

Abstract
Marinomonas posidonica IVIA-Po-181T Lucas-Elío et al. 2011 belongs to the family Oceanospirillaceae within the phylum Proteobacteria. Different species of the genus Marinomonas can be readily isolated from the seagrass Posidonia oceanica. M. posidonica is among the most abundant species of the genus detected in the cultured microbiota of P. oceanica, suggesting a close relationship with this plant, which has a great ecological value in the Mediterranean Sea, covering an estimated surface of 38,000 Km2. Here we describe the genomic features of M. posidonica. The 3,899,940 bp long genome harbors 3,544 protein-coding genes and 107 RNA genes and is a part of the Genomic Encyclopedia of Bacteria and Archaea project.
Keywords: Aerobic, Gram-negative, marine, plant-associated
Introduction
Strain IVIA-Po-181T is the type strain of Marinomonas posidonica, which belongs to the order Oceanospirillales within the class Gammaproteobacteria. Microorganisms belonging to the genus Marinomonas can be readily isolated from different parts of the marine angiosperm Posidonia oceanica, and many different Marinomonas species have been isolated from it [1,2]. The data obtained so far indicate that among the culturable microbiota in the phylloplane and rhizoplane of this plant, the closely related M. posidonica and M. aquiplantarum are the most abundant species [2]. P. oceanica (known commonly as Mediterranean tapeweed or Neptune Grass), is of great ecological value in the Mediterranean Sea. Recently, it has been estimated that large clones of this slow-growing plant can spread over several kilometers and can be several thousand years old [3]. The long lifespan of this plant and the abundance of M. posidonica in its microbiota suggest the possibility of an ancient relationship between them. In fact, M. posidonica has been shown to have a positive effect on the growth of P. oceanica seeds [4]. Here we describe the complete genomic sequencing and annotation of M. posidonica IVIA-Po-181T, along with some relevant features of this microorganism.
Classification and features
M. posidonica IVIA-Po-181T contains 8 copies of the 16S rRNA gene. The only observed difference among these is that the nucleotide at position 454 can be either T (in 6 of the copies) or C (two copies). The phylogenetic neighborhood of M. posidonica IVIA-Po-181T in a tree based on 16S rRNA sequences is shown in Figure 1.
Figure 1.

Phylogenetic tree highlighting the position of Marinomonas posidonica IVIA-Po-181T in relation to other type and non-type strains within the genus Marinomonas. The species that have been isolated from Posidonia oceanica are in bold. The tree was generated using the program MEGA version 4 [5]. The sequences were aligned using the CLUSTAL W program within MEGA software. The tree was generated using the neighbor-joining method. Asterisks indicate branches that were also identified by the maximum-parsimony method. Numbers at branches indicate bootstrap values from 1,000 replicates. P. haloplanktis (X67024) was used as an outgroup.
The cells of M. posidonica IVIA-Po-181T are helical with rounded ends. Cell lengths and widths are approximately 1.3-2.1 and 0.4 μm respectively (Figure 2, Table 1). Strain IVIA-Po-181T is motile by a single polar flagellum [2] (Figure 2). Colonies in complex medium, such as marine 2216 agar, do not show pigmentation. A Na+ concentration greater than 0.5% is required for growth of M. posidonica IVIA-Po-181T, which can tolerate NaCl concentrations up to 10%. It grows in the range of 5-30 ºC, is strictly aerobic and chemoorganotrophic. It does not hydrolyze starch, agar, gelatin or Tween 80. It utilizes D-glucose, D-galactose, D-fructose, sucrose, maltose, D-sorbitol, L-glutamate, malate and acetate as carbon sources.
Figure 2.

Electron micrograph of M. posidonica IVIA-Po-181T.
Table 1. Classification and general features of Marinomonas posidonica IVIA-Po-181T according to the MIGS recommendations [6].
| MIGS ID | Property | Term | Evidence codea |
|---|---|---|---|
| Current classification | Domain Bacteria | TAS [7] | |
| Phylum Proteobacteria | TAS [8] | ||
| Class Gammaproteobacteria | TAS [9,10] | ||
| Order Oceanospirillales | TAS [9,11] | ||
| Family Oceanospirillaceae | TAS [9,12] | ||
| Genus Marinomonas | TAS [13-15] | ||
| Species Marinomonas posidonica | TAS [2] | ||
| Type strain IVIA-Po-181T, CECT 7376 T | TAS [2] | ||
| Gram stain | Negative | TAS [2] | |
| Cell shape | Helical | TAS [2] | |
| Motility | Single polar flagellum | TAS [2] | |
| Sporulation | none | TAS [2] | |
| Temperature range | 5 ºC-30º C | TAS [2] | |
| Optimum temperature | 25º C | ||
| Carbon source | Carbohydrates, amino acids | TAS [2] | |
| Energy source | Chemoheterotroph | TAS [2] | |
| Terminal electron receptor | Oxygen | TAS [2] | |
| MIGS-6 | Habitat | Sea water | TAS [2] |
| MIGS-6.3 | Salinity | 0.5-10% NaCl | TAS [2] |
| MIGS-22 | Oxygen | Aerobic | TAS [2] |
| MIGS-15 | Biotic relationship | Microbiota of the rhizome of Posidonia oceanica | TAS [2] |
| MIGS-14 | Pathogenicity | None | TAS [2] |
| MIGS-4 | Geographic location | Pollença Bay, Balearic Islands in the Mediterranean Sea | TAS [2] |
| MIGS-5 | Sample collection time | May 2005 | IDA |
| MIGS-4.1 | Latitude | N 39,89 | IDA |
| MIGS-4.2 | Longitude | E 3,09 | IDA |
| MIGS-4.3 | Depth | 4 m | IDA |
| MIGS-4.4 | Altitude | not reported |
Evidence codes - IDA: Inferred from Direct Assay; TAS: Traceable Author Statement (i.e., a direct report exists in the literature); NAS: Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are from the Gene Ontology project [16]. For the purposes of this specific publication, if the evidence code is IDA, then the property was observed on a live isolate by one of the authors, or an expert mentioned in the acknowledgements.
Genome sequencing information
Genome project history
The genus Marinomonas comprises an increasing number (21 as of March 2012) of recognized bacterial species isolated from different marine habitats, including the seagrass Posidonia oceanica [1,2]. Marinomonas species show important differences in terms of substantive phenotypic characteristics. For instance, the cells of some species are helical, while others are either straight or curved rods [2]. M. mediterranea, which can be also isolated from P. oceanica [1], is the only known species within the genus that expresses a tyrosinase involved in melanin synthesis [17]. Another unique feature of that strain is the synthesis of a multicopper oxidase with laccase activity [18]. A different strain, Marinomonas sp. MWYL1, can catabolize dimethylsulfoniopropionate (DMSP), releasing a gas, dimethyl sulfide (DMS), which has a range of environmental effects, including changes in animal behavior and sulfur biogeochemical cycling [19,20]. M. posidonica is an abundant component of the culturable microbiota associated with P. oceanica. Since this seagrass is of enormous importance in Mediterranean ecosystems, it would be interesting to determine the metabolic capacities of the associated microorganisms. In addition, the genomic comparison with other genome-sequenced Marinomonas strains, namely M. mediterranea MMB-1T [21] and Marinomonas sp. MWYL1 (accession number NC-009654), could offer clues about the diverse properties of this marine genus.
The M. posidonica IVIA-Po-181T genome was sequenced under the Community Sequencing Program, CSP-2010 of DOE Joint Genome Institute who performed the sequencing, finishing and annotation. The genome has been deposited in GenBank with accession number NC_015559. Table 2 presents the project information and its association with MIGS version 2.0 compliance [6].
Table 2. Project information.
| MIGS ID | Property | Term |
|---|---|---|
| MIGS-31 | Finishing quality | Finished |
| MIGS-28 | Libraries used | Three genomic libraries: one Illumina GAii shotgun library, one 454 Titanium standard library, one paired-end 454 library |
| MIGS-29 | Sequencing platforms | Illumina GAii, 454 GS FLX Titanium |
| MIGS-31.2 | Fold coverage | 746.4 × Illumina, 37.9 × 454 |
| MIGS-30 | Assemblers | Newbler version 2.3, Velvet version .7.63, phrap version SPS 4.24 |
| MIGS-32 | Gene calling method | Prodigal, GenePRIMP |
| Genome Database release | February 19, 2011 | |
| Genbank ID | NC_015559 | |
| Genbank Date of Release | May 24, 2011 | |
| GOLD ID | Gc01770 | |
| NCBI project ID | 52545 | |
| MIGS-13 | Source material identifier | CECT 7376T, NCIMB 14433T |
| Project relevance | Comparative analysis, Environmental |
Growth conditions and DNA isolation
In order to isolate quality genomic DNA for sequencing, Marinomonas posidonica IVIA-Po-181T was grown from a -70 ºC stock in MMC agar medium [22]. A single colony was inoculated into the same broth medium and incubated at 25 ºC overnight. This culture was used to inoculate 200 ml of MMC at OD600 0.05. The culture was grown at 25 ºC, shaking at 130 rpm to early stationary phase (OD600 0.75), when it was kept at 4 ºC for 20 minutes to halt replication. DNA isolation from this culture was performed using the CTAB method (Ausubel et al., 1994) with some modifications. The cells were harvested by centrifugation (6000 × g) and the pellet resuspended in T10E1 pH 8 to an OD600 of 1.0. The cell suspension was treated at 37 ºC for 30 minutes with 0.53% SDS (Sigma) and 0.1 mg/ml Proteinase K (Fermentas). After the addition of RNase A (DNase-free from Qiagen) at 0.01 mg/ml, the cells were incubated at 37 ºC for another 30 min. To remove cell wall debris, denatured proteins and polysaccharides, the NaCl concentration was raised to 0.6 M, and 28.5 mM of CTAB (preheated to 65 ºC) was added to the cell extract, followed by incubation for 10 minutes at 65 ºC. CTAB-protein and CTAB-polysaccharide complexes were removed by chloroform/isoamyl alcohol (24:1), followed by phenol/chloroform/isoamyl alcohol (25:24:1) extractions. To precipitate the nucleic acids in the aqueous phase, 0.6 vol of isopropanol was added and the sample was incubated for 30 minutes at room temperature. The DNA precipitate was recovered by centrifugation, followed by a 70% ethanol wash to remove residual CTAB. The DNA pellet was resuspended in TE with 0.1 mg/ml RNaseA and stored at -80 ºC until further used.
Genome sequencing and assembly
The draft genome of Marinomonas posidonica IVIA-Po-181T was generated at the DOE Joint Genome Institute (JGI) using a combination of Illumina [23] and 454 technologies [24]. For this genome, we constructed and sequenced an Illumina GAii shotgun library which generated 58,265,190 reads totaling 4,428.2 Mb, a 454 Titanium standard library which generated 252,750 reads and one paired end 454 library with an average insert size of 7 kb which generated 709,174 reads totaling 203.5 Mb of 454 data. All general aspects of library construction and sequencing performed at the JGI can be found at the JGI website [25]. The initial draft assembly contained 35 contigs in 1 scaffold. The 454 Titanium standard data and the 454 paired end data were assembled together with Newbler, version 2.3. The Newbler consensus sequences were computationally shredded into 2 kb overlapping fake reads (shreds). Illumina sequencing data were assembled with VELVET, version 0.7.63 [26], and the consensus sequences were computationally shredded into 1.5 kb overlapping fake reads (shreds). We integrated the 454 Newbler consensus shreds, the Illumina VELVET consensus shreds and the read pairs in the 454 paired end library using parallel phrap, version SPS - 4.24 (High Performance Software, LLC). The software Consed [27-29] was used in the following finishing process. Illumina data were used to correct potential base errors and increase consensus quality using the software Polisher developed at JGI (Alla Lapidus, unpublished). Possible mis-assemblies were corrected using gapResolution (Cliff Han, unpublished), Dupfinisher [30], or by sequencing cloned bridging PCR fragments with sub-cloning. Gaps between contigs were closed by editing in Consed, by PCR and by Bubble PCR (J-F Cheng, unpublished) primer walks. A total of 110 additional reactions were necessary to close gaps and to raise the quality of the finished sequence. The total size of the genome is 3,899,940 bp and the final assembly is based on 130.3 Mb of 454 draft data, which provide an average 33.4× coverage of the genome and 4,327.5 Mb of Illumina draft data, which provide an average 1,109.6× coverage of the genome.
Genome annotation
Genes were identified using Prodigal [31] as part of the Oak Ridge National Laboratory genome annotation pipeline, followed by a round of manual curation using the JGI GenePRIMP pipeline [32]. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) non-redundant database, UniProt, TIGRFam, Pfam, PRIAM, KEGG, COG, and InterPro databases. These data sources were combined to assert a product description for each predicted protein. Non-coding genes and miscellaneous features were predicted using tRNAscan-SE [33], RNAMMer [34], Rfam [35], TMHMM [36], and signalP [37]. Further comparative analysis was performed using the IMG-ER system [38].
Genome properties
The genome has no plasmids, and the 3,899,940 bp circular chromosome has a GC content of 44.29% (Table 3 and Figure 3). Of the 3,651 predicted genes, 3,544 were protein-coding genes, and 107 RNAs. There were 53 pseudogenes. The majority of the protein-coding genes (78.09%) were assigned with a putative function; the others were annotated as hypothetical proteins. The distribution of genes into COGs functional categories is presented in Table 4.
Table 3. Genome statistics.
| Attribute | Value | % of totala |
|---|---|---|
| Genome size (bp) | 3,899,940 | 100 |
| DNA coding region (bp) | 3,492,581 | 89.55 |
| DNA G+C content (bp) | 1,727,094 | 44.29 |
| Number of replicons | 1 | |
| Extrachromosomal elements | 0 | |
| Total genes | 3,651 | 100.00 |
| Pseudogenesb | 53 | 1.45 |
| RNA genes | 107 | 2.93 |
| rRNA operons | 8 | |
| Protein-coding genes | 3,544 | 97.07 |
| Genes with function prediction | 2,851 | 78.09 |
| Genes in paralog clusters | 1,734 | 47.49 |
| Genes assigned to COGs | 3,062 | 83.87 |
| Genes with Pfam | 3,096 | 84.80 |
| Genes with signal peptides | 702 | 19.23 |
| Genes with transmembrane helices | 852 | 23.34 |
| CRISPR repeats | 0 |
a) The total is based on either the size of the genome in base pairs or the total number of protein coding genes in the annotated genome.
b) Pseudogenes may also be counted as protein-coding or RNA genes, so are not additive under total gene count.
Figure 3.

Graphical circular map of the chromosome. From outside to the center: genes on forward strand (color by COG categories), genes on reverse strand (color by COG categories) RNA genes (tRNAs green, rRNAs red, other RNAs black), GC content, GC skew.
Table 4. Number of genes associated with the 25 general COG functional categories.
| Code | Value | % age | Description |
|---|---|---|---|
| J | 181 | 5.26 | Translation, ribosomal structure and biogenesis |
| A | 1 | 0.03 | RNA processing and modification |
| K | 289 | 8.4 | Transcription |
| L | 119 | 3.46 | Replication, recombination and repair |
| B | 2 | 0.06 | Chromatin structure and dynamics |
| D | 31 | 0.9 | Cell cycle control, cell division and chromosome partitioning |
| Y | 0 | 0 | Nuclear structure |
| V | 22 | 0.64 | Defense mechanisms |
| T | 254 | 7.38 | Signal transduction mechanisms |
| M | 157 | 4.56 | Cell wall/membrane/envelope biogenesis |
| N | 97 | 2.82 | Cell motility |
| Z | 0 | 0.0 | Cytoskeleton |
| W | 0 | 0.0 | Extracellular structures |
| U | 75 | 2.18 | Intracellular trafficking, secretion and vesicular transport |
| O | 129 | 3.75 | Post-translational modification, protein turnover, chaperones |
| C | 175 | 5.09 | Energy production and conversion |
| G | 239 | 6.95 | Carbohydrate transport and metabolism |
| E | 366 | 10.64 | Amino acid transport and metabolism |
| F | 77 | 2.24 | Nucleotide transport and metabolism |
| H | 165 | 4.8 | Coenzyme transport and metabolism |
| I | 109 | 3.17 | Lipid transport and metabolism |
| P | 195 | 5.67 | Inorganic ion transport and metabolism |
| Q | 86 | 2.5 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 372 | 10.81 | General function prediction only |
| S | 299 | 8.69 | Function unknown |
| - | 589 | 16.13 | Not in COGs |
Insights from genome sequence
The genome of M. posidonica IVIA-Po-181T (3.9 Mb) is 16.75 and 23.54% smaller than those of M. mediterranea (4.6 Mb) and Marinomonas sp. MWYL1 (5.1 Mb) respectively. Comparison of the protein sets of these three Marinomonas strains revealed that the genome of IVIA-Po-181T encodes a much smaller number of unique proteins (Figure 4), indicating that most of the proteins encoded in its genome are also present in the other two strains. Among these is a cluster involved in the catabolism of DMSP resulting in the production of DMS, an environmentally important gas. This capacity was first described in Marinomonas sp. MWYL1, where the genes involved are Mmwyl1_4041- Mmwyl1_4045 [20]. Highly similar clusters of genes are detected in M. mediterranea MMB-1T (Marme_2354-Marme_2350) and M. posidonica IVIA-Po-181T (Mar181_0285-Mar181_0290). Although DMSP is an intracellular compatible solute synthesized by marine phytoplankton, as far as we know, its synthesis by P. oceanica has not been reported.
Figure 4.
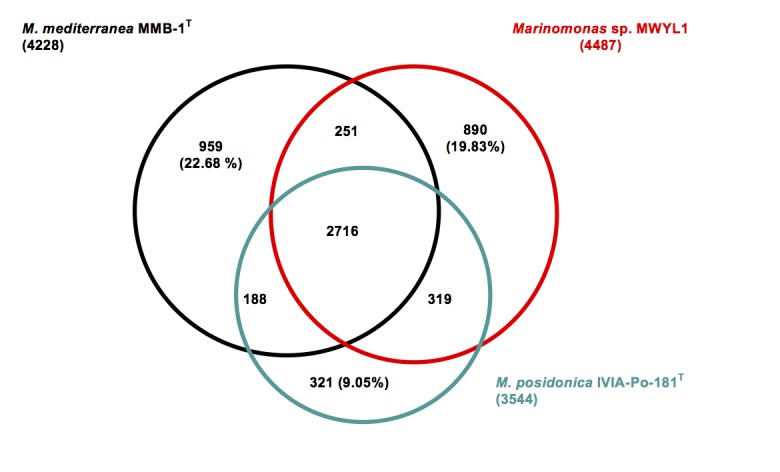
Venn-diagram showing the intersection of proteins sets (total number in parenthesis) of the three completed Marinomonas genomes. The intersections were calculated using the IMG tool phylogenetic profiler with default values (E value 1 e-5 and 30% minimum identity). All intersections concerning M. posidonica IVIA-Po-181T are gene counts in this strain. The remaining intersection between M. mediterranea and Marinomonas sp. MWYL1 represents gene counts in M. mediterranea. In some cases, the intersections do not add to total numbers of genes, due to the dependence of the results on the strain used as query.
The analysis of the set of unique genes in M. posidonica IVIA-Po-181T revealed that 57% (183 out of 321) of such genes are annotated as encoding hypothetical proteins. From Mar181_0471 to Mar181_0507 there is a cluster of genes whose sequence suggests that they are involved in the synthesis of exopolysaccharide. It is tempting to speculate that such a polymer might be involved in cellular attachment to the plant. Two other genes of interest are the adjacent Mar181_1314, annotated as coding for a insecticidal toxin complex/plasmid virulence protein, and Mar181_1315 annotated as encoding RHS repeat-associated core domain, both of these gene products being very large polypeptides, of 2,746 and 2,149 amino acids respectively. BLAST-based searches using each of these as the query, showed marked similarities to proteins that are in the insecticidal protein complex (Tc) synthesized by different entomopathogenic bacteria, including endosymbionts of nematodes [39].
The product of Mar181_1315 seems to be the result of a fusion of different proteins. The first 1,500 amino acids show 29.4% identity and 45.3% similarity to Photorhabdus luminescens TcdB1 (accession number AAL18467) [40]. Both proteins also show some similarity to the pathogenic factor SpvB of Salmonella enterica. However, the similarity is limited to the N terminal region and does not include the region from amino acids 375-391 of SpvB which encodes its ADP-ribosyl transferase activity [41].
In addition, the product of Mar181_1315 shows, from amino acid 1,400 to 2,141, similarity to both, the toxin protein TccC2 and Rhs elements, consistent with the similarity of Rhs proteins to some toxin proteins, such as TccC2, which was also previously noticed [40]. For instance, the product of Mar181_1315 shows the typical conserved sequences limiting the core of Rhs proteins [42]. The lower limit is R(1412)VxxxxxxxG and the upper limit is P(2033)XXXXDPXGL. rhs-like genes are distributed among many different bacteria, the genetic acronym “rhs” being originally based on “recombination hot spots” since they were initially related to genomic rearrangements [43]. However, they are now considered to be involved in many different cellular processes. For example, it has been recently shown that they show high similarity to the pairs of proteins CdiA/CdiB involved in contact-dependent growth inhibition and toxin/immunity function [44]. In relation to the similarity to TccC2 (accession number AAL18492), part of the Mar181_1315 product (from amino acid N(1467) to G(2141)) displays 34% identity and 49% similarity to the first 663 amino acids of P. lumincescens TccC2. Finally, the C-terminal 250 amino acids of the Mar181_1315 do not show similarity to any protein in the NCBI database. The variability of the C-terminal region is also a property of Rhs proteins [42].
It is noteworthy that many different proteins showing an organization similar to the product of Mar181_1315 occur in a wide range of taxonomically distinct bacteria such as Wolbachia , an endosymbiont of Cadra cautella (BAH22314), the myxobacterium Sorangium cellulosum (YP001619210) or the Gram-positive Paenibacillus curdlanolyticus (ZP_07386246). There are even homologues in some ascomycete fungi such as the plant pathogenic fungus Verticillum dahliae (EGY15726) and Podospora anserina (XP_001907253).
The locus Mar181_1314, encoding a protein of 2,746 amino acids is also similar to proteins that form part of insecticidal complexes such as TcdA1 (AAL18486) from Photorhabdus luminescens [40]. Genes encoding proteins similar to Mar181_1314 are detected in diverse microorganisms, and interestingly, in many cases they are close to the genes encoding proteins similar to Mar181_1315, for instance in Wolbachia (BAH22316), S. cellulosum (YP001619211), P. curdlanolyticus (ZP_07386247) and even the fungus P. anserina (XP_001907253), as well as in Photorhabdus luminescens. The expression of both proteins in M. posidonica would determine that all the components necessary for the toxicity of the toxin complexes are present [40]. It is remarkable that there are no detectable homologues of either gene product in any other Marinomonas or indeed, in any genome-sequenced strain of the Oceanospirillales.
The small number of unique genes in M. posidonica IVIA-Po-181T could be related to the observation that this strain has fewer mobile genetic elements than the other genome-sequenced Marinomonas strains, as deduced by the numbers of genes encoding predicted transposases or inactive derivatives; there are 44 in M. mediterranea MMB-1T, 33 in Marinomonas sp. MWYL1, but only 6 in M. posidonica IVIA-Po-181T. Furthermore, the genome of strain IVIA-Po-181T does not apparently contain any prophages, but there are at least two in both strain MMB-1T and strain MWYL1. In relation to CRISPR repeats, which are involved in phage resistance, there are two complete CRISPR sequences in Marinomonas sp. MWYL1 and four such elements in M. mediterranea, two of which are complete and two that lack Cas associated proteins. On the contrary, no CRISPR elements are detected in M. posidonica.
The genome size of M. posidonica IVIA-Po-181T is in the lower range of those observed in marine bacteria of the copiotrophs/opportunitroph type, which are characterized by a high growth rate when nutrients become available [45,46]. The smaller size of the genome of M. posidonica compared to the other Marinomonas strains could be a consequence of a close and stable relationship with Posidonia oceanica, making unnecessary a high genomic potential for the adaptation to different environments.
The availability of the genome sequence of this strain may facilitate direct examination of the functions that are involved in its association with one of the more important marine plant species.
Acknowledgments
The work conducted by the U.S. Department of Energy Joint Genome Institute is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. Work on Marinomonas posidonica was supported by several grants from the Fundación Séneca (CARM, Spain) (Current project 11867/PI/09).
References
- 1.Espinosa E, Marco-Noales E, Gomez D, Lucas-Elio P, Ordax M, Garcias-Bonet N, Duarte CM, Sanchez-Amat A. Taxonomic study of Marinomonas strains isolated from the seagrass Posidonia oceanica, with descriptions of Marinomonas balearica sp. nov. and Marinomonas pollencensis sp. nov. Int J Syst Evol Microbiol 2010; 60:93-98 10.1099/ijs.0.008607-0 [DOI] [PubMed] [Google Scholar]
- 2.Lucas-Elío P, Marco-Noales E, Espinosa E, Ordax M, Lopez MM, Garcias-Bonet N, Marba N, Duarte CM, Sanchez-Amat A. Marinomonas alcarazii sp. nov., M. rhizomae sp. nov., M. foliarum sp. nov., M. posidonica sp. nov. and M. aquiplantarum sp. nov., isolated from the microbiota of the seagrass Posidonia oceanica. Int J Syst Evol Microbiol 2011; 61:2191-2196 10.1099/ijs.0.027227-0 [DOI] [PubMed] [Google Scholar]
- 3.Arnaud-Haond S, Duarte CM, Díaz-Almela E, Marbà N, Sintes T, Serräo EA. Implications of extreme life span in clonal organisms: millenary clones in meadows of the threatened seagrass Posidonia oceanica. PLoS ONE 2012; 7:e30454 10.1371/journal.pone.0030454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Celdran D, Espinosa E, Sanchez-Amat A, Atucha A. Effects of epibiotic bacteria on leaf growth and epiphytes of seagrass. Posidonia oceanica. Mar Ecol Prog Ser 2012; 456:21-27; 10.3354/meps09672 [DOI] [Google Scholar]
- 5.Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 2007; 24:1596-1599 10.1093/molbev/msm092 [DOI] [PubMed] [Google Scholar]
- 6.Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. I, Wilson G, and Wipat A: The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008; 26:541-547 10.1038/nbt1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA 1990; 87:4576-4579 10.1073/pnas.87.12.4576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garrity GM, Bell JA, Lilburn T. Phylum XIV. Proteobacteria phyl. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT (eds), Bergey's Manual of Systematic Bacteriology, Second Edition, Volume 2, Part B, Springer, New York, 2005, p. 1. [Google Scholar]
- 9.Validation of publication of new names and new combinations previously effectively published outside the IJSEM. List no. 106. Int J Syst Evol Microbiol 2005; 55:2235-2238 10.1099/ijs.0.64108-0 [DOI] [PubMed] [Google Scholar]
- 10.Garrity GM, Bell JA, Lilburn T. Class III. Gammaproteobacteria class. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT (eds), Bergey's Manual of Systematic Bacteriology, Second Edition, Volume 2, Part B, Springer, New York, 2005, p. 1. [Google Scholar]
- 11.Garrity GM, Bell JA, Lilburn T. Order VIII. Oceanospirillales ord. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT (eds), Bergey's Manual of Systematic Bacteriology, Second Edition, Volume 2, Part B, Springer, New York, 2005, p. 270. [Google Scholar]
- 12.Garrity GM, Bell JA, Lilburn T. Family I. Oceanospirillaceae fam. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT (eds), Bergey's Manual of Systematic Bacteriology, Second Edition, Volume 2, Part B, Springer, New York, 2005, p. 271. [Google Scholar]
- 13.Validation List no. 13. Validation of the publication of new names and new combinations previously effectively published outside the lJSB. Int J Syst Bacteriol 1984; 34:91-92 10.1099/00207713-34-1-91 [DOI] [Google Scholar]
- 14.van Landschoot A, De Ley J. Intra- and intergeneric similarities of the rRNA cistrons of Alteromonas, Marinomonas, gen. nov. and some other Gram-negative bacteria. J Gen Microbiol 1983; 129:3057-3074 [Google Scholar]
- 15.Espinosa E, Marco-Noales E, Gómez D, Lucas-Elío P, Ordax M, Garcías-Bonet N, Duarte CM, Sanchez-Amat A. Taxonomic study of Marinomonas strains isolated from the seagrass Posidonia oceanica, with descriptions of Marinomonas balearica sp. nov. and Marinomonas pollencensis sp. nov. Int J Syst Evol Microbiol 2010; 60:93-98 10.1099/ijs.0.008607-0 [DOI] [PubMed] [Google Scholar]
- 16.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000; 25:25-29 10.1038/75556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.López-Serrano D, Solano F, Sanchez-Amat A. Identification of an operon involved in tyrosinase activity and melanin synthesis in Marinomonas mediterranea. Gene 2004; 342:179-187 10.1016/j.gene.2004.08.003 [DOI] [PubMed] [Google Scholar]
- 18.Sanchez-Amat A, Lucas-Elio P, Fernández E, Garcia-Borrón JC, Solano F. Molecular cloning and functional characterization of a unique multipotent polyphenol oxidase from Marinomonas mediterranea. Biochim Biophys Acta 2001; 1547:104-116 10.1016/S0167-4838(01)00174-1 [DOI] [PubMed] [Google Scholar]
- 19.Curson ARJ, Todd JD, Sullivan MJ, Johnston AWB. Catabolism of dimethylsulphoniopropionate: microorganisms, enzymes and genes. Nat Rev Microbiol 2011; 9:849-859 10.1038/nrmicro2653 [DOI] [PubMed] [Google Scholar]
- 20.Todd JD, Rogers R, Li YG, Wexler M, Bond PL, Sun L, Curson AR, Malin G, Steinke M, Johnston AWB. Structural and regulatory genes required to make the gas dimethyl sulfide in bacteria. Science 2007; 315:666-669 10.1126/science.1135370 [DOI] [PubMed] [Google Scholar]
- 21.Lucas-Elío P, Goodwin L, Woyke T, Pitluck S, Nolan M, Kyrpides N, Detter JC, Copeland A, Teshima H, Bruce D, et al. Complete genome sequence of the melanogenic marine bacterium Marinomonas mediterranea type strain (MMB-1T). Stand Genomic Sci 2012; 6:63-73 10.4056/sigs.2545743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fernández E, Sanchez-Amat A, Solano F. Location and catalytic characteristics of a multipotent bacterial polyphenol oxidase. Pigment Cell Res 1999; 12:331-339 10.1111/j.1600-0749.1999.tb00767.x [DOI] [PubMed] [Google Scholar]
- 23.Bennett S. Solexa Ltd. Pharmacogenomics 2004; 5:433-438 10.1517/14622416.5.4.433 [DOI] [PubMed] [Google Scholar]
- 24.Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005; 437:376-380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DOE Joint Genome Institute http://www.jgi.doe.gov
- 26.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 2008; 18:821-829 10.1101/gr.074492.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res 1998; 8:175-185 [DOI] [PubMed] [Google Scholar]
- 28.Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res 1998; 8:186-194 [PubMed] [Google Scholar]
- 29.Gordon D, Abajian C, Green P. Consed: a graphical tool for sequence finishing. Genome Res 1998; 8:195-202 [DOI] [PubMed] [Google Scholar]
- 30.Han C, Chain P. Finishing repeat regions automatically with Dupfinisher. Arabnia, H. R. and Valafar, H. Proceeding of the 2006 International Conference on bioinformatics and computational biology, 141-146. 2006. CSREA Press. [Google Scholar]
- 31.Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 2010; 11:119 10.1186/1471-2105-11-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pati A, Ivanova NN, Mikhailova N, Ovchinnikova G, Hooper SD, Lykidis A, Kyrpides NC. GenePRIMP: a gene prediction improvement pipeline for prokaryotic genomes. Nat Methods 2010; 7:455-457 10.1038/nmeth.1457 [DOI] [PubMed] [Google Scholar]
- 33.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 1997; 25:955-964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lagesen K, Hallin P, Rodland EA, Staerfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 2007; 35:3100-3108 10.1093/nar/gkm160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR. Rfam: an RNA family database. Nucleic Acids Res 2003; 31:439-441 10.1093/nar/gkg006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 2001; 305:567-580 10.1006/jmbi.2000.4315 [DOI] [PubMed] [Google Scholar]
- 37.Bendtsen JD, Nielsen H, von Heinje G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol 2004; 340:783-795 10.1016/j.jmb.2004.05.028 [DOI] [PubMed] [Google Scholar]
- 38.Markowitz VM, Mavromatis K, Ivanova NN, Chen IM, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics 2009; 25:2271-2278 10.1093/bioinformatics/btp393 [DOI] [PubMed] [Google Scholar]
- 39.ffrench-Constant RH, Dowling A, Waterfield NR. Insecticidal toxins from Photorhabdus bacteria and their potential use in agriculture. Toxicon 2007; 49:436-451 10.1016/j.toxicon.2006.11.019 [DOI] [PubMed] [Google Scholar]
- 40.Waterfield NR, Bowen DJ, Fetherston JD, Perry RD, French-Constant RH. The tc genes of Photorhabdus: a growing family. Trends Microbiol 2001; 9:185-191 10.1016/S0966-842X(01)01978-3 [DOI] [PubMed] [Google Scholar]
- 41.Barth H, Aktories K. New insights into the mode of action of the actin ADP-ribosylating virulence factors Salmonella enterica SpvB and Clostridium botulinum C2 toxin. Eur J Cell Biol 2011; 90:944-950 10.1016/j.ejcb.2010.11.007 [DOI] [PubMed] [Google Scholar]
- 42.Jackson AP, Thomas GH, Parkhill J, Thomson NR. Evolutionary diversification of an ancient gene family (rhs) through C-terminal displacement. BMC Genomics 2009; 10:584 10.1186/1471-2164-10-584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin RJ, Capage M, Hill CW. A repetitive DNA sequence, rhs, responsible for duplications within the Escherichia coli K-12 chromosome. J Mol Biol 1984; 177:1-18 10.1016/0022-2836(84)90054-8 [DOI] [PubMed] [Google Scholar]
- 44.Poole SJ, Diner EJ, Aoki SK, Braaten BA. t'Kint dR, Low DA, and Hayes CS. Identification of functional toxin/immunity genes linked to contact-dependent growth inhibition (CDI) and rearrangement hotspot (Rhs) systems. PLoS Genet 2011; 7:e1002217 10.1371/journal.pgen.1002217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Polz MF, Hunt DE, Preheim SP, Weinreich DM. Patterns and mechanisms of genetic and phenotypic differentiation in marine microbes. Philos Trans R Soc Lond B Biol Sci 2006; 361:2009-2021 10.1098/rstb.2006.1928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lauro FM, McDougald D, Thomas T, Williams TJ, Egan S, Rice S, DeMaere MZ, Ting L, Ertan H, Johnson J, et al. The genomic basis of trophic strategy in marine bacteria. Proc Natl Acad Sci USA 2009; 106:15527-15533 10.1073/pnas.0903507106 [DOI] [PMC free article] [PubMed] [Google Scholar]