

Abstract
Protein kinase A (PKA) phosphorylation of myofibrillar proteins constitutes an important pathway for β-adrenergic modulation of cardiac contractility. In myofilaments PKA targets troponin I (cTnI), myosin binding protein-C (cMyBP-C) and titin. We studied how this affects the sarcomere length (SL) dependence of force–pCa relations in demembranated cardiac muscle. To distinguish cTnI from cMyBP-C/titin phosphorylation effects on the force–pCa relationship, endogenous troponin (Tn) was exchanged in rat ventricular trabeculae with either wild-type (WT) Tn, non-phosphorylatable cTnI (S23/24A) Tn or phosphomimetic cTnI (S23/24D) Tn. PKA cannot phosphorylate either cTnI S23/24 variant, leaving cMyBP-C/titin as PKA targets. Force was measured at 2.3 and 2.0 μm SL. Decreasing SL reduced maximal force (Fmax) and Ca2+ sensitivity of force (pCa50) similarly with WT and S23/24A trabeculae. PKA treatment of WT and S23/24A trabeculae reduced pCa50 at 2.3 but not at 2.0 μm SL, thus eliminating the SL dependence of pCa50. In contrast, S23/24D trabeculae reduced pCa50 at both SL values, primarily at 2.3 μm, also eliminating SL dependence of pCa50. Subsequent PKA treatment moderately reduced pCa50 at both SLs. At each SL, Fmax was unaffected by either Tn exchange and/or PKA treatment. Low-angle X-ray diffraction was performed to determine whether pCa50 shifts were associated with changes in myofilament spacing (d1,0) or thick–thin filament interaction. PKA increased d1,0 slightly under all conditions. The ratios of the integrated intensities of the equatorial X-ray reflections (I1,1/I1,0) indicate that PKA treatment increased crossbridge proximity to thin filaments under all conditions. The results suggest that phosphorylation by PKA of either cTnI or cMyBP-C/titin independently reduces the pCa50 preferentially at long SL, possibly through reduced availability of thin filament binding sites (cTnI) or altered crossbridge recruitment (cMyBP-C/titin). Preferential reduction of pCa50 at long SL may not reduce cardiac output during periods of high metabolic demand because of increased intracellular Ca2+ during β-adrenergic stimulation.
Key points
β-Adrenergic stimulation is an important control mechanism, matching cardiac output to venous return during increased metabolic demand.
β-Adrenergic signalling leads to protein kinase A (PKA) phosphorylation of myofilament proteins cardiac troponin I (cTnI), cardiac myosin binding protein-C (cMyBP-C) and titin, but their specific effects on the sarcomeric length (SL) dependence of contraction – which underlies the Frank–Starling Law of the Heart – is debated.
Recombinant cTnI phosphomimetics were exchanged into cardiac muscle to isolate the effects of cTnI from those of cMyBP-C/titin phosphorylation on SL-dependent force–Ca2+ relations and sarcomeric structure.
Results suggest cTnI or cMyBP-C/titin phosphorylation, separately or together, eliminate the SL dependence of Ca2+ sensitivity of force, but not maximal force. The reduction occurs particularly at long SL, suggesting effects on thin filament access and crossbridge recruitment.
The net effect of PKA phosphorylation is to blunt SL dependence of force at submaximal [Ca2+] to maintain elevated systolic function.
Introduction
Increasing sarcomere length (SL) of cardiac myocytes with increased ventricular filling results in a steep increase in force generation that underlies the heart's ability to match ventricular output to venous return; i.e. the Frank–Starling relationship. Elevated force from increasing SL results in part from an accompanying increase in sensitivity of the contractile regulatory apparatus to myoplasmic Ca2+, which is particularly important as Ca2+ activation is sub-maximal during a cardiac twitch (Gordon et al. 2000; Regnier et al. 2004). During increased metabolic demand β-adrenergic stimulation of the heart and subsequent activation of protein kinase A (PKA) phosphorylates several myofilament and Ca2+ handling proteins, allowing an adaptive increase of cardiac contractility and ventricular output. β-Adrenergic stimulation enhances myoplasmic Ca2+ transients during cardiac contractions, decreases the sensitivity of the myofilament contractile apparatus to Ca2+ and increases the rate of myoplasmic Ca2+ return to sub-threshold levels to initiate diastole (Bers, 2002). While cardiac contraction is enhanced by β-adrenergic stimulation, it is not clear whether (or how) the myofilament basis for the Frank–Starling mechanism is altered by β-adrenergic activation via PKA. Literature reports are contradictory, with reports that PKA treatment either increased (Konhilas et al. 2000, 2003; Hanft & McDonald, 2009), decreased (Kajiwara et al. 2000) or did not change (Cazorla et al. 2006) the influence of SL on the Ca2+ sensitivity of force. Our approach to resolve this disparity is based on previous studies suggesting that the actin-binding inhibitory subunit of cardiac troponin (cTnI) may play an important role in determining the SL dependence of the Ca2+ sensitivity of contractile force in demembranated (skinned) cardiac muscle (Arteaga et al. 2000; Konhilas et al. 2003; Tachampa et al. 2007) via phosphorylation of the N-terminal serines (Ser 23, 24) by PKA (Layland et al. 2005).
The influence of SL on the Ca2+ sensitivity of force results from complex interactions between both thin and thick filament-associated regulatory proteins. These interactions include (1) Ca2+ binding to the troponin complex (Tn) to initiate contractile activation of thin filaments, (2) strong myosin crossbridge (CB) binding to further activate cardiac thin filaments and (3) the intrinsic properties of the troponin regulatory subunits (Gordon et al. 2000; Fuchs & Martyn, 2005). The interactions between these contractile regulatory proteins are additionally modulated by phosphorylation of specific amino acids by a host of protein kinases, including PKA (Burkart et al. 2003; Sumandea et al. 2004; Kobayashi & Solaro, 2005; Layland et al. 2005). Central to these interactions is the phosphorylation of cTnI (Moir et al. 1980; Zhang et al. 1995b; Chandra et al. 1997; Kentish et al. 2001). By itself, PKA phosphorylation of cTnI decreases the Ca2+ sensitivity of force in skinned cardiac trabeculae and increases the rate of myofibril relaxation (Zhang et al. 1995a; Kentish et al. 2001). N-terminal phosphorylation of cTnI weakens interaction between the C-terminus of cTnI and the N-terminus of cardiac troponin C (cTnC) during Ca2+ activation (Chandra et al. 1997; Finley et al. 1999; Burkart et al. 2003; Li et al. 2003; Dong et al. 2007) and increases the Ca2+ dissociation rate from cTnC (Robertson et al. 1982; Zhang et al. 1995b). This results in more rapid dissociation of Ca2+ from cTnC to begin diastole, increasing the rate of relaxation to subsequently enhance ventricular filling.
The interactions of CBs with thin filaments may also be influenced by the myosin-associated proteins including myosin-binding protein C (cMyBP-C) (Moolman-Smook et al. 2002; Flashman et al. 2004) and regulatory light chain 2 (RLC2) (Olsson et al. 2004; Stelzer et al. 2006), as well as the giant sarcomeric protein titin (Cazorla et al. 2001; Granzier et al. 2002; Granzier & Labeit, 2002). Thick filament targets for PKA phosphorylation include the cardiac-specific motif of cMyBP-C (Moolman-Smook et al. 2002; Flashman et al. 2004) and the N2B domain of titin (Yamasaki et al. 2002; Fukuda et al. 2005). PKA phosphorylation within the motif region of cMyBP-C reduces binding to the S2 domain of myosin and increases the proximity of CBs to thin filaments (Moolman-Smook et al. 2002; Colson et al. 2008, 2010; Shaffer et al. 2009). PKA phosphorylation of the cardiac N2B titin domain reduces passive tension in skinned cardiac preparations (Yamasaki et al. 2002; Fukuda et al. 2005; Kruger & Linke, 2006).
Because PKA targets cTnI, cMyBP-C and titin it has been difficult to determine the precise mechanistic role that phosphorylation of each protein plays in myofilament force regulation and the Frank–Starling mechanism. The goal of this study was to determine the specific role of PKA phosphorylation of cTnI (vs. cMyBP-C and titin). This was accomplished by exchanging endogenous Tn in skinned right ventricular trabeculae from rats with recombinant Tn containing either a cTnI variant in which serines 23 and 24 represent a non-phosphorylatable state, cTnI (S23/24A) (Noland et al. 1995), or a constitutively phosphorylated state cTnI (S23/24D) (Dohet et al. 1995). Neither of these cTnI variants can be phosphorylated by PKA. Thus, with either cTnI variant, PKA treatment primarily targets both cMyBP-C and titin, so that any additional effects could be attributed to phosphorylation at sites on those proteins. However, it is important to understand that these experiments do not allow separation of the individual influences of cMyBP-C or titin phosphorylation by PKA.
Using a combination of phosphomimetic cTnI mutants and PKA treatment, we found that the SL dependence of the pCa50 of force (but not maximal force) could be reduced by phosphorylation of cTnI Ser23/24 or cMyBP-C/titin. Combined with X-ray diffraction studies of myofilament lattice structure these results point to separate length-dependent effects of PKA on cTnI and cMyBP-C/titin that are most evident at long SL where the probability of CB formation increases. PKA phosphorylation of cTnI may reduce cTnC–cTnI interaction, leading to decreased Ca2+ sensitivity and reduced thin filament access for CBs, particularly at the longer SL. In contrast, PKA phosphorylation of cMyBP-C may reduce the SL dependence of contraction by promoting weak myosin binding to the thin filament. The net effect of PKA phosphorylation is to attenuate the effect of SL on force generation over the physiologically relevant range of myoplasmic [Ca2+].
