

Abstract
Background
The detection of significant compensatory mutation signals in multiple sequence alignments (MSAs) is often complicated by noise. A challenging problem in bioinformatics is remains the separation of significant signals between two or more non-conserved residue sites from the phylogenetic noise and unrelated pair signals. Determination of these non-conserved residue sites is as important as the recognition of strictly conserved positions for understanding of the structural basis of protein functions and identification of functionally important residue regions. In this study, we developed a new method, the Coupled Mutation Finder (CMF) quantifying the phylogenetic noise for the detection of compensatory mutations.
Results
To demonstrate the effectiveness of this method, we analyzed essential sites of two human proteins: epidermal growth factor receptor (EGFR) and glucokinase (GCK). Our results suggest that the CMF is able to separate significant compensatory mutation signals from the phylogenetic noise and unrelated pair signals. The vast majority of compensatory mutation sites found by the CMF are related to essential sites of both proteins and they are likely to affect protein stability or functionality.
Conclusions
The CMF is a new method, which includes an MSA-specific statistical model based on multiple testing procedures that quantify the error made in terms of the false discovery rate and a novel entropy-based metric to upscale BLOSUM62 dissimilar compensatory mutations. Therefore, it is a helpful tool to predict and investigate compensatory mutation sites of structural or functional importance in proteins. We suggest that the CMF could be used as a novel automated function prediction tool that is required for a better understanding of the structural basis of proteins. The CMF server is freely accessible at http://cmf.bioinf.med.uni-goettingen.de.
Background
A multiple sequence alignment (MSA) of proteins contains a set of aligned amino acid sequences in which homologous residues of different sequences are placed in same columns. Therefore, functionally or structurally important amino acids and their positions both of which are often strictly conserved are easily detectable with MSAs [1-3]. On the other hand, detection of important non-conserved residue positions related to several essential conserved residues requires a more sophisticated approach. The usage of methods such as correlation analysis allow the identification of important non-conserved residue positions based on their correlated mutation manners [4,5] due to functional coupling of mutation positions. This coupling might stem from one mutation in a certain site affecting a compensating mutation at another site, even if both related residue sites are distantly positioned in the protein structure. Moreover, these coupled mutations can result from spatial, physical, or chemical restrictions or signaling of allostery [4,5]. Thus, determination of these positions is as crucial as the recognition of strictly conserved positions for the understanding of the structural basis of protein functions, and for the identification of functionally important residue regions which might be disease associated, responsible for the maintenance of internal protein volume, or possibly form key sites for interactions within or between proteins [6-9].
Until now, a variety of studies have employed Pearson’s correlation coefficient methods [10-12], perturbation based methods [9,13] and mutual information (MI) based methods [6,14-17] because of their simplicity and efficiency for the detection of coupled mutations in MSAs. However, due to background noise, all of these methods interfere with the identification of compensatory mutation signals [14,18,19]. Hence, the significant compensatory mutation signals must be separated from the background noise that might occur as a result of: i) false signals arising from insufficient data; ii) sites with low or high conservation biasing the signal; iii) phylogenetic noise. While the first two types of noise can be easily overcome by appropriately filtering the data [16], phylogenetic noise can only be eliminated to some extent by excluding highly similar sequences from the MSA [19].
Recently, several methods such as bootstrapping, simulation or randomization methods have been utilized in order to minimize the influence of phylogenetic linkage and stochastic noise [15,20,21]. Dunn et al. [19] have introduced the average product correction (APC), to adjust MI for background effects. Merkl and Zwick, in their study, [16] have used a normalized MI (see Equation 1) and focused on only 75 residue pairs with the highest normalized MI values as significant for each MSA. Gao et al. [17] have pursued a similar approach, where they have replaced the metric used in [16] with the amino acid background distribution (MIB). While the reduction of background noise in the model of Dunn et al. is not quantified, the approaches of Gao et al. and Merkl and Zwick appear to be over-conservative, yet specific.
Despite the presence of a variety of different methods as mentioned above, to date there is still need for a method to deal with the noise as well as for powerful metrics to improve the sensitivity. In this study, we have developed such a method called Coupled Mutation Finder (CMF). The CMF includes an MSA-specific statistical model based on multiple testing procedures described in [22,23] and a novel entropy-based metric to upscale dissimilar compensatory mutations and also to complement the normalized MI metric used in [16]. Unlike previous normalized MI based studies [16,17], we have separated metric-based significant compensatory mutation signals from background noise with respect to our MSA specific statistical model that quantifies the error made in terms of the false discovery rate.
To demonstrate the performance and functionality of the CMF, we analyzed the structurally or functionally important positions of two human proteins, namely epidermal growth factor receptor (EGFR) and glucokinase (GCK). The proteins have been chosen because their functionally and structurally important positions have been experimentally well studied previously [24-35]. As a result, the CMF detects in these two proteins disease associated amino acid mutations (non-synonymous single nucleotide polymorphisms (nsSNPs)), not strictly conserved catalytic or binding sites, and residues that are nearby one of these sites or in the close neighborhood of a strictly conserved positions, which are likely to affect protein stability or functionality [36-38].
Results
Our method to predict functionally or structurally important residue positions is composed of two steps. First, we have devised a new MSA-specific statistical method for the identification of significant MSA column pairs with respect to either of the two metrics and . Assume that M is the MSA under study, these pairs are annotated as -significant and -significant, respectively. Second, we utilized the connectivity degree of a residue site with respect to a metric introduced in [16]. The connectivity degree of a residue site indicates the number of its significant coupled mutation pairs. In this case, a site is called (U,M)-significant when the frequency of occurrence of this site in the set of -significant pairs exceeds the 90-th percentile. Having defined the concept of a -significant site analogously, a site is defined as CMF-significant with respect to M, when it is either -significant or -significant.
In this study, we analyzed human EGFR (pdb entry 2J6M) and GCK (pdb entry 1V4S) proteins with a false discovery rate (FDR) of 1%. For the preceding one, we defined a total of 14339 out of 26079 non-conserved column pairs as significant. 11365 of these significant pairs are detected as -significant and 3798 pairs are observed as -significant. Only 824 EGFR pairs are significant with respect to both metrics. On the other hand, for GCK, we identified a total of 32654 out of 69645 non-conserved column pairs as significant where 18106 of them are -significant and 16241 are -significant. Moreover, 1693 pairs are defined as significant for both and -significant.
Applying the connectivity degree technique, we identified 22 and 36 residue positions as -significant for human EGFR and GCK proteins, respectively. Additionally, 21 positions of EGFR and 36 positions of GCK were detected as -significant. Finally, a total of 43 sites of EGFR and 72 of GCK were found as CMF-significant, and predicted as of structural or functional importance. However, there have been no residue sites defined as significant with respect to either metric.
