organisms.
TLR signaling in the airway
The pattern recognition receptors on the surface of exposed airway epithelial cells readily respond to the surface components of S. aureus. The airway epithelium expresses the full complement of Toll-like receptors (TLRs). High levels of gene transcription are observed for TLRs 2–6, while the expression for 7/8 is variable, depending upon the cell type [186–189]. Adaptors such as MyD88 and MD2 are also present in addition to RNA receptors like MDA5 ad RIG-I [186, 190].
TLR2, which recognizes lipoteichoic acid (LTA) in the staphylococcal cell wall, is important in the response to S. aureus airway infection (Fig. 2). The instillation of LTA into human airways results in a significant inflammation and neutrophil recruitment, an observation mimicked in murine models [191, 192]. TLR2 is upregulated in response to S. aureus [193]. The virulence factor PVL also directly activates a small group of genes via TLR2 signaling that incorporates the adaptors CD14 and MyD88, leading to NF-κBsignaling [114]. TLR2 is important in some models of systemic S. aureus infections [194, 195]. Consistent with this, strains lacking lipoproteins avoid immune recognition and cause increased disease [196]. However, in respiratory infection, MyD88 is dispensable for bacterial clearance, even with a significantly abrogated NF-κB-dependent cytokine response [182], consistent with the observations that several other pathways (TNF and type I IFN) are also critical for the immune response to S. aureus. The airway epithelium responds to this TLR–S. aureus interaction with a variety of cytokines, including GM-CSF, G-CSF, CXCL8, and TGF-α, as well as the antimicrobial peptides beta-defensins [180, 193, 197, 198]. The activation of G-CSF and GM-CSF is important in the airway as this promotes neutrophil survival [197]. S. aureus is capable of activating CXCL8 production via the MAPK p38 and ERK1/2, which leads to the activation of the NK-κB transcription factor [199].
Type I interferons
Intracellular receptors, such as those linked to type I interferon (IFN), can also recognize staphylococci in the airway as bacterial cells often secrete and shed components that may be internalized by airway epithelial cells. Type I IFN signaling has long been known as an important viral defense pathway in the lung [200] resulting in the transcription of hundreds of genes [201]. S. aureus, as well as several other extracellular bacterial pathogens, activate this pathway (Fig. 2) [202, 203]. The role type I IFN signaling plays in bacterial infection is variable, depending upon the organisms [204]. Bacteria activate type I IFN signaling through a number of TLR, NOD-like receptors (NLR), and cytosolic receptors that sense products such as cell wall components, DNA, and RNA [205–208]. The activation of type I IFN signaling leads to the production of IFN-β via the phosphorylation of an interferon regulatory factor (IRF), namely, IRF3, IRF5, and IRF7 [209–212]. IFN-β interacts with its receptor, IFNAR (interferon alpha/beta receptor), leading to the phosphorylation of STAT1/2 via the kinases Jak1 and Tyk2 [213]. The activation of type I IFN signaling also activates MAPK [214] and NF-κB responses [215].
S. aureus activates type I IFN signaling in airway epithelial cells as well as in in vivo models of pneumonia [173]. Incubation of epithelial cells with live organisms activates Ifnb as well as a number of downstream genes, including Lif and Mx-1, as early as 2 h after stimulation via the expected pathways [173]. In vivo, the activation of type I IFN signaling in response to S. aureus is detrimental to the host; Ifnar−/− mice were significantly less likely to succumb to pulmonary infection than their wild-type counterparts [173]. Consistent with this phenotype being host-derived, Ifnar−/− mice had less TNF and IL-6 in their bronchoalveolar lavage (BAL) fluid while having similar bacterial loads when inoculated with non-lethal doses.
The amplification of potentially damaging inflammation that is initiated through type I IFN signaling is mediated by the CXCR3 chemokines. The CXCR3 chemokines—CXCL9 (MIG), CXCL10 (IP-10), and CXCL11 (I-TAC)—perform a variety of functions including the recruitment of Th1 and Th17 cells [21–218], increasing PMN accumulation and inflammation [216, 219]. Depletion of macrophages had a detrimental effect on the outcome of MRSA pneumonia in mice, which in part was due to the increased expression of the CXCR3 ligands [219]. Blocking CXCR3 resulted in reduced CD4+ cells in the BAL and reduced pulmonary pathology [219]. Given the multiple pro-inflammatory pathogen-associated molecular patterns (PAMPs) associated with virulent staphylococci, strategies to modulate the host responses to these effectors may be useful to control pulmonary damage. While it is not fully understood how S. aureus causes lung damage, certainly a portion of the pathology is due to the host itself, as activated by these organisms. Moreover, from the murine models, it seems that PMNs, despite their damaging release of reactive oxygen intermediates and elastase, are not the sole cause of pulmonary destruction due to staphylococcal pneumonia.
The inflammasome
Much of the pathology caused by infection with S. aureus is a result of the intense immune response. A known cause of pulmonary pathology, the cytokine IL-1β, is produced in response to TLR2 recognition of staphylococcal lipoproteins and membrane perturbation by α-hemolysin as part of pyroptosis, a highly inflammatory form of cell death (Fig. 3) [84, 220]. Pyroptosis involves the activation of the inflammasome, characterized by the activation of caspase-1 and the production of IL-1β and IL-18 [221, 222]. Activation of the inflammasome is a two-step process that typically involves the activation of a TLR (such as via microbial PAMPs) to induce the production of pro-IL-1β then membrane perturbation (e.g., via a toxin to generate K+ efflux and the proteins NLRP3/NALP3/cryopyrin and ASC) to produce active caspase-1 that cleaves pro-IL-1β to enable the secretion of mature IL-1β [223]. This is accomplished in immune cells by S. aureus expressing α-hemolysin and TLR2-MyD88 signaling from lipoproteins [83, 84, 224, 225]. An additional S. aureus component that activates the production of IL-1β is peptidogylcan and cell wall degradation products [226]. Inflammasome activation in known to occur in monocytic cells; the IL-1β generated from this process is likely to contribute to pulmonary pathology. The role epithelial cells play in this process is an area of active investigation.
Staphylococcal superinfection following influenza and the Th17 response
The importance of host immune signaling in the successful clearance of S. aureus from the respiratory tract is well illustrated in the common clinical presentation of bacterial superinfection following influenza. The high mortality associated with staphylococcal pneumonia following influenza was first documented during the 1918 influenza pandemic in which many young, previously health individuals died of staphylococcal pneumonia, having previously recovered from influenza [227, 228]. Infecting mice with S. aureus after influenza exposure significantly increases mortality and lung pathology [18, 229–231]. Several components of immune signaling may contribute to this excess mortality. Influenza-infected mice were observed to have impaired NK cell responses upon S. aureus infection [230], which were essential in producing TNF to mediate bacterial clearance.
A major component of the immune response to influenza is the activation of type I IFN signaling, which increases susceptibility to S. aureus pneumonia [173]. It seems likely that excessive IFN-β signaling contributes to this pathology [229]. Kudva et al. [231] reinforced the negative impact of type I IFN signaling in staphylococcal pneumonia, linking the significantly higher levels of type I IFNs that resulted in a reduced Th17 response, contributing to a reduced bacterial clearance. When the co-infection was repeated in Ifnar−/− mice, an improved clearance of S. aureus was observed.
Airway epithelial cells are highly dependent on typical pro-inflammatory as well as Th17 cytokines to produce anti-staphylococcal factors [232]. It has been observed that S. aureus can activate IL-17 in DCs and blood monocytes [233, 234]. Deficiency in the Th17 pathway increases susceptibility to cutaneous staphylococcal infections [235] and is also the mechanism attributed to the frequent staphylococcal infection in patients with hyper-IgE syndrome [236]. Hyper-IgE patients possess mutations in the STAT3 transcription factor, resulting in a failure of CD4 T cells to differentiate into Th17 cells. Influenza-infected mice have reduced Th17 cytokines. A decrease in Th17 cytokines (IL-17, 22, and 23) is observed in influenza-infected mice [231] in part due to increased type I interferons. Consistent with this observation, Th17 knockout mice exhibited impaired clearance to S. aureus [231]. Transgenic mice overexpressing IL-23 and co-infected with influenza and S. aureus had improved outcomes. These studies provide examples of how viral infections (namely influenza) and their profound immunological consequences can predispose individuals to potentially lethal secondary pneumonia.
Conclusion
S. aureus has evolved into the consummate respiratory pathogen able to not only persist in the respiratory tract but also thrive as an invasive pulmonary pathogen. Through the horizontal acquisition of genetic material and positive selective pressure, successful clones of staphylococci, as typified by the USA300 strains, express virulence factors that enable the organisms to successfully exploit the immune response that is evoked in the lung. Strategies to prevent invasive infection will not only need to target the virulence determinants that mediate colonization, iron acquisition, biofilm production, and immune evasion but will also need to be effective against organisms that can disseminate intracellularly. Approaches to the management of staphylococcal pneumonia, perhaps by modulating the host immune response, could be considered as an adjunct to the conventional antimicrobial therapy.
Acknowledgments
Work on S. aureus in the laboratory is supported by 5R01HL079395 (to AP) from the NIH.