Methods
Ethical approval and tissue preparation
All animal procedures were conducted in accordance with the US National Institutes of Health Policy on Humane Care and Use of Laboratory Animals and were approved by the University of Washington (UW) Institutional Animal Care and Use Committee (IACUC). Rats were housed in the Department of Comparative Medicine at UW and cared for in accordance with UW IACUC procedures. Forty male Sprague–Dawley rats (150–250 g) were anaesthetized with an intraperitoneal injection of pentobarbital (50 mg kg−1) after initial exposure to isoflurane (3–5% in oxygen). When the animal had no reflexive response, the heart was rapidly excised and dissected in oxygenated physiological salt solution containing (in mm): 100 NaCl, 24 NaHCO3, 2.5 KCl, 1 MgSO4.7H2O, 1 Na2HPO4 and 1 CaCl2 (Adhikari et al. 2004). Animals were prepared for either mechanics (n= 20) or X-ray diffraction studies (n= 20). Spliced left ventricles were demembranated overnight at 4°C in relaxing solutions containing (in mm): 100 KCl, 9.0 MgCl2, 4.0 Na2ATP, 5.0 K2EGTA, 10 Mops, 1% non-ionic detergent Triton X-100, pH 7.0, and 50% (v/v) glycerol. Right ventricular trabeculae (150 ± 10 μm diameter, 1.4 ± 0.2 mm length) were dissected and stored in the same solution without Triton X-100 at −20°C for experiments within 1 week.
Recombinant protein isolation and exchange
Site-directed mutagenesis was performed using the QuikChange II Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA) to substitute serines 23, 24 in cTnI with either alanine, to mimic the N-terminal unphosphorylated state of cTnI (cTnI (S23/24A)), or with aspartic acid (cTnI (S23/24D)) to mimic the N-terminal phosphorylated cTnI state (Supplemental Fig. S1). A pET-24 (Novagen, Madison, WI, USA) vector containing the T7 promoter, lac operator and a kanamycin resistant gene was used for expression of wild-type (WT) and mutant proteins in Escherichia coli (BL21). The DNA sequences of the expressing constructs were verified by DNA sequencing. The expressed protein was extracted from bacterial cells as previously described (Kohler et al. 2003) and purified on DE 52 or CM 52 (GE Healthcare Life Sciences, Piscataway, NJ, USA) columns equilibrated by 6 m urea, 25 mm Tris at pH 8.0, 1 mm EDTA and 15 mm 2-mercaptoethanol. Proteins were eluted with a salt gradient washing in the same buffer from 0 to 0.3 m NaCl. The fractions containing the desired protein and their concentrations were monitored by SDS-PAGE and a DU 800 spectrophotometer. Proteins were stored at −80°C before use. Troponin complex (Tn) was reconstituted from isolated recombinant subunits (1:1:1), as previously described (Potter, 1982) and stored in buffer containing (in mm) 200 potassium chloride, 20 Mops, 5 EGTA, 5 MgCl2, 15 mercaptoethanol, pH 7.0; Tn concentration was ∼1.0 mg ml−1. For exchange of endogenous Tn with Tn containing WT or variant cTnI, detergent skinned trabeculae were placed into Tn solution containing 4 mm ATP for overnight passive exchange on a slow rocker at 4°C. Following exchange protocols, trabeculae were incubated twice for 30 min with gentle mixing in relaxing solution containing 1 mg ml−1 bovine serum albumin to remove any non-specifically bound exogenous Tn.
Solutions
Solution composition was determined by an iterative computer program that calculates the equilibrium concentration of ligands and ions based on published affinity constants (Fabiato, 1988). Relaxing solutions contained (in mm): 80 Mops, 15 EGTA, 1 Mg2+, 5 MgATP, 135 (Na++ K+), 15 creatine phosphate and 20 units ml-1 creatine phosphokinase, pH 7.0; solution ionic strength was 170 mm. Experimental temperature was 15°C. For activation solutions, the Ca2+ level (expressed as pCa =–log[Ca2+]) was set by adjusting with CaCl2. For PKA treatment skinned trabeculae were exposed to 200 μl relaxing solution containing 100 units of the catalytic subunit of PKA (Sigma-Aldrich, St Louis, MO, USA) and 6 mm dithiothreitol for 45 min at 20°C.
Mechanical measurements
Chemically ‘skinned’ (1% Triton X-100) trabeculae were attached to a force transducer (Aurora Model 400A) and a Model 312B servo motor (Aurora Scientific, Aurora, Ontario, Canada) tuned for a 350 μs step response via T-clips. Trabeculae were placed in 200 μl temperature-controlled wells (15°C) that could be moved to expose the preparation to different solutions. SL was measured by image analysis using a MyoCam (Ion Optix, Inc., Boston, MA, USA) and set at either 2.3 or 2.0 μm in relaxing solution (pCa 9.0). The baseline for isometric force was measured by transiently shortening the fibre to slack length; relaxed passive force was subtracted from active force.
X-ray diffraction
Low-angle X-ray diffraction measurements were performed on the small-angle BioCAT instrument on beamline 18-D at the Advanced Photon Source, Argonne National Laboratory (Fischetti et al. 2004), using trabeculae mounted in a simple plexiglass X-ray chamber containing pCa 9.0 relaxing solution at an SL of 2.3 μm. X-ray exposures were 1 s at an incident flux of ∼1 × 1012 photons s-1 with 12 keV photon energy. Camera length was 2.8 m. Diffraction patterns were collected on a CCD-based X-ray detector (Mar 165; Rayonix Inc., Evanston, IL, USA) and the spacings of the 1,0 and 1,1 equatorial reflections were acquired (Irving et al. 2000; Colson et al. 2010). The distance between the 1,0 and 1,1 reflections were converted to the d1,0 lattice spacing using Bragg's law. This may be converted to the inter-thick filament spacing (d1,0) by multiplying by 2/√3 (Irving & Millman, 1989). Intensities of the 1,0 and 1,1 equatorial reflections were determined from non-linear least square fits to one-dimensional projections of the integrated intensity along the equator (Colson et al. 2010). All data were analysed independently by three individuals and the results were averaged.
SDS-PAGE and Western blots
To monitor the extent of mutant cTn incorporation into trabeculae, we used Tn in which cardiac troponin T (cTnT) contained a 9 amino acid myc-tag at the N terminus, similar to previous studies (Tachampa et al. 2008). Exchange efficiency was determined through Western blot analysis after the proteins were extracted by SDS sample buffer and separated by 12.5% SDS-PAGE. The presence of the myc-tag allowed us to visibly separate the exchanged protein from endogenous protein (see Fig. 1A). Exchange efficiency was determined by calculating the percentage of myc-tagged cTnT (top band) and endogenous cTnT (bottom band) present in the sample.
Figure 1. Exchange efficiency and phosphoprotein analysis.
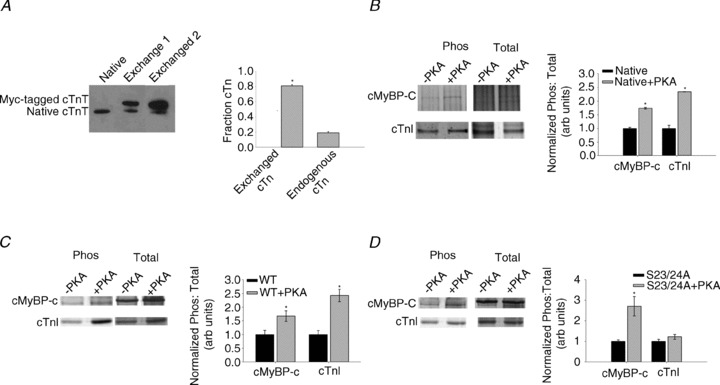
A, efficiency of exchange of recombinant cardiac troponin complex (Tn) into rat cardiac trabeculae. Representative Western blot against cTnT antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) in native trabeculae and two exchanged samples (left) and relative levels of exchanged Tn containing cTnT + myc-tag and endogenous, unexchanged Tn (right), n = 3 (Western blots). B, phosphorylation analysis of native cardiac trabeculae, ± PKA. Pro-Q Diamond phosphoprotein and total protein (Coomassie blue) stain of cMyBP-C and cTnI, ± PKA (left) and phosphorylation: total protein staining ratios of native (filled bars) and native+PKA (grey bars) samples (right), n= 3 (gels), ten exchanged trabeculae per lane. C, phosphorylation analysis of wild-type (WT) trabeculae, ± PKA. Pro-Q Diamond phosphoprotein and total protein stain of cMyBP-C and cTnI, ± PKA (left) and phosphorylation: total protein staining ratios of WT (filled bars) and WT+PKA (grey bars) samples (right), n= 3. D, phosphorylation analysis of S23/24A trabeculae ± PKA. Pro-Q Diamond phosphoprotein and total protein stain of cMyBP-C and cTnI, ± PKA (left) and phosphorylation: total protein staining ratios of S23/24A (filled bars) and S23/24A+PKA (grey bars) samples (right), n= 3. Data are means ± SEM; *P < 0.05 compared to no PKA treatment.
SDS-PAGE and concurrent phosphoprotein and total protein staining were performed on exchanged trabeculae (n = 3 gels, ten trabeculae per lane collected from eight hearts) with and without PKA treatment. Each gel represents data collected from 2–3 hearts. Trabeculae populations were mixed (i.e. each lane represents trabeculae from multiple hearts) to reduce bias from a single animal. Gels were stained with Pro-Q Diamond (Invitrogen, Carlsbad, CA, USA) phosphoprotein staining solution and imaged using a BioSpectrum AC Imaging System (UVP, Upland, CA, USA). Following phosphoprotein staining and imaging, gels were stained for total protein with Coomassie Blue. Densitometry analysis was performed using the ImageJ gel analysis toolkit. Relative phosphorylation was based on the ratio of phosphoprotein to total protein for the same band (see Fig. 1B–D).
Data processing and statistical analysis
Force–pCa data were fit by the Hill equation (eqn 1), where Fmax is the maximal Ca2+ activated force, nH is the Hill coefficient, or slope of the relationship, and pCa50 is the pCa at which force is half-maximal:
| (1) |
The reported pCa50 and nH values represent the means of the values from the individual fits, ± standard error of the means (SEM). Means are compared with Student's t test with significance at the 95% confidence level (P < 0.05). Statistical analysis was performed using Excel (Microsoft, Redmond, WA, USA), SigmaPlot (Systat, Richmond, CA, USA) and Fityk (Wojdyr, 2010).
Results
Recombinant troponin exchange and phosphorylation profiles
To quantify the extent of Tn exchange, recombinant Tn complex containing cTnT labelled at the N terminus with a c-myc tag (Fig. 1A) was exchanged into skinned right ventricular trabeculae from rat hearts. Densitometry analysis of Western blots using cTnT-specific antibodies indicated that 80% of endogenous Tn was replaced by Tn containing the c-myc-tagged cTnT, similar to exchange efficiency values reported previously (Chandra et al. 1997; Tachampa et al. 2008). This suggests the exchange protocol was efficient and that the resulting changes in contractility can primarily be attributed to the exchanged Tn containing either WT cTnI or cTnI variants. Consistent with a previously published report from our group (Gillis et al. 2007), no evidence was found for extraction of myosin regulatory light chains (Supplemental Fig. S2A).