Essential sites of human EGFR and GCK proteins
To evaluate the CMF-significant residue sites, we have investigated essential sites of human EGFR (pdb entry 2J6M) and GCK (pdb entry 1V4S) proteins. The essential sites of both proteins have been assigned into three main categories: i) the nsSNP positions and their adjacent sites; ii) residue positions which are located at or near active sites, allosteric sites, or binding sites; iii) residue positions which are nearby strictly conserved sites. Here, we have used “nearby” definition of Nussinov et al. [39] and defined two residues as in contact or adjacent when the distance between their major carbon atoms is less than 6 Å. We have defined positions which are nearby nsSNPs as essential, because several of them are also adjacent to active sites, allosteric sites, binding sites, or strictly conserved sites. Thus, we refer to a CMF-significant residue site as “functionally or structurally important” if it falls into one of these categories of essential sites.
Position analysis of the Human Epidermal Growth Factor Receptor (EGFR) protein
The epidermal growth factor receptor (EGFR) is a member of the ErbB (Erythroblastic Leukemia Viral Oncogene Homolog) family receptors. Signaling through this receptor is a highly conserved mechanism from nematode to humans involved in numerous processes, including proliferation, cell fate determination, and tissue specification [40]. Furthermore, several studies have implicated that mutations resulting in misregulation of the activity or action of EGFR led to multiple cancers, including those of the brain, lung, mammary gland, and ovary [24-27]. Here, in order to detect essential mutation positions in corresponding sequence of human EGFR protein, we determined altogether 43 CMF-significant residue sites (see Additional file 1). 15 of these significant residue sites have been verified as nsSNP sites through the Ensembl database annotation and they are illustrated in Figure 1.
Figure 1.
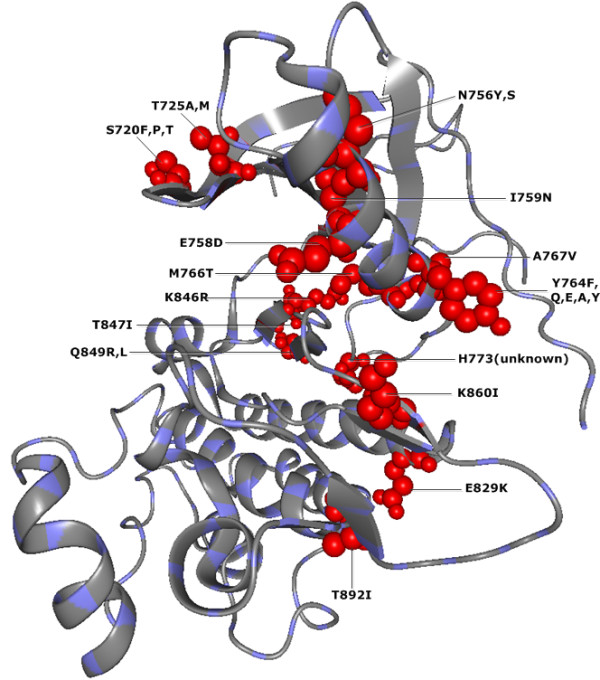
CMF-significant nsSNP positions in human EGFR protein (PDB-Entry 2J6M). The red spheres correspond to structural localization of 15 different nsSNP positions found by CMF as significant in the EGFR protein.
Additionally, the significant sites E746, Q791, and four of the nsSNP positions (I759,Y764,M766 and K846) are also in contact with critical active site regions for gefitinib binding site in the wild type EGFR kinase [25,28] (see Figure 2).
Figure 2.
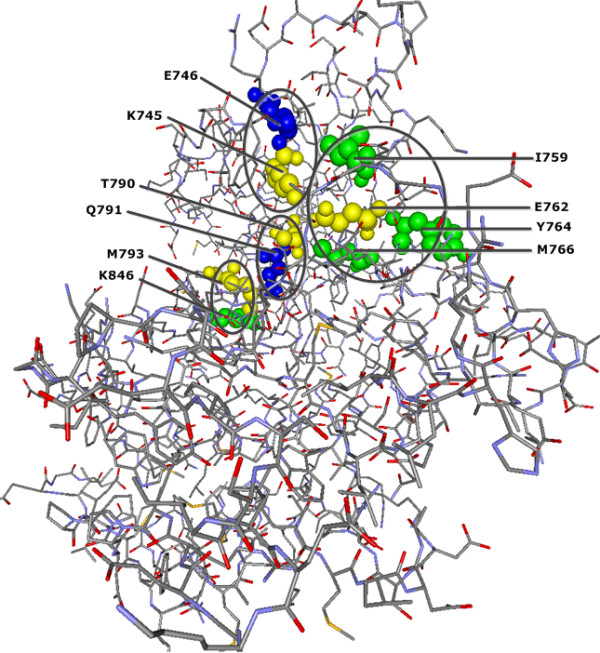
CMF-significant residue positions are in contact with gefitinib binding sites in human EGFR protein (PDB-Entry 2J6M). Yellow spheres denote positions of the gefitinib binding sites in the wild type kinase. Blue spheres show the localization of significant adjacent residue positions found by CMF which are in contact with these binding sites. Moreover, the CMF-significant sites I759, Y764, M766 and K846, shown with green spheres, are already described as nsSNP positions and they are also in contact with gefitinib binding sites E762 and M793, respectively. The circles indicate clusters of gefitinib binding sites and their significant adjacent sites.
Moreover, we observed that 17 further CMF-significant positions are essential sites (see Table 1). In total, we have established here for EGFR protein the importance of 34 out of 43 CMF-significant residue sites via different resources [25,28,35].
Table 1.
CMF-significant essential sites in human EGFR protein, which are nearby either nsSNPs or strictly conserved sites
| CMF-significantessential sites | Nearby nsSNPs, or strictlyconserved sites | Reference |
|---|---|---|
| Y727 |
726c 743c |
- |
|
H755 |
756s, 758s |
[35] |
|
D800 |
798c |
- |
|
G824 |
773s |
[35] |
|
D830 |
829s |
[35] |
|
E868 |
892s |
[35] |
|
E872 |
873s |
[34] |
|
V876 |
877c |
- |
|
K879 |
877c, 880c |
- |
|
Y891 |
892s, 895c |
[35] |
|
S899 |
880c, 896c, 898c, 901c |
- |
|
Y900 |
898c, 901c |
- |
|
T909 |
906c, 936c |
- |
|
S912 |
906c, 936c |
- |
|
K913 |
914c |
- |
|
D916 |
914c |
- |
| M947 | 901c, 950c | - |
s: non-synonymous snp site,c: strictly conserved site.