References
- 1.Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, et al. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA. 2007;298:1763–1771. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 2.Deleo FR, Otto M, Kreiswirth BN, Chambers HF. Community-associated meticillin-resistant Staphylococcus aureus. Lancet. 2010;375:1557–1568. doi: 10.1016/S0140-6736(09)61999-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Montgomery CP, Boyle-Vavra S, Adem PV, Lee JC, Husain AN, Clasen J, et al. Comparison of virulence in community-associated methicillin-resistant Staphylococcus aureus pulso-types USA300 and USA400 in a rat model of pneumonia. J Infect Dis. 2008;198:561–570. doi: 10.1086/590157. [DOI] [PubMed] [Google Scholar]
- 4.Oswald NC, Shooter RA, Curwen MP. Pneumonia complicating Asian influenza. Br Med J. 1958;2:1305–1311. doi: 10.1136/bmj.2.5108.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hageman JC, Uyeki TM, Francis JS, Jernigan DB, Wheeler JG, Bridges CB, et al. Severe community-acquired pneumonia due to Staphylococcus aureus, 2003–04 influenza season. Emerg Infect Dis. 2006;12:894–899. doi: 10.3201/eid1206.051141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McDougal LK, Steward CD, Killgore GE, Chaitram JM, McAllister SK, Tenover FC. Pulsed-field gel electrophoresis typing of oxacillin-resistant Staphylococcus aureus isolates from the United States: establishing a national database. J Clin Microbiol. 2003;41:5113–5120. doi: 10.1128/JCM.41.11.5113-5120.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Centers for Disease Control and Prevention Outbreaks of community-associated methicillin-resistant Staphylococcus aureus skin infections—Los Angeles County, California, 2002–2003. MMWR Morb Mortal Wkly Rep. 2003;52:88. [PubMed] [Google Scholar]
- 8.Begier EM, Frenette K, Barrett NL, Mshar P, Petit S, Boxrud DJ, et al. A high-morbidity outbreak of methicillin-resistant Staphylococcus aureus among players on a college football team, facilitated by cosmetic body shaving and turf burns. Clin Infect Dis. 2004;39:1446–1453. doi: 10.1086/425313. [DOI] [PubMed] [Google Scholar]
- 9.Kazakova SV, Hageman JC, Matava M, Srinivasan A, Phelan L, Garfinkel B, et al. A clone of methicillin-resistant Staphylococcus aureus among professional football players. N Engl J Med. 2005;352:468–475. doi: 10.1056/NEJMoa042859. [DOI] [PubMed] [Google Scholar]
- 10.Gilbert M, MacDonald J, Gregson D, Siushansian J, Zhang K, Elsayed S, et al. Outbreak in Alberta of community-acquired (USA300) methicillin-resistant Staphylococcus aureus in people with a history of drug use, homelessness or incarceration. CMAJ. 2006;175:149–154. doi: 10.1503/cmaj.051565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Centers for Disease Control and Prevention Methicillin-resistant Staphylococcus aureus infections in correctional facilities–Georgia, California, and Texas, 2001–2003. MMWR Morb Mortal Wkly Rep. 2003;52:992–996. [PubMed] [Google Scholar]
- 12.Carrillo-Marquez MA, Hulten KG, Hammerman W, Lamberth L, Mason EO, Kaplan SL. Staphylococcus aureus pneumonia in children in the era of community-acquired methicillin-resistance at Texas Children's Hospital. Pediatr Infect Dis J. 2011;30:545–550. doi: 10.1097/INF.0b013e31821618be. [DOI] [PubMed] [Google Scholar]
- 13.Herold BC, Immergluck LC, Maranan MC, Lauderdale DS, Gaskin RE, Boyle-Vavra S, et al. Community-acquired methicillin-resistant Staphylococcus aureus in children with no identified predisposing risk. JAMA. 1998;279:593–598. doi: 10.1001/jama.279.8.593. [DOI] [PubMed] [Google Scholar]
- 14.Boucher HW, Corey GR. Epidemiology of methicillin-resistant Staphylococcus aureus. Clin Infect Dis. 2008;46(Suppl 5):S344–S349. doi: 10.1086/533590. [DOI] [PubMed] [Google Scholar]
- 15.Finelli L, Fiore A, Dhara R, Brammer L, Shay DK, Kamimoto L, et al. Influenza-associated pediatric mortality in the United States: increase of Staphylococcus aureus coinfection. Pediatrics. 2008;122:805–811. doi: 10.1542/peds.2008-1336. [DOI] [PubMed] [Google Scholar]
- 16.Garnier F, Tristan A, Francois B, Etienne J, Delage-Corre M, Martin C, et al. Pneumonia and new methicillin-resistant Staphylococcus aureus clone. Emerg Infect Dis. 2006;12:498–500. doi: 10.3201/eid1203.051040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Risson DC, O'Connor ED, Guard RW, Schooneveldt JM, Nimmo GR. A fatal case of necrotising pneumonia due to community-associated methicillin-resistant Staphylococcus aureus. Med J Aust. 2007;186:479–480. doi: 10.5694/j.1326-5377.2007.tb01002.x. [DOI] [PubMed] [Google Scholar]
- 18.Iverson AR, Boyd KL, McAuley JL, Plano LR, Hart ME, McCullers JA. Influenza virus primes mice for pneumonia from Staphylococcus aureus. J Infect Dis. 2011;203:880–888. doi: 10.1093/infdis/jiq113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li M, Diep BA, Villaruz AE, Braughton KR, Jiang X, DeLeo FR, et al. Evolution of virulence in epidemic community-associated methicillin-resistant Staphylococcus aureus. Proc Natl Acad Sci USA. 2009;106:5883–5888. doi: 10.1073/pnas.0900743106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diep BA, Gill SR, Chang RF, Phan TH, Chen JH, Davidson MG, et al. Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet. 2006;367:731–739. doi: 10.1016/S0140-6736(06)68231-7. [DOI] [PubMed] [Google Scholar]
- 21.Diep BA, Carleton HA, Chang RF, Sensabaugh GF, Perdreau-Remington F. Roles of 34 virulence genes in the evolution of hospital- and community-associated strains of methicillin-resistant Staphylococcus aureus. J Infect Dis. 2006;193:1495–1503. doi: 10.1086/503777. [DOI] [PubMed] [Google Scholar]
- 22.Diep BA, Stone GG, Basuino L, Graber CJ, Miller A, des Etages SA, et al. The arginine catabolic mobile element and staphylococcal chromosomal cassette mec linkage: convergence of virulence and resistance in the USA300 clone of methicillin-resistant Staphylococcus aureus. J Infect Dis. 2008;197:1523–1530. doi: 10.1086/587907. [DOI] [PubMed] [Google Scholar]
- 23.Montgomery CP, Boyle-Vavra S, Daum RS. The arginine catabolic mobile element is not associated with enhanced virulence in experimental invasive disease caused by the community-associated methicillin-resistant Staphylococcus aureus USA300 genetic background. Infect Immun. 2009;77:2650–2656. doi: 10.1128/IAI.00256-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Diep BA, Otto M. The role of virulence determinants in community-associated MRSA pathogenesis. Trends Microbiol. 2008;16:361–369. doi: 10.1016/j.tim.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bubeck Wardenburg J, Patel RJ, Schneewind O. Surface proteins and exotoxins are required for the pathogenesis of Staphylococcus aureus pneumonia. Infect Immun. 2007;75:1040–1044. doi: 10.1128/IAI.01313-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bubeck Wardenburg J, Bae T, Otto M, Deleo FR, Schneewind O. Poring over pores: alpha-hemolysin and Panton–Valentine leukocidin in Staphylococcus aureus pneumonia. Nat Med. 2007;13:1405–1406. doi: 10.1038/nm1207-1405. [DOI] [PubMed] [Google Scholar]
- 27.Bartlett AH, Foster TJ, Hayashida A, Park PW. Alpha-toxin facilitates the generation of CXC chemokine gradients and stimulates neutrophil homing in Staphylococcus aureus pneumonia. J Infect Dis. 2008;198:1529–1535. doi: 10.1086/592758. [DOI] [PubMed] [Google Scholar]
- 28.Pishchany G, McCoy AL, Torres VJ, Krause JC, Crowe JE, Jr, Fabry ME, et al. Specificity for human hemoglobin enhances Staphylococcus aureus infection. Cell Host Microbe. 2010;8:544–550. doi: 10.1016/j.chom.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Otto M. A MRSA-terious enemy among us: end of the PVL controversy? Nat Med. 2011;17:169–170. doi: 10.1038/nm0211-169. [DOI] [PubMed] [Google Scholar]
- 30.Kapral FA, Shayegani MG. Intracellular survival of staphylococci. J Exp Med. 1959;110:123–138. doi: 10.1084/jem.110.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Melly MA, Thomison JB, Rogers DE. Fate of staphylococci within human leukocytes. J Exp Med. 1960;112:1121–1130. doi: 10.1084/jem.112.6.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.da Silva MC, Zahm JM, Gras D, Bajolet O, Abely M, Hinnrasky J, et al. Dynamic interaction between airway epithelial cells and Staphylococcus aureus. Am J Physiol Lung Cell Mol Physiol. 2004;287:L543–L551. doi: 10.1152/ajplung.00256.2003. [DOI] [PubMed] [Google Scholar]
- 33.Belay N, Rasooly A. Staphylococcus aureus growth and enterotoxin A production in an anaerobic environment. J Food Prot. 2002;65:199–204. doi: 10.4315/0362-028x-65.1.199. [DOI] [PubMed] [Google Scholar]
- 34.Recsei P, Kreiswirth B, O'Reilly M, Schlievert P, Gruss A, Novick RP. Regulation of exoprotein gene expression in Staphylococcus aureus by agar. Mol Gen Genet. 1986;202:58–61. doi: 10.1007/BF00330517. [DOI] [PubMed] [Google Scholar]