Phosphoprotein analysis confirmed that recombinant Tn had no measurable levels of cTnI or cTnT phosphorylation prior to exchange (Supplemental Fig. S1). Endogenous cTnI phosphorylation levels in native trabeculae were relatively low according to Pro-Q signal intensity (un-normalized Pro-Q signal: total protein signal = 1.2), probably reflecting low overall cTnI phosphorylation. Following exchange of unphosphorylated recombinant WT Tn or S23/24A Tn, phosphorylation levels of Tn from trabeculae were further reduced (WT Tn = 0.6 ± 0.2 and S23/24A Tn = 0.8 ± 0.3). PKA treatment of trabeculae significantly increased cTnI phosphorylation in native (Fig. 1B) and WT trabeculae (Fig. 1C), exhibiting an ∼3-fold increase in cTnI phosphorylation signal over endogenous levels. S23/24A trabeculae had no change in the cTnI phosphorylation signal following treatment with PKA, demonstrating that this variant was effective in suppressing PKA phosphorylation of cTnI, and confirming that Tn exchange was nearly complete (Fig. 1D). cMyBP-C phosphorylation levels were relatively low and were significantly increased following PKA treatment in all experimental conditions, consistent with previous observations by others (Olsson et al. 2004). PKA treatment increased cMyBP-C phosphorylation by ∼50% (Fig. 1B) for native trabeculae, approximately 2-fold for WT trabeculae (Fig. 1C) and ∼2.5-fold for S23/24A trabeculae (Fig. 1D). Although PKA is known to phosphorylate titin in skinned cardiac myocytes (Yamasaki et al. 2002; Fukuda et al. 2005; Kruger & Linke, 2006) we did not attempt to determine titin phosphorylation levels in this study.
Because exchange of recombinant Tn for native Tn was not 100%, some residual cTnI phosphorylation was evident in every exchange condition. This residual phosphorylation can be attributed to N-terminal sites on cTnI (S23 and S24), as well as the cTnI protein kinase C sites (S43, S45 and T144). Furthermore, phosphorylation of serines 23 and 24 would represent both mono- and bisphosphorylated cTnI; the bisphosphorylated N-terminal form is required to decrease Ca2+ sensitivity of force (pCa50) (Zhang et al. 1995b). Regulatory light chain (RLC; Supplemental Fig. S2A), cTnT (Supplemental Fig. S2B) and tropomyosin (Supplemental Fig. S2C) phosphorylation levels were also assessed by densitometry (normalized for protein loading) and did not change significantly following PKA treatment for any condition tested.
PKA effects on SL-dependent activation in WT Tn exchanged trabeculae
To determine how phosphorylation of myofibrillar proteins by PKA influences SL dependence of force generation of cardiac muscle, we measured force–pCa relations at long (2.3 μm) and short (2.0 μm) SL in WT trabeculae before and following treatment with the catalytic subunit of PKA. Data from each experiment was fit by the Hill equation (eqn 1) to obtain the Ca2+ sensitivity of force (pCa50) and slope (nH) of the force–pCa relationship (see Table 1). For comparison with previous studies, EC50 values were also calculated and are reported in Table 1. Prior to PKA treatment, increasing SL increased pCa50 and EC50 compared to the short SL (Fig. 2A, Table 1). Following PKA treatment (in relaxing solution), pCa50 (and EC50) at long SL was significantly decreased, while pCa50 at short SL was not altered. As a result, the SL dependence of pCa50 was greatly reduced. In contrast, PKA treatment did not affect maximum force (Fmax; pCa 4.5) at either SL (Fig. 2C). Put another way, PKA treatment resulted in a loss of SL influence on pCa50, but at any given [Ca2+] increasing SL still resulted in a corresponding increase in force (Table 1). Thus the Frank–Starling effect was reduced by PKA but not eliminated.
Table 1.
Hill fit parameters of force–pCa relationships obtained from rat cardiac trabeculae before and after PKA treatment at sarcomere lengths (SLs) of 2.3 and 2.0 μm
| Condition | SL (μm) | pCa50 | ΔpCa50 | EC50 (μm) | ΔEC50 (μm) | Fmax (mN mm−2) | Fpass (mN mm−2) | nH |
|---|---|---|---|---|---|---|---|---|
| Native (n= 5) | 2.3 | 5.45 ± 0.05*† | 0.13 | 3.55 ± 0.36*† | 1.2 | 56 ± 8* | 7.5 ± 1.1* | 4.4 ± 0.7 |
| 2.0 | 5.32 ± 0.04 | 4.75 ± 0.42 | 34 ± 6 | 1.7 ± 0.4 | 5.1 ± 0.8 | |||
| Native + PKA (n= 5) | 2.3 | 5.32 ± 0.04 | 0.04 | 4.74 ± 0.38 | 0.49 | 53 ± 4* | 5.9 ± 1.7* | 3.3 ± 0.3 |
| 2.0 | 5.28 ± 0.05 | 5.22 ± 0.58 | 31 ± 6 | 1.8 ± 0.6 | 5.0 ± 0.5 | |||
| Wild-type (n= 8) | 2.3 | 5.17 ± 0.04*† | 0.13 | 6.77 ± 0.55*† | 2.5 | 56 ± 8* | 9.1 ± 3.1* | 2.9 ± 0.4 |
| 2.0 | 5.04 ± 0.05 | 9.22 ± 1.00 | 31 ± 5 | 1.7 ± 0.4 | 2.8 ± 0.3 | |||
| Wild-type + PKA (n= 8) | 2.3 | 5.04 ± 0.04 | 0.01 | 9.83 ± 1.00 | 0.16 | 56 ± 8* | 5.8 ± 0.8* | 2.4 ± 0.4 |
| 2.0 | 5.03 ± 0.05 | 10.3 ± 1.10 | 30 ± 4 | 1.6 ± 0.4 | 2.6 ± 0.3 | |||
| S23/24A (n= 7) | 2.3 | 5.16 ± 0.02*† | 0.10 | 6.95 ± 0.31*† | 1.8 | 44 ± 5* | 10 ± 2.4* | 3.9 ± 0.3 |
| 2.0 | 5.06 ± 0.03 | 8.76 ± 0.54 | 31 ± 3 | 2.5 ± 0.6 | 3.5 ± 0.2 | |||
| S23/24A + PKA (n= 7) | 2.3 | 5.07 ± 0.03 | — | 8.42 ± 0.55 | 0.16 | 51 ± 5* | 5.7 ± 1.5* | 3.2 ± 0.3 |
| 2.0 | 5.07 ± 0.04 | 8.59 ± 0.73 | 34 ± 4 | 2.4 ± 0.7 | 3.4 ± 0.2 | |||
| S23/24D (n= 8) | 2.3 | 4.98 ± 0.04 | 0.03 | 10.5 ± 0.91 | 0.77 | 54 ± 6* | 9.7 ± 2.6* | 2.3 ± 0.2 |
| 2.0 | 4.95 ± 0.04 | 11.3 ± 0.94 | 32 ± 3 | 3.6 ± 1.1 | 2.8 ± 0.4 | |||
| S23/24D + PKA (n= 8) | 2.3 | 4.91 ± 0.03 | — | 12.4 ± 0.81 | — | 50 ± 6* | 8.3 ± 1.8* | 2.5 ± 0.3 |
| 2.0 | 4.91 ± 0.02 | 12.4 ± 0.54 | 31 ± 4 | 3.5 ± 1.1 | 2.6 ± 0.2 |
P < 0.05 compared to SL 2.0 μm, indicating length-dependent effects. †P < 0.05 compared to paired samples after PKA treatment.
Figure 2. Force-pCa relationship of WT trabeculae before and after PKA treatment.
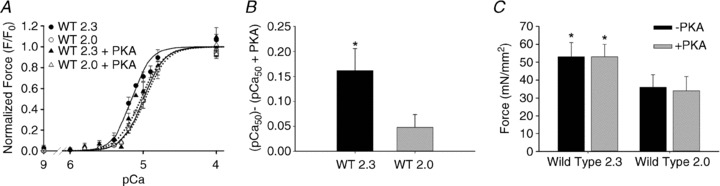
A, force–pCa relations are illustrated for WT trabeculae. Data (means ± SEM) were 2.3 (closed symbols) and 2.0 (open symbols) μm SL before (○; solid lines) and after treatment with the catalytic subunit of PKA (Δ; dotted lines). B, pCa50 for WT trabeculae before and after PKA treatment at SL 2.3 μm (filled bars) and 2.0 μm (grey bars), respectively. C, maximal Ca2+-activated (pCa 4.5) force at 2.3 and 2.0 μm SL before (filled bars) and after PKA treatment (grey bars). Data (means ± SEM) were obtained from eight trabeculae. *P < 0.05 compared to SL 2.0 μm.
To verify our WT Tn exchanged results, we measured the force–pCa relationship in native cardiac trabeculae at long and short SL, before and after PKA treatment. Consistent with other groups and our WT trabeculae results, the pCa50 of native trabeculae increased with increasing SL (Supplemental Fig. S3). Also similar to WT trabeculae, the length dependence of maximum force (Fmax) production was maintained before and after PKA treatment. In a subset of experiments, measurements were made with native trabeculae only following PKA treatment, to check for biases related to the order of treatment. No differences were found between these post-PKA treatment groups. It is worth noting that we observed a 0.28 pCa unit decrease at both long and short SL following exchange of recombinant Tn into skinned trabeculae. Despite this decrease in the absolute value of pCa50, physiological effects were maintained as the length-dependent difference in Ca2+ sensitivity, ΔpCa50, and force levels were equal to native samples before and after PKA treatment.
