

Abstract
Hypoxia–reoxygenation induces loss of endothelial barrier function and oedema formation, which presents a major impediment for recovery of the organ. The integrity of the endothelial barrier is highly dependent on its contractile machinery and actin dynamics, which are precisely regulated by Rho GTPases. Perturbed activities of these Rho-GTPases under hypoxia–reoxygenation lead to derangement of the actin cytoskeleton and therefore may affect the integrity of the endothelial barrier. The aim of the present study was to analyse the role of these GTPases in regulating endothelial barrier function during hypoxia–reoxygenation in cultured porcine aortic endothelial cells and isolated perfused rat hearts. Hypoxia–reoxygenation induced an increase in albumin permeability of endothelial monolayers accompanied by an activation of the endothelial contractile machinery, derangement of the actin cytoskeleton and loss of VE-cadherin from cellular junctions. Inhibition of contractile activation with ML-7 partially protected against hypoxia–reoxygenation-induced hyperpermeability. Likewise, reoxygenation caused an increase in RhoA and a reduction in Rac1 activity accompanied by enhanced stress fibre formation and loss of peripheral actin. Inhibition of RhoA/rho kinase (Rock) signalling with RhoA or Rock inhibitors led to a complete depolymerisation and derangement of the actin cytoskeleton and worsened hypoxia–reoxygenation-induced hyperpermeability. Activation of Rac1 using a cAMP analogue, 8-CPT-O′-Me-cAMP, which specifically activates Epac/Rap1 signalling, restored peripheral localisation of actin and VE-cadherin at cellular junctions and abrogated reoxygenation-induced hyperpermeability. Similar results were reproduced in isolated saline-perfused rat hearts. These data show that activation of Rac1 but not the inhibition of RhoA preserves endothelial integrity against reoxygenation-induced loss of barrier function.
Key points
Hypoxia–reoxygenation induces loss of endothelial barrier function and oedema formation accompanied by a rise in intracellular Ca2+, an increase in myosin light chain (MLC) phosphorylation, and RhoA/Rho kinase (Rock) signalling and an inactivation of Rac1.
Neither inhibition of RhoA/Rock signalling nor antagonising Ca2+ increase could protect against this hypoxia–reoxygenation-induced loss of barrier function.
Inhibition of MLC kinase (MLCK) abrogates hypoxia–reoxygenation-induced MLC phosphorylation and partially protects against hypoxia–reoxygenation-induced endothelial hyperpermeability.
Activation of Rac1 using a cAMP analogue, 8-CPT-O′-Me-cAMP, which specifically activates Epac/Rap1 signalling abrogated reoxygenation-induced hyperpermeability. The data help us to better understand the role of Rho GTPases and contractile machinery in the regulation of endothelial barrier function during hypoxia–reoxygenation.
Introduction
The primary function of the vascular endothelium is to form a selective barrier and regulate trafficking of macromolecules and blood cells across the vessel wall (Mehta & Malik, 2006). Hypoxia–reoxygenation induces a disruption of this endothelial barrier, leading to tissue oedema, which impedes the functional recovery of vital organs such as the heart, brain or lung during reoxygenation and may jeopardise survival of the tissue. Additionally, an increased endothelial permeability to plasma constituents during chronic hypoxia may also contribute to the pathogenesis of atherosclerosis (Stocker & Keaney, 2004), as impaired endothelial barrier function facilitates the deposition of lipid molecules in the vessel wall and accelerates the development of inflammation and coagulation, processes that are involved in the genesis of atherosclerotic plaques (Stocker & Keaney, 2005).