- 35.Novick RP. Autoinduction and signal transduction in the regulation of staphylococcal virulence. Mol Microbiol. 2003;48:1429–1449. doi: 10.1046/j.1365-2958.2003.03526.x. [DOI] [PubMed] [Google Scholar]
- 36.Cheung GY, Wang R, Khan BA, Sturdevant DE, Otto M. Role of the accessory gene regulator agr in community-associated methicillin-resistant Staphylococcus aureus pathogenesis. Infect Immun. 2011;79:1927–1935. doi: 10.1128/IAI.00046-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Traber KE, Lee E, Benson S, Corrigan R, Cantera M, Shopsin B, et al. agr function in clinical Staphylococcus aureus isolates. Microbiology. 2008;154:2265–2274. doi: 10.1099/mic.0.2007/011874-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heyer G, Saba S, Adamo R, Rush W, Soong G, Cheung A, et al. Staphylococcus aureus agr and sarA functions are required for invasive infection but not inflammatory responses in the lung. Infect Immun. 2002;70:127–133. doi: 10.1128/IAI.70.1.127-133.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Montgomery CP, Boyle-Vavra S, Daum RS. Importance of the global regulators Agr and SaeRS in the pathogenesis of CA-MRSA USA300 infection. PLoS One. 2010;5:e15177. doi: 10.1371/journal.pone.0015177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qazi SN, Counil E, Morrissey J, Rees CE, Cockayne A, Winzer K, et al. agr expression precedes escape of internalized Staphylococcus aureus from the host endosome. Infect Immun. 2001;69:7074–7082. doi: 10.1128/IAI.69.11.7074-7082.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shompole S, Henon KT, Liou LE, Dziewanowska K, Bohach GA, Bayles KW. Biphasic intracellular expression of Staphylococcus aureus virulence factors and evidence for Agr-mediated diffusion sensing. Mol Microbiol. 2003;49:919–927. doi: 10.1046/j.1365-2958.2003.03618.x. [DOI] [PubMed] [Google Scholar]
- 42.Giese B, Glowinski F, Paprotka K, Dittmann S, Steiner T, Sinha B, et al. Expression of delta-toxin by Staphylococcus aureus mediates escape from phago-endosomes of human epithelial and endothelial cells in the presence of beta-toxin. Cell Microbiol. 2011;13:316–329. doi: 10.1111/j.1462-5822.2010.01538.x. [DOI] [PubMed] [Google Scholar]
- 43.Jarry TM, Cheung AL. Staphylococcus aureus escapes more efficiently from the phagosome of a cystic fibrosis bronchial epithelial cell line than from its normal counterpart. Infect Immun. 2006;74:2568–2577. doi: 10.1128/IAI.74.5.2568-2577.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bayles KW, Wesson CA, Liou LE, Fox LK, Bohach GA, Trumble WR. Intracellular Staphylococcus aureus escapes the endosome and induces apoptosis in epithelial cells. Infect Immun. 1998;66:336–342. doi: 10.1128/iai.66.1.336-342.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kubica M, Guzik K, Koziel J, Zarebski M, Richter W, Gajkowska B, et al. A potential new pathway for Staphylococcus aureus dissemination: the silent survival of S. aureus phagocytosed by human monocyte-derived macrophages. PLoS One. 2008;3:e1409. doi: 10.1371/journal.pone.0001409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gresham HD, Lowrance JH, Caver TE, Wilson BS, Cheung AL, Lindberg FP. Survival of Staphylococcus aureus inside neutrophils contributes to infection. J Immunol. 2000;164:3713–3722. doi: 10.4049/jimmunol.164.7.3713. [DOI] [PubMed] [Google Scholar]
- 47.Voyich JM, Braughton KR, Sturdevant DE, Whitney AR, Said-Salim B, Porcella SF, et al. Insights into mechanisms used by Staphylococcus aureus to avoid destruction by human neutrophils. J Immunol. 2005;175:3907–3919. doi: 10.4049/jimmunol.175.6.3907. [DOI] [PubMed] [Google Scholar]
- 48.Konetschny-Rapp S, Jung G, Meiwes J, Zahner H, Staphyloferrin A. A structurally new siderophore from staphylococci. Eur J Biochem. 1990;191:65–74. doi: 10.1111/j.1432-1033.1990.tb19094.x. [DOI] [PubMed] [Google Scholar]
- 49.Courcol RJ, Trivier D, Bissinger MC, Martin GR, Brown MR. Siderophore production by Staphylococcus aureus and identification of iron-regulated proteins. Infect Immun. 1997;65:1944–1948. doi: 10.1128/iai.65.5.1944-1948.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dale SE, Doherty-Kirby A, Lajoie G, Heinrichs DE. Role of siderophore biosynthesis in virulence of Staphylococcus aureus: identification and characterization of genes involved in production of a siderophore. Infect Immun. 2004;72:29–37. doi: 10.1128/IAI.72.1.29-37.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Torres VJ, Pishchany G, Humayun M, Schneewind O, Skaar EP. Staphylococcus aureus IsdB is a hemoglobin receptor required for heme iron utilization. J Bacteriol. 2006;188:8421–8429. doi: 10.1128/JB.01335-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beasley FC, Marolda CL, Cheung J, Buac S, Heinrichs DE. Staphylococcus aureus transporters Hts, Sir, and Sst capture iron liberated from human transferrin by Staphyloferrin A, Staphyloferrin B, and catecholamine stress hormones, respectively, and contribute to virulence. Infect Immun. 2011;79:2345–2355. doi: 10.1128/IAI.00117-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Torres VJ, Attia AS, Mason WJ, Hood MI, Corbin BD, Beasley FC, et al. Staphylococcus aureus fur regulates the expression of virulence factors that contribute to the pathogenesis of pneumonia. Infect Immun. 2010;78:1618–1628. doi: 10.1128/IAI.01423-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Datta R, Huang SS. Risk of infection and death due to methicillin-resistant Staphylococcus aureus in long-term carriers. Clin Infect Dis. 2008;47:176–181. doi: 10.1086/589241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lederer SR, Riedelsdorf G, Schiffl H. Nasal carriage of meticillin resistant Staphylococcus aureus: the prevalence, patients at risk and the effect of elimination on outcomes among outclinic haemodialysis patients. Eur J Med Res. 2007;12:284–288. [PubMed] [Google Scholar]
- 56.Diller R, Sonntag AK, Mellmann A, Grevener K, Senninger N, Kipp F, et al. Evidence for cost reduction based on pre-admission MRSA screening in general surgery. Int J Hyg Environ Health. 2008;211:205–212. doi: 10.1016/j.ijheh.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 57.Patti JM, Allen BL, McGavin MJ, Hook M. MSCRAMM-mediated adherence of microorganisms to host tissues. Annu Rev Microbiol. 1994;48:585–617. doi: 10.1146/annurev.mi.48.100194.003101. [DOI] [PubMed] [Google Scholar]
- 58.Hienz SA, Schennings T, Heimdahl A, Flock JI. Collagen binding of Staphylococcus aureus is a virulence factor in experimental endocarditis. J Infect Dis. 1996;174:83–88. doi: 10.1093/infdis/174.1.83. [DOI] [PubMed] [Google Scholar]
- 59.Rhem MN, Lech EM, Patti JM, McDevitt D, Hook M, Jones DB, et al. The collagen-binding adhesin is a virulence factor in Staphylococcus aureus keratitis. Infect Immun. 2000;68:3776–3779. doi: 10.1128/iai.68.6.3776-3779.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.de Bentzmann S, Tristan A, Etienne J, Brousse N, Vandenesch F, Lina G. Staphylococcus aureus isolates associated with necrotizing pneumonia bind to basement membrane type I and IV collagens and laminin. J Infect Dis. 2004;190:1506–1515. doi: 10.1086/424521. [DOI] [PubMed] [Google Scholar]
- 61.Mongodin E, Bajolet O, Cutrona J, Bonnet N, Dupuit F, Puchelle E, et al. Fibronectin-binding proteins of Staphylococcus aureus are involved in adherence to human airway epithelium. Infect Immun. 2002;70:620–630. doi: 10.1128/iai.70.2.620-630.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schaffer AC, Solinga RM, Cocchiaro J, Portoles M, Kiser KB, Risley A, et al. Immunization with Staphylococcus aureus clumping factor B, a major determinant in nasal carriage, reduces nasal colonization in a murine model. Infect Immun. 2006;74:2145–2153. doi: 10.1128/IAI.74.4.2145-2153.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McElroy MC, Cain DJ, Tyrrell C, Foster TJ, Haslett C. Increased virulence of a fibronectin-binding protein mutant of Staphylococcus aureus in a rat model of pneumonia. Infect Immun. 2002;70:3865–3873. doi: 10.1128/IAI.70.7.3865-3873.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Higgins J, Loughman A, van Kessel KP, van Strijp JA, Foster TJ. Clumping factor A of Staphylococcus aureus inhibits phagocytosis by human polymorphonuclear leucocytes. FEMS Microbiol Lett. 2006;258:290–296. doi: 10.1111/j.1574-6968.2006.00229.x. [DOI] [PubMed] [Google Scholar]
- 65.Palmqvist N, Patti JM, Tarkowski A, Josefsson E. Expression of staphylococcal clumping factor A impedes macrophage phagocytosis. Microbes Infect. 2004;6:188–195. doi: 10.1016/j.micinf.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 66.Josefsson E, Hartford O, o'Brien L, Patti JM, Foster T. Protection against experimental Staphylococcus aureus arthritis by vaccination with clumping factor A, a novel virulence determinant. J Infect Dis. 2001;184:1572–1580. doi: 10.1086/324430. [DOI] [PubMed] [Google Scholar]