PKA effects on SL-dependent activation with cTnI phosphorylation mutants
To investigate the role of cTnI phosphorylation independent of PKA-mediated effects on cMyBP-C and titin, we substituted endogenous Tn with non-phos-phorylatable cTnI (S23/24A) Tn or phosphomimetic cTnI (S23/24D) Tn into demembranated rat cardiac trabeculae. S23/24A trabeculae exhibited similar SL-dependent effects as WT preparations, with a greater pCa50 and Fmax at long SL (Fig. 3A). To determine the influence of cMyBP-C/titin phosphorylation, S23/24A trabeculae were treated with PKA. This significantly decreased pCa50 at the long SL but had no effect at the short SL (Fig. 3C), effectively eliminating the SL dependence of pCa50, similar to results for WT trabeculae (Table 1). Because Tn exchange efficiency was high, and cTnI (S23/24A) cannot be phosphorylated by PKA, these results imply that PKA phosphorylation of cMyBP-C/titin was probably the main contributor to both decreased Ca2+ sensitivity of force and elimination of its SL dependence. However, the possible contribution of a small fraction of remaining native, phosphorylatable cTnI cannot be ruled out.
Figure 3. Force-pCa relationship of S23/24A and S23/24D trabeculae before and after PKA treatment.
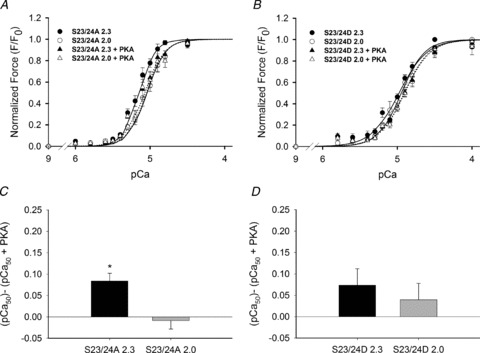
A, force–pCa relationships for S23/24A trabeculae. Data (means ± SEM; n= 8) were obtained at 2.3 (closed symbols) and 2.0 (open symbols) μm SL before (○; solid lines) and after treatment with the catalytic subunit of PKA (Δ; dotted lines). B, force–pCa relationships for S23/24D trabeculae. Data (means ± SEM; n= 8) were obtained at 2.3 (closed symbols) and 2.0 (open symbols) μm SL before (○; solid lines) and after treatment with the catalytic subunit of PKA (Δ; dotted lines). ΔpCa50 for S23/24A (C) and S23/24D (D) trabeculae before and after PKA treatment at SL 2.3 μm (filled bars) and 2.0 μm (grey bars), respectively. *P < 0.05 compared to SL 2.0 μm.
When trabeculae were exchanged with the N-terminal cTnI phosphomimetic S23/24D, we observed a significantly decreased pCa50 at both SL, with a greater effect at long (−0.18 pCa units) vs. short SL (−0.11 units) when compared to S23/24A trabeculae. This effectively eliminated the SL dependence of pCa50 (Fig. 3B). Together with the maintained SL dependence of pCa50 for S23/24A trabeculae, the data strongly indicate that phosphorylation of cTnI serines 23 and 24 alone can eliminate the SL dependence of myofilament calcium sensitivity. PKA treatment of S23/24D trabeculae caused a small but statistically insignificant further decrease in pCa50 (Fig. 3B; Table 1). This implies the possible contribution of cMyBP-C/titin phosphorylation to decreasing the Ca2+ sensitivity of force that we observed with S23/24A trabeculae. In total, these data suggest potential overlapping roles of both cMyBP-C/titin and cTnI in modulating length dependence of force during PKA phosphorylation. It also worth noting that there was little to no phosphatase activity in our demembranated preparations. As a result, the exchange of S23/24D or phosphorylation by PKA treatment was assumed to be irreversible. Physiologically, we would expect that phosphorylation turnover (i.e. balance of kinase and phosphatase activity) would impact how cTnI and/or cMyBP-C phosphorylation influences SL dependence.
Effects of cTnI variants and PKA treatment on myofilament structure by low-angle X-ray diffraction
To determine if changes in the SL dependence of the force–pCa relationship were correlated with myofilament structural changes, we performed low-angle X-ray diffraction experiments in resting trabeculae (pCa 9.0) under all conditions for which mechanical data were obtained. Measurements included the inter-thick filament spacing (d1,0) and the ratio of the integrated intensities of the 1,0 and 1,1 equatorial X-ray reflections (I1,1/I1,0) at 2.3 μm SL. Representative low-angle X-ray diffraction patterns from a skinned WT trabecula before and after PKA treatment are illustrated in Fig. 4A, along with a scan of the integrated intensity along the equator from the diffraction patterns (Fig. 4B). The value of d1,0 was not significantly different for any experimental conditions. Notably, d1,0 was not different between S23/24A trabeculae and S23/24D trabeculae, suggesting that altered myofilament lattice spacing did not contribute to the loss of SL dependence of pCa50 with cTnI (S23/24D) Tn. In all cases PKA treatment cause a small (∼1 nm) increase in d1,0 (Fig. 5A), as observed by others (Colson et al. 2008, 2010).
Figure 4. Representative low-angle X-ray diffraction patterns of WT Trabeculae.
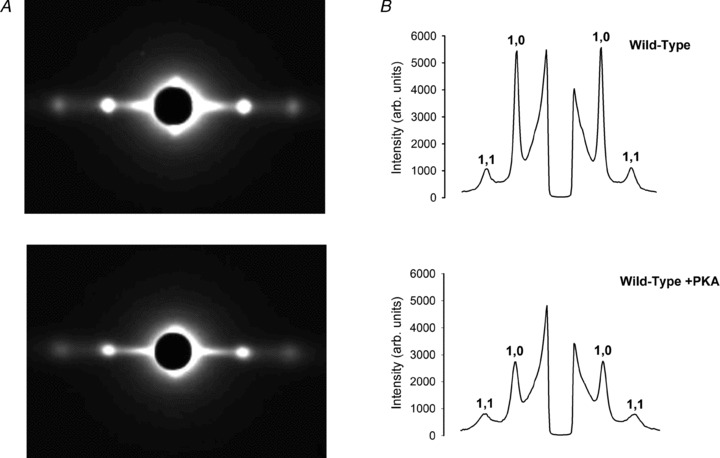
A, representative equatorial patterns from low-angle X-ray diffraction were obtained from WT trabeculae before and after treatment with PKA. B, the corresponding traces of diffraction intensities integrated along the equator vs. spacing in A are illustrated. The 1,0 and 1,1 diffraction peaks are labelled.
Figure 5. Interfilament spacing and equatorial intensity ratios before and after PKA treatment.
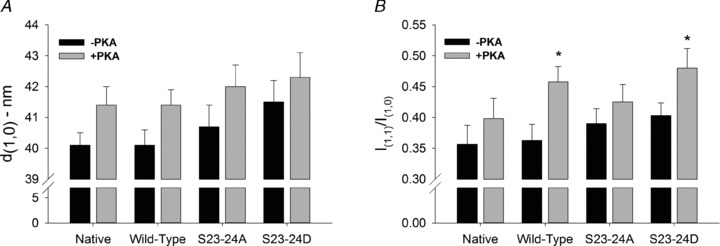
A and B, inter-thick filament spacing (d1,0) (A) and the ratio of 1,1 to 1,0 peak intensities (I1,1/I1,0) (B) obtained from native (n= 9, n= 7 after PKA), WT (n= 15, n= 12 after PKA), S23/24A (n= 13, n= 10 after PKA) or S23/24D (n= 12, n= 9 after PKA) trabeculae. Values (means + SEM) are shown before (filled bars) and after (grey bars) treatment with PKA for each condition. Data were analysed by fitting the diffraction intensity profiles with Fityk to determine the position of the centroid of the peaks and the values of the integrated intensities. *P < 0.05 compared to before PKA treatment.
The equatorial intensity ratio, I1,1/I1,0, provides information on the distribution of CB mass between the thick and thin filaments (Millman, 1998), with increasing I1,1/I1,0 indicating a redistribution of CB mass from thick towards thin filaments. Recent evidence from low-angle X-ray diffraction experiments shows an increase in I1,1/I1,0 following PKA phosphorylation of cMyBP-C (Colson et al. 2008, 2010). In the current study, prior to PKA treatment, equatorial intensity ratios were not significantly different for any condition (Fig. 5B). PKA treatment increased I1,1/I1,0 for WT and S23/24D trabeculae, while the intensity ratio appeared to increase slightly (but not significantly) following PKA treatment of S23/24A and native trabeculae (Fig. 5B).
Discussion
The goal of this study was to determine the particular role of PKA phosphorylation of the cardiac thin and thick filament proteins in modulating the SL dependence of myocardial force generation, i.e. the sarcomere level basis of the Frank–Starling relationship. PKA phosphorylates both the thin filament cardiac troponin subunit cTnI (Kobayashi & Solaro, 2005) and thick filament associated proteins, cMyBP-C (Winegrad, 2000; Flashman et al. 2004) and titin (Granzier & Labeit, 2004), all of which have been shown to modulate cardiac contractility. By replacing endogenous Tn with recombinant WT Tn, a non-phosphorylatable cTnI (S23/24A) Tn or the phosphomimetic cTnI (S23/24D) Tn, we determined that phosphorylation of cTnI or cMyBP-C/titin, individually or in combination, significantly reduced the SL dependence of calcium sensitivity (pCa50). This occurred primarily through a reduction of pCa50 at the long SL where the propensity for strong CB formation is greater. However, PKA phosphorylation did not alter the SL dependence of Fmax, suggesting the SL dependence of CB binding and force was maintained. X-ray diffraction studies showed that CB mass movement under relaxing conditions was increased by PKA treatment. Taken together, our findings point to cTnC–cTnI interactions as a switch that sets the length dependence of calcium sensitivity in cardiac muscle, in conjunction with PKA-induced changes in CB structure.