The integrity of the endothelial barrier is maintained by an equilibrium of competing contractile and adhesive forces generated by the acto-myosin-based endothelial contractile machinery and adhesive molecules located at cell–cell and cell–matrix contacts, respectively. Endothelial cells are tightly connected to each other via interactions of adherens junction (AJ) proteins, such as VE-cadherin, of adjacent cells which are linked to the peripheral actin cytoskeleton present directly under the cell membrane. Activation of the endothelial contractile machinery is regulated by the phosphorylation status of the regulatory myosin light chain (MLC), through an interplay of MLC kinase (MLCK) and MLC phosphatase (MLCP). Therefore, changes in the activation state of endothelial contractile machinery or actin cytoskeleton dynamics affect the stability of endothelial AJs. The Rho-family of GTPases (RhoA, Rac1 and cdc42) are considered to be important regulators of endothelial contraction, actin cytoskeleton dynamics and AJs and hence play a pivotal role in the maintenance of endothelial integrity (Wojciak-Stothard & Ridley, 2002). Perturbed activities of these Rho-GTPases under hypoxia–reoxygenation lead to a derangement of the actin cytoskeleton and therefore may affect the integrity of the endothelial barrier. Similarly, hypoxia–reoxygenation may also lead to a rise in intracellular Ca2+ levels (Gündüz et al. 2006), which can activate Ca2+–calmodulin-dependent MLCK, leading to activation of the endothelial contractile machinery. However, a precise role of these signalling mechanisms in the control of endothelial barrier function during reoxygenation is not well understood. Therefore, the main goal of the present study was to analyse the role of Rho-GTPases (RhoA and Rac1) and contractile activation. The study was carried out in a well-established in vitro model of porcine aortic endothelial cells and a model of isolated saline perfused rat hearts.
Methods
Materials
Horseradish peroxidase (HRP)-conjugated anti-mouse IgG, and rabbit IgG antibodies were from Amersham Biosciences (Heidelberg, Germany); anti VE-cadherin antibody was from Beckman Coulter (Krefeld, Germany); anti-HIF1α (polyclonal) antibody was from Bethyl Laboratories (Montgomery, TX, USA); 8-CPT-O′-Me-cAMP (8-CPT) was from Biolog (Bremen, Germany); BAPTA-AM, benzonase, ML-7 and Y-27632 were from Calbiochem (Darmstadt, Germany); anti-phospho cofilin, anti-phospho MLC and anti-GAPDH were from Cell Signaling (Danvers, MA, USA); C3-transferase, RhoA and Rac1 activation assay kits were from Cytoskeleton (Denver, CO, USA); Pierce ECL solution was from Fischer Scientific (Niederlassung Nidderau, Germany); Alexa-Fluor-labelled anti-mouse IgG and anti-rabbit IgG antibodies were from Invitrogen (Karlsruhe, Germany); M199 medium was from Biochrom AG (Berlin, Germany); Complete protease inhibitor cocktail was from Roche (Mannheim, Germany); bovine serum albumin (BSA), Phalloidin-TRITC and human thrombin were from Sigma (Steinheim, Germany); anti-phospho-MYPT1 (Thr850) antibody was from Upstate (UK); Costar Transwell polycarbonate membrane filters (24 mm in circumference) were from VWR (Darmstadt, Germany); all other chemicals were of the best available quality, usually analytical grade.
Cell culture
The study conforms to the principles outlined in the Declaration of Helsinki. Porcine aortic endothelial cells (PAECs) were isolated from porcine aortas obtained freshly from a local slaughter house and cultured in basal medium 199 with Earle's salts, supplemented with 100 IU ml−1 penicillin G, 100 μg ml−1 streptomycin and 20% (v/v) newborn calf serum (NCS). Human umbilical vein endothelial cells (HUVECs) were isolated from umbilical cords derived from normal healthy uncomplicated pregnancies obtained from the University Hospital Giessen after approval from the hospital ethics committee and cultured as described previously (Aslam et al. 2010). All experiments were performed with passage 1.
Experimental protocols
The basal medium used in incubations was modified Tyrode solution (composition in mm: 150 NaCl, 2.7 KCl, 1.2 KH2PO4, 1.2 MgSO4, 1.0 CaCl2, 5.0 glucose and 30.0 Hepes; pH 7.4, 37°C). Agents were added as indicated. Stock solutions of 8-CPT-cAMP, C3-transferase and Y27632 were prepared in basal medium, while that of BAPTA-AM and ML-7 were prepared in DMSO. Appropriate volumes of these solutions were added to the cells yielding final solvent concentrations < 0.1% (v/v). The same final concentrations of basal medium were included in all respective control experiments. Unless stated otherwise, the drugs were used in following optimal concentrations as previously deterimed by us and others: BAPTA-AM (25 μm; Sandoval et al. 2001), C3-transferase (1 μg ml−1; Aslam et al. 2011), 8-CPT-cAMP (200 μm; Aslam et al. 2010), ML-7 (10 μm; Huang et al. 2003), xestospongin C (3 μm; Peters & Piper, 2007) and Y27632 (10 μm; Ishizaki et al. 2000).