- 67.Vernachio J, Bayer AS, Le T, Chai YL, Prater B, Schneider A, et al. Anti-clumping factor A immunoglobulin reduces the duration of methicillin-resistant Staphylococcus aureus bacteremia in an experimental model of infective endocarditis. Antimicrob Agents Chemother. 2003;47:3400–3406. doi: 10.1128/AAC.47.11.3400-3406.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moreillon P, Entenza JM, Francioli P, McDevitt D, Foster TJ, Francois P, et al. Role of Staphylococcus aureus coagulase and clumping factor in pathogenesis of experimental endocarditis. Infect Immun. 1995;63:4738–4743. doi: 10.1128/iai.63.12.4738-4743.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Weems JJ, Jr, Steinberg JP, Filler S, Baddley JW, Corey GR, Sampathkumar P, et al. Phase II, randomized, double-blind, multicenter study comparing the safety and pharmacokinetics of tefibazumab to placebo for treatment of Staphylococcus aureus bacteremia. Antimicrob Agents Chemother. 2006;50:2751–2755. doi: 10.1128/AAC.00096-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.O'Brien LM, Walsh EJ, Massey RC, Peacock SJ, Foster TJ. Staphylococcus aureus clumping factor B (ClfB) promotes adherence to human type I cytokeratin 10: implications for nasal colonization. Cell Microbiol. 2002;4:759–770. doi: 10.1046/j.1462-5822.2002.00231.x. [DOI] [PubMed] [Google Scholar]
- 71.Mazmanian SK, Liu G, Ton-That H, Schneewind O. Staphylococcus aureus sortase, an enzyme that anchors surface proteins to the cell wall. Science. 1999;285:760–763. doi: 10.1126/science.285.5428.760. [DOI] [PubMed] [Google Scholar]
- 72.Wertheim HF, Walsh E, Choudhurry R, Melles DC, Boelens HA, Miajlovic H, et al. Key role for clumping factor B in Staphylococcus aureus nasal colonization of humans. PLoS Med. 2008;5:e17. doi: 10.1371/journal.pmed.0050017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Song L, Hobaugh MR, Shustak C, Cheley S, Bayley H, Gouaux JE. Structure of staphylococcal alpha-hemolysin, a heptameric transmembrane pore. Science. 1996;274:1859–1866. doi: 10.1126/science.274.5294.1859. [DOI] [PubMed] [Google Scholar]
- 74.Burlak C, Hammer CH, Robinson MA, Whitney AR, McGavin MJ, Kreiswirth BN, et al. Global analysis of community-associated methicillin-resistant Staphylococcus aureus exoproteins reveals molecules produced in vitro and during infection. Cell Microbiol. 2007;9:1172–1190. doi: 10.1111/j.1462-5822.2006.00858.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yun YS, Min YG, Rhee CS, Jung IH, Koh YY, Jang TY, et al. Effects of alpha-toxin of Staphylococcus aureus on the ciliary activity and ultrastructure of human nasal ciliated epithelial cells. Laryngoscope. 1999;109:2021–2024. doi: 10.1097/00005537-199912000-00024. [DOI] [PubMed] [Google Scholar]
- 76.Rose F, Dahlem G, Guthmann B, Grimminger F, Maus U, Hanze J, et al. Mediator generation and signaling events in alveolar epithelial cells attacked by S. aureus alpha-toxin. Am J Physiol Lung Cell Mol Physiol. 2002;282:L207–L214. doi: 10.1152/ajplung.00156.2001. [DOI] [PubMed] [Google Scholar]
- 77.Wilke GA, Bubeck Wardenburg J. Role of a disintegrin and metalloprotease 10 in Staphylococcus aureus alpha-hemolysin-mediated cellular injury. Proc Natl Acad Sci USA. 2010;107:13473–13478. doi: 10.1073/pnas.1001815107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Seeger W, Birkemeyer RG, Ermert L, Suttorp N, Bhakdi S, Duncker HR. Staphylococcal alpha-toxin-induced vascular leakage in isolated perfused rabbit lungs. Lab Invest. 1990;63:341–349. [PubMed] [Google Scholar]
- 79.Phillips JR, Tripp TJ, Regelmann WE, Schlievert PM, Wangensteen OD. Staphylococcal alpha-toxin causes increased tracheal epithelial permeability. Pediatr Pulmonol. 2006;41:1146–1152. doi: 10.1002/ppul.20501. [DOI] [PubMed] [Google Scholar]
- 80.McElroy MC, Harty HR, Hosford GE, Boylan GM, Pittet JF, Foster TJ. Alpha-toxin damages the air–blood barrier of the lung in a rat model of Staphylococcus aureus-induced pneumonia. Infect Immun. 1999;67:5541–5544. doi: 10.1128/iai.67.10.5541-5544.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bantel H, Sinha B, Domschke W, Peters G, Schulze-Osthoff K, Janicke RU. alpha-Toxin is a mediator of Staphylococcus aureus-induced cell death and activates caspases via the intrinsic death pathway independently of death receptor signaling. J Cell Biol. 2001;155:637–648. doi: 10.1083/jcb.200105081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jonas D, Walev I, Berger T, Liebetrau M, Palmer M, Bhakdi S. Novel path to apoptosis: small transmembrane pores created by staphylococcal alpha-toxin in T lymphocytes evoke internucleosomal DNA degradation. Infect Immun. 1994;62:1304–1312. doi: 10.1128/iai.62.4.1304-1312.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Craven RR, Gao X, Allen IC, Gris D, Bubeck Wardenburg J, McElvania-Tekippe E, et al. Staphylococcus aureus alpha-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PLoS One. 2009;4:e7446. doi: 10.1371/journal.pone.0007446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Munoz-Planillo R, Franchi L, Miller LS, Nunez G. A critical role for hemolysins and bacterial lipoproteins in Staphylococcus aureus-induced activation of the Nlrp3 inflammasome. J Immunol. 2009;183:3942–3948. doi: 10.4049/jimmunol.0900729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Grimminger F, Rose F, Sibelius U, Meinhardt M, Potzsch B, Spriestersbach R, et al. Human endothelial cell activation and mediator release in response to the bacterial exotoxins Escherichia coli hemolysin and staphylococcal alpha-toxin. J Immunol. 1997;159:1909–1916. [PubMed] [Google Scholar]
- 86.Liang X, Ji Y. Involvement of alpha5beta1-integrin and TNF-alpha in Staphylococcus aureus alpha-toxin-induced death of epithelial cells. Cell Microbiol. 2007;9:1809–1821. doi: 10.1111/j.1462-5822.2007.00917.x. [DOI] [PubMed] [Google Scholar]
- 87.Bubeck Wardenburg J, Schneewind O. Vaccine protection against Staphylococcus aureus pneumonia. J Exp Med. 2008;205:287–294. doi: 10.1084/jem.20072208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ragle BE, Bubeck Wardenburg J. Anti-alpha-hemolysin monoclonal antibodies mediate protection against Staphylococcus aureus pneumonia. Infect Immun. 2009;77:2712–2718. doi: 10.1128/IAI.00115-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ragle BE, Karginov VA, Bubeck Wardenburg J. Prevention and treatment of Staphylococcus aureus pneumonia with a beta-cyclodextrin derivative. Antimicrob Agents Chemother. 2010;54:298–304. doi: 10.1128/AAC.00973-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Aarestrup FM, Larsen HD, Eriksen NH, Elsberg CS, Jensen NE. Frequency of alpha- and beta-haemolysin in Staphylococcus aureus of bovine and human origin. A comparison between pheno- and genotype and variation in phenotypic expression. APMIS. 1999;107:425–430. [PubMed] [Google Scholar]
- 91.Doery HM, Magnusson BJ, Cheyne IM, Sulasekharam J. A phospholipase in staphylococcal toxin which hydrolyses sphingomyelin. Nature. 1963;198:1091–1092. doi: 10.1038/1981091a0. [DOI] [PubMed] [Google Scholar]
- 92.Marshall MJ, Bohach GA, Boehm DF. Characterization of Staphylococcus aureus beta-toxin induced leukotoxicity. J Nat Toxins. 2000;9:125–138. [PubMed] [Google Scholar]
- 93.Walev I, Weller U, Strauch S, Foster T, Bhakdi S. Selective killing of human monocytes and cytokine release provoked by sphingomyelinase (beta-toxin) of Staphylococcus aureus. Infect Immun. 1996;64:2974–2979. doi: 10.1128/iai.64.8.2974-2979.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Huseby M, Shi K, Brown CK, Digre J, Mengistu F, Seo KS, et al. Structure and biological activities of beta toxin from Staphylococcus aureus. J Bacteriol. 2007;189:8719–8726. doi: 10.1128/JB.00741-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kim CS, Jeon SY, Min YG, Rhyoo C, Kim JW, Yun JB, et al. Effects of beta-toxin of Staphylococcus aureus on ciliary activity of nasal epithelial cells. Laryngoscope. 2000;110:2085–2088. doi: 10.1097/00005537-200012000-00021. [DOI] [PubMed] [Google Scholar]
- 96.Hayashida A, Bartlett AH, Foster TJ, Park PW. Staphylococcus aureus beta-toxin induces lung injury through syndecan-1. Am J Pathol. 2009;174:509–518. doi: 10.2353/ajpath.2009.080394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mehlin C, Headley CM, Klebanoff SJ. An inflammatory polypeptide complex from Staphylococcus epidermidis: isolation and characterization. J Exp Med. 1999;189:907–918. doi: 10.1084/jem.189.6.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang R, Braughton KR, Kretschmer D, Bach TH, Queck SY, Li M, et al. Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat Med. 2007;13:1510–1514. doi: 10.1038/nm1656. [DOI] [PubMed] [Google Scholar]