Effects of cTnI and cMyBP-C/titin phosphorylation state on the Ca2+-sensitivity of cardiac force
Decreased pCa50 in cardiac muscle occurs in response to β-adrenergic stimulation (Li et al. 2004; Kobayashi & Solaro, 2005) and following PKA treatment (Fig. 2, Supplementary Fig. S3) and has been attributed primarily to phosphorylation of serines 23 and 24 (S23/24) in the N terminus of cTnI (Zhang et al. 1995a,b). At the protein level, cTnI S23/24 phosphorylation decreases the Ca2+ affinity of cTnC when complexed with cTnI and weakens the interaction between cTnI and the regulatory domain of cTnC (Finley et al. 1999; Howarth et al. 2007; Sadayappan et al. 2008). Structural data suggest that in the unphosphorylated state the cardiac-specific N-terminal extension of cTnI interacts with the N-terminal regulatory domain of cTnC, stabilizing the ‘open’ or Ca2+-activated cTnC state (Howarth et al. 2007). Our finding that S23/24D trabeculae have reduced Ca2+ sensitivity of force (Fig. 3), relative to non-phosphorylatable S23/24A trabeculae, supports previous observations that alterations in cTnC–cTnI interaction by PKA phosphorylation can decrease the Ca2+ sensitivity of force (Zhang et al. 1995b). Furthermore, because PKA treatment of S23/24D trabeculae, which allows phosphorylation primarily of cMyBP-C/titin (and not cTnI), had no significant additional effect on pCa50, the data confirm a significant role for cTnI S23/24 phosphorylation in regulating the Ca2+ sensitivity of force (Wattanapermpool et al. 1995).
PKA phosphorylation of the cardiac-specific motif of cMyBP-C reduces cMyBP-C interaction with the S2 domain of myosin (Harris et al. 2004) and is associated with a redistribution of myosin CB mass towards thin filaments (Colson et al. 2008, 2010). This implies the potential for increased myosin–thin filament interaction, which is supported by observations that increased force development kinetics following PKA treatment is specific to cMyBP-C phosphorylation (Tong et al. 2008; Chen et al. 2010). In this study, PKA treatment of S23/24A trabeculae increased cMyBP-C phosphorylation 3-fold (Fig. 1D) and was associated with reduced Ca2+ sensitivity of force (Fig. 3C); there was no corresponding PKA-induced elevation of cTnI phosphorylation. Reduction of pCa50 by cMyBP-C phosphorylation is consistent with previous studies where RLC2 phosphorylation was controlled (Chen et al. 2010). In addition to effects on cMyBP-C, PKA phosphorylation increases the compliance of titin and is associated with increased d1,0 in skinned cardiac trabeculae and reduced passive force, at least at longer SL (Fukuda et al. 2005). Here we found that PKA treatment induced a small (∼1.0 nm) increase of d1,0 under all conditions tested (Fig. 5A). By itself increasing d1,0 would be expected to reduce pCa50 (Fuchs & Martyn, 2005), although this should have contributed similarly to pCa50 reduction under all conditions tested. However, it must be considered that the relationship between PKA treatment, Ca2+ sensitivity and lattice spacing may not be unique. For example, PKA treatment does not always result in increased lattice spacing and changes in lattice spacing do not always have an inverse relationship with pCa50 (Konhilas et al. 2003). PKA treatment of skinned trabeculae from transgenic mice that expressed the slow skeletal isoform of cTnI decreased myofilament lattice spacing, with no change in pCa50. In contrast, PKA treatment increased lattice spacing and reduced pCa50 in trabeculae of non-transgenic animals. These observations emphasize the complexity of the molecular interactions that underlie the coupling between lattice spacing and Ca2+ sensitivity in cardiac muscle.
Effects of cTnI and cMyBP-C phosphorylation state on the SL dependence of force–[Ca2+] relations
Previous studies to determine the effects of PKA treatment on the length-dependent force activation in cardiac muscle have produced complex and often contradictory results. PKA treatment of skinned cardiac muscle preparations was found to either increase (Konhilas et al. 2000, 2003; Hanft & McDonald, 2009) or decrease (Kajiwara et al. 2000) the SL dependence of Ca2+ sensitivity. Furthermore, PKA treatment was found either not to change or to decrease the SL dependence of Ca2+ sensitivity (ΔpCa50) when measured in younger or older mice, respectively (Cazorla et al. 2006). Similar to our studies, Kajiwara et al. (2000) found that PKA treatment reduced ΔpCa50, primarily by a greater reduction of Ca2+ sensitivity at longer SL. In contrast, others (Konhilas et al. 2003) have observed an increase in ΔEC50 following PKA treatment of skinned trabeculae from non-transgenic mice, although ΔpCa50 was unaffected. Recent studies have also shown that PKA phosphorylation of cMyBP-C and cTnI increases the SL dependence of power output (Hanft & McDonald, 2009). Additional complexity is indicated by observations that in younger transgenic cMyBP-C knock-out mice ΔpCa50 was increased by PKA, relative to younger non-transgenic preparations in which PKA had no significant effect on ΔpCa50 (Cazorla et al. 2006). In the same study ΔpCa50 was reduced by PKA treatment in older non-transgenic animals, while in older cMyBP-C knock-out mice both ΔpCa50 and the effect of PKA on ΔpCa50 was reduced relative to non-transgenic controls.
The source of these disparities between multiple studies is not clear, but a possible explanation is that the effect of PKA on length-dependent activation is a complex function of differences in the relative phosphorylation levels of RLC, cTnI and cMyBP-C. For example, RLC phosphorylation was not controlled in these earlier studies, and has subsequently been shown to impact the contractile response to PKA activation. Verduyn et al. (2007) and Chen et al. (2010) observed a larger decrease in Ca2+ sensitivity following PKA treatment in skinned myocardium when RLC and cMyBP-C phosphorylation were reduced. Chen et al. (2010) found that following PKA treatment Ca2+ sensitivity decreased most when both cTnI and cMyBP-C could be phosphorylated. Additionally, Colson et al. (2010) found that while phosphorylation of both RLC-2 and cMyBP-C increase myosin CB proximity to thin filaments, elevation of RLC-2 phosphorylation alone increased Ca2+ sensitivity, while PKA treatment and phosphorylation of cTnI and cMyBP-C decreased the Ca2+ sensitivity of force. Unfortunately, the isolated effects of RLC and cMyBP-C phosphorylation levels on length-dependent activation have not been determined alone or in combination. In our study RLC phosphorylation level was unaffected by Tn exchange or by PKA treatment (Supplemental Fig. S2A), and we found that PKA treatment eliminated the SL dependence of pCa50 in native and WT trabeculae, in which both cTnI and cMyBP-C were phosphorylated. As exchange with S23/24D Tn also eliminated the SL dependence of pCa50, the results indicate that phosphorylation of the cTnI N terminus alone reduces cardiac length-dependent activation. Additionally, because PKA treatment reduced ΔpCa50 in S23/24A trabeculae, in which mutant cTnI cannot be phosphorylated by PKA, the evidence suggests that phosphorylation of cMyBP-C likewise reduced or eliminated the length-dependent Ca2+ sensitivity.
In addition to reducing ΔpCa50 in native, WT and S23/24A trabeculae, our data (Fig. 5) indicate that PKA treatment increases myofilament lattice spacing and promotes CB mass distribution towards thin filaments, as found in previous studies (Colson et al. 2007, 2008, 2010). Changes in lattice spacing can alter pCa50 independent of changes in SL (Fuchs & Martyn, 2005). However, our data indicate that decreases in pCa50 and reduction of the SL dependence of pCa50 by PKA do not result directly from alterations in d1,0. For example, the SL dependence of pCa50 is lost in S23/24D trabeculae, even though d1,0 at longer SL is similar to that of S23/24A trabeculae (Fig. 5), where pCa50 was dependent on SL (Fig. 4A). The value of d1,0 did not differ for S23/24A trabeculae compared with PKA-treated native and WT trabeculae, which also exhibit reduced SL dependence of pCa50. Assuming the d1,0 relationship between the conditions tested is not altered during contraction, these data suggest that the loss of SL dependence of pCa50 resulted primarily from the influences of cTnI phosphorylation state and cMyBP-C/titin phosphorylation, and not from PKA-induced changes in myofilament lattice spacing.
How does cTnI and cMyBP-C phosphorylation alter cardiac length-dependent activation?
A potential explanation for the disproportionate decrease in pCa50 at the longer SL (Table 1) may lie in the ability of strong-binding, force-generating CBs to displace tropomyosin into the activated position and the unique ability of CBs to enhance the Ca2+ affinity of cTnC in cardiac muscle (Gordon et al. 2000; Fuchs & Martyn, 2005). We previously suggested that enhanced strong-CB binding with low [ATP] increased pCa50 more at the shorter SL because the CB population available for recruitment and contribution to activation was greater at short SL (Adhikari et al. 2004). Using this same reasoning, if phosphorylation of cTnI and/or cMyBP-C/titin reduced coupling between strong CBs and thin filament activation, the effect should be more evident at longer SL as myosin S1 proximity to actin and potential for binding is greater with reduced lattice spacing (Martyn et al. 2004). Supporting this idea are observations that strong CB binding decreases the distance between cTnI and cTnC (Robinson et al. 2004) and is required to achieve the fully ‘open’ conformation of the cTnC N terminus (Dong et al. 2007). Furthermore, cTnI interaction with actin decreases with strong CB binding, independent of the effects of Ca2+ (Xing et al. 2008). Importantly, in the unphosphorylated state the N terminus of cTnI interacts with the N-terminal regulatory domain of cTnC and stabilizes the Ca2+-bound state (Finley et al. 1999; Gaponenko et al. 1999). As cTnI N-terminal phosphorylation weakens cTnI–cTnC interaction, CB-mediated increases in cTnC Ca2+ affinity may be reduced, consistent with a PKA-mediated reduction of pCa50 at longer SL. However, this speculation remains to be tested directly. It should be noted that our Tn exchange was not complete (∼80%), and thus it is possible, however unlikely, that reduced Ca2+ sensitivity and loss of pCa50 SL dependence resulted from phosphorylation of the small of amount of un-exchanged (native) cTnI. This would require that the full effects of PKA on pCa50 and ΔpCa50 through cTnI S23/24 phosphorylation were caused by phosphorylation of 10–20% of the cTnI pool. Moreover, evidence from others also argues that there is a significant contribution of cMyBP-C phosphorylation in reducing calcium sensitivity at the long SL. PKA treatment following alanine substitution at PKA phosphorylation sites of either cTnI or cMyBP-C results in a similar reduction of pCa50 at long SL (∼2.2 μm) (Chen et al. 2010). In cMyBP-C knock-out mice, pCa50 at the long SL was reduced by a lesser degree after PKA treatment (Cazorla et al. 2006).