Hypoxia–reoxygenation
Hypoxia reoxygenation was given by two different approaches.
Hypoxic incubator
Hypoxia (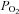 < 10 mmHg) was simulated by exposing endothelial cells to hypoxia in a hypoxic incubator (atmosphere of 5% CO2 and 95% N2) in Tyrode medium without glucose to simulate ischaemic conditions. Reoxygenation was given by transferring the cells to a normal cell culture incubator (5% CO2 and atmospheric air) and replacing the medium with Tyrode buffer containing glucose to simulate reperfusion conditions. As normoxic controls, cells were incubated in a normal cell culture incubator. Incubation was performed in Hank's balance saline solution (pH 7.4) supplemented with 1.2 mm MgCl2 and 1.3 CaCl2.
< 10 mmHg) was simulated by exposing endothelial cells to hypoxia in a hypoxic incubator (atmosphere of 5% CO2 and 95% N2) in Tyrode medium without glucose to simulate ischaemic conditions. Reoxygenation was given by transferring the cells to a normal cell culture incubator (5% CO2 and atmospheric air) and replacing the medium with Tyrode buffer containing glucose to simulate reperfusion conditions. As normoxic controls, cells were incubated in a normal cell culture incubator. Incubation was performed in Hank's balance saline solution (pH 7.4) supplemented with 1.2 mm MgCl2 and 1.3 CaCl2.
Oxygen scavenging
As an alternative, we also used an EC-Oxyrase obtained from Oxyrase, Inc. (Mansfield, OH, USA) to scavenge O2 from the medium. In this method O2 was scavenged from the incubation medium by adding sodium lactate (5 mm) and the oxygen scavenger EC-Oxyrase (1 IU ml−1) for 30 min. After 30 min the medium was replaced with normal normoxic medium for different time periods and endothelial permeability and other parameters were recorded. Parallel controls used heat-inactivated oxyrase. This method has been employed successfully by several researchers to produce hypoxic/anoxic conditions in variety of cell cultures (Ho et al. 2003; Robin et al. 2007).
To ensure that a sufficient hypoxic environment was achieved in both of the hypoxic systems used, HIF1α stabilisation as a marker of hypoxia was analysed by Western blot analysis. Pharmacological inhibitors were added during reoxygenation and were present until the end of the experiment.
Macromolecule permeability measurement
The permeability of trypan blue-labelled albumin across PAEC monolayers was studied in a two-compartment system separated by a filter membrane as described previously (Pfeil et al. 2009). Briefly, both compartments contained as basal medium modified Tyrode solution supplemented with 2% (v/v) NCS. There was no hydrostatic pressure gradient between the two compartments. The ‘luminal’ compartment containing the monolayer had a volume of 2.5 ml, and the ‘abluminal’ compartment of 6.5 ml. The fluid in the ‘abluminal’ compartment was constantly stirred. Trypan blue-labelled albumin (60 μm) was added to the luminal compartment. The appearance of the labelled albumin in the abluminal compartment was continuously monitored by pumping the liquid through a spectrophotometer (Specord 10; Zeiss, Jena, Germany). Increase in the concentration of labelled albumin was detected with a time delay of less than 15 s. For measurement of reoxygenation-induced hyperpermeability, cells were first treated with oxyrase (1 IU ml−1) for 30 min after which medium was replaced with fresh Tyrode medium containing Trypan blue-labelled albumin and the permeability was measured as described above.
Western blotting
Western blotting was performed as described previously (Aslam et al. 2010). Briefly, cells were harvested in lysis buffer (composition: 2% SDS, 50 mm Tris-HCl, pH6.8, 10% glycerol, 10 mm dithiothreitol, 5%β-mercaptoethanol) containing phosphatase and protease inhibitors. Equal amounts of lysates (30 μg) were subjected to SDS-PAGE (12.5% gel) and blotted. Membranes were incubated with respective primary antibodies overnight at 4°C followed by HRP-labelled secondary antibodies for 1 h at room temperature. Proteins were detected by using a Bio-Rad luminescence imaging system (Discovery series).