Although the vast majority of CMF-significant sites are verified to be structurally or functionally important in human EGFR protein, nine CMF-significant sites do not overlap with essential sites. The reason for the high connectivity degree of these unconfirmed significant sites and their role in the EGFR protein is unclear.
Position analysis of the Human Glucokinase (GCK) protein
Glucokinase (GCK) is a monomeric enzyme catalyzing phosphorylation of glucose to glucose-6-phosphate, which is the first step in the utilization of glucose, at physiological glucose concentration in pancreas and liver. Given the fact that GCK displays low affinity for glucose, it acts as a glucose sensor playing an important role in the regulation of carbohydrate metabolism. Mutations of the GCK gene can lead to maturity onset diabetes of the young (MODY) characterized by an autosomal dominant mode of inheritance and onset early adulthood [32], or familial hyperinsulinemic hypoglycemia type 3 (HHF), common cause of persistent hypoglycemia in infancy [41].
Applying our method, we found 72 CMF-significant sites to be structurally or functionally important in human GCK protein (see Additional file 2). 16 of these significant residue positions are related to disease associated nsSNP positions [29-31,34,35] (see Figure 3).
Figure 3.
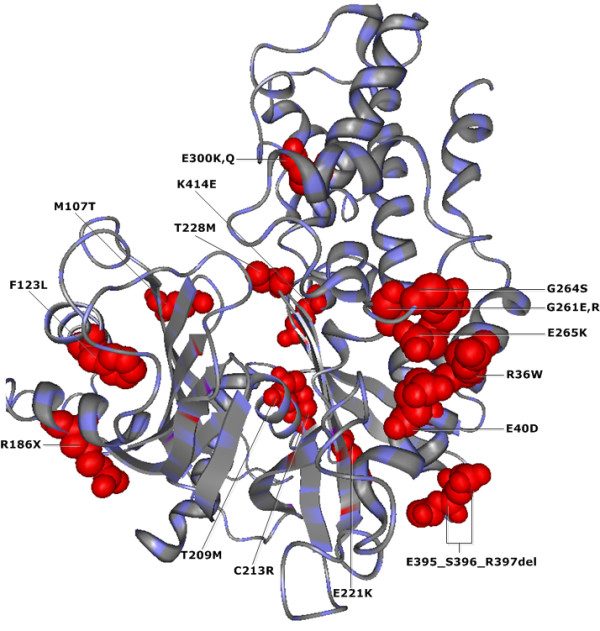
CMF-significant nsSNP positions in human GCK protein (PDB-Entry 1V4S). Red spheres show the structural localization of 16 different nsSNP positions found byCMF as significant in the GCK protein.
Furthermore, nine significant sites are found to be in contact with allosteric sites in the GCK protein structure. Among these sites, the R63 is also allosteric site by itself [32] and T209, C213 and E221 overlap with nsSNP regions (see Figure 4B). Moreover, the five significant sites T149, F171, T206, Q287, and G294 interact with glucose binding sites K169, D204, N205, and E290 [32] (see Figure 4A).
Figure 4.
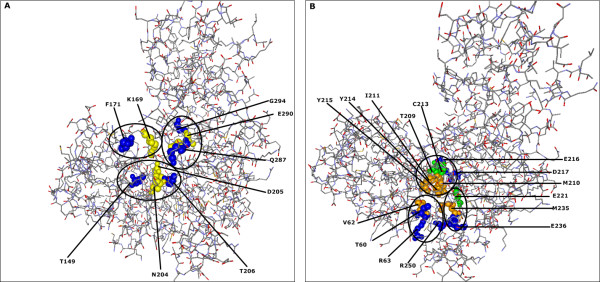
CMF-significant residue positions are in contact with glucose binding site and allosteric site in human GCK protein (PDB-Entry 1V4S). (A) Yellow spheres show the structural positions of the glucose binding sites (active sites). Blue spheres correspond to localization of significant adjacent residue positions found by CMF which are in contact with these active sites. (B) Orange spheres denote the allosteric sites. Blue spheres correspond to localization of just significant adjacent residue positions and green spheres indicate the significant residue positions which are already described as nsSNP position and in contact with these allosteric site. Additionally, the significant position R63 is allosteric site by itself and it is also in contact with an other allosteric site. The circles indicate clusters of glucose binding sites (A), allosteric sites (B), and their significant adjacent sites.
Besides this, there are further 30 CMF-significant essential sites which are nearby nsSNPs or strictly conserved residue positions (see Table 2). Altogether, we showed the functionality of 57 positions out of 72 CMF-significant residue sites via different resources [29-35].
Table 2.
CMF-significant essential sites in human GCK protein, which are nearby either nsSNPs or strictly conserved sites
| CMF-significant essential sites | Nearby nsSNPs or strictly conserved sites | Reference |
|---|---|---|
| M34 |
36s |
[34] |
|
T65 |
66c |
|
|
E67 |
66c, 68c |
- |
|
T82 |
81c |
- |
|
N83 |
81c, 108s, 110s |
[34] |
|
H105 |
106s |
[29] |
|
C129 |
131s, 132s |
[29,34] |
|
F133 |
131s, 132s |
[29,34] |
|
F148 |
147c, 150c,s |
[34] |
|
F152 |
150c,s, 151c |
[34] |
|
H156 |
162s |
[29] |
|
N180 |
162s, 182s |
[29,34] |
|
F260 |
257s, 258c, 259s, 261s |
[34] |
|
D262 |
259s, 261s, 264s |
[34,42] |
|
L266 |
261s, 264s, 265s |
[29,34,42] |
|
D267 |
264s, 265s |
[29,42] |
|
L271 |
274c |
- |
|
S281 |
278c, 279s |
[34] |
|
Q286 |
259s |
[34] |
|
E331 |
299c,s |
[34] |
|
T332 |
295c, 299c,s |
[34] |
|
R333 |
336s |
[34] |
|
Q337 |
336s |
[34] |
|
E339 |
336s |
[34] |
|
N391 |
392s |
[29,34] |
|
S411 |
227c,s, 410c, 414s |
[34] |
|
S418 |
416s |
[30] |
|
F419 |
416s |
[30] |
|
E442 |
444c |
- |
| E443 | 444c, 445c | - |
s: non-synonymous snp site,c: strictly conserved site.
While we are able to establish the large number ofCMF-significant sites as structurally or functionally important in human GCK protein, 15CMF-significant sites do not overlap with essential sites. Their importance in the GCK protein and the reason of high connectivity degree of these unconfirmed significant sites has not been explicitly determined yet.