While phosphorylation-mediated weakening of the cTnC-cTnI interaction may reduce the SL dependence of pCa50 (as argued above), the mechanism by which cMyBP-C phosphorylation could decrease pCa50 and its SL dependence is less clear. In agreement with others (Bardswell et al. 2010; Chen et al. 2010) cMyBP-C phosphorylation reduced pCa50 in our study, independent of RLC or cTnI phosphorylation. Several studies suggest that phosphorylated cMyBP-C interacts with cardiac thin filaments (Whitten et al. 2008; Shaffer et al. 2009; Kensler et al. 2011), while our study suggests more generally that phosphorylated cMyBP-C reduces thin filament activation at sub-maximal [Ca2+] (Fig. 3A; Table 1). The increase in I1,1/I1,0 with cMyBP-C phosphorylation (Fig. 5) indicates CBs are closer to the surface of thin filaments and potentially available for weak-binding interaction with thin filaments. We previously demonstrated that SL- and/or lattice spacing-induced changes in weak myosin binding to cardiac thin filaments were an important component regulating subsequent strong CB binding and thin filament activation (Martyn et al. 2004). Supporting this idea are observations that lowering ionic strength (Smith & Fuchs, 1999) or temperature (Martyn & Smith, 2005), which both favour weak CB binding (Martyn et al. 2004), reduce the SL dependence of pCa50. Thus, it is possible that cMyBP-C phosphorylation mediated reduction of the SL dependence of pCa50 results because changes in SL or myofilament lattice spacing induce relatively smaller changes in the enhanced population of weak binding CBs.
Others have found that cMyBP-C phosphorylation state can determine the level of maximal Ca2+-activated force, possibly through an influence of cMyBP-C on myosin head orientation (Kulikovskaya et al. 2003). In fact, recent studies have reported that RLC phosphorylation influences myosin head orientation and force production (Farman et al. 2011). However, we observed that Fmax was unaffected either by phosphomimetic, S23/24D, exchange or by PKA treatment (Table 1). A potential explanation may be that at higher [Ca2+], thin filament state is dominated by strong CB displacement of tropomyosin into its ‘on’ position, such that weakening of cTnI–cTnC interaction (by cTnI phosphorylation) or increased myosin S1 proximity to thin filaments (cMyBP-C phosphorylation) would have reduced influence on force. Furthermore, the lack of a PKA effect on Fmax at 2.3 or 2.0 μm SL under all conditions tested indicates that the effect of SL on force at sub-maximal pCa was present, but attenuated. Thus, the Frank–Starling effect was not eliminated, just reduced.
The attenuation of SL dependence of force at all levels of thin filament activation is illustrated by the solid curves in Fig. 6, which plots the difference in force at 2.3 vs. 2.0 μm SL at each activating pCa. As in our previous study (Adhikari et al. 2004), the sensitivity of force to decreasing SL reached a maximum over the physiologically relevant range of sub-maximal pCa for native (Fig. 6A), WT (Fig. 6B) and S23/24A trabeculae (Fig. 6C). The peak in each force difference curve corresponds to increased force sensitivity to altered SL. We have suggested this peak results from optimal ‘tuning’ of the interaction between Ca2+ binding to Tn and CB binding to activate cardiac thin filaments (Adhikari et al. 2004). Following treatment with PKA (corresponding dashed lines in Fig. 6) these peaks were eliminated for native, WT and S23/24A preparations and the SL dependence of force was reduced over the physiologically relevant pCa range. It is also noteworthy that S23/24D trabeculae (Fig. 6D) produced changes in these force difference curves that mimic the effects of PKA treatment, including loss of the peak (compare Fig. 6D with dashed lines in Fig. 6A–C).
Figure 6. Difference in force–[Ca2+] relationships.
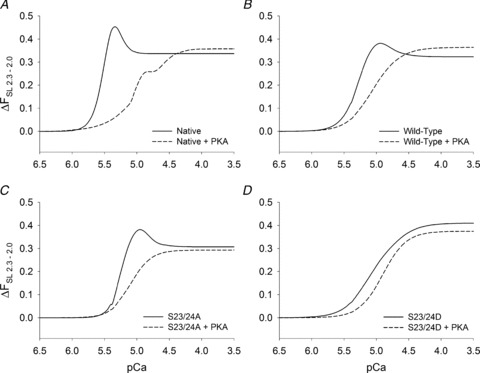
Difference between Hill fit curves to the data obtained at 2.3 and 2.0 μm SL and data illustrated in Figs 2, 3 and 4 are shown for native (no exchange, A), WT (B), S23/24A (C) and S23/24D (D) trabeculae. Data are shown before (solid curves) and after (dashed curves) treatment with PKA.
Physiological consequence of PKA phosphorylation for the Frank–Starling relationship
If the net effect of PKA-mediated phosphorylation of cTnI, cMyBP-C and titin is to decrease the Ca2+ sensitivity of force and its SL dependence, what is the physiological relevance? At normal levels of activity the steep SL dependence of myocardial force generation enables the heart to respond automatically to small alterations in venous return, without nervous or endocrine input. However, at heightened levels of activity, β-adrenergic stimulation increases heart rate, elevates myoplasmic [Ca2+] and force, increases the kinetics for force development and shortening, and also increases the rate of ventricular relaxation and diastolic filling to maintain high cardiac output (Bers, 2002). Furthermore, myofilament Ca2+ sensitivity is reduced to support rapid resequestration of myoplasmic Ca2+ by the sarcoplasmic reticulum, allowing faster relaxation for increased diastolic filling. Under these conditions a steep dependence of cardiac force on sarcomere length or ventricular volume could limit ventricular output during a cardiac contractile cycle by reducing force and ventricular ejection fraction, as shortening proceeds in systole. Our results indicate that following PKA treatment, pCa50 at long SL is reduced (Fig. 7, grey dashed line), with no effects on maximum force production, so that pCa50 at the long and short SL are identical and the difference in force production between long and short SL is reduced. Reduction of the effect of shortening on the Ca2+ sensitivity of contractile activation by PKA-mediated myofilament protein phosphorylation, in combination with elevated myoplasmic [Ca2+] during β-adrenergic stimulation, should help maintain ventricular force generation throughout systole. Thus, at a given [Ca2+] the ability of PKA to blunt a relative reduction of force from shortening during systole could increase the ventricular ejection fraction and help maintain cardiac output per contraction cycle during periods of metabolic demand.
Figure 7. Physiological relevance of PKA phosphorylation of myofibrillar proteins.
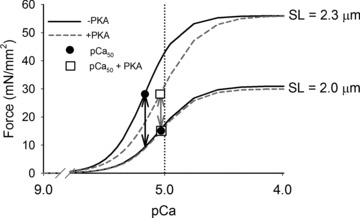
The effects of PKA treatment on the fitted force–pCa curves for WT trabeculae from Fig. 2 are described. Prior to PKA treatment (black lines), pCa50 (filled circles) at the long SL is greater than at the short SL. The difference in force production between long and short SL (black arrows) at the long SL pCa50 is illustrated. Following PKA treatment (grey, dashed lines), pCa50 at the long SL (open box) is reduced, with no effects on maximum force production. The pCa50 values at the long and short SL (open boxes) are identical and the difference in force production (grey arrows) between long and short SL is reduced not only at long SL pCa50, but also over the range of submaximal activating [Ca2+].
Acknowledgments
We would like to thank Drs An-Yue Tu and Charles Luo for preparations of cTnI mutant proteins and protein isolation, and Dr Jin Dai for preparation of trabeculae and protein gels for characterization of protein content and phosphorylation. We would also like to thank Mengjie Zhang and Luping Xie for help with analysing X-ray diffraction patterns. We are indebted to Martha Mathiason for development data acquisition and analysis software. This research was supported by NIH RO1 HL-67071 (D.A.M.), NIH R01 HL-65497 (M.R.) and AHA 11POST7400069 (V.S.R.). Use of the Advanced Photon Source, an Office of Science User Facility operated for the US Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the US DOE under Contract No. DE-AC02–06CH11357. BioCAT is a National Institutes of Health-supported Research Centre RR-08630. The content is solely the responsibility of the authors and does not necessarily reflect the official views of the National Centre for Research Resources or the National Institutes of Health.
Glossary
- CB
crossbridge
- cMyBP-C
cardiac myosin binding protein-C
- cTnC
cardiac troponin C
- cTnI
cardiac troponin I
- cTnT
cardiac troponin T
- d1,0
inter-thick filament spacing
- ΔEC50
difference in EC50 between SL 2.3 and 2.0 μm
- ΔpCa50
difference in pCa50 between SL 2.3 and 2.0 μm
- Fmax
maximum steady-state force (mN mm−2)
- Fpass
resting tension (mN mm−2)
- I1.1/I1,0
equatorial intensity ratio of 1,1 and 1,0 peaks of X-ray diffraction pattern
- nH
Hill coefficient
- pCa50
calcium sensitivity of force
- PKA
protein kinase A
- RLC
myosin regulatory light chain 2
- S23/24A
recombinant troponin complex containing S23/24A cTnI
- S23/24D
recombinant troponin complex containing S23/24D cTnI
- SL
sarcomere length
- Tn
troponin complex
- WT
recombinant wild-type troponin complex
Author contributions
V.S.R. contributed to experimental design, performed the experiments at the University of Washington and Argonne National Laboratory, analysed and interpreted the data, and contributed to writing the manuscript. F.S.K. contributed to the design of experiments and critically reviewed the manuscript. M.V.R. contributed to the design of experiments, performed mechanics experiments, analysed the results and critically reviewed the manuscript. E.R.F. contributed to the design and performed the experiments at the University of Washington and Argonne National Laboratory. H.M.H. supported experiments at Argonne National Laboratory and analysed low-angle X-ray diffraction data. T.I. contributed to the design of X-ray diffraction studies and critically reviewed the manuscript. M.R. contributed to overall study design, interpretation of results and contributed to writing the manuscript. D.A.M. contributed to overall study design, interpretation of results and contributed to writing of the manuscript. All authors approved the final version of this manuscript for publication.