RhoA and Rac1 pulldown assay
The activation of RhoA and Rac1 was assessed by pulldown assay in the cells subjected to normoxia or hypoxia–reoxygenation. PAECs were washed with ice-cold PBS and lysed with 600 μl of lysis buffer on ice for 10 min. Lysate was centrifuged at 14,000 g for 1 min at 4°C. The, 500 μg of cell lysates was incubated with 10 μg of RBD-beads (RhoA) or GST-PAK beads (Rac1) at 4°C for 40 min. The beads were washed four times with wash buffer, heated to 95°C for 5 min with 40 μl of Laemelli buffer and loaded on a 12.5% SDS gel. Bound RhoA or Rac1 protein was then detected by immunoblotting using polyclonal antibodies against RhoA or Rac1, respectively. The total amount of RhoA or Rac1 in cell lysates was used as a control for the cross-comparison of RhoA or Rac1 activity (level of GTP-bound RhoA or Rac1).
Immunocytochemistry and confocal microscopy
PAECs were grown until confluence on glass cover slips. After treatment cells were washed with PBS and fixed with 4% paraformaldehyde at 37°C for 20 min. Non-specific binding was blocked by incubating cells with blocking solution (5% BSA + 5% NCS for 1 h). Cells were incubated with the primary antibody (1:100) overnight at 4°C, washed three times with PBS, and subsequently incubated with the secondary antibody (1:200) for 1 h at room temperature. For actin cells were stained with phalloidine-TRITC (1:50) for 1 h at room temperature. The cover slips were embedded in fluorescent mounting medium (buffered glycerol, pH 8.4) and put onto glass slides. Images were obtained using a Zeiss LSM 510 META confocal microscope.
Myocardial water content
Hearts from 250-g male Wistar rats were mounted immediately after isolation on a Langendorff perfusion system in a temperature-controlled chamber (37°C), as previously described (Noll et al. 1999) with some modifications. Hearts were perfused with Krebs–Hanseleit buffer (composition in mm: 120.0 NaCl, 25.0 NaHCO3, 4.7 KCl, 1.18 KH2PO4, 1.0 MgSO4, 1.5 CaCl2, 5.0 glucose, pH 7.4) for 30 min before each experiment and then exposed to one of the following protocols: (1) normoxic conditions for 90 min, (2) 45 min of no-flow ischaemia followed by 45 min of reperfusion and (3) 45 min of no-flow ischaemia followed by 45 min of reperfusion with indicated drugs. The normoxic perfusion (10 ml min−1) was with Krebs–Hanseleit buffer. Flow was adjusted to a perfusion pressure of 80 mmHg and held constant with the exception of the time during no-flow ischaemia. At the end of each experiment, wet weight and after 24 h (heated at 100°C), dry weights of the perfused hearts were measured. In each group four hearts were used.
Statistical analysis
Data are given as means ± SEM of three to five experiments using independent cell preparations. The comparison of means between groups was performed by one-way analysis of variance (ANOVA) followed by a Student–Newman–Keuls post hoc test. Probability (P) values of less than 0.05 were considered significant.
Results
Hypoxia–reoxygenation disrupts endothelial AJs and induces hyperpermeability
In the first step, the effect of hypoxia–reoxygenation on VE-cadherin dynamics was analysed using both the hypoxic incubator and the oxyrase system. As shown in Fig. 1A (left panel), after 45 min of hypoxia (using hypoxic incubator), VE-cadherin had already started to disperse from cell–cell adhesions. A comparable effect was seen when endothelial cells were exposed to oxyrase for 30 min (Fig. 1A, right panel). To ensure that a sufficient hypoxic environment was achieved in both of the hypoxic systems used, HIF1α stabilisation as a marker of hypoxia (Jung et al. 2002) was analysed by Western blot analysis. As shown in Fig. 1B, exposure of endothelial cells to hypoxia either in the N2 incubator or by oxyrase resulted in HIF1α stabilisation. Figure 1C shows a comparison of the effect of reoxygenation using both hypoxic systems on disruption of VE-cadherin. Next, using the oxyrase system, the effect of reoxygenation on endothelial permeability was analysed. As both systems were comparable in inducing hypoxia–reoxygenation injury, the oxyrase system was used for all the permeability experiments. As shown in Fig. 1D, exposure of endothelial cells to hypoxia–reoxygenation caused a constant increase in permeability compared to normoxic controls which almost doubled in 60 min.
Figure 1. Reoxygenation disrupts endothelial adherens junctions.