- 99.Kretschmer D, Gleske AK, Rautenberg M, Wang R, Koberle M, Bohn E, et al. Human formyl peptide receptor 2 senses highly pathogenic Staphylococcus aureus. Cell Host Microbe. 2010;7:463–473. doi: 10.1016/j.chom.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Queck SY, Khan BA, Wang R, Bach TH, Kretschmer D, Chen L, et al. Mobile genetic element-encoded cytolysin connects virulence to methicillin resistance in MRSA. PLoS Pathog. 2009;5:e1000533. doi: 10.1371/journal.ppat.1000533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Joo HS, Cheung GY, Otto M. Antimicrobial activity of community-associated methicillin-resistant Staphylococcus aureus is caused by phenol-soluble modulin derivatives. J Biol Chem. 2011;286:8933–8940. doi: 10.1074/jbc.M111.221382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang R, Khan BA, Cheung GY, Bach TH, Jameson-Lee M, Kong KF, et al. Staphylococcus epidermidis surfactant peptides promote biofilm maturation and dissemination of biofilm-associated infection in mice. J Clin Invest. 2011;121:238–248. doi: 10.1172/JCI42520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Janzon L, Lofdahl S, Arvidson S. Identification and nucleotide sequence of the delta-lysin gene, hld, adjacent to the accessory gene regulator (agr)of Staphylococcus aureus. Mol Gen Genet. 1989;219:480–485. doi: 10.1007/BF00259623. [DOI] [PubMed] [Google Scholar]
- 104.Kreger AS, Kim KS, Zaboretzky F, Bernheimer AW. Purification and properties of staphylococcal delta hemolysin. Infect Immun. 1971;3:449–465. doi: 10.1128/iai.3.3.449-465.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Prevost G, Cribier B, Couppie P, Petiau P, Supersac G, Finck-Barbancon V, et al. Panton-Valentine leucocidin and gamma-hemolysin from Staphylococcus aureus ATCC 49775 are encoded by distinct genetic loci and have different biological activities. Infect Immun. 1995;63:4121–4129. doi: 10.1128/iai.63.10.4121-4129.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gladstone GP, Van Heyningen WE. Staphylococcal leucocidins. Br J Exp Pathol. 1957;38:123–137. [PMC free article] [PubMed] [Google Scholar]
- 107.Gauduchon V, Werner S, Prevost G, Monteil H, Colin DA. Flow cytometric determination of Panton–Valentine leucocidin S component binding. Infect Immun. 2001;69:2390–2395. doi: 10.1128/IAI.69.4.2390-2395.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Miles G, Movileanu L, Bayley H. Subunit composition of a bicomponent toxin: staphylococcal leukocidin forms an octameric transmembrane pore. Protein Sci. 2002;11:894–902. doi: 10.1110/ps.4360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Finck-Barbancon V, Duportail G, Meunier O, Colin DA. Pore formation by a two-component leukocidin from Staphylococcus aureus within the membrane of human polymorphonuclear leukocytes. Biochim Biophys Acta. 1993;1182:275–282. doi: 10.1016/0925-4439(93)90069-d. [DOI] [PubMed] [Google Scholar]
- 110.Li M, Cheung GY, Hu J, Wang D, Joo HS, Deleo FR, et al. Comparative analysis of virulence and toxin expression of global community-associated methicillin-resistant Staphylococcus aureus strains. J Infect Dis. 2010;202:1866–1876. doi: 10.1086/657419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Loffler B, Hussain M, Grundmeier M, Bruck M, Holzinger D, Varga G, et al. Staphylococcus aureus Panton–Valentine leukocidin is a very potent cytotoxic factor for human neutrophils. PLoS Pathog. 2010;6:e1000715. doi: 10.1371/journal.ppat.1000715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Diep BA, Chan L, Tattevin P, Kajikawa O, Martin TR, Basuino L, et al. Polymorphonuclear leukocytes mediate Staphylococcus aureus Panton–Valentine leukocidin-induced lung inflammation and injury. Proc Natl Acad Sci USA. 2010;107:5587–5592. doi: 10.1073/pnas.0912403107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Genestier AL, Michallet MC, Prevost G, Bellot G, Chalabreysse L, Peyrol S, et al. Staphylococcus aureus Panton–Valentine leukocidin directly targets mitochondria and induces Bax-independent apoptosis of human neutrophils. J Clin Invest. 2005;115:3117–3127. doi: 10.1172/JCI22684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zivkovic A, Sharif O, Stich K, Doninger B, Biaggio M, Colinge J, et al. TLR 2 and CD14 mediate innate immunity and lung inflammation to staphylococcal Panton–Valentine leukocidin in vivo. J Immunol. 2011;186:1608–1617. doi: 10.4049/jimmunol.1001665. [DOI] [PubMed] [Google Scholar]
- 115.Ward PD, Turner WH. Identification of staphylococcal Panton–Valentine leukocidin as a potent dermonecrotic toxin. Infect Immun. 1980;28:393–397. doi: 10.1128/iai.28.2.393-397.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lina G, Piemont Y, Godail-Gamot F, Bes M, Peter MO, Gauduchon V, et al. Involvement of Panton–Valentine leukocidin-producing Staphylococcus aureus in primary skin infections and pneumonia. Clin Infect Dis. 1999;29:1128–1132. doi: 10.1086/313461. [DOI] [PubMed] [Google Scholar]
- 117.Francis JS, Doherty MC, Lopatin U, Johnston CP, Sinha G, Ross T, et al. Severe community-onset pneumonia in healthy adults caused by methicillin-resistant Staphylococcus aureus carrying the Panton–Valentine leukocidin genes. Clin Infect Dis. 2005;40:100–107. doi: 10.1086/427148. [DOI] [PubMed] [Google Scholar]
- 118.Gillet Y, Issartel B, Vanhems P, Fournet JC, Lina G, Bes M, et al. Association between Staphylococcus aureus strains carrying gene for Panton–Valentine leukocidin and highly lethal necrotising pneumonia in young immunocompetent patients. Lancet. 2002;359:753–759. doi: 10.1016/S0140-6736(02)07877-7. [DOI] [PubMed] [Google Scholar]
- 119.Vardakas KZ, Matthaiou DK, Falagas ME. Incidence, characteristics and outcomes of patients with severe community acquired-MRSA pneumonia. Eur Respir J. 2009;34:1148–1158. doi: 10.1183/09031936.00041009. [DOI] [PubMed] [Google Scholar]
- 120.Labandeira-Rey M, Couzon F, Boisset S, Brown EL, Bes M, Benito Y, et al. Staphylococcus aureus Panton–Valentine leukocidin causes necrotizing pneumonia. Science. 2007;315:1130–1133. doi: 10.1126/science.1137165. [DOI] [PubMed] [Google Scholar]
- 121.Brown EL, Dumitrescu O, Thomas D, Badiou C, Koers EM, Choudhury P, et al. The Panton–Valentine leukocidin vaccine protects mice against lung and skin infections caused by Staphylococcus aureus USA300. Clin Microbiol Infect. 2009;15:156–164. doi: 10.1111/j.1469-0691.2008.02648.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Vandenesch F, Couzon F, Boisset S, Benito Y, Brown EL, Lina G, et al. The Panton–Valentine leukocidin is a virulence factor in a murine model of necrotizing pneumonia. J Infect Dis. 2010;201:967–969. doi: 10.1086/651026. author reply 969-970. [DOI] [PubMed] [Google Scholar]
- 123.Villaruz AE, Bubeck Wardenburg J, Khan BA, Whitney AR, Sturdevant DE, Gardner DJ, et al. A point mutation in the agr locus rather than expression of the Panton–Valentine leukocidin caused previously reported phenotypes in Staphylococcus aureus pneumonia and gene regulation. J Infect Dis. 2009;200:724–734. doi: 10.1086/604728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mollick JA, McMasters RL, Grossman D, Rich RR. Localization of a site on bacterial superantigens that determines T cell receptor beta chain specificity. J Exp Med. 1993;177:283–293. doi: 10.1084/jem.177.2.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Li H, Llera A, Malchiodi EL, Mariuzza RA. The structural basis of T cell activation by superantigens. Annu Rev Immunol. 1999;17:435–466. doi: 10.1146/annurev.immunol.17.1.435. [DOI] [PubMed] [Google Scholar]
- 126.Marrack P, Kappler J. The staphylococcal enterotoxins and their relatives. Science. 1990;248:705–711. doi: 10.1126/science.2185544. [DOI] [PubMed] [Google Scholar]
- 127.Lambris JD, Ricklin D, Geisbrecht BV. Complement evasion by human pathogens. Nat Rev Microbiol. 2008;6:132–142. doi: 10.1038/nrmicro1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bekeredjian-Ding I, Inamura S, Giese T, Moll H, Endres S, Sing A, et al. Staphylococcus aureus protein A triggers T cell-independent B cell proliferation by sensitizing B cells for TLR2 ligands. J Immunol. 2007;178:2803–2812. doi: 10.4049/jimmunol.178.5.2803. [DOI] [PubMed] [Google Scholar]
- 129.Silverman GJ, Nayak JV, Warnatz K, Hajjar FF, Cary S, Tighe H, et al. The dual phases of the response to neonatal exposure to a VH family-restricted staphylococcal B cell superantigen. J Immunol. 1998;161:5720–5732. [PubMed] [Google Scholar]
- 130.Smith EJ, Visai L, Kerrigan SW, Speziale P, Foster TJ. The Sbi protein: a multifunctional immune evasion factor of Staphylococcus aureus. Infect Immun. 2011;79:3801–3809. doi: 10.1128/IAI.05075-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Moks T, Abrahmsen L, Nilsson B, Hellman U, Sjoquist J, Uhlen M. Staphylococcal protein A consists of five IgG-binding domains. Eur J Biochem. 1986;156:637–643. doi: 10.1111/j.1432-1033.1986.tb09625.x. [DOI] [PubMed] [Google Scholar]