A comparison between -metric and -metric
Similarities in physical or biochemical properties of amino acids are likely to be crucial for the detection of functionally or structurally important positions of a protein. In contrast to the -metric, which is a normalized mutual information that uses only the frequencies of occurrences of amino acids in the MSA columns, the novel -metric includes dissimilarities according to the BLOSUM62 matrix when calculating normalized mutual information. As a result the positions which have undergone dissimilar compensatory mutations are upscaled.
Having applied the -metric as well as the -metric to human EGFR and GCK proteins, the -metric has shown better sensitivity and specificity. However, only when we use the both metrics together, the sensitivity is significantly increased, whereas the specificity is only moderately decreased. The details are presented in Table 3.
Table 3.
Comparison between-metric and-metric
| Sensitivity | Specificity | |
|---|---|---|
|
-significance |
9.7% |
91.5% |
|
-significance |
12.4% |
97.2% |
| CMF-significance | 22.1% | 88.7% |
It is important to note that the two metrics complement each other. Thus, we propose to use them together.
CMF as a Web service
We have implemented a CMF Web service ( http://cmf.bioinf.med.uni-goettingen.de) that takes an MSA in multiple FASTA format and a real number from (0,1) interpreted as false discovery rate as input. It reports the results via email.
Discussion
To predict sites of structural or functional importance, we combine the known -metric of normalized mutual information [16] with a novel metric to enhance the influence of dissimilar compensatory mutations when measuring covariation of two sites. We discuss how we devised in Methods section.
To learn the frequency of compensatory mutations, we took -significant site pairs as training data. We did that for reasons of computation time regardless of the fact that these data are biased. To deal with this bias, one could carry through the training in an iterative process, with our training being the first iteration. For i > 0, in the (i + 1)-th iteration of this modified training, a doubly stochastic matrix is calculated based on -significant site pairs. This is done until the training data are stable.
According to Birkhoff’s Theorem [43], every doubly stochastic matrix is a convex combination of permutation matrices. Moreover, from the Hardy-Littlewood-Pólya majorization theorem [44] follows that transforming the probability mass function by a doubly stochastic matrix increases entropy. Consequently, by linearly transforming the empirical amino acid pair distribution of a site pair by D(1) before calculating the -value, we penalized those site pairs whose original distribution does not match the frequency pattern of formal dissimilar compensatory mutations in the training data described in the Methods section.
The challenge was to separate the signal caused by structural and functional constraints from the background. To address this issue, we studied only metrics μ that satisfy the following condition. The larger the μ(k,l)-value, the larger the probability that the two sites k and l have co-evolved. Our critical assumptions were: i) the μ(k,l)-values follow three different distributions, one for the signal, one for the noise, and one for pairs of completely unrelated sites; ii) there is an MSA-dependent threshold below which the metric μ does not fall with overwhelming probability, when it is applied to the site pairs of functional or structural importance to which μ is sensitive; iii) there is an MSA-dependent threshold significantly smaller then the one in (ii) such that with overwhelming probability there are no μ(k,l)-values of pairs (k,l) of unrelated sites exceeding it.
In order to near-completely eliminate the noise, we filtered both our training and input data. We calculated the significant pairs such that the preassigned false discovery rate was guaranteed by generalizing the Storey-Tibshirani procedure devised for multiple testing problems [22].
Our method to eliminate noise is orthogonal to the technique developed in [19]. Therein, for every pair of sites the so-called average product correction (APC) is calculated as an explicit noise measure, by which the mutual information is then decreased. Furthermore, it generalizes the way Merkl and Zwick [16] as well as Gao et al. [17] cope with noise. According to our judgment, taking only the top 75 high-scoring pairs or the top 25 pairs into account as done in [16,17], respectively, is too conservative.
We based our noise separation technique on rather weak distribution assumptions that are standard practice in multiple hypothesis testing, instead of explicitly model the noise in terms of a metric. We applied the connectivity degree technique due to Merkl and Zwick [16] to significant site pairs with respect to our metrics. The cut-off for the connectivity degree was set to the 90-th percentile. That way we defined significant sites. Finally, a site was defined to be CMF-significant, if it was μ-significant, where μ is either or .
Why did we set the cut-off value for the connectivity degree to the 90-th percentile? Going through all possiblen-th percentiles for n = 80,81,…,99, the Matthews correlation coefficient (MCC) of a joint prediction for human EGFR and GCK proteins is maximal if n = 90.
It is plausible that the number of functionally or structurally important sites does not only depend on the length of the protein. Therefore, the 90-th percentile cut-off should be replaced by an MSA-dependent threshold in future studies.
Our results for human EGFR and GCK proteins suggest that the large majority of significant compensatory mutation sites found by CMF are in agreement with previous experimental studies regarding the functions and stability of these proteins. 15 and 16 CMF-significant sites in human EGRF and GCK proteins, respectively, are verified as disease associated nsSNP positions (see Figures 1 and 2) where most amino acid substitutions in protein sequences damage structural stability of proteins [36,37,45]. Moreover, we have observed that in both proteins some of CMF-significant nsSNP positions are nearby allosteric sites, binding sites or catalytic sites each of which are considered to be functionally important [46,47] (see Figures 2 and 4). Disease associated mutations at these nearby positions are likely to affect protein function [38,48].
Despite the large number of CMF-significant sites demonstrated to be structurally or functionally important for both of the proteins, 9 and 15 significant sites in human EGFR and GCK proteins, respectively, are not included in essential sites. However, we hypothesize that most of the novel significant sites may play a critical role in both proteins notwithstanding the absence of previous experimental data. Therefore, further progress from the molecular and structural biology end is required not only to assess the importance of these sites, but also for a future perspective on a deeper understanding of protein structure.
Because we have also used the -metric, we compared our tool with H2r presented in [16] rather than with those methods developed in [17]. This way, we studied the impact of applying the Storey-Tibshirani procedure in combination with the effect of using the 90-th percentile cut-off for the connectivity degree. We have applied H2r without adding pseudo counts to the human EGFR and GCK protein. For EGFR, the 14 sites T725, A755, N756, A767, Q791, V802, N816, V819, K846, V876, M881, K913, D916, and E931 are identified as significant. Out of these significant sites, ten of these residue sites T725, A755, N756, A767, Q791,K846, V876, M881, K913, and D916 are essential sites. On the other hand, for GCK, H2r identified the 15 residue positions L25, R36, R63, M107, C213, V226, G261, D262, G264, L266, D267, E268, T405, K414, and H416 as significant. Twelve of these sites, namely R36, R63, M107, C213, V226, G261,D262, G264, L266, D267, K414, and H416, are essential sites. However, when using the H2r Web service ( http://www-bioinf.uni-regensburg.de/) to analyze EGFR and GCK proteins, sensitivity is decreased, while precision is increased. By this service only eight sites for EGFR and nine sites for GCK were found to be significant. Moreover, only five and eight of them are verified as functionally or structurally important for EGFR and GCK proteins, respectively. This difference stems from the fact that the H2r Web service tightens the filtering of the columns. In addition to this, statistically evaluating H2r for EGFR and GCK proteins, we observed a sensitivity of 5.4%, specificity of 96.7%, precision of 75.9%, and a Matthews correlation coefficient value of 0.047. On the other hand, the CMF reaches precision of 79.1%, and a Matthews correlation coefficiant value of 0.133. For sensitivity and specificity of the CMF refer to the last row of Table 3.
