

Abstract
In high-throughput cancer genomic studies, markers identified from the analysis of single data sets often suffer a lack of reproducibility because of the small sample sizes. An ideal solution is to conduct large-scale prospective studies, which are extremely expensive and time consuming. A cost-effective remedy is to pool data from multiple comparable studies and conduct integrative analysis. Integrative analysis of multiple data sets is challenging because of the high dimensionality of genomic measurements and heterogeneity among studies. In this article, we propose a sparse boosting approach for marker identification in integrative analysis of multiple heterogeneous cancer diagnosis studies with gene expression measurements. The proposed approach can effectively accommodate the heterogeneity among multiple studies and identify markers with consistent effects across studies. Simulation shows that the proposed approach has satisfactory identification results and outperforms alternatives including an intensity approach and meta-analysis. The proposed approach is used to identify markers of pancreatic cancer and liver cancer.
Keywords: Cancer genomics, Marker identification, Sparse boosting
1. INTRODUCTION
Cancer research has entered the -omics era. High-throughput profiling studies have been extensively conducted, searching for genomic markers associated with the development, progression, and variation in response to treatment of cancer (Knudsen, 2006). In this article, we focus on gene profiling studies with microarrays, but note that the proposed approach is also applicable to other high-throughput profiling studies. In cancer gene profiling studies, markers identified from the analysis of single data sets often suffer a lack of reproducibility (Choi and others, 2007, Shen and others, 2004). Consider, for example, the liver cancer microarray studies in Ma and Huang (2009). A gradient thresholding approach was used to analyze the 4 data sets and identified 27, 10, 20, and 6 genes, respectively. Among the identified genes, one was identified in 3 data sets, another one was identified in 2 data sets, and the remainder were identified in only 1 data set. Similar low reproducibility has been observed with other gene signatures. Among the possible causes, the most important one is the small sample sizes and hence lack of power of individual studies. A typical cancer microarray study profiles genes on samples. The “large d, small n” problem is even worse in other -omics, for example, genome-wide association, studies. For many cancer outcomes and phenotypes (e.g. diagnosis, prognosis, and treatment selection of breast cancer, ovarian cancer, lymphoma, and lung cancer), there are multiple independent studies with comparable designs (Knudsen, 2006). A cost-effective solution to the small sample size and lack of reproducibility problem is to pool data from multiple studies and increase power.
Available multidata sets approaches include meta-analysis and integrative analysis approaches. In meta-analysis, multiple data sets are first analyzed separately, and then, summary statistics such as p values or lists of identified genes are pooled (Guerra and Goldstein, 2009). In contrast, in integrative analysis, raw data from multiple studies are pooled and analyzed. Multiple studies have shown that integrative analysis may be more effective than meta-analysis and analysis of individual data sets (Shen and others, 2004, Choi and others, 2007, Ma and Huang, 2009). This result is reconfirmed by our numerical study in Section 4. With microarray gene expression data, a family of integrative analysis approaches called “intensity approaches” search for transformations that make gene expressions comparable across different studies and platforms (Shabalin and others, 2008). Such approaches may be limited in that they need to be conducted on a case-by-case basis, and there is no guarantee that the desired transformations always exist.
In this article, we investigate marker selection in integrative analysis of multiple cancer diagnosis studies. The marker selection approach we propose belongs to the family of boosting approaches. Boosting provides an effective way for combining a family of weak learners into a strong one. Dettling (2004), Dettling and Buhlmann (2003), and others show the satisfactory performance of boosting approaches with gene expression data. We provide a brief introduction of the most relevant boosting techniques in Section 3 and refer to Buhlmann and Hothorn (2007), Buhlmann and Yu (2010), and references therein for comprehensive reviews.
The contributions of this paper are as follows. First, compared with the existing methods that are based on analysis of single data sets, the proposed approach can analyze multiple heterogeneous data sets in an integrative manner. Second, compared with existing multidata sets studies, a more effective marker identification approach is developed. Third, the proposed approach can naturally accommodate the heterogeneity among multiple data sets. A significant advantage of the proposed approach is that it does not require full comparability of measurements in different studies. Thus, with gene expression data, although normalization is still needed for each data set separately, the proposed approach does not require additional cross-platform normalization or transformation. As analysis of multiple data sets is commonly encountered, the proposed approach may have more applications beyond cancer gene expression studies.
The remainder of this article is organized as follows. In Section 2, we describe the data and model setup. In Section 3, we describe marker selection using the sparse boosting approach. In Section 4, we conduct simulation and comparison with penalization and boosting-based meta-analysis and intensity approaches to investigate finite sample performance of the proposed approach. We also identify markers for pancreatic cancer and liver cancer. The article concludes with discussion in Section 5.
2. INTEGRATIVE ANALYSIS OF MULTIPLE HETEROGENEOUS DATA SETS
In integrative analysis of multiple cancer genomic studies, we make the working assumption of sparsity. Specifically, we postulate that of the thousands of genes surveyed, only a small number of them are associated with cancer. In genome-wide studies, it is safe to assume that only a fraction of genes surveyed are cancer associated (Knudsen, 2006). To date, only over 300 genes have been identified as “cancer genes” (all types of cancers combined; Greenman and others, 2007). Based on our current knowledge, for a specific type of cancer, the number of cancer genes is expected to be small. Variable selection approaches, including the proposed one, make the sparsity assumption. Dimension reduction approaches, such as principal component analysis, do not make the sparsity assumption. However, the findings from such approaches may suffer a lack of interpretability. For the purpose of prediction, published studies have shown that performance of variable selection and dimension reduction approaches is data dependent, with no one dominating the other. We analyze multiple studies that share comparable designs and common biological ground. With such data, we search for genes with consistent effects across multiple studies. For a single data set, it is possible that multiple gene sets have comparable prediction performance. In our study, we reinforce that the same set of genes are identified in all studies, as such genes may be more likely to represent the essential genomic features of cancer (Rhodes and Chinnaiyan, 2004, Rhodes and others, 2004). Even though the same cancer outcome or phenotype is investigated, different studies may use different platforms for profiling. They may also differ in other experimental setup. Thus, for a specific gene, its strengths of association with the response variables in different studies, which are measured using regression coefficients, may differ. It is desirable to allow for different regression coefficients for different data sets (Stevens and Doerge, 2005, Ma and Huang, 2009).