- 132.Uhlen M, Guss B, Nilsson B, Gatenbeck S, Philipson L, Lindberg M. Complete sequence of the staphylococcal gene encoding protein A. A gene evolved through multiple duplications. J Biol Chem. 1984;259:1695–1702. [PubMed] [Google Scholar]
- 133.Rajagopalan G, Sen MM, Singh M, Murali NS, Nath KA, Iijima K, et al. Intranasal exposure to staphylococcal enterotoxin B elicits an acute systemic inflammatory response. Shock. 2006;25:647–656. doi: 10.1097/01.shk.0000209565.92445.7d. [DOI] [PubMed] [Google Scholar]
- 134.Zen K, Masuda J, Ogata J. Monocyte-derived macrophages prime peripheral T cells to undergo apoptosis by cell–cell contact via ICAM-1/LFA-1-dependent mechanism. Immunobiology. 1996;195:323–333. doi: 10.1016/S0171-2985(96)80049-0. [DOI] [PubMed] [Google Scholar]
- 135.Miller EJ, Nagao S, Carr FK, Noble JM, Cohen AB. Interleukin-8 (IL-8) is a major neutrophil chemotaxin from human alveolar macrophages stimulated with staphylococcal enterotoxin A (SEA) Inflamm Res. 1996;45:386–392. doi: 10.1007/BF02252933. [DOI] [PubMed] [Google Scholar]
- 136.Fuller AF, Jr, Swartz MN, Wolfson JS, Salzman R. Toxic-shock syndrome. N Engl J Med. 1980;303:881. doi: 10.1056/nejm198010093031512. [DOI] [PubMed] [Google Scholar]
- 137.Shands KN, Schmid GP, Dan BB, Blum D, Guidotti RJ, Hargrett NT, et al. Toxic-shock syndrome in menstruating women: association with tampon use and Staphylococcus aureus and clinical features in 52 cases. N Engl J Med. 1980;303:1436–1442. doi: 10.1056/NEJM198012183032502. [DOI] [PubMed] [Google Scholar]
- 138.Osterholm MT, Davis JP, Gibson RW, Forfang JC, Stolz SJ, Vergeront JM. Toxic shock syndrome: relation to catamenial products, personal health and hygiene, and sexual practices. Ann Intern Med. 1982;96:954–958. doi: 10.7326/0003-4819-96-6-954. [DOI] [PubMed] [Google Scholar]
- 139.Reingold AL, Dan BB, Shands KN, Broome CV. Toxicshock syndrome not associated with menstruation. A review of 54 cases. Lancet. 1982;1:1–4. doi: 10.1016/s0140-6736(82)92552-1. [DOI] [PubMed] [Google Scholar]
- 140.Wilkins EG, Nye F, Roberts C, de Saxe M. Probable toxic shock syndrome with primary staphylococcal pneumonia. J Infect. 1985;11:231–232. doi: 10.1016/s0163-4453(85)93195-0. [DOI] [PubMed] [Google Scholar]
- 141.MacDonald KL, Osterholm MT, Hedberg CW, Schrock CG, Peterson GF, Jentzen JM, et al. Toxic shock syndrome. A newly recognized complication of influenza and influenzalike illness. JAMA. 1987;257:1053–1058. doi: 10.1001/jama.257.8.1053. [DOI] [PubMed] [Google Scholar]
- 142.Hirsch B, Stair T, Horowitz BZ, Brooks C. Toxic shock syndrome from staphylococcal pharyngitis. Ear Nose Throat J. 1984;63:494–497. [PubMed] [Google Scholar]
- 143.Dann EJ, Weinberger M, Gillis S, Parsonnet J, Shapiro M, Moses AE. Bacterial laryngotracheitis associated with toxic shock syndrome in an adult. Clin Infect Dis. 1994;18:437–439. doi: 10.1093/clinids/18.3.437. [DOI] [PubMed] [Google Scholar]
- 144.Zhang WJ, Sarawar S, Nguyen P, Daly K, Rehg JE, Doherty PC, et al. Lethal synergism between influenza infection and staphylococcal enterotoxin B in mice. J Immunol. 1996;157:5049–5060. [PubMed] [Google Scholar]
- 145.Dinges MM, Orwin PM, Schlievert PM. Exotoxins of Staphylococcus aureus. Clin Microbiol Rev. 2000;13:16–34. doi: 10.1128/cmr.13.1.16-34.2000. (table of contents) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Martin WJ, Marcus S. Relation of pyrogenic and emetic properties of enterobacteriaceal endotoxin and of staphylococcal enterotoxin. J Bacteriol. 1964;87:1019–1026. doi: 10.1128/jb.87.5.1019-1026.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Bachert C, Gevaert P, van Cauwenberge P. Staphylococcus aureus enterotoxins: a key in airway disease? Allergy. 2002;57:480–487. doi: 10.1034/j.1398-9995.2002.02156.x. [DOI] [PubMed] [Google Scholar]
- 148.Mariano NS, de Mello GC, Ferreira T, Schenka A, Camargo EA, de Nucci G, et al. Pre-exposure to staphylococcal enterotoxin A exacerbates the pulmonary allergic eosinophil recruitment in rats. Int Immunopharmacol. 2010;10:43–49. doi: 10.1016/j.intimp.2009.09.017. [DOI] [PubMed] [Google Scholar]
- 149.Bachert C, Gevaert P, Howarth P, Holtappels G, van Cauwenberge P, Johansson SG. IgE to Staphylococcus aureus enterotoxins in serum is related to severity of asthma. J Allergy Clin Immunol. 2003;111:1131–1132. [PubMed] [Google Scholar]
- 150.Rossi RE, Monasterolo G. Prevalence of serum IgE antibodies to the Staphylococcus aureus enterotoxins (SAE, SEB, SEC, SED, TSST-1) in patients with persistent allergic rhinitis. Int Arch Allergy Immunol. 2004;133:261–266. doi: 10.1159/000076833. [DOI] [PubMed] [Google Scholar]
- 151.O'Brien GJ, Riddell G, Elborn JS, Ennis M, Skibinski G. Staphylococcus aureus enterotoxins induce IL-8 secretion by human nasal epithelial cells. Respir Res. 2006;7:115. doi: 10.1186/1465-9921-7-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Patou J, Gevaert P, Van Zele T, Holtappels G, van Cauwenberge P, Bachert C. Staphylococcus aureus enterotoxin B, protein A, and lipoteichoic acid stimulations in nasal polyps. J Allergy Clin Immunol. 2008;121:110–115. doi: 10.1016/j.jaci.2007.08.059. [DOI] [PubMed] [Google Scholar]
- 153.Herz U, Ruckert R, Wollenhaupt K, Tschernig T, Neuhaus-Steinmetz U, Pabst R, et al. Airway exposure to bacterial superantigen (SEB) induces lymphocyte-dependent airway inflammation associated with increased airway responsiveness—a model for non-allergic asthma. Eur J Immunol. 1999;29:1021–1031. doi: 10.1002/(SICI)1521-4141(199903)29:03<1021::AID-IMMU1021>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 154.Desouza IA, Franco-Penteado CF, Camargo EA, Lima CS, Teixeira SA, Muscara MN, et al. Acute pulmonary inflammation induced by exposure of the airways to staphylococcal enterotoxin type B in rats. Toxicol Appl Pharmacol. 2006;217:107–113. doi: 10.1016/j.taap.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 155.Rajagopalan G, Iijima K, Singh M, Kita H, Patel R, David CS. Intranasal exposure to bacterial superantigens induces airway inflammation in HLA class II transgenic mice. Infect Immun. 2006;74:1284–1296. doi: 10.1128/IAI.74.2.1284-1296.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Shinbori T, Matsuki M, Suga M, Kakimoto K, Ando M. Induction of interstitial pneumonia in autoimmune mice by intratracheal administration of superantigen staphylococcal enterotoxin B. Cell Immunol. 1996;174:129–137. doi: 10.1006/cimm.1996.0302. [DOI] [PubMed] [Google Scholar]
- 157.Hellings PW, Hens G, Meyts I, Bullens D, Vanoirbeek J, Gevaert P, et al. Aggravation of bronchial eosinophilia in mice by nasal and bronchial exposure to Staphylococcus aureus enterotoxin B. Clin Exp Allergy. 2006;36:1063–1071. doi: 10.1111/j.1365-2222.2006.02527.x. [DOI] [PubMed] [Google Scholar]
- 158.Yu RL, Dong Z. Proinflammatory impact of Staphylococcus aureus enterotoxin B on human nasal epithelial cells and inhibition by dexamethasone. Am J Rhinol Allergy. 2009;23:15–20. doi: 10.2500/ajra.2009.23.3252. [DOI] [PubMed] [Google Scholar]
- 159.Nguyen T, Ghebrehiwet B, Peerschke EI. Staphylococcus aureus protein A recognizes platelet gC1qR/p33: a novel mechanism for staphylococcal interactions with platelets. Infect Immun. 2000;68:2061–2068. doi: 10.1128/iai.68.4.2061-2068.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Atkins KL, Burman JD, Chamberlain ES, Cooper JE, Poutrel B, Bagby S, et al. S. aureus IgG-binding proteins SpA and Sbi: host specificity and mechanisms of immune complex formation. Mol Immunol. 2008;45:1600–1611. doi: 10.1016/j.molimm.2007.10.021. [DOI] [PubMed] [Google Scholar]
- 161.Burman JD, Leung E, Atkins KL, O'Seaghdha MN, Lango L, Bernado P, et al. Interaction of human complement with Sbi, a staphylococcal immunoglobulin-binding protein: indications of a novel mechanism of complement evasion by Staphylococcus aureus. J Biol Chem. 2008;283:17579–17593. doi: 10.1074/jbc.M800265200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Rooijakkers SH, van Wamel WJ, Ruyken M, van Kessel KP, van Strijp JA. Anti-opsonic properties of staphylokinase. Microbes Infect. 2005;7:476–484. doi: 10.1016/j.micinf.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 163.Jin T, Bokarewa M, Foster T, Mitchell J, Higgins J, Tarkowski A. Staphylococcus aureus resists human defensins by production of staphylokinase, a novel bacterial evasion mechanism. J Immunol. 2004;172:1169–1176. doi: 10.4049/jimmunol.172.2.1169. [DOI] [PubMed] [Google Scholar]
- 164.Verkaik NJ, Benard M, Boelens HA, de Vogel CP, Nouwen JL, Verbrugh HA, et al. Immune evasion cluster-positive bacteriophages are highly prevalent among human Staphylococcus aureus strains, but they are not essential in the first stages of nasal colonization. Clin Microbiol Infect. 2011;17:343–348. doi: 10.1111/j.1469-0691.2010.03227.x. [DOI] [PubMed] [Google Scholar]