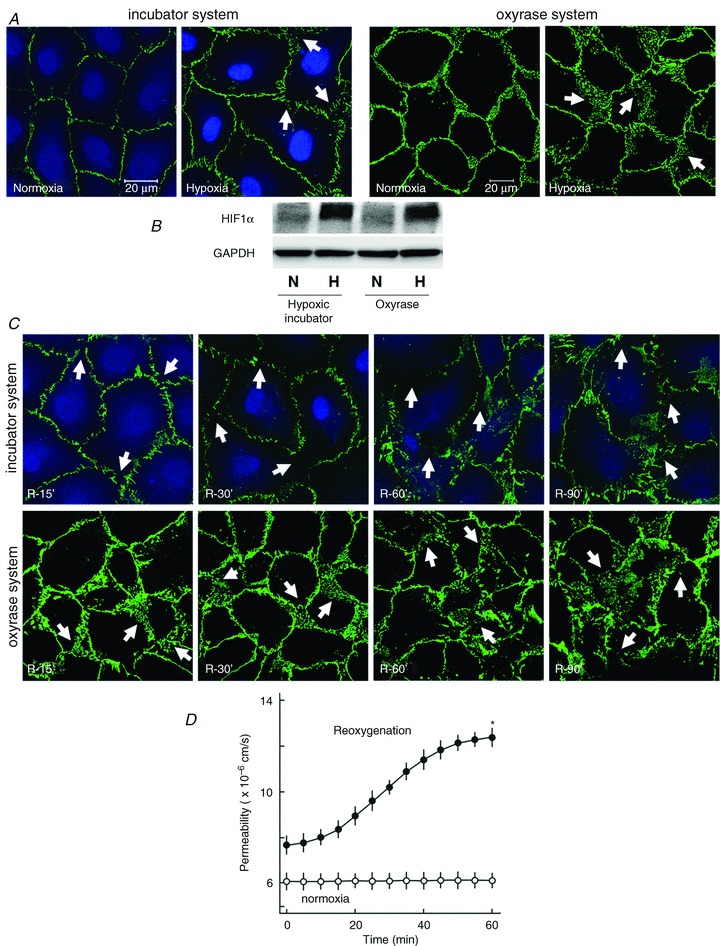
A, PAEC monolayers were subjected to hypoxia in an hypoxic incubator for 45 min (left) or exposed to oxyrase (1 IU ml−1) for 30 min (right) or normoxia and immunostained for VE-cadherin. Arrows denote dispersed VE-cadherin and gaps between cells (scale bar 20 μm; representative immunostaining of three experiments of independent cell preparations). B, HIF1α stabilisation during hypoxia. Endothelial cells were exposed to normoxia or hypoxia as indicated and HIF1α stabilisation was analysed by Western blot analysis. C, PAEC monolayers were subjected to hypoxia in an hypoxic incubator for 45 min or exposed to oxyrase for 30 min followed by reoxygenation for the indicated time periods and immunostained for VE-cadherin. D, effect of reoxygenation on endothelial permeability. PAEC monolayers were exposed to oxyrase (1 IU ml−1) for 30 min followed by reoxygenation for the indicated time periods or normoxia. The maximum effect was observed after 60 min while time to half-maximum response was 28 min. Mean ± SEM of three experiments of independent cell preparations; *P < 0.05 vs. normoxia.
Hypoxia–reoxygenation causes an increase in cytosolic Ca2+ and activation of the contractile machinery
We have previously shown that exposure of coronary microvascular endothelial cells to ischaemia/reperfusion causes a robust increase in cytosolic Ca2+ levels (Gündüz et al. 2006). Therefore, we tested whether inhibition of Ca2+ release or an increase in intracellular Ca2+ levels can protect against reoxygenation-induced hyperpermeability. As shown in Fig. 2A, inhibition of either inositol trisphosphate (IP3) receptors by xestospongin C or chelating intracellular Ca2+ by BAPTA-AM did not abrogate but rather worsened reoxygenation-induced hyperpermeability. Similarly, BAPTA-AM caused a more severe disruption of the actin cytoskeleton and loss of VE-cadherin from cell–cell junctions (Fig. 2B). As a rise in intracellular Ca+2 can activate endothelial MLCK and thus the contractile machinery, next the effect of reoxygenation on the endothelial contractile machinery was analysed. As shown in Fig. 2C, reoxygenation caused an increase in the phosphorylation state of regulatory MLC. Inhibition of MLCK using ML-7, a specific pharmacological inhibitor, completely abrogated reoxygenation-induced increase in MLC phosphorylation (Fig. 2C). In contrast to Ca2+ antagonists (BAPTA-AM and xestospongin C), inhibition of MLCK with ML-7 during reoxygenation significantly attenuated reoxygenation-induced actin cytoskeleton derangement, loss of VE-cadherin (Fig. 2B) and hyperpermeability (Fig. 2D). Both BAPTA-AM and ML-7 at the concentration used in the study did not affect basal permeability of endothelial monolayers under normoxic conditions (Supplementary Fig. S1)
Figure 2. Role of Ca2+ and contractile machinery.