The results of this comparison show that a vast majority of functionally or structurally important residue positions cannot be detected without using our novel MSA specific model and both metrics ( and ) together.
Conclusions
The CMF is a new method which includes an MSA-specific statistical model based on multiple testing procedures that quantifies the error made in terms of the false discovery rate and a novel entropy-based metric to upscale BLOSUM62 dissimilar compensatory mutations. Hence, it shows how dissimilar compensatory mutations have affected genomic sequences in the course of evolution. The method is able to predict significant compensatory mutation positions in protein sequences. We suggest that CMF could be used as a novel automated function prediction tool that is required for a better understanding of the structural basis of proteins.
Methods
In this section we describe the training data used and the methods applied and partly developed. Our descriptions follows the structure of Figure 5, i.e. we start with the data and the preprocessing and systemically work towards the CMF-significant site prediction.
Figure 5.
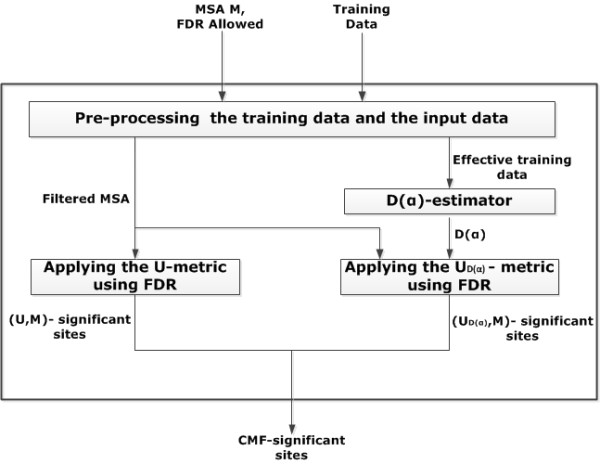
Flowchart of the CMF-analysis.
Training data set and pre-processing
We used a redundancy free set of more than 35000 protein structures computed in Rainer Merkl’s Lab at the University of Regensburg in the following way. The protein structures were taken from the protein data base ( http://www.pdb.org/). The PISCES services [49] was applied to assess proteins on sequence similarity and equality of 3D-data. The related MSAs were gathered from the HSSP data base ( http://swift.cmbi.ru.nl/gv/hssp/).
Taking pattern from [16], we filtered every MSA obtained as follows. First, highly similar and dissimilar sequences were deleted to ensure that the sequence identity between any two sequences is at least 20% and no more than 90%. Second, we removed strictly conserved residue columns, where the percentage of identical residues is greater than 95%. Third, we eliminated the residue columns which contain more than 25% gaps. Finally, we discarded all MSAs with less than 125 sequences. More than 17000 MSAs survived the last filtering step. We used approximately 1700 MSAs as training data which we randomly chose from this set. The pdb entries of the corresponding protein structures are listed in Additional file 3.
Detecting compensatory mutations by the -metric
In [16] a normalized measure of mutual information ranging over the interval [0,1] is successfully used to detect important residues. It is defined as
| (1) |
where and are the entropy of the empirical amino acid distributions of the columns i and j, and is their joint entropy.
We determine an MSA-dependent threshold τ above which -values are defined as significant. Let M be the MSA for the protein under investigation. We extend a standard approach of multiple testing theory [22,50,51] with the following assumptions in mind. M’s -values follow three different distributions. The null distribution F0 represents background signals. The distributions G1 and G2 model the unrelated pairs and the signal pairs, respectively.
We assume F0 to be a β -distribution, and M’s -values U1,U2,…,Uμ to be an independent and identically distributed (iid) sample.
Let Xι:= 1−F0(Uι) be thep-value of Uι with respect toF0. If Uι is F0-distributed, then Xι is uniform over [0,1]. However, if Uι is G1-distributed or G2-distributed, then Xι is skewed to 1 or to 0 (see Figure 6). According to [22,23], the fraction γ of the Uι’s that are F0-distributed is estimated by
Figure 6.
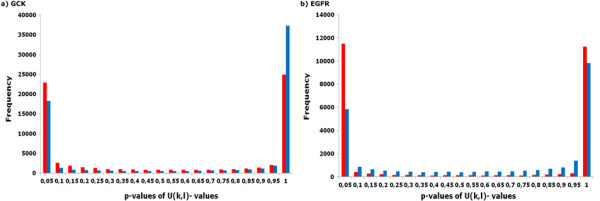
Four p-value distributions of (transformed) normalized mutual information values for human GCK and EFGR proteins having PDB-ID 1V4S and 2J6M, respectively. The bar charts illustrate the two steps of our model: i) blue bars show the p-value distribution of the -scores; ii) red bars display the p-value distribution of the -values. The p-values close to zero represent the significant pairs by means of which we assess the individual residue position. As one can see, within [0.25,0.70] these four distributions are approximately uniform.
The tuning parameters λ1 and λ2 are chosen such that the fraction of not uniformly distributed p-values that fall into [λ1,λ2] is negligible.
We call a pair of sites (i,j) of the protein under study ( -significant if and only if thep-value is less than or equal to τ, for a threshold τ ≤ λ1 that ensures the input false discovery rate FDR, which in turn can be estimated by
In order to determine the parameters of the β-distribution F0, it is sufficient to estimate the expected value and the variance. The expected value is estimated by the sample mean of all -values of M. As for the variance, we take pattern from [52]. Having drawn an iid sample of random column pairs of a sufficient size whose -values fall in a preassigned subinterval of [0,1], we calculate D1,D2,…,Dν by randomly shuffling for every ι =1,2,…,ν. The variance is then estimated as the sample variance of (C1,D1),(C2,D2),…,(Cν,Dν).
The connectivity degree of a site i with respect to the metric and the MSAM is defined as number of sitesj such that (i,j) is -significant [16]. Site i is defined to be -significant, if i’s connectivity degree with respect to and M is greater than or equal to the 90-th percentile. The -significant sites of a protein do not coincide with those predicted by H2r [16]. The connectivity degrees attained and the threshold used substantially differ. In particular, the latter one is data-dependent rather than constant.