Assume that data from M independent studies are available. For simplicity of notation, assume that the same set of d genes are measured in all studies. Let be the response variables and be the gene expressions. For , assume that is associated with via , where is the length d regression coefficient, is the transpose of , and ϕ is the link function. To accommodate the heterogeneity across studies, we allow for different regression coefficients for different m.
Diagnosis studies have binary outcomes. In study , let and 0 denote the presence and absence of cancer, respectively. We assume the commonly adopted logistic regression model, where . Here, is the unknown intercept. Assume i.i.d. observations. Let . Denote as the regression coefficient matrix.
3. INTEGRATIVE SPARSE LOGIT BOOSTING
3.1. Boosting
As a generic machine learning technique, boosting assembles a stronger learner using a family of weak learners. Compared with other methods, boosting may be preferred because of its flexibility, low computational cost, and satisfactory empirical performance. We refer to Berk (2008), Hastie and others (2009), and others for comprehensive reviews. Among the numerous boosting approaches, the proposed approach is most closely related to the LogitBoost (Dettling and Buhlmann, 2003) and sparse boosting (Buhlmann and Yu, 2006).
There are different choices of weak learners. With cancer genomic data, it is possible to use complex functions such as spline functions of gene expressions as weak learners. However, such a choice may lead to high computational cost and a lack of interpretability. Following Dettling and Buhlmann (2003) and others, we take linear functions of gene expressions as weak learners.
LogitBoost was developed for the analysis of a single data set with a binary response variable (Friedman and others, 2000). It has been applied to diagnosis studies with gene expression measurements and shown to have satisfactory performance (Dettling and Buhlmann, 2003). For the sake of completeness, we describe the LogitBoost algorithm below and refer to Dettling and Buhlmann (2003) and others for more details. Let be the length d covariate (gene expressions). Suppose that a random sample of size n is available. The LogitBoost proceeds as follows.
Step 1: Initialization. Set , , and . Here, is an estimate of ;
Step 2: Fit and update. .
Compute the weights and pseudo-response variables as 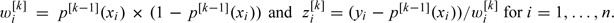 .
.
Compute . Here, is the sth component of .
Update , where . ν is the step size. As suggested in Buhlmann and Yu (2006) and references therein, the choice of ν is not crucial as long as it is small. In our numerical study, we set .
Update 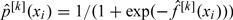 ;
;
Step 3: Iteration. Repeat Step 2 until a certain stopping rule is reached.
With LogitBoost, the negative log likelihood is taken as the loss function. With the logistic model, we are able to rewrite the negative log likelihood as a weighted least squares criterion. In Step 3, there are multiple choices for the stopping rule, including, for example, Akaike information criterion (AIC), Bayesian information criterion (BIC), cross-validation, and minimum description length (MDL). Our literature review suggests that with high-dimensional data, there is no consensus on the relative performance of different stopping rules. The resulted , also referred to as the “committee function,” is an estimate of the log odds ratio . For a subject with covariate , the LogitBoost classifier classifies it as . Here, the sign function is defined as for .
Sparse boosting is developed by Buhlmann and Yu (2006) for linear regression models. A different sparse boosting approach is proposed by Zhang and Ramadge (2009). When the weak learners and stopping rule are properly chosen, ordinary boosting may enjoy a certain degree of sparsity. However, numerical studies suggest that with high-dimensional data, it may not be “sparse enough.” The sparse boosting introduces further sparsity by modifying the loss function. More specifically, at each iteration, the sparse boosting loss function consists of 2 parts. The first part is the same as that with ordinary boosting, which, for example, can be the negative log-likelihood function. The second part is a penalty term measuring the sparsity of model. The weak learners are chosen in a manner that balances goodness-of-fit and sparsity, which can lead to sparser classifiers.
3.2. Integrative sparse logit boosting
In this section, we describe the integrative LogitBoost (iLB) and integrative sparse LogitBoost (iSLB) algorithms. They are related to LogitBoost in that they all deal with binary response variables and logistic regression models and use the negative likelihood as a measure of goodness-of-fit. In addition, as high-dimensional data are analyzed, sparsity is desirable. iSLB belongs to the family of sparse boosting approaches and enjoys more sparsity than ordinary boosting. On the other hand, the proposed algorithms significantly differ from existing approaches. First of all, the existing approaches focus on the analysis of single data sets, whereas both iLB and iSLB analyze multiple data sets and automatically accommodate the heterogeneity among them. In addition, iSLB differs from LogitBoost by introducing further sparsity, differs from the sparse boosting in Buhlmann and Yu (2006) by analyzing data with binary responses under the logistic regression models, and differs from the sparse boosting in Zhang and Ramadge (2009) by achieving sparsity using a different computationally simpler penalty.
iLB algorithm.
Consider the data and model settings described in Section 2. Denote as the committee function in study m. Following the discussions in Section 2, we allow for study-specific to accommodate heterogeneity. Let be the empirical measure and let be the normalized empirical log-likelihood function in study m. The iLB algorithm proceeds as follows.
Step 1: Initialization. Set . For , set ;
Step 2: Fit and update. .
Compute 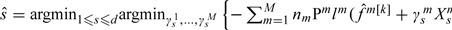 , where is the sth component of and is the unknown regression coefficient of .
, where is the sth component of and is the unknown regression coefficient of .
Compute 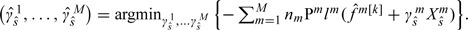
Update , where ν is the step size as in the LogitBoost;
Step 3: Iteration. Repeat Step 2 for K iterations;
Step 4: Stopping. At iteration k, denote . Define . Denote and . Compute the objective function
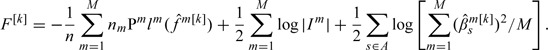 |
(3.1) |
Here,  is the value of for the ith subject in study m. is an estimate of . Estimate the stopping iteration by
is the value of for the ith subject in study m. is an estimate of . Estimate the stopping iteration by
In integrative analysis, multiple data sets need to be analyzed simultaneously. Thus, all quantities in the proposed algorithm are summations over multiple data sets. In Step 2, we adopt the sum of negative log-likelihood functions as the loss function (boosting criterion), which makes the proposed algorithm easily extendable to other models. When choosing the index of weak learner, we evaluate the “overall effects” of genes across M studies. The selected gene (weak learner) has the strongest overall effect and may be different from genes with the strongest effects in individual studies. When a gene is selected, it is included in all M classifiers, leading to the same set of genes and the same sparsity structure for all M classifiers. However, we allow the regression coefficients to be different for different m, which is in line with discussions in Section 2. In boosting, a tuning parameter is the step size. Multiple studies have shown that the choice of step size is not crucial. We follow the suggestion of Buhlmann and Yu (2010) and set the step size = 0.1. In boosting, another tuning is the stopping rule. In the proposed algorithm, the stopping criterion in (3.1) has 2 parts. The first part measures the goodness-of-fit. The second part is the MDL criterion (Rissanen, 1989), which has been advocated in Buhlmann and Yu (2006). The MDL bridges between AIC and BIC and can be more flexible (Hansen and Yu, 2003). It has significantly lower computational cost than cross-validation and may be preferred in high-dimensional data analysis. With logistic models, we adopt the computationally efficient approximated MDL criterion (Qian and Field, 2000; Zou and Hastie, 2005). Our numerical study demonstrates satisfactory performance of the MDL criterion. We conjecture that other stopping rules may also be applicable. A thorough investigation of stopping rules is beyond scope of this article. For study m, is the resulted classifier. For a subject with gene expression in study m, the resulted classifier classifies its status as . Genes with index in A at iteration are identified as cancer markers.
iSLB algorithm.
In Step 2 of iLB, only the goodness-of-fit is considered in the selection of weak learner. As suggested in Buhlmann and Yu (2006), such a strategy may have the following limitation. Consider, for example, 2 genes A and B. Suppose that by the end of iteration , gene A has already been selected, with its corresponding weaker learner included in . Suppose that gene B has not been selected. Consider the scenario where the weak learner corresponding to gene B can improve model fitting slightly better than that with gene A. With iLB, as the goodness-of-fit is the only selection criterion, gene B will be selected in iteration k. In cancer genomic studies, a shorter list of identified markers, which can lead to more focused hypotheses and reduce workload for downstream analysis, is desirable. Thus, we are willing to slightly sacrifice goodness-of-fit in exchange for sparsity. Under the scenario described above, it is desirable to select gene A (instead of gene B) in iteration k. To achieve such a goal, we modify the loss function in Step 2. Specifically, we propose
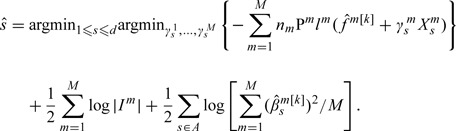 |
Other steps remain the same. With iSLB, we choose weak learners that can balance goodness-of-fit and sparsity in each iteration. This differs from iLB, which only considers sparsity in the stopping rule. As a different criterion is used, selected by iSLB can be different from those selected by iLB. Specifically, iSLB tends to select fewer genes.
4. NUMERICAL STUDIES
4.1. Practical considerations
In practical data analysis, preprocessing is needed. Particularly, normalization of gene expressions needs to be conducted for each data set separately. For Affymetrix data, a floor and a ceiling may be added, and then, measurements are log2 transformed. We fill in missing expressions with means across samples. We then standardize each gene expression to have zero mean and unit variance. There are many different data preprocessing approaches. We choose the one described above for its simplicity. A significant advantage of the proposed approach is that it does not require the direct comparability of measurements from different studies. Thus, cross-study normalization or transformations are not needed.
For simplicity of notation, we have assumed that multiple studies have matched gene sets. When different sets of genes are measured in different studies, we employ a simple rescaling approach. Assume that gene s is only measured in the first studies. Consider, for example, Step 2 of iLB. We modify it as
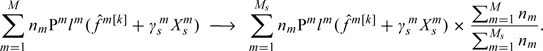 |
Other quantities can be rescaled in a similar manner. The power of integrative analysis decreases when the overlap among gene sets measured in multiple studies decreases. Numerical study is carried out using R (www.r-project.org). Research code is available from the authors.
4.2. Simulation
We simulate data for 4 independent studies. In each study, there are 30 or 100 subjects. We simulate 50 or 100 gene clusters with 20 genes in each cluster. Gene expressions have marginally normal distributions. Genes in different clusters have independent expressions. For genes within the same clusters, their expressions have the following correlation structures: (a) autoregressive correlation, where expressions of genes j and k have correlation coefficient , or 0.7; (b) banded correlation, where expressions of genes j and k have correlation coefficient , or 0.33; (c) compound symmetry, where expressions of genes j and k have correlation coefficient ρ when , or 0.7. Within each of the first 3 clusters, the first 10 genes are associated with the responses. There are a total of 30 cancer-associated genes and the rest are noises. For cancer-associated genes, we generate their regression coefficients from . Thus, some genes have large effects, and others have small to moderate effects. Six (20%) cancer-associated genes and 10% noisy genes are only measured in 2 studies, with their indices randomly generated in each replicate. We generate the binary response variables from the logistic regression models and Bernoulli distributions. The simulation settings closely mimic practical pharmacogenomic studies, where genes have the pathway structures. Genes within the same pathways tend to have correlated expressions, whereas genes within different pathways tend to have weakly correlated or uncorrelated expressions. Among a large number pathways, only a few are associated with the responses. Within those important pathways, some genes are cancer associated and others are noises.
To better gauge performance of the proposed approach, we also consider the following alternatives. (a) An intensity approach: Since the 4 data sets are generated under similar settings, we adopt an intensity approach (Shabalin and others, 2008), make transformations of gene expressions, combine 4 data sets, and analyze as if they were from a single study. The combined data set is analyzed using 2 approaches. The first is the LogitBoost described in Section 3.1. An MDL criterion similar to that in Step 4 of iLB is used for stopping. The second is the Enet (elastic net; Zou and Hastie, 2005), which is an extensively adopted penalization approach. The tuning parameters in Enet are chosen using cross-validation. (b) Meta-analysis: We analyze each data set separately using the LogitBoost (with an MDL criterion as the stopping rule) and Enet (with tunings selected by cross-validation). Genes that are identified in at least one study are identified in meta-analysis. An alternative is to consider genes identified in all studies. However, we have examined all simulation scenarios and found that there are very few such genes.
In simulation, we are interested in evaluating both identification and prediction performance. For identification, we evaluate the number of genes identified and number of true positives. In prediction evaluation, for each set of 4 data sets (training), we simulate 4 additional data sets (testing). We generate estimates using the training data, make prediction for subjects in the testing data, and compute prediction errors. Summary statistics based on 200 replicates are shown in Table 1. Among the 3 families of approaches, meta-analysis approaches have the least satisfactory performance. Particularly, a large number of false positives are identified. This observation is reasonable, considering that individual data sets have small sample sizes, and hence, the identification results can be unsatisfactory. By pooling raw data from multiple data sets, intensity approaches significantly outperform meta-analysis approaches. Such a result is also reasonable considering that the 4 data sets are generated under comparable settings, which favors intensity approaches. Both iLB and iSLB are capable of identifying the majority of true positives with a reasonable number of false positives. They outperform the meta-analysis and intensity approaches by identifying more true positives and/or fewer false positives. iSLB sometimes identifies fewer true positives. However, it identifies much fewer false positives. This observation is in line with Buhlmann and Yu (2006). Examination of the prediction performance also suggests that iLB and iSLB can outperform alternatives. Simulation clearly demonstrates the power of integrative analysis. More specifically, with meta-analysis and intensity approaches, the Enet penalization approach has better performance than boosting under certain simulation scenarios. However, by conducting integrative analysis, the proposed boosting approaches are able to outperform Enet in both identification and prediction accuracy.
Table 1.
Simulation study: summary statistics based on 200 replicates
| nm | # clus | ρ | Meta-analysis |
Intensity approach |
iLB | iSLB | ||
| Enet | LB | Enet | LB | |||||
| Correlation structure: autoregressive | ||||||||
| 30 | 50 | 0.3 | 80, 13, 46 | 70, 10, 44 | 41, 10, 36 | 33, 9, 31 | 32, 15, 20 | 28, 16, 14 |
| 0.7 | 74, 17, 40 | 75, 12, 41 | 29, 14, 20 | 25, 12, 19 | 34, 17, 15 | 24, 17, 12 | ||
| 100 | 0.3 | 97, 8, 47 | 96, 9, 47 | 31, 5, 40 | 44, 6, 41 | 30, 14, 29 | 29, 14, 29 | |
| 0.7 | 93, 10, 43 | 88, 10, 41 | 29, 9, 24 | 32, 10, 20 | 34, 16, 22 | 25, 17, 13 | ||
| 100 | 50 | 0.3 | 105, 23, 34 | 69, 14, 32 | 36, 23, 21 | 39, 12, 25 | 38, 21, 18 | 27, 22, 10 |
| 0.7 | 79, 25, 19 | 65, 15, 21 | 25, 22, 13 | 37, 17, 14 | 42, 23, 12 | 26, 23, 11 | ||
| 100 | 0.3 | 122, 19, 40 | 72, 15, 35 | 24, 17, 25 | 41, 12, 20 | 40, 19, 13 | 29, 21, 10 | |
| 0.7 | 110, 23, 25 | 71, 15, 23 | 30, 21, 14 | 40, 16, 18 | 48, 24, 18 | 29, 22, 12 | ||
| Correlation structure: banded | ||||||||
| 30 | 50 | 0.3 | 85, 13, 36 | 70, 9, 38 | 29, 13, 20 | 32, 9, 23 | 38, 17, 19 | 28, 15, 17 |
| 0.7 | 74, 10, 40 | 91, 10, 38 | 35, 13, 27 | 40, 11, 29 | 35, 20, 16 | 29, 21, 15 | ||
| 100 | 0.3 | 85, 5, 46 | 76, 10, 40 | 40, 11, 23 | 44, 8, 24 | 37, 14, 22 | 25, 18, 20 | |
| 0.7 | 65, 7, 45 | 85, 8, 43 | 25, 8, 34 | 44, 10, 31 | 31, 20, 20 | 27, 15, 19 | ||
| 100 | 50 | 0.3 | 87, 23, 18 | 66, 10, 20 | 32, 20, 12 | 42, 15, 14 | 41, 18, 15 | 29, 19, 9 |
| 0.7 | 98, 26, 26 | 62, 12, 24 | 37, 22, 15 | 40, 13, 17 | 45, 24, 16 | 29, 20, 8 | ||
| 100 | 0.3 | 124, 25, 24 | 68, 13, 29 | 37, 20, 15 | 43, 16, 16 | 43, 18, 17 | 32, 23, 9 | |
| 0.7 | 171, 20, 32 | 72, 18, 28 | 36, 20, 16 | 42, 19, 18 | 51, 21, 19 | 32, 19, 13 | ||
| Correlation structure: compound symmetric | ||||||||
| 30 | 50 | 0.3 | 61, 11, 40 | 51, 12, 31 | 34, 13, 23 | 40, 8, 25 | 34, 19, 19 | 31, 18, 20 |
| 0.7 | 69, 12, 27 | 74, 10, 29 | 33, 12, 14 | 33, 11, 14 | 40, 20, 12 | 20, 15, 8 | ||
| 100 | 0.3 | 84, 7, 43 | 88, 8, 40 | 41, 13, 35 | 44, 10, 36 | 36, 12, 20 | 30, 14, 19 | |
| 0.7 | 58, 7, 36 | 70, 8, 38 | 30, 10, 13 | 34, 9, 15 | 30, 16, 18 | 27, 13, 20 | ||
| 100 | 50 | 0.3 | 101, 26, 22 | 67, 12, 21 | 32, 24, 14 | 41, 10, 17 | 35, 19, 16 | 29, 24, 8 |
| 0.7 | 128, 26, 14 | 62, 13, 19 | 27, 19, 9 | 40, 20, 19 | 43, 24, 23 | 30, 25, 7 | ||
| 100 | 0.3 | 143, 25, 27 | 75, 12, 30 | 36, 23, 14 | 38, 13, 21 | 38, 17, 15 | 26, 19, 12 | |
| 0.7 | 104, 22, 14 | 74, 18, 21 | 28, 18, 10 | 44, 12, 17 | 49, 27, 15 | 26, 22, 9 | ||
nm, sample size of each data set; # clus, number of gene clusters; ρ, correlation coefficient parameter; LB, LogitBoost.
In cells with “a, b, c,” a is the number of genes identified, b is the number of true positives, and c is the number of prediction errors per 100 subjects.
Under some simulation scenarios, the iLB is less satisfactory with a considerable number of false positives. One possible cause is that the boosting stops “too late.” Numerically, we have experimented with the following modification of Step 4 of iLB
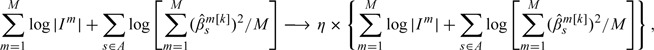 |
where . We have experimented with different η values and found that with a moderate η, the number of true positives remains almost the same, whereas the number of false positives may decrease. Such a modification may demand tuning an additional parameter or revising the MDL criterion and will not be pursued.
4.3. Pancreatic cancer studies
Pancreatic ductal adenocarcinoma (PDAC) is a major cause of malignancy-related death. Apart from surgery, there is still no effective therapy, and even resected patients usually die within 1 year postoperatively. We collect and analyze 4 data sets, which have been described in Iacobuzio-Donahue and others (2003), Logsdon and others (2003), Crnogorac-Jurcevic and others (2003), and Friess and others (2002). We provide brief data description in Table 2. Two studies used complementary DNA arrays, and the other 2 used oligonucleotide arrays. The 2 sample groups were the PDAC and normal pancreatic tissues. We remove genes with more than 30% missingness. A total of 1204 genes are included in analysis. For each data set, we conduct the preprocessing described in Section 4.1.
Table 2.
Pancreatic cancer studies
| Data set | D1 | D2 | D3 | D4 |
| Author | Logsdon and others (2003) | Friess and others (2002) | Iacobuzio-Donahue and others (2003) | Crnogorac-Jurcevic and others (2003) |
| PDAC | 10 | 8 | 9 | 8 |
| Normal | 5 | 3 | 8 | 5 |
| Array | Affymetrix HuGeneFL | Affymetrix HuGeneFL | cDNA Stanford | cDNA Sanger |
| UG | 5521 | 5521 | 29 621 | 5794 |
PDAC, number of PDAC samples; Normal, number of normal samples; Array, type of array used; UG, number of unique UniGene clusters; cDNA, complementary DNA.
The lists of genes identified using the proposed approaches are presented in Supplementary Table 6 available at Biostatistics online (iLB) and Table 3 (iSLB), respectively. Properties of the estimates are in line with discussions in Section 2. Although sharing common genes, the sets of selected genes using iLB and iSLB are considerably different. This is not surprising considering the difference in algorithms, difference observed in simulation study, and extremely noisy nature of gene expression data. We search NCBI for the pathological implications of identified genes and find that some of those genes have been established as cancer markers in multiple independent studies. Examples include genes FGL1, CRAT, GSTs, FN1, PTPN12, PDIA2, NBL1, and others.
Table 3.
Pancreatic cancer markers identified using iSLB and their estimated regression coefficients
| UniGene | Gene name | D1 | D2 | D3 | D4 |
| Hs.155418 | TRIB2 | 0.020 | 0.020 | 0.036 | −0.031 |
| Hs.169900 | PABPC4 | −0.504 | −0.535 | −0.364 | −0.585 |
| Hs.287820 | FN1 | 1.191 | 1.228 | 1.297 | 0.964 |
| Hs.433434 | PSMB7 | −0.008 | −0.028 | 0.013 | 0.019 |
| Hs.5591 | MKNK1 | −0.128 | −0.237 | −0.227 | −0.354 |
| Hs.66581 | PDIA2 | −0.177 | −0.178 | −0.204 | −0.093 |
| Hs.83942 | CTSK | 0.021 | 0.029 | 0.007 | 0.013 |
We evaluate prediction performance using a 4-fold cross-validation–based approach. Besides being of interest itself, the prediction evaluation also provides an indirect evaluation of the identification accuracy: If the identified markers are more meaningful, prediction using these markers is expected to be more accurate. We first randomly cut each data set into 4 subsets with equal sizes. We then remove one subset from each data set. With the reduced data, we apply the proposed approach. We then use the resulted classifiers to make prediction for subjects in the removed subsets. We repeat this procedure over all 4 subsets of each data. The overall prediction error can then be computed. With this approach, 2 and 3 subjects cannot be correctly classified under iLB and iSLB, respectively.
We also analyze data using the alternative approaches described in the above section. In meta-analysis, we first analyze the 4 data sets separately using the LogitBoost approach and identify 10, 12, 9, and 7 genes in data sets D1–D4, respectively. A total of 29 genes are identified in at least one data set, with no gene identified in all 4 data sets. The numbers of overlapped genes with iLB and iSLB are 5 and 2, respectively. With the cross-validation–based evaluation, a total of 4 subjects cannot be properly classified. We also analyze the 4 data sets separately using Enet and identify 10, 7, 11, and 6 genes in data sets D1–D4, respectively. A total of 21 genes are identified in at least one data set, with one gene identified in all 4 data sets. The numbers of overlapped genes with iLB and iSLB are 3 and 2, respectively. With the cross-validation–based evaluation, a total of 3 subjects cannot be properly classified. When adopting the intensity approach, we analyze the combined data using LogitBoost and identify 25 genes. The numbers of overlapped genes with iLB and iSLB are 9 and 3, respectively. With the cross-validation–based approach, 4 subjects are not properly classified. We also analyze the combined data using Enet and identify 16 genes. The numbers of overlapped genes with iLB and iSLB are 5 and 1, respectively. With the cross-validation–based approach, 3 subjects are not properly classified.
4.4. Liver cancer studies
Gene profiling studies have been conducted on hepatocellular carcinoma, which is among the leading causes of cancer death in the world. Four microarray studies are described in Table 4. Data sets D1–D4 were collected in 3 different hospitals in South Korea. Although the studies were conducted in a controlled setting, the researchers “failed to directly merge the data even after normalization of each data set” (Choi and others, 2004). A total of 9984 genes were measured in all 4 studies, among which 3122 genes have less than 30% missingness and are analyzed. We remove 8 subjects that have more than 30% gene expression measurements missing. Compared with the pancreatic cancer data, the liver cancer data have a larger sample size, are more difficult to classify, and hence may demonstrate performance of the proposed approach from a different perspective.
Table 4.
Liver cancer studies
| Data set | D1 | D2 | D3 | D4 |
| Experimenter | Hospital A | Hospital B | Hospital C | Hospital C |
| Tumor | 16 (14) | 23 | 29 | 12 (10) |
| Normal | 16 (14) | 23 | 5 | 9 (7) |
| Chip type | cDNA (Ver. 1) | cDNA (Ver. 1) | cDNA (Ver. 1) | cDNA (Ver. 2) |
| (Cy5:Cy3) | Sample:normal liver | Sample:placenta | Sample:placenta | Sample:sample |
Tumor, number of tumor samples; Normal, number of normal samples.
Numbers in the brackets are the number of subjects used in analysis. Ver. 2 chips have different spot locations from Ver. 1 chips.
With iLB and iSLB, we identify 33 (see Supplementary Table 7 available at Biostatistics online) and 16 (Table 5) genes, respectively. Similar estimation properties as with the pancreatic cancer studies are observed. With the cross-validation–based approach, 19 (iLB) and 17 (iSLB) subjects are not properly classified. We also analyze data using the alternative approaches described in Section 4.2. In meta-analysis, we analyze the 4 data sets using the LogitBoost approach and identify 17, 14, 16, and 10 genes in data sets D1–D4, respectively. A total of 51 genes are identified in at least one data set. The numbers of overlapped genes with iLB and iSLB are 11 and 6, respectively. With the cross-validation–based approach, a total of 36 subjects are not properly classified. We also analyze using Enet and identify 12, 8, 11, and 10 genes in data sets D1–D4, respectively. A total of 30 genes are identified in at least one data set. The numbers of overlapped genes with iLB and iSLB are 9 and 4, respectively. With the cross-validation–based approach, a total of 33 subjects are not properly classified. When adopting the intensity approach, we analyze the combined data using LogitBoost and identify 38 genes. The numbers of overlapped genes with iLB and iSLB are 13 and 7, respectively. With the cross-validation–based approach, a total of 24 subjects are not properly classified. We also analyze the combined data using Enet and identify 35 genes. The numbers of overlapped genes with iLB and iSLB are 10 and 5, respectively. With the cross-validation–based approach, a total of 27 subjects are not properly classified.
Table 5.
Liver cancer markers identified using iSLB and their estimated regression coefficients
| Gene Name | D1 | D2 | D3 | D4 |
| EST387826 cDNA | −0.085 | −0.057 | −0.011 | −0.047 |
| Podocalyxin-like (PODXL), mRNA | −0.129 | −0.126 | −0.020 | −0.045 |
| Protocadherin 10 (PCDH10), transcript variant 1, mRNA | −0.050 | −0.055 | −0.006 | −0.082 |
| Nomatch | −0.339 | 0.127 | 0.019 | 0.087 |
| Human G protein-coupled receptor V28 mRNA | −0.011 | −0.026 | −0.010 | −0.015 |
| Stromal cell-derived factor 1 (SDF1), mRNA | −0.280 | −0.215 | −0.179 | −0.255 |
| 6-phosphofructo-2-kinase | −0.503 | −0.194 | −0.132 | 0.036 |
| Polymerase (DNA directed), delta 1, catalytic subunit (POLD1), mRNA | −0.101 | −0.109 | −0.068 | −0.125 |
| Homer, neuronal immediate early gene, 3 (HOMER-3), mRNA | −0.005 | −0.133 | −0.040 | −0.016 |
| Tryptophan 2,3-dioxygenase (TDO2), mRNA | −0.041 | −0.062 | −0.050 | −0.094 |
| Epididymal secretory protein (19.5kD) (HE1), mRNA | 0.010 | 0.026 | −0.003 | 0.005 |
| CD33 antigen (gp67) (CD33), mRNA | 0.218 | 0.042 | 0.050 | 0.113 |
| Tubulin, beta polypeptide (TUBB), mRNA | 0.090 | 0.039 | 0.014 | 0.065 |
| mRNA; cDNA DKFZp586B1824 (from clone DKFZp586B1824) | 0.009 | 0.033 | 0.039 | 0.091 |
| Betaine-homocysteine methyltransferase (BHMT), mRNA | −0.322 | −0.008 | −0.166 | −0.280 |
| RNA helicase-related protein (RNAHP), mRNA | −0.326 | −0.614 | −0.496 | −0.746 |
mRNA, messenger RNA.
5. DISCUSSION
In the identification of cancer genomic markers, integrative analysis provides an effective way of pooling data from multiple studies, increasing statistical power, and improving reproducibility of identified markers. When pooling data from multiple studies, it is critical to select studies with comparable designs. Another important issue is the interpretation and practical usage of identified markers. We acknowledge the importance and difficulty of these issues. In this study, we focus on the marker identification aspect and refer to Guerra and Goldstein (2009) for established guidelines on data selection and other issues related to the analysis of multiple data sets.
We propose an integrative sparse boosting approach for the analysis of multiple cancer diagnosis studies and marker identification. The negative log likelihood is chosen as the loss function. It is possible to extend the proposed approach to other types of loss functions and to other types of response variables. It is also possible to extend to other types of weak learners, for example, nonparametric functions of gene expressions. We use an MDL criterion for stopping with both iLB and iSLB and boosting with iSLB. Such a choice has been motivated by the satisfactory performance of MDL shown in Buhlmann and Yu (2006) and Zou and Hastie (2005). We conjecture that it is possible to replace the MDL criterion with other criteria that can measure the complexity of learners. Possible choices may include AIC, BIC, and others. With ultrahigh dimensional data, to the best of our knowledge, there is still no definitive result on the optimality of tuning parameter selection approaches. The MDL is intuitively reasonable and has low computational cost and satisfactory numerical performance. It is beyond the scope of this study to establish its relative performance over AIC or BIC. The proposed approach applies the same number of boosting steps to all data sets. Different studies may not be equally informative. It is expected that by allowing different regression coefficients, such difference can be at least partially accounted for. It is of interest to explore whether it is necessary to allow different number of boosting steps.
Simulation study suggests that the proposed approach outperforms penalization and boosting-based meta-analysis and intensity approaches. Comparison with Enet-based analysis is especially interesting, as it shows the power of integrative analysis. We are aware that there are other alternatives. The boosting-based approaches we compare with have a statistical framework closest to that of the proposed approach. The Enet approach is one of the most popular penalization approaches. The proposed approach is shown to have satisfactory performance with the pancreatic and liver cancer data sets. In particular, the sparse boosting algorithms identify shorter lists of genes, which can lead to more focused hypothesis and reduced downstream workload, and have better prediction performance. The pancreatic and liver cancer studies are representative of what is encountered in practice, with one being relatively easy and the other being more difficult to classify. In the most recent studies on other types of cancers (e.g. breast cancer), the sample sizes may be considerably larger. We choose to analyze data with relatively small sample sizes as it can be more critical to conduct integrative analysis with such data. The cross-validation–based approach is used for prediction evaluation. Although it does not use completely independent data, it compares all approaches on the same ground and is reasonably fair. Despite the satisfactory empirical performance of the proposed approach, it appears difficult to establish its theoretical property. In Buhlmann and Yu (2006), the theoretical properties of sparse boosting with simple linear regression models are established under the strong assumption of orthogonality, which does not hold in general. Logistic regression models are more complicated than linear regression models. Further work is needed to understand the theoretical properties of the proposed approach.
SUPPLEMENTARY MATERIAL
Supplementary material is available at http://biostatistics.oxfordjournals.org.
FUNDING
This study is supported in part by National Institutes of Health (CA120988, CA142774, CA152301, LM009828); National Science Foundation (DMS0904181).
Supplementary Material
Acknowledgments
We thank the Editor, Associate Editor, and 2 referees for careful review and insightful comments, which led to significant improvement of the paper. Conflict of Interest: None declared.
References
- Berk RA. Statistical Learning from a Regression Perspective. Springer; 2008. [Google Scholar]
- Buhlmann P, Hothorn T. Boosting algorithms: regularization, prediction and model fitting (with discussion) Statistical Science. 2007;22:477–505. [Google Scholar]
- Buhlmann P, Yu B. Sparse boosting. Journal of Machine Learning Research. 2006;7:1001–1024. [Google Scholar]
- Buhlmann P, Yu B. Boosting. WIREs Computational Statistics. 2010;2:69–74. [Google Scholar]
- Choi H, Shen R, Chinnaiyan AM, Ghosh D. A latent variable approach for meta analysis of gene expression data from multiple microarray experiments. BMC Bioinformatics. 2007;8:364. doi: 10.1186/1471-2105-8-364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Choi J, Kim D, Choi D, Kim B, Lee K, Yeom Y, Yoo H, Yoo O, Kim S. Integrative analysis of multiple gene expression profiles applied to liver cancer study. FEBS Letters. 2004;565:93–100. doi: 10.1016/j.febslet.2004.03.081. [DOI] [PubMed] [Google Scholar]
- Crnogorac-Jurcevic T, Missiaglia E, Blaveri E, Gangeswaran R, Jones M, Terris B, Costello E, Neoptolemos JP, Lemoine NR. Molecular alterations in pancreatic carcinoma: expression profiling shows that dysregulated expression of S100 genes is highly prevalent. Journal of Pathology. 2003;201:63–74. doi: 10.1002/path.1418. [DOI] [PubMed] [Google Scholar]
- Dettling M. BagBoosting for tumor classification with gene expression data. Bioinformatics. 2004;20:3583–3593. doi: 10.1093/bioinformatics/bth447. [DOI] [PubMed] [Google Scholar]
- Dettling M, Buhlmann P. Boosting for tumor classification with gene expression data. Bioinformatics. 2003;19:1061–1069. doi: 10.1093/bioinformatics/btf867. [DOI] [PubMed] [Google Scholar]
- Friedman J, Hastie T, Tibshirani R. Additive logistic regression: a statistical view of boosting. Annals of Statistics. 2000;28:337–407. [Google Scholar]
- Friess H, Ding J, Kleeff J, Fenkell L, Rosinski JA, Guweidhi A, Reidhaar-Olson JF, Korc M, Hammer J, Buchler MW. Microarray-based identification of differentially expressed growth- and metastasis-associated genes in pancreatic cancer. Cellular and Molecular Life Sciences. 2003;60:1180–1199. doi: 10.1007/s00018-003-3036-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C and others. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra R, Goldstein DR. Meta-Analysis and Combining Information in Genetics and Genomics. Chapman and Hall/CRC; 2009. [Google Scholar]
- Hansen MH, Yu B. Minimum description length model selection criteria for generalized linear models. In: Goldstein DR, editor. Statistics and Science: A Festschrift for Terry Speed. Beachwood, OH: Institute of Mathematical Statistics; 2003. pp. 145–163. [Google Scholar]
- Hastie T, Tibshirani R, Friedman J. The Elements of Statistical Learning. Springer; 2009. [Google Scholar]
- Iacobuzio-Donahue CA, Ashfaq R, Maitra A, Adsay NV, Shen-Ong GL, Berg K, Hollingsworth MA, Cameron JL, Yeo CJ, Kern SE and others. Highly expressed genes in pancreatic ductal adenocarcinomas: a comprehensive characterization and comparison of the transcription profiles obtained from three major technologies. Cancer Research. 2003;63:8614–8622. [PubMed] [Google Scholar]
- Knudsen S. Cancer Diagnostics with DNA Microarrays. Wiley; 2006. [Google Scholar]
- Logsdon CD, Simeone DM, Binkley C, Arumugam T, Greenson J, Giordano TJ, Misek D, Hanash S. Molecular profiling of pancreatic adenocarcinoma and chronic pancreatitis identifies multiple genes differentially regulated in pancreatic cancer. Cancer Research. 2003;63:2649–2657. [PubMed] [Google Scholar]
- Ma S, Huang J. Regularized gene selection in cancer microarray meta-analysis. BMC Bioinformatics. 2009;10:1. doi: 10.1186/1471-2105-10-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian G, Field C. Using MCMC for logistic regression model selection involving large number of candidate models. Proceedings of the 4th International Conference on Monte Carlo and Quasi-Monte Carlo Methods in Scientific Computing, Hong Kong. 2000 [Google Scholar]
- Rhodes D, Chinnaiyan AM. Bioinformatics strategies for translating genome-wide expression analyses into clinically useful cancer markers. Annals of the New York Academy of Sciences. 2004;1020:32–40. doi: 10.1196/annals.1310.005. [DOI] [PubMed] [Google Scholar]
- Rhodes D, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM. Large-scale meta-analysis of cancer microarray data identified common transcriptional profiles of neoplastic transformation and progression. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:9309–9314. doi: 10.1073/pnas.0401994101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rissanen J. Stochastic Complexity in Statistical Inquiry. World Scientific Publishing Company; 1989. [Google Scholar]
- Shabalin AA, Tjelmeland H, Fan C, Perou CM, Nobel AB. Merging two gene expression studies via cross platform normalization. Bioinformatics. 2008;24:1154–1160. doi: 10.1093/bioinformatics/btn083. [DOI] [PubMed] [Google Scholar]
- Shen R, Ghosh D, Chinnaiyan AM. Prognostic meta signature of breast cancer developed by two-stage mixture modeling of microarray data. BMC Genomics. 2004;5:94. doi: 10.1186/1471-2164-5-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens JR, Doerge RW. Meta-analysis combines Affymetrix microarray results across laboratories. Comparative and Functional Genomics. 2005;6:116–122. doi: 10.1002/cfg.460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ramadge PJ. 2009 International Conference on Acoustics, Speech and Signal Processing. 2009. Sparse boosting. [Google Scholar]
- Zou H, Hastie T. Regularization and variable selection via the elastic net. Journal of Royal Statistical Society. Series B, Statistical Methodology. 2005;67:301–320. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.