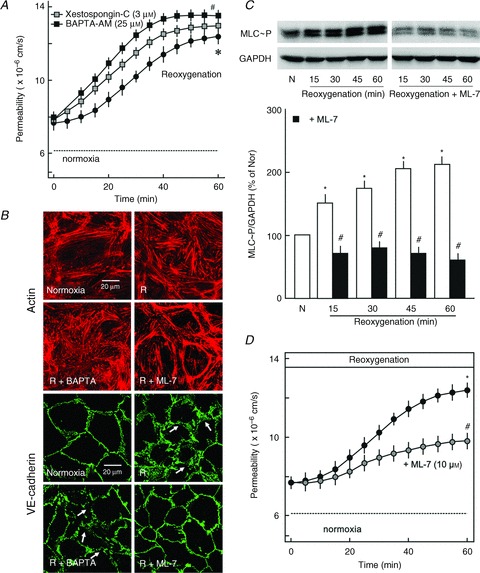
A, effect of xestospongin C and BAPTA-AM on reoxygenation-induced hyperpermeability. PAEC monolayers were exposed to oxyrase (1 IU ml−1) for 30 min followed by reoxygenation in the presence or absence of xestospongin C (3 μm) or BAPTA-AM (25 μm). Mean ± SEM of three experiments of independent cell preparations; *P < 0.05 vs. normoxia, #P < 0.05 vs. reoxygenation alone. B, VE-cadherin and actin staining. PAEC monolayers were subjected to normoxia (N) or hypoxia in an hypoxic incubator for 45 min followed by reoxygenation (R) in the presence or absence of BAPTA-AM (25 μm) or ML-7 (10 μm). Cellular VE-cadherin was visualised by immunostaining and actin with phalloidin-TRITC. Arrows denote dispersed VE-cadherin and gaps between cells (scale bar 20 μm; representative immunostaining of three experiments of independent cell preparations). C, effect of reoxygenation on MLC phosphorylation. PAEC monolayers were subjected to normoxia (N) or hypoxia in hypoxic incubators for 45 min followed by reoxygenation in the absence (open bars) or presence (filled bars) of ML-7 (10 μm) for the indicated time periods and phosphorylation of MLC at Ser18/19 was analysed by Western blotting. GAPDH was used as loading control. Top, representative Western blots; bottom, densitometric analysis of the Western blots n= 3; *P < 0.05 vs. N, #P < 0.05 vs. reoxygenation alone. D, effect of ML-7 on reoxygenation-induced hyperpermeability. PAEC monolayers were exposed to oxyrase (1 IU ml−1) for 30 min followed by reoxygenation in the presence or absence of ML-7 (10 μm). The maximum inhibitory effect was observed after 60 min while time to half-maximum response was 33 min. Mean ± SEM of three experiments of independent cell preparations; *P < 0.05 vs. normoxia, #P < 0.05 vs. reoxygenation.
Reoxygenation activates RhoA/rho kinase (Rock) signalling in endothelial cells
The RhoA/Rock pathway is an important regulator of endothelial actin cytoskeleton dynamics, and therefore the effect of reoxygenation on RhoA/Rock signalling was analysed. Activation of the RhoA/Rock pathway was analysed by measuring RhoA activity and analysing the phosphorylation state of MYPT1, an endogenous direct substrate of Rock. As shown in Fig. 3A and B, reoxygenation caused an increase in RhoA activity and phosphorylation of MYPT1 in a time-dependent manner, indicating an activation of RhoA/Rock signalling. Rock activation was accompanied by increased actin stress fibre formation (Fig. 2B). Rock induces actin polymerisation by phosphorylating and thus inhibiting the actin severing protein cofilin. As shown in Fig. 3C, reoxygenation caused a hyper-phosphorylation of cofilin, which was completely blocked by the Rock inhibitor Y27632.