Enhancing prediction by the -metric that models dissimilar compensatory mutations
A pair ((ai,aj),(ak,al)) of amino acid pairs is defined to be a formal dissimilar compensatory mutation, if the BLOSUM62 score both of (ai,ak) and (aj,al) is negative.
We use the training data set of approximately 1700 MSAs described above to estimate a 400 × 400 doubly stochastic matrix DCompMut. This matrix is our mathematical model of how dissimilar compensatory mutations have affected genomic sequences in the course of evolution. Its training consists of five phases.
Phase 1. We calculate a signal and a null set of column pairs. The signal set consists of all -significant column pairs, where M ranges over all training MSA. The null set consists of sufficiently many column pairs randomly chosen from every training MSA. For both the signal set and the null set we compute a symmetric 400 × 400 integer-valued matrix of frequencies of pair substitutions Calt and Cnull. To this end, the method used to compute BLOSUM62 matrices [53] is applied to count residue pair substitutions in MSA column pairs rather than residue substitution in columns.
Phase 2. Using Calt and Cnull, we define the matrix Csig by
where φ((ai,aj),(ak,al)) = 1 if and only if (ai,aj) = (ak,al) or
Phase 3. We set all entries of the matrix Csig outside the main diagonal that do not represent a formal dissimilar compensatory mutation to zero. This results in the matrix CCompMut. By normalizing CCompMut, we obtain a symmetric matrix PCompMut. For ai,aj,ak,al ranging over all amino acids,PCompMut((ai,aj),(ak,al)) represents an empirical probability distribution on pairs of amino acid pairs.
Phase 4. We calculate the symmetric 400 × 400-matrix
where is the marginal distribution of PCompMut.
Phase 5. We set all negative entries of SCompMut to zero. Then we compute the doubly stochastic matrix DCompMut by means of the canonical iterated row-column normalization procedure [54].
Now we define our new -metric based on DCompMut. For every column pair (i,j) of the input MSA M, we linearly transform the associated empirical pair distribution with the doubly stochastic matrix
where 1 is the 400 × 400 unit matrix, DCompMut is the result of training phase 5, and α ∈(0,1] is a preassigned real number. is then defined to be the -value (see Equation 1) of this transform.
Having canonically carried over the definition of a significant site pair and of the connectivity degree of a site to this case, a site i is called -significant, if i’s connectivity degree with respect to the metric is greater than or equal to the 90-th percentile.
Finally, a site is defined to be CMF-significant with respect to the MSAM, if it is -significant or -significant. TheCMF-significant sites are predicted as functionally or structurally important ones.
Principally, the controlling parameter α ∈ (0,1] can be adjusted by the user. We set α to 1 to allow the two sets of -significant and -significant positions to complement each other.
Note, that the matrix SCompMut could be replaced with another scoring matrix meaningful in this context.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SW developed the model underlying CMF. MG developed the model together with SW, designed and implemented the tool, and interpreted the results together with NT and MH. All authors read and approved the manuscript.
Supplementary Material
EGFR significant sites. CMF-significant residue sites of the human epidermal growth factor receptor (EGFR) protein.
GCK significant sites. CMF-significant residue sites of the human glucokinase (GCK) protein.
Pdb entries of training MSAs. Pdb entries of redundancy free data set.
Contributor Information
Mehmet Gültas, Email: gueltas@cs.uni-goettingen.de.
Martin Haubrock, Email: martin.haubrock@bioinf.med.uni-goettingen.de.
Nesrin Tüysüz, Email: n.tuysuz@erasmusmc.nl.
Stephan Waack, Email: waack@cs.uni-goettingen.de.
Acknowledgements
We thank Rainer Merkl for placing a redundancy free set of more than 35000 protein structures at our disposal. Special thank goes to Torsten Schöps for setting up the CMF Web server. Mehmet Gltas acknowledges financial support by the Deutsche Forschungsgemeinschaft (DFG: WA 766/7-1).
References
- Jeon J, Yang JS, Kim S. Integration of Evolutionary Features for the Identification of Functionally Important Residues in Major Facilitator Superfamily Transporters. PLoS Comput Biol. 2009;5(10):e1000522. doi: 10.1371/journal.pcbi.1000522. [ http://dx.doi.org/10.13712Fjournal.pcbi.1000522] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadovsky E, Yifrach O. Principles underlying energetic coupling along an allosteric communication trajectory of a voltage-activated K+ channel. Proc Nat Acad Sci. 2007;104(50):19813–19818. doi: 10.1073/pnas.0708120104. [ http://www.pnas.org/content/104/50/19813.abstract] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pupko T, Bell RE, Mayrose I, Glaser F, Ben-Tal N. Rate4Site: an algorithmic tool for the identification of functional regions in proteins by surface mapping of evolutionary determinants within their homologues. Bioinformatics. 2002;18(suppl 1):S71—S77. doi: 10.1093/bioinformatics/18.suppl_1.s71. [ http://bioinformatics.oxfordjournals.org/content/18/suppl_1/S71.abstract] [DOI] [PubMed] [Google Scholar]
- Wilson K, Walker J. Principles and Techniques of Biochemistry and Molecular Biology. New York: Cambridge University Press; 2010. [Google Scholar]
- Altschuh D, Lesk AM, Bloomer AC, Klug A. Correlation of co-ordinated amino acid substitutions with function in viruses related to tobacco mosaic virus. J Mol Biol. 1987;193(4):693–707. doi: 10.1016/0022-2836(87)90352-4. [DOI] [PubMed] [Google Scholar]
- Martin LC, Gloor GB, Dunn SD, Wahl LM. Using information theory to search for co-evolving residues in proteins. Bioinformatics. 2005;21(22):4116–4124. doi: 10.1093/bioinformatics/bti671. [DOI] [PubMed] [Google Scholar]
- Yeang CH, Haussler D. Detecting Coevolution in and among Protein Domains. PLoS Comput Biol. 2007;3(11):e211. doi: 10.1371/journal.pcbi.0030211. [ http://dx.plos.org/10.13712Fjournal.pcbi.0030211] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh CS, Bogan AA, Joachimiak M, Walther D, Cohen FE. Co-evolution of proteins with their interaction partners. J Mol Biol. 2000;299(2):283–293. doi: 10.1006/jmbi.2000.3732. [ http://www.sciencedirect.com/science/article/pii/S002228360093732X] [DOI] [PubMed] [Google Scholar]
- Lockless SW, Ranganathan R. Evolutionarily Conserved Pathways of Energetic Connectivity in Protein Families. Science. 1999;286(5438):295–299. doi: 10.1126/science.286.5438.295. [ http://www.sciencemag.org/content/286/5438/295.abstract] [DOI] [PubMed] [Google Scholar]
- Göbel U, Sander C, Schneider R, Valencia A. Correlated mutations and residue contacts in proteins. Proteins-Struct Funct Genet. 1994;18(4):309–317. doi: 10.1002/prot.340180402. [DOI] [PubMed] [Google Scholar]
- Neher E. How frequent are correlated changes in families of protein sequences? Proc Nat AcadSci. 1994;91:98–102. doi: 10.1073/pnas.91.1.98. [ http://www.pnas.org/content/91/1/98.abstract] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock DD, Taylor WR. Effectiveness of correlation analysis in identifying protein residues undergoing correlated evolution. Protein Eng. 1997;10(6):647–657. doi: 10.1093/protein/10.6.647. [ http://peds.oxfordjournals.org/content/10/6/647.abstract] [DOI] [PubMed] [Google Scholar]
- Dekker JP, Fodor A, Aldrich RW, Yellen G. A perturbation-based method for calculating explicit likelihood of evolutionary co-variance in multiple sequence alignments. Bioinformatics. 2004;20(10):1565–1572. doi: 10.1093/bioinformatics/bth128. [DOI] [PubMed] [Google Scholar]
- Atchley WR, Wollenberg KR, Fitch WM, Terhalle W, Dress AW. Correlations among amino acid sites in bHLH protein domains: an information theoretic analysis. Mol Biol Evol. 2000;17:164. doi: 10.1093/oxfordjournals.molbev.a026229. [DOI] [PubMed] [Google Scholar]
- Tillier ER, Lui TW. Using multiple interdependency to separate functional from phylogenetic correlations in protein alignments. Bioinformatics. 2003;19(6):750–755. doi: 10.1093/bioinformatics/btg072. [ http://bioinformatics.oxfordjournals.org/content/19/6/750.abstract] [DOI] [PubMed] [Google Scholar]
- Merkl R, Zwick M. H2r: Identification of evolutionary important residues by means of an entropy based analysis of multiple sequence alignments. BMC Bioinformatics. 2008;9:151. doi: 10.1186/1471-2105-9-151. [ http://www.biomedcentral.com/1471-2105/9/151] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H, Dou Y, Yang J, Wang J. New methods to measure residues coevolution in proteins. BMC Bioinformatics. 2011;12:206. doi: 10.1186/1471-2105-12-206. [ http://www.biomedcentral.com/1471-2105/12/206] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codoner FM, Fares M. Why Should We Care About Molecular Coevolution? Evolutionary c. 2008;4:29–38. [PMC free article] [PubMed] [Google Scholar]
- Dunn SD, Wahl LM, Gloor GB. Mutual information without the influence of phylogeny or entropy dramatically improves residue contact prediction. Bioinformatics. 2008;24(3):333–340. doi: 10.1093/bioinformatics/btm604. [DOI] [PubMed] [Google Scholar]
- Noivirt O, Eisenstein M, Horovitz A. Detection and reduction of evolutionary noise in correlated mutation analysis. Protein Eng Design and Sel. 2005;18(5):247–253. doi: 10.1093/protein/gzi029. [ http://peds.oxfordjournals.org/content/18/5/247.abstract] [DOI] [PubMed] [Google Scholar]
- Wollenberg KR, Atchley WR. Separation of phylogenetic and functional associations in biological sequences by using the parametric bootstrap. Proc Nat Acad Sci. 2000;97(7):3288–3291. doi: 10.1073/pnas.97.7.3288. [ http://www.pnas.org/content/97/7/3288.abstract] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Acad Sci. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh B. Multiple comparisons: Bonferroni Corrections and False Discovery Rates. Lecture Notes EEB 581, Department of Ecology and Evolutionary Biology, University of Arizona 2004.
- Dixit A, Yi L, Gowthaman R, Torkamani A, Schork NJ, Verkhivker GM. Sequence and Structure Signatures of Cancer Mutation Hotspots in Protein Kinases. PLoS ONE. 2009;4(10):e7485. doi: 10.1371/journal.pone.0007485. [ http://dx.doi.org/10.13712Fjournal.pone.0007485] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun CH, Boggon TJ, Li Y, Woo MS, Greulich H, Meyerson M, Eck MJ. Structures of Lung Cancer-Derived EGFR Mutants and Inhibitor Complexes: Mechanism of Activation and Insights into Differential Inhibitor Sensitivity. Cancer Cell. 2007;11(3):217–227. doi: 10.1016/j.ccr.2006.12.017. [ http://www.sciencedirect.com/science/article/pii/S1535610807000281] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Berezov A, Wang Q, Zhang G, Drebin J, Murali R, Greene MI. ErbB receptors: from oncogenes to targeted cancer therapies. J Clin Invest. 2007;117(8):2051–2058. doi: 10.1172/JCI32278. [ http://www.jci.org/articles/view/32278] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. Activating Mutations in the Epidermal Growth Factor Receptor Underlying Responsiveness of Non-Small-Cell Lung Cancer to Gefitinib. New England J Med. 2004;350(21):2129–2139. doi: 10.1056/NEJMoa040938. [ http://www.nejm.org/doi/full/10.1056/NEJMoa040938] [DOI] [PubMed] [Google Scholar]
- Balius TE, Rizzo RC. Quantitative Prediction of Fold Resistance for Inhibitors of EGFR. Biochemistry. 2009;48(35):8435–8448. doi: 10.1021/bi900729a. [PMID: 19627157] [ http://pubs.acs.org/doi/abs/10.1021/bi900729a] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinto N, Zagari A, Capuano M, De Simone A, Capobianco V, Daniele G, Giugliano M, Spadaro R, Franzese A, Sacchetti L. Glucokinase Gene Mutations: Structural and Genotype-Phenotype Analyses in MODY Children from South Italy. PLoS ONE. 2008;3(4):e1870. doi: 10.1371/journal.pone.0001870. [ http://dx.plos.org/10.13712Fjournal.pone.0001870] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capuano M, Garcia-Herrero CM, Tinto N, Carluccio C, Capobianco V, Coto I, Cola A, Iafusco D, Franzese A, Zagari A, Navas MA, Sacchetti L. Glucokinase (GCK) Mutations and Their Characterization in MODY2 Children of Southern Italy. PLoS ONE. 2012;7(6):e38906. doi: 10.1371/journal.pone.0038906. [ http://dx.doi.org/10.13712Fjournal.pone.0038906] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Herrero CM, Rubio-Cabezas O, Azriel S, Gutierrez-Nogues A, Aragones A, Vincent O, Campos-Barros A, Argente J, Navas MA. Functional Characterization of MODY2 Mutations Highlights the Importance of the Fine-Tuning of Glucokinase and Its Role in Glucose Sensing. PLoS ONE. 2012;7:e30518. doi: 10.1371/journal.pone.0030518. [ http://dx.doi.org/10.13712Fjournal.pone.0030518] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamata K, Mitsuya M, Nishimura T, ichi Eiki J, Nagata Y. Structural Basis for Allosteric Regulation of the Monomeric Allosteric Enzyme Human Glucokinase. Structure. 2004;12(3):429–438. doi: 10.1016/j.str.2004.02.005. [ http://www.sciencedirect.com/science/article/pii/S0969212604000474] [DOI] [PubMed] [Google Scholar]
- Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005;33(suppl 1):D514—D517. doi: 10.1093/nar/gki033. [ http://nar.oxfordjournals.org/content/33/suppl_1/D514.abstract] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichert J, Sühnel J. The IMB Jena Image Library of Biological Macromolecules: 2002 update. Nucleic Acids Res. 2002;30:253–254. doi: 10.1093/nar/30.1.253. [ http://nar.oxfordjournals.org/content/30/1/253.abstract] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flicek P, Amode MR, Barrell D, Beal K, Brent S, Chen Y, Clapham P, Coates G, Fairley S, Fitzgerald S, Gordon L, Hendrix M, Hourlier T, Johnson N, Kähäri A, Keefe D, Keenan S, Kinsella R, Kokocinski F, Kulesha E, Larsson P, Longden I, McLaren W, Overduin B, Pritchard B, Riat HS, Rios D, Ritchie GRS, Ruffier M. et al. Ensembl 2011. Nucleic Acids Res. 2011;39(suppl 1):D800—D806. doi: 10.1093/nar/gkq1064. [ http://nar.oxfordjournals.org/content/39/suppl_1/D800.abstract] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunyaev S, Ramensky V, Bork P. Towards a structural basis of human non-synonymous single nucleotide polymorphisms. Trends in Genet. 2000;16(5):198–200. doi: 10.1016/S0168-9525(00)01988-0. [ http://www.sciencedirect.com/science/article/pii/S0168952500019880] [DOI] [PubMed] [Google Scholar]
- Wang Z, Moult J. SNPs, protein structure, and disease. Human Mutation. 2001;17(4):263–270. doi: 10.1002/humu.22. [ http://dx.doi.org/10.1002/humu.22] [DOI] [PubMed] [Google Scholar]
- Burke D, Worth C, Priego EM, Cheng T, Smink L, Todd J, Blundell T. Genome bioinformatic analysis of nonsynonymous SNPs. BMC Bioinformatics. 2007;8:301. doi: 10.1186/1471-2105-8-301. [ http://www.biomedcentral.com/1471-2105/8/301] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keskin O, Tsai CJ, Wolfson H, Nussinov R. A new, structurally nonredundant, diverse data set of protein-protein interfaces and its implications. Protein Science. 2004;13(4):1043–1055. doi: 10.1110/ps.03484604. [ http://dx.doi.org/10.1110/ps.03484604] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst RS. Review of epidermal growth factor receptor biology. Int J Radiat Oncol Biol Phys. 2004;59(Supplement 2):S21—S26. doi: 10.1016/j.ijrobp.2003.11.041. [ http://www.sciencedirect.com/science/article/pii/S0360301604003311] [DOI] [PubMed] [Google Scholar]
- Thornton PS, Satin-Smith MS, Herold K, Glaser B, Chiu KC, Nestorowicz A, Permutt M, Baker L, Stanley CA. Familial hyperinsulinism with apparent autosomal dominant inheritance: Clinical and genetic differences from the autosomal recessive variant. J Pediatrics. 1998;132:9–14. doi: 10.1016/S0022-3476(98)70477-9. [ http://www.sciencedirect.com/science/article/pii/S0022347698704779] [DOI] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [ http://nar.oxfordjournals.org/content/29/1/308.abstract] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkhoff G. Tres observationes sobre et algebra lineal. Univ Nac Tucaman Rev. 1946;A(5):147–151. [Google Scholar]
- Hardy G, Littlewood J, Pólya G. Inequalities. Oxford: Oxford University Press; 1952. [Google Scholar]
- Cheng TMK, Lu YE, Vendruscolo M, Blundell TL, Lio’ P. Prediction by Graph Theoretic Measures of Structural Effects in Proteins Arising from Non-Synonymous Single Nucleotide Polymorphisms. PLoS Comput Biol. 2008;4(7):e1000135. doi: 10.1371/journal.pcbi.1000135. [ http://dx.doi.org/10.13712Fjournal.pcbi.1000135] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao L, Cui Y. Functional impacts of non-synonymous single nucleotide polymorphisms: Selective constraint and structural environments. FEBS Letters. 2006;580(5):1231–1234. doi: 10.1016/j.febslet.2006.01.035. [ http://www.sciencedirect.com/science/article/pii/S0014579306000755] [DOI] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31(13):3812–3814. doi: 10.1093/nar/gkg509. [ http://nar.oxfordjournals.org/content/31/13/3812.abstract] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30(17):3894–3900. doi: 10.1093/nar/gkf493. [ http://nar.oxfordjournals.org/content/30/17/3894.abstract] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Dunbrack Jr RLD. PISCES: recent improvements to a PDB sequence culling server. Nucleic Acids Res. 2005;33(Web-Server-Issue):94–98. doi: 10.1093/nar/gki402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J R Stat Soc Ser B (Methodological) 1995;57:289–300. [ http://www.jstor.org/stable/2346101] [Google Scholar]
- Ferreira JA, Zwinderman AH. On the Benjamini-Hochberg Method. Ann Stat. 2006;34(4):1827–1849. doi: 10.1214/009053606000000425. [ http://www.jstor.org/stable/25463486] [DOI] [Google Scholar]
- Bremm S, Schreck T, Boba P, Held S, Hamacher K. Computing and visually analyzing mutual information in molecular co-evolution. BMC Bioinformatics. 2010;11:330. doi: 10.1186/1471-2105-11-330. [ http://www.biomedcentral.com/1471-2105/11/330] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc Nat Acad Sci. 1992;89(22):10915–10919. doi: 10.1073/pnas.89.22.10915. [ http://www.pnas.org/content/89/22/10915.abstract] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappellini V, Sommer HJ, Bruzda W, Zyczkowski K. Random bistochastic matrices. J Phys A: Math Theor. 2009;42:23. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
EGFR significant sites. CMF-significant residue sites of the human epidermal growth factor receptor (EGFR) protein.
GCK significant sites. CMF-significant residue sites of the human glucokinase (GCK) protein.
Pdb entries of training MSAs. Pdb entries of redundancy free data set.