- 165.Rooijakkers SH, Ruyken M, Roos A, Daha MR, Presanis JS, Sim RB, et al. Immune evasion by a staphylococcal complement inhibitor that acts on C3 convertases. Nat Immunol. 2005;6:920–927. doi: 10.1038/ni1235. [DOI] [PubMed] [Google Scholar]
- 166.Postma B, Poppelier MJ, van Galen JC, Prossnitz ER, van Strijp JA, de Haas CJ, et al. Chemotaxis inhibitory protein of Staphylococcus aureus binds specifically to the C5a and formylated peptide receptor. J Immunol. 2004;172:6994–7001. doi: 10.4049/jimmunol.172.11.6994. [DOI] [PubMed] [Google Scholar]
- 167.de Haas CJ, Veldkamp KE, Peschel A, Weerkamp F, Van Wamel WJ, Heezius EC, et al. Chemotaxis inhibitory protein of Staphylococcus aureus, a bacterial antiinflammatory agent. J Exp Med. 2004;199:687–695. doi: 10.1084/jem.20031636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Jongerius I, Kohl J, Pandey MK, Ruyken M, van Kessel KP, van Strijp JA, et al. Staphylococcal complement evasion by various convertase-blocking molecules. J Exp Med. 2007;204:2461–2471. doi: 10.1084/jem.20070818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Movitz J. Formation of extracellular protein A by Staphylococcus aureus. Eur J Biochem. 1976;68:291–299. doi: 10.1111/j.1432-1033.1976.tb10788.x. [DOI] [PubMed] [Google Scholar]
- 170.Schneewind O, Model P, Fischetti VA. Sorting of protein A to the staphylococcal cell wall. Cell. 1992;70:267–281. doi: 10.1016/0092-8674(92)90101-h. [DOI] [PubMed] [Google Scholar]
- 171.Shopsin B, Gomez M, Montgomery SO, Smith DH, Waddington M, Dodge DE, et al. Evaluation of protein A gene polymorphic region DNA sequencing for typing of Staphylococcus aureus strains. J Clin Microbiol. 1999;37:3556–3563. doi: 10.1128/jcm.37.11.3556-3563.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Koreen L, Ramaswamy SV, Graviss EA, Naidich S, Musser JM, Kreiswirth BN. spa typing method for discriminating among Staphylococcus aureus isolates: implications for use of a single marker to detect genetic micro- and macrovariation. J Clin Microbiol. 2004;42:792–799. doi: 10.1128/JCM.42.2.792-799.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Martin FJ, Gomez MI, Wetzel DM, Memmi G, O'Seaghdha M, Soong G, et al. Staphylococcus aureus activates type I IFN signaling in mice and humans through the Xr repeated sequences of protein A. J Clin Invest. 2009;119:1931–1939. doi: 10.1172/JCI35879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Kahl BC, Mellmann A, Deiwick S, Peters G, Harmsen D. Variation of the polymorphic region X of the protein A gene during persistent airway infection of cystic fibrosis patients reflects two independent mechanisms of genetic change in Staphylococcus aureus. J Clin Microbiol. 2005;43:502–505. doi: 10.1128/JCM.43.1.502-505.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Hakoda M, Hayashimoto S, Yamanaka H, Terai C, Kamatani N, Kashiwazaki S. Molecular basis for the interaction between human IgM and staphylococcal protein A. Clin Immunol Immunopathol. 1994;72:394–401. doi: 10.1006/clin.1994.1158. [DOI] [PubMed] [Google Scholar]
- 176.Kim HK, Cheng AG, Kim HY, Missiakas DM, Schneewind O. Nontoxigenic protein A vaccine for methicillin-resistant Staphylococcus aureus infections in mice. J Exp Med. 2010;207:1863–1870. doi: 10.1084/jem.20092514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Gomez MI, Lee A, Reddy B, Muir A, Soong G, Pitt A, et al. Staphylococcus aureus protein A induces airway epithelial inflammatory responses by activating TNFR1. Nat Med. 2004;10:842–848. doi: 10.1038/nm1079. [DOI] [PubMed] [Google Scholar]
- 178.Palmqvist N, Foster T, Tarkowski A, Josefsson E. Protein A is a virulence factor in Staphylococcus aureus arthritis and septic death. Microb Pathog. 2002;33:239–249. doi: 10.1006/mpat.2002.0533. [DOI] [PubMed] [Google Scholar]
- 179.MacEwan DJ. TNF receptor subtype signalling: differences and cellular consequences. Cell Signal. 2002;14:477–492. doi: 10.1016/s0898-6568(01)00262-5. [DOI] [PubMed] [Google Scholar]
- 180.Gomez MI, Seaghdha MO, Prince AS. Staphylococcus aureus protein A activates TACE through EGFR-dependent signaling. EMBO J. 2007;26:701–709. doi: 10.1038/sj.emboj.7601554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Gomez MI, O'Seaghdha M, Magargee M, Foster TJ, Prince AS. Staphylococcus aureus protein A activates TNFR1 signaling through conserved IgG binding domains. J Biol Chem. 2006;281:20190–20196. doi: 10.1074/jbc.M601956200. [DOI] [PubMed] [Google Scholar]
- 182.Skerrett SJ, Liggitt HD, Hajjar AM, Wilson CB. Cutting edge: myeloid differentiation factor 88 is essential for pulmonary host defense against Pseudomonas aeruginosa but not Staphylococcus aureus. J Immunol. 2004;172:3377–3381. doi: 10.4049/jimmunol.172.6.3377. [DOI] [PubMed] [Google Scholar]
- 183.Shaykhiev R, Behr J, Bals R. Microbial patterns signaling via Toll-like receptors 2 and 5 contribute to epithelial repair, growth and survival. PLoS One. 2008;3:e1393. doi: 10.1371/journal.pone.0001393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Basbaum C, Li D, Gensch E, Gallup M, Lemjabbar H. Mechanisms by which Gram-positive bacteria and tobacco smoke stimulate mucin induction through the epidermal growth factor receptor (EGFR) Novartis Found Symp. 2002;248:171–176. 176–180, 277–182. [PubMed] [Google Scholar]
- 185.Miller LS, Cho JS. Immunity against Staphylococcus aureus cutaneous infections. Nat Rev Immunol. 2011;11:505–518. doi: 10.1038/nri3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Muir A, Soong G, Sokol S, Reddy B, Gomez MI, Van Heeckeren A, et al. Toll-like receptors in normal and cystic fibrosis airway epithelial cells. Am J Respir Cell Mol Biol. 2004;30:777–783. doi: 10.1165/rcmb.2003-0329OC. [DOI] [PubMed] [Google Scholar]
- 187.Mayer AK, Muehmer M, Mages J, Gueinzius K, Hess C, Heeg K, et al. Differential recognition of TLR-dependent microbial ligands in human bronchial epithelial cells. J Immunol. 2007;178:3134–3142. doi: 10.4049/jimmunol.178.5.3134. [DOI] [PubMed] [Google Scholar]
- 188.Greene CM, Carroll TP, Smith SG, Taggart CC, Devaney J, Griffin S, et al. TLR-induced inflammation in cystic fibrosis and non-cystic fibrosis airway epithelial cells. J Immunol. 2005;174:1638–1646. doi: 10.4049/jimmunol.174.3.1638. [DOI] [PubMed] [Google Scholar]
- 189.Sha Q, Truong-Tran AQ, Plitt JR, Beck LA, Schleimer RP. Activation of airway epithelial cells by Toll-like receptor agonists. Am J Respir Cell Mol Biol. 2004;31:358–364. doi: 10.1165/rcmb.2003-0388OC. [DOI] [PubMed] [Google Scholar]
- 190.Xing Z, Harper R, Anunciacion J, Yang Z, Gao W, Qu B, et al. Host immune and apoptotic responses to avian influenza virus H9N2 in human tracheobronchial epithelial cells. Am J Respir Cell Mol Biol. 2011;44:24–33. doi: 10.1165/rcmb.2009-0120OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.von Aulock S, Morath S, Hareng L, Knapp S, van Kessel KP, van Strijp JA, et al. Lipoteichoic acid from Staphylococcus aureus is a potent stimulus for neutrophil recruitment. Immunobiology. 2003;208:413–422. doi: 10.1078/0171-2985-00285. [DOI] [PubMed] [Google Scholar]
- 192.Hoogerwerf JJ, de Vos AF, Bresser P, van der Zee JS, Pater JM, de Boer A, et al. Lung inflammation induced by lipoteichoic acid or lipopolysaccharide in humans. Am J Respir Crit Care Med. 2008;178:34–41. doi: 10.1164/rccm.200708-1261OC. [DOI] [PubMed] [Google Scholar]
- 193.Quinn GA, Cole AM. Suppression of innate immunity by a nasal carriage strain of Staphylococcus aureus increases its colonization on nasal epithelium. Immunology. 2007;122:80–89. doi: 10.1111/j.1365-2567.2007.02615.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Takeuchi O, Hoshino K, Akira S. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J Immunol. 2000;165:5392–5396. doi: 10.4049/jimmunol.165.10.5392. [DOI] [PubMed] [Google Scholar]
- 195.Mullaly SC, Kubes P. The role of TLR2 in vivo following challenge with Staphylococcus aureus and prototypic ligands. J Immunol. 2006;177:8154–8163. doi: 10.4049/jimmunol.177.11.8154. [DOI] [PubMed] [Google Scholar]
- 196.Bubeck Wardenburg J, Williams WA, Missiakas D. Host defenses against Staphylococcus aureus infection require recognition of bacterial lipoproteins. Proc Natl Acad Sci USA. 2006;103:13831–13836. doi: 10.1073/pnas.0603072103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Saba S, Soong G, Greenberg S, Prince A. Bacterial stimulation of epithelial G-CSF and GM-CSF expression promotes PMN survival in CF airways. Am J Respir Cell Mol Biol. 2002;27:561–567. doi: 10.1165/rcmb.2002-0019OC. [DOI] [PubMed] [Google Scholar]