Figure 3. Reoxygenation activates RhoA/Rock signalling.
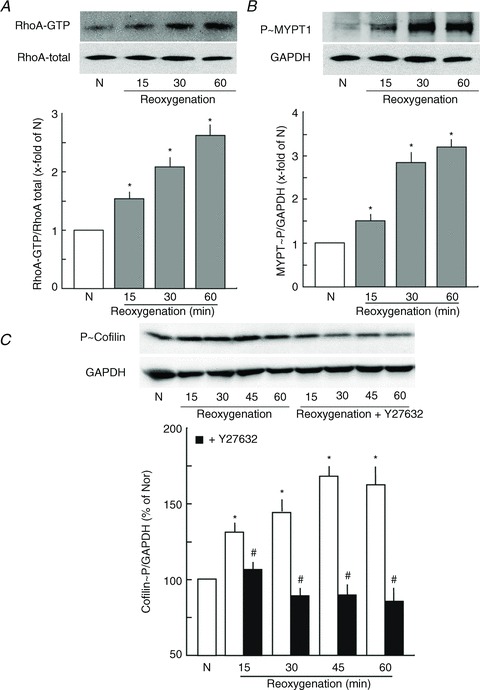
A, effect of reoxygenation on RhoA activation. PAEC monolayers were subjected to normoxia (N) or hypoxia in an hypoxic incubator for 45 min followed by reoxygenation for the indicated time periods and active RhoA was pulled down by a pulldown assay. Top, representative Western blots; bottom, densitometric analysis of the Western blots, n= 3; *P < 0.05 vs. N. B, effect of reoxygenation on MYPT1 phosphorylation at Thr850. Top, representative Western blots; bottom, densitometric analysis of the Western blots, n= 3; *P < 0.05 vs. N. C, effect of reoxygenation on cofilin phosphorylation at Ser3. PAEC monolayers were subjected to normoxia (N) or hypoxia in hypoxic incubators for 45 min followed by reoxygenation in the absence (open bars) or presence (filled bars) of Y27632 (Rock inhbitor; 10 μm) for the indicated time periods and phosphorylation of cofilin at Ser3 was analysed by Western blotting. GAPDH was used as a loading control. Top, representative Western blots; bottom, densitometric analysis of the Western blots, n= 3; *P < 0.05 vs. N, #P < 0.05 vs. reoxygenation alone.
In the next step we analysed whether inhibition of RhoA/Rock signalling can protect against reoxygenation-induced hyperpermeability. RhoA/Rock signalling was inhibited pharmacologically using C3 transferase (RhoA inhibitor) and Y27632 (Rock inhibitor). Surprisingly, inhibition of either RhoA with C3-transferase or Rock with Y27632 (10 μm) did not abrogate but rather worsened reoxygenation-induced hyperpermeability (Fig. 4A). The worsening of reoxygenation-induced hyperpermeability by Rock inhibitors was accompanied by enhanced loss of VE-cadherin from cell–cell junctions (Fig. 4B). However, Y27632 (10 μm) had no effect on basal permeability of the endothelial monolayer (Supplementary Fig. S1). In contrast, Y27632 abrogated thrombin-induced stress fibre formation, loss of VE-cadherin and attenuated thrombin-induced hyperpermeability in HUVEC monolayers (Supplementary Fig. S2A and B).
Figure 4.