Supplementary material
Supplemental Fig. S1
Supplemental Fig. S2A
Supplemental Fig. S3
References
- Adhikari BB, Regnier M, Rivera AJ, Kreutziger KL, Martyn DA. Cardiac length dependence of force and force redevelopment kinetics with altered crossbridge cycling. Biophys J. 2004;87:1784–1794. doi: 10.1529/biophysj.103.039131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arteaga GM, Palmiter KA, Leiden JM, Solaro RJ. Attenuation of length dependence of calcium activation in myofilaments of transgenic mouse hearts expressing slow skeletal troponin I. J Physiol. 2000;526:541–549. doi: 10.1111/j.1469-7793.2000.t01-1-00541.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardswell SC, Cuello F, Rowland AJ, Sadayappan S, Robbins J, Gautel M, Walker JW, Kentish JC, Avkiran M. Distinct sarcomeric substrates are responsible for protein kinase D-mediated regulation of cardiac myofilament Ca2+ sensitivity and cross-bridge cycling. J Biol Chem. 2010;285:5674–5682. doi: 10.1074/jbc.M109.066456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM. Cardiac excitation–contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- Burkart EM, Sumandea MP, Kobayashi T, Nili M, Martin AF, Homsher E, Solaro RJ. Phosphorylation or glutamic acid substitution at protein kinase C sites on cardiac troponin I differentially depress myofilament tension and shortening velocity. J Biol Chem. 2003;278:11 265–11 272. doi: 10.1074/jbc.M210712200. [DOI] [PubMed] [Google Scholar]
- Cazorla O, Szilagyi S, Vignier N, Salazar G, Kramer E, Vassort G, Carrier L, Lacampagne A. Length and protein kinase A modulations of myocytes in cardiac mysoin binding protein C-deficient mice. Cardiovasc Res. 2006;69:370–380. doi: 10.1016/j.cardiores.2005.11.009. [DOI] [PubMed] [Google Scholar]
- Cazorla O, Wu Y, Irving TC, Granzier H. Titin-based modulation of calcium sensitivity of active tension in mouse skinned cardiac myocytes. Circ Res. 2001;88:1028–1035. doi: 10.1161/hh1001.090876. [DOI] [PubMed] [Google Scholar]
- Chandra M, Dong WJ, Pan BS, Cheung HC, Solaro RJ. Effects of protein kinase A phosphorylation on signaling between cardiac troponin I and the N-terminal domain of cardiac troponin C. Biochem. 1997;36:13,305–13,311. doi: 10.1021/bi9710129. [DOI] [PubMed] [Google Scholar]
- Chen PP, Patel JR, Rybakova IN, Walker JW, Moss RL. Protein kinase A-induced myofilament desensitization to Ca2+ as a result of phosphorylation of cardiac myosin-binding protein C. J Gen Physiol. 2010;186:615–627. doi: 10.1085/jgp.201010448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colson BA, Bekyarova T, Fitzsimons DP, Irving TC, Moss RL. Radial displacement of myosin-crossbridges in mouse myocardium due to ablation of myosin binding protein C. J Mol Biol. 2007;376:36–41. doi: 10.1016/j.jmb.2006.12.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colson BA, Bekyarova T, Locher MR, Fitzsimmons DP, Irving TC, Moss RL. Protein kinase A-mediated phosphorylation of cMyBP-C increases proximity of myosin heads to actin in resting myocardium. Circ Res. 2008;103:244–251. doi: 10.1161/CIRCRESAHA.108.178996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colson BA, Locher MR, Bekyarova T, Patel JR, Fitzsimons DP, Irving TC, Moss RL. Differential roles of regulatory light chain and myosin binding protein-C phosphorylation in the modulation of cardiac force development. J Physiol. 2010;588:981–993. doi: 10.1113/jphysiol.2009.183897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohet C, Al-Hillawi E, Trayer IP, Ruegg JC. Reconstitution of skinned cardiac fibres with human recombinant cardiac troponin-I mutants and troponin-C. FEBS Lett. 1995;377:131–134. doi: 10.1016/0014-5793(95)01319-9. [DOI] [PubMed] [Google Scholar]
- Dong W-J, Jayasundar JJ, An J, Xing J, Cheung HC. Effects of PKA phosphorylation of cardiac troponin I and strong crossbridge on conformational transitions of the N-domain of cardiac troponin C in regulated thin filaments. Biochemistry. 2007;46:9752–9761. doi: 10.1021/bi700574n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabiato A. Computer programs for calculating total from specified free or free from specified total ionic concentrations in aqueous solutions containing multiple metals and ligands. Methods Enzymol. 1988;157:378–417. doi: 10.1016/0076-6879(88)57093-3. [DOI] [PubMed] [Google Scholar]
- Farman GP, Gore D, Allen E, Schoenfelt K, Irving TC, de Tombe PP. Myosin head orientation: a structural determinant for the Frank–Starling relationship. Am J Physiol Heart Circ Physiol. 2011;300:H2155–H2160. doi: 10.1152/ajpheart.01221.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley N, Abbott MB, Abusamhadneh E, Gaponenko V, Dong W, Gasmi-Seabrook G, Howarth JW, Rance M, Solaro RJ, Cheung HC, Rosevear PR. NMR analysis of cardiac troponin C-troponin I complexes: effects of phosphorylation. FEBS Lett. 1999;453:107–112. doi: 10.1016/s0014-5793(99)00693-6. [DOI] [PubMed] [Google Scholar]
- Fischetti R, Stepanov S, Rosenbaum G, Barrea R, Black E, Gore D, Heurich R, Kondrashkina E, Kropf AJ, Wang S, Zhang K, Irving TC, Bunker GB. The BioCAT undulator beamline 18ID: a facility for biological non-crystalline diffraction and X-ray absorption spectroscopy at the Advanced Photon Source. J Synchrotron Radiat. 2004;11:399–405. doi: 10.1107/S0909049504016760. [DOI] [PubMed] [Google Scholar]
- Flashman E, Redwood C, Moolman-Smook J, Watkins H. Cardiac myosin binding protein C: its role in physiology and disease. Circ Res. 2004;94:1279–1289. doi: 10.1161/01.RES.0000127175.21818.C2. [DOI] [PubMed] [Google Scholar]
- Fuchs F, Martyn DA. Length-dependent activation in cardiac muscle: some remaining questions. J Musc Res Cell Motil. 2005;26:199–212. doi: 10.1007/s10974-005-9011-z. [DOI] [PubMed] [Google Scholar]
- Fukuda N, Wu Y, Nair P, Granzier H. Phosphorylation of titin modulates passive stiffness of cardiac muscle in a titin isoform-dependent manner. J Gen Physiol. 2005;125:257–271. doi: 10.1085/jgp.200409177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaponenko V, Abusamhadneh E, Abbott MB, Finley N, Gasmi-Seabrook G, Solaro RJ, Rance M, Rosevear PR. Effects of troponin I phosphorylation on conformational exchange in the regulatory domain of cardiac troponin C. J Biol Chem. 1999;274:16,681–16,684. doi: 10.1074/jbc.274.24.16681. [DOI] [PubMed] [Google Scholar]
- Gillis TE, Martyn DA, Rivera AJ, Regnier M. Investigation of thin filament near-neighbour regulatory unit interactions during force development in skinned cardiac and skeletal muscle. J Physiol. 2007;580:561–576. doi: 10.1113/jphysiol.2007.128975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon AM, Homsher E, Regnier M. Regulation of contraction in striated muscle. Physiol Rev. 2000;80:853–924. doi: 10.1152/physrev.2000.80.2.853. [DOI] [PubMed] [Google Scholar]
- Granzier H, Labeit D, Wu Y, Labeit S. Titin as a modular spring: emerging mechanisms for elasticity control by titin in cardiac physiology and pathophysiology. J Musc Res Cell Motil. 2002;23:457–471. doi: 10.1023/a:1023458406346. [DOI] [PubMed] [Google Scholar]
- Granzier H, Labeit S. Cardiac titin: an adjustable multi-functional spring. J Physiol. 2002;541:335–342. doi: 10.1113/jphysiol.2001.014381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granzier H, Labeit S. The giant protein Titin: a major player in myocardial mechanics, signalling and disease. Circ Res. 2004;94:284–295. doi: 10.1161/01.RES.0000117769.88862.F8. [DOI] [PubMed] [Google Scholar]
- Hanft LM, McDonald KS. Sarcomere length dependence of power output is increased after PKA treatment in rat cardiac myocytes. Am J Physiol Heart Circ Physiol. 2009;296:H1524–H1531. doi: 10.1152/ajpheart.00864.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris SP, Rostkova EV, Gautel M, Moss RL. Binding of myosin binding protein-C to myosin subfragment S2 affects contractility independent of a tether mechanism. Circ Res. 2004;95:930–936. doi: 10.1161/01.RES.0000147312.02673.56. [DOI] [PubMed] [Google Scholar]
- Howarth J, Meller J, Solaro RJ, Trewhella J, Rosevear PR. Phosphorylation-dependent conformational transition of the cardiac specific N-extension of troponin-I in cardiac troponin. J Mol Biol. 2007;373:706–722. doi: 10.1016/j.jmb.2007.08.035. [DOI] [PubMed] [Google Scholar]
- Irving TC, Konhilas J, Perry D, Fischetti R, de Tombe PP. Myofilament lattice spacing as a function of sarcomere length in isolated rat myocardium. Am J Physiol Heart Circ Physiol. 2000;279:H2568–H2573. doi: 10.1152/ajpheart.2000.279.5.H2568. [DOI] [PubMed] [Google Scholar]
- Irving TC, Millman BM. Changes in thick filament structure during compression of the filament lattice in relaxed frog sartorius muscle. J Muscle Res Cell Motil. 1989;10:385–394. doi: 10.1007/BF01758435. [DOI] [PubMed] [Google Scholar]
- Kajiwara H, Morimoto S, Fukuda N, Ohtsuki I, Kurihara S. Effects of troponin I phosphorylation by protein kinase A on length-dependence of tension in rat skinned myocardium. Biochem Biophys Res Comm. 2000;272:104–110. doi: 10.1006/bbrc.2000.2741. [DOI] [PubMed] [Google Scholar]
- Kensler RW, Shaffer JF, Harris SP. Binding of N-terminal fragment C0–C2 of cardiac MyBP-C to cardiac F-actin. J Struct Biol. 2011;174:44–51. doi: 10.1016/j.jsb.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kentish JC, McCloskey DT, Layland J, Palmer S, Leiden JM, Martin AF, Solaro RJ. Phosphorylation of troponin I by protein kinase A accelerates relaxation and crossbridge cycle kinetics in mouse ventricular muscle. Circ Res. 2001;88:1059–1065. doi: 10.1161/hh1001.091640. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Solaro RJ. Calcium, thin filaments and the integrative biology of cardiac contractility. Annu Rev Physiol. 2005;67:39–67. doi: 10.1146/annurev.physiol.67.040403.114025. [DOI] [PubMed] [Google Scholar]
- Kohler J, Chen Y, Brenner B, Gordon AM, Kraft T, Martyn DA, Regnier M, Rivera AJ, Wang CK, Chase PB. Familial hypertrophic cardiomyopathy mutations in troponin I (K183D, G203S, K206Q) enhance filament sliding. Physiol Genomics. 2003;14:117–128. doi: 10.1152/physiolgenomics.00101.2002. [DOI] [PubMed] [Google Scholar]
- Konhilas J, Irving TC, Wolska B, Jweied EE, Martin A, Solaro RJ, de Tombe PP. Troponin I in the murine myocardium: influence on length dependent activation and interfilament spacing. J Physiol. 2003;547:951–961. doi: 10.1113/jphysiol.2002.038117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konhilas JP, Wolska BM, Martin AF, Solaro RJ, de Tombe PP. PKA modulates length-dependent activation in murine myocardium. Biophys J. 2000;78:108A. doi: 10.1113/jphysiol.2002.038117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger M, Linke WA. Protein kinase-A phosphorylates titin in human heart muscle and reduces myofibrillar passive tension. J Muscle Res Cell Motil. 2006;27:435–444. doi: 10.1007/s10974-006-9090-5. [DOI] [PubMed] [Google Scholar]
- Kulikovskaya I, McClellan G, Flavigny J, Carrier L, Winegrad S. Effect of MyBP-C binding to actin on contractility in heart muscle. J Gen Physiol. 2003;122:761–774. doi: 10.1085/jgp.200308941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layland J, Solaro RJ, Shah AM. Regulation of cardiac contractile function by troponin I phosphorylation. Cardiovasc Res. 2005;66:12–21. doi: 10.1016/j.cardiores.2004.12.022. [DOI] [PubMed] [Google Scholar]
- Li MX, Wang X, Lindhout DA, Buscemi N, van Eyk J, Sykes BD. Phosphorylation and mutation of cardiac troponin I deferentially destabilize the interaction of the functional regions of troponin I with troponin C. Biochemistry. 2003;42:14 460–14 468. doi: 10.1021/bi035408y. [DOI] [PubMed] [Google Scholar]
- Li MX, Wang X, Sykes BD. Structure based insights into the role of troponin in cardiac muscle pathophysiology. J Musc Res Cell Motil. 2004;25:559–579. doi: 10.1007/s10974-004-5879-2. [DOI] [PubMed] [Google Scholar]
- Martyn DA, Adhikari BB, Regnier M, Gu J, Xu S, Yu L. Response of equatorial x-ray reflections and stiffness to altered sarcomere length and myofilament lattice spacing in relaxed skinned cardiac muscle. Biophys J. 2004;86:1002–1011. doi: 10.1016/S0006-3495(04)74175-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martyn DA, Smith L. The temperature dependence of length-dependent activation in cardiac muscle. Biophys J. 2005;88:120a. [Google Scholar]
- Millman B. The filament lattice of striated muscle. Physiol Rev. 1998;78:360–391. doi: 10.1152/physrev.1998.78.2.359. [DOI] [PubMed] [Google Scholar]
- Moir AJG, Solaro RJ, Perry SV. The site of phosphorylation of troponin I in the perfused rabbit heart. Biochem J. 1980;185:505–513. doi: 10.1042/bj1850505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moolman-Smook J, Flashman E, de Lange W, Li Z, Corfield V, Redwood C, Watkins H. Identification of novel interactions between domains of myosin binding protein-C that are modulated by hypertrophic missense mutations. Circ Res. 2002;91:704–711. doi: 10.1161/01.res.0000036750.81083.83. [DOI] [PubMed] [Google Scholar]
- Noland TA, Jr, Guo X, Raynor RL, Jideama NM, Averyhart-Fullard V, Solaro RJ, Kuo JF. Cardiac troponin I mutants. Phosphorylation by protein kinases C and A and regulation of Ca2+-stimulated MgATPase of reconstituted actomyosin S-1. J Biol Chem. 1995;270:25,445–25,454. doi: 10.1074/jbc.270.43.25445. [DOI] [PubMed] [Google Scholar]
- Olsson MC, Patel JR, Fitzsimmons DP, Walker JW, Moss RL. Basal myosin light chain phosphorylation is a determinant of Ca2+-sensitivity of force and activation dependence of the kinetics of myocardial force development. Am J Physiol Heart Circ Physiol. 2004;287:H2712–H2718. doi: 10.1152/ajpheart.01067.2003. [DOI] [PubMed] [Google Scholar]
- Potter JD. Preparation of troponin and its subunits. Methods Enzymol. 1982;85:241–263. doi: 10.1016/0076-6879(82)85024-6. [DOI] [PubMed] [Google Scholar]
- Regnier M, Martin H, Barsotti RJ, Martyn DA, Clemmens EW. Cross-bridge versus thin filament contributions to the level and rate of force development in cardiac muscle. Biophys J. 2004;87:1815–1824. doi: 10.1529/biophysj.103.039123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson SP, Johnson JD, Holroyde MJ, Kranias EG, Potter JD, Solaro RJ. The effect of troponin I phosphorylation on the Ca2+-binding properties of the Ca2+-regulatory site of bovine cardiac troponin. J Biol Chem. 1982;257:260–263. [PubMed] [Google Scholar]
- Robinson JM, Dong WJ, Xing J, Cheung HC. Switching of troponin I: Ca2+ and myosin-induced activation of heart muscle. J Mol Biol. 2004;340:295–305. doi: 10.1016/j.jmb.2004.04.046. [DOI] [PubMed] [Google Scholar]
- Sadayappan S, Finley N, Howarth J, Osinka H, Klevitsky R, Lorenz JN, Rosevear PR, Robbins J. Role of the acidic N¢ region of cardiac troponin I in regulating myocardial function. FASEB J. 2008;22:1246–1257. doi: 10.1096/fj.07-9458com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer JF, Kensler RW, Harris SP. The myosin-binding Protein C motif binds to f-actin in a phosphorylation sensitive manner. J Biol Chem. 2009;284:12,318–12,327. doi: 10.1074/jbc.M808850200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SH, Fuchs F. Effect of ionic strength on length-dependent Ca2+ activation in skinned cardiac muscle. J Mol Cell Cardiol. 1999;31:2115–2125. doi: 10.1006/jmcc.1999.1043. [DOI] [PubMed] [Google Scholar]
- Stelzer JE, Larsson L, Fitzsimmons DP, Moss RL. Activation dependence of stretch activation in mouse skinned myocardium: implications for ventricular function. J Gen Physiol. 2006;127:95–107. doi: 10.1085/jgp.200509432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumandea MP, Burkart EM, Kobayashi T, de Tombe PP, Solaro RJ. Molecular and integrated biology of thin filament protein phosphorylation in heart muscle. Ann NY Acad Sci. 2004;1015:39–52. doi: 10.1196/annals.1302.004. [DOI] [PubMed] [Google Scholar]
- Tachampa K, Kobayashi T, Wang H, Martin AF, Biesiadecki BJ, Solaro RJ, de Tombe PP. Increased cross-bridge cycling kinetics after exchange of C-terminal truncated troponin I in skinned rat cardiac muscle. J Biol Chem. 2008;283:15 114–15 121. doi: 10.1074/jbc.M801636200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachampa K, Wang H, Farman GP, de Tombe PP. Cardiac troponin I threonine 144: role in myofilament length dependent activation. Circ Res. 2007;101:1081–1083. doi: 10.1161/CIRCRESAHA.107.165258. [DOI] [PubMed] [Google Scholar]
- Tong CW, Stelzer JE, Greaser ML, Powers PA, Moss RL. Acceleration of crossbridge kinetics by protein kinase A phosphorylation of cardiac myosin binding protein C modulates cardiac function. Circ Res. 2008;103:974–982. doi: 10.1161/CIRCRESAHA.108.177683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verduyn SC, Zaremba R, van der Velden J, Steinen GJM. Effects of contractile protein phosphorylation on force development in permeabilized rat cardiac myocytes. Basic Res Cardiol. 2007;102:476–487. doi: 10.1007/s00395-007-0663-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wattanapermpool J, Guo X, Solaro RJ. The unique amino-terminal peptide of cardiac troponin I regulates myofibrillar activity only when it is phosphorylated. J Mol Cell Cardiol. 1995;27:1383–1391. doi: 10.1006/jmcc.1995.0131. [DOI] [PubMed] [Google Scholar]
- Whitten AE, Jeffries CM, Harris SP, Trewhella J. Cardiac myosin-binding protein C decorates F-actin: implications for cardiac function. Proc Natl Acad Sci USA. 2008;105:18 360–18 365. doi: 10.1073/pnas.0808903105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winegrad S. Myosin binding protein C, a potential regulator of cardiac contractility. Circ Res. 2000;86:6–7. doi: 10.1161/01.res.86.1.6. [DOI] [PubMed] [Google Scholar]
- Wojdyr M. Fityk: a general-purpose peak fitting program. J Appl Crystallogr. 2010;43:1126–1128. [Google Scholar]
- Xing J, Chinnaraj M, Zhang Z, Cheung HC, Dong W-J. Structural studies of interactions between cardiac troponin I and actin in regulated thin filament using Forster Resonance Energy Transfer. Biochemistry. 2008;47:13 383–13 393. doi: 10.1021/bi801492x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki R, Wu Y, McNabb M, Greaser M, Labeit S, Granzier H. Protein kinase A phosphorylates titin's cardiac-specific N2B domain and reduces passive tension in rat cardiac myocytes. Circ Res. 2002;90:1181–1188. doi: 10.1161/01.res.0000021115.24712.99. [DOI] [PubMed] [Google Scholar]
- Zhang R, Zhao J, Mandveno A, Potter JD. Cardiac troponin I phosphorylation increases the rate of cardiac muscle relaxation. Circ Res. 1995a;76:1028–1035. doi: 10.1161/01.res.76.6.1028. [DOI] [PubMed] [Google Scholar]
- Zhang R, Zhao J, Potter JD. Phosphorylation of both serine residues in cardiac troponin I is required to decrease the Ca2+ affinity of cardiac troponin C. J Biol Chem. 1995b;270:30,773–30,780. doi: 10.1074/jbc.270.51.30773. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.