- 198.Sorrentino R, de Souza PM, Sriskandan S, Duffin C, Paul-Clark MJ, Mitchell JA. Pattern recognition receptors and interleukin-8 mediate effects of Gram-positive and Gram-negative bacteria on lung epithelial cell function. Br J Pharmacol. 2008;154:864–871. doi: 10.1038/bjp.2008.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Ratner AJ, Bryan R, Weber A, Nguyen S, Barnes D, Pitt A, et al. Cystic fibrosis pathogens activate Ca2+-dependent mitogen-activated protein kinase signaling pathways in airway epithelial cells. J Biol Chem. 2001;276:19267–19275. doi: 10.1074/jbc.M007703200. [DOI] [PubMed] [Google Scholar]
- 200.Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, et al. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 201.Der SD, Zhou A, Williams BR, Silverman RH. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc Natl Acad Sci USA. 1998;95:15623–15628. doi: 10.1073/pnas.95.26.15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Parker D, Martin FJ, Soong G, Harfenist BS, Aguilar JL, Ratner AJ, et al. Streptococcus pneumoniae DNA initiates type I interferon signaling in the respiratory tract. MBio. 2011;2:e00016–11. doi: 10.1128/mBio.00016-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Parker D, Cohen TS, Alhede M, Harfenist BS, Martin FJ, Prince A. Induction of type I interferon signaling by Pseudomonas aeruginosa is diminished in cystic fibrosis epithelial cells. Am J Respir Cell Mol Biol. 2011 doi: 10.1165/rcmb.2011-0080OC. Jul 21. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Decker T, Muller M, Stockinger S. The yin and yang of type I interferon activity in bacterial infection. Nat Rev Immunol. 2005;5:675–687. doi: 10.1038/nri1684. [DOI] [PubMed] [Google Scholar]
- 205.Charrel-Dennis M, Latz E, Halmen KA, Trieu-Cuot P, Fitzgerald KA, Kasper DL, et al. TLR-independent type I interferon induction in response to an extracellular bacterial pathogen via intracellular recognition of its DNA. Cell Host Microbe. 2008;4:543–554. doi: 10.1016/j.chom.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Toshchakov V, Jones BW, Perera PY, Thomas K, Cody MJ, Zhang S, et al. TLR4, but not TLR2, mediates IFN-beta-induced STAT1alpha/beta-dependent gene expression in macrophages. Nat Immunol. 2002;3:392–398. doi: 10.1038/ni774. [DOI] [PubMed] [Google Scholar]
- 207.Mancuso G, Gambuzza M, Midiri A, Biondo C, Papasergi S, Akira S, et al. Bacterial recognition by TLR7 in the lysosomes of conventional dendritic cells. Nat Immunol. 2009;10:587–594. doi: 10.1038/ni.1733. [DOI] [PubMed] [Google Scholar]
- 208.Chiu YH, Macmillan JB, Chen ZJ. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell. 2009;138:576–591. doi: 10.1016/j.cell.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, et al. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity. 2000;13:539–548. doi: 10.1016/s1074-7613(00)00053-4. [DOI] [PubMed] [Google Scholar]
- 210.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 211.Schoenemeyer A, Barnes BJ, Mancl ME, Latz E, Goutagny N, Pitha PM, et al. The interferon regulatory factor, IRF5, is a central mediator of Toll-like receptor 7 signaling. J Biol Chem. 2005;280:17005–17012. doi: 10.1074/jbc.M412584200. [DOI] [PubMed] [Google Scholar]
- 212.Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, et al. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 213.Schindler C, Plumlee C. Inteferons pen the JAK-STAT pathway. Semin Cell Dev Biol. 2008;19:311–318. doi: 10.1016/j.semcdb.2008.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.David M, Petricoin E, 3rd, Benjamin C, Pine R, Weber MJ, Larner AC. Requirement for MAP kinase (ERK2) activity in interferon alpha- and interferon beta-stimulated gene expression through STAT proteins. Science. 1995;269:1721–1723. doi: 10.1126/science.7569900. [DOI] [PubMed] [Google Scholar]
- 215.Lenardo MJ, Fan CM, Maniatis T, Baltimore D. The involvement of NF-kappa B in beta-interferon gene regulation reveals its role as widely inducible mediator of signal transduction. Cell. 1989;57:287–294. doi: 10.1016/0092-8674(89)90966-5. [DOI] [PubMed] [Google Scholar]
- 216.Manicone AM, Burkhart KM, Lu B, Clark JG. CXCR3 ligands contribute to Th1-induced inflammation but not to homing of Th1 cells into the lung. Exp Lung Res. 2008;34:391–407. doi: 10.1080/01902140802221987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Qian C, An H, Yu Y, Liu S, Cao X. TLR agonists induce regulatory dendritic cells to recruit Th1 cells via preferential IP-10 secretion and inhibit Th1 proliferation. Blood. 2007;109:3308–3315. doi: 10.1182/blood-2006-08-040337. [DOI] [PubMed] [Google Scholar]
- 218.Debes GF, Dahl ME, Mahiny AJ, Bonhagen K, Campbell DJ, Siegmund K, et al. Chemotactic responses of IL-4-, IL-10-, and IFN-gamma-producing CD4+ T cells depend on tissue origin and microbial stimulus. J Immunol. 2006;176:557–566. doi: 10.4049/jimmunol.176.1.557. [DOI] [PubMed] [Google Scholar]
- 219.Martin FJ, Parker D, Harfenist BS, Soong G, Prince A. Participation of CD11c(+) leukocytes in methicillin-resistant Staphylococcus aureus clearance from the lung. Infect Immun. 2011;79:1898–1904. doi: 10.1128/IAI.01299-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Lappalainen U, Whitsett JA, Wert SE, Tichelaar JW, Bry K. Interleukin-1beta causes pulmonary inflammation, emphysema, and airway remodeling in the adult murine lung. Am J Respir Cell Mol Biol. 2005;32:311–318. doi: 10.1165/rcmb.2004-0309OC. [DOI] [PubMed] [Google Scholar]
- 221.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 222.Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. 2004;430:213–218. doi: 10.1038/nature02664. [DOI] [PubMed] [Google Scholar]
- 223.Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7:99–109. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose-Girma M, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 225.Bhakdi S, Muhly M, Korom S, Hugo F. Release of interleukin-1 beta associated with potent cytocidal action of staphylococcal alpha-toxin on human monocytes. Infect Immun. 1989;57:3512–3519. doi: 10.1128/iai.57.11.3512-3519.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Shimada T, Park BG, Wolf AJ, Brikos C, Goodridge HS, Becker CA, et al. Staphylococcus aureus evades lysozyme-based peptidoglycan digestion that links phagocytosis, inflammasome activation, and IL-1beta secretion. Cell Host Microbe. 2010;7:38–49. doi: 10.1016/j.chom.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Chien YW, Klugman KP, Morens DM. Bacterial pathogens and death during the 1918 influenza pandemic. N Engl J Med. 2009;361:2582–2583. doi: 10.1056/NEJMc0908216. [DOI] [PubMed] [Google Scholar]
- 228.Morens DM, Taubenberger JK, Fauci AS. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J Infect Dis. 2008;198:962–970. doi: 10.1086/591708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Lee MH, Arrecubieta C, Martin FJ, Prince A, Borczuk AC, Lowy FD. A postinfluenza model of Staphylococcus aureus pneumonia. J Infect Dis. 2010;201:508–515. doi: 10.1086/650204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Small CL, Shaler CR, McCormick S, Jeyanathan M, Damjanovic D, Brown EG, et al. Influenza infection leads to increased susceptibility to subsequent bacterial superinfection by impairing NK cell responses in the lung. J Immunol. 2010;184:2048–2056. doi: 10.4049/jimmunol.0902772. [DOI] [PubMed] [Google Scholar]
- 231.Kudva A, Scheller EV, Robinson KM, Crowe CR, Choi SM, Slight SR, et al. Influenza A inhibits Th17-mediated host defense against bacterial pneumonia in mice. J Immunol. 2011;186:1666–1674. doi: 10.4049/jimmunol.1002194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Minegishi Y, Saito M, Nagasawa M, Takada H, Hara T, Tsuchiya S, et al. Molecular explanation for the contradiction between systemic Th17 defect and localized bacterial infection in hyper-IgE syndrome. J Exp Med. 2009;206:1291–1301. doi: 10.1084/jem.20082767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Frodermann V, Chau TA, Sayedyahossein S, Toth JM, Heinrichs DE, Madrenas J. A modulatory interleukin-10 response to staphylococcal peptidoglycan prevents Th1/Th17 adaptive immunity to Staphylococcus aureus. J Infect Dis. 2011;204:253–262. doi: 10.1093/infdis/jir276. [DOI] [PubMed] [Google Scholar]
- 234.Niebuhr M, Gathmann M, Scharonow H, Mamerow D, Mommert S, Balaji H, et al. Staphylococcal alpha-toxin is a strong inducer of interleukin-17 in humans. Infect Immun. 2011;79:1615–1622. doi: 10.1128/IAI.00958-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, Komiyama Y, et al. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity. 2009;30:108–119. doi: 10.1016/j.immuni.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 236.Ma CS, Chew GY, Simpson N, Priyadarshi A, Wong M, Grimbacher B, et al. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J Exp Med. 2008;205:1551–1557. doi: 10.1084/jem.20080218. [DOI] [PMC free article] [PubMed] [Google Scholar]