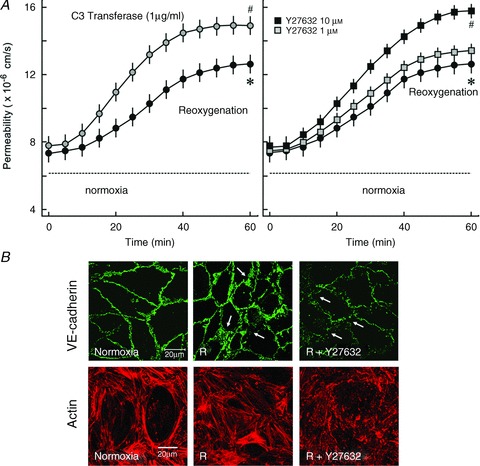
A, effect of RhoA and Rock inhibition on reoxygenation-induced hyperpermeability. PAEC monolayers were exposed to oxyrase (1 IU ml−1) for 30 min followed by reoxygenation in the presence or absence of C3 transferase (1 μg ml−1) or Y27632 (10 μm). Mean ± SEM of three experiments of independent cell preparations; *P < 0.05 vs. normoxia, #P < 0.05 vs. reoxygenation alone. B, VE-cadherin and actin staining. PAEC monolayers were exposed to normoxia (N) or oxyrase (1 IU ml−1) for 30 min followed by reoxygenation (R) in the presence or absence of Y27632 (10 μm). Cellular VE-cadherin was visualised by immunostaining and actin with phalloidin-TRITC. Arrows denote dispersed VE-cadherin and gaps between cells (scale bar 20 μm; representative immunostaining of three experiments of independent cell preparations).
Rac1 is inactivated during reoxygenation
As Rac1 controls actin dynamics at the cell periphery (Hall, 1998), a change in Rac1 activity during reoxygenation were analysed. As shown in Fig. 5A and B, exposure of endothelial cells to hypoxia–reoxygenation caused a strong reduction in Rac1 activity. In the next step, we tested whether activation of Rac1 during reoxygenation can protect against reoxygenation-induced hyperpermeability. To activate Rac1, a cAMP analogue, 8-CPT, was used. 8-CPT specifically activates Rac1 via Epac/Rap1 signalling (Baumer et al. 2008) without interfering with RhoA/Rock signalling (Aslam et al. 2010). As shown in Fig. 5C, exposure of endothelial cells to 8-CPT caused a robust activation of Rac1 and when endothelial cells were exposed to 8-CPT during reoxygenation, it abolished the reoxygenation-induced hyperpermeability (Fig. 5D) as well as loss of peripheral actin cytoskeleton and VE-cadherin from cell–cell junctions (Fig. 5E). In contrast to ML-7 and Y27632, which had no effect on basal endothelial permeability, 8-CPT significantly reduced endothelial permeability under normoxic conditions (Supplementary Fig. S1).
Figure 5. Reoxygenation inactivates Rac1 signalling.
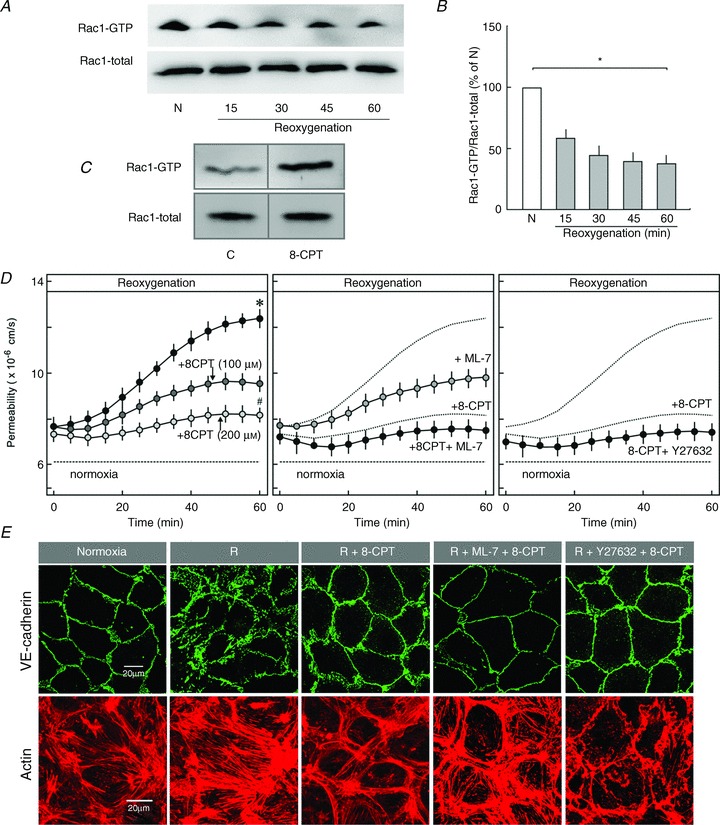
A, representative Western blots of Rac1 activity. PAEC monolayers were subjected to normoxia (N) or hypoxia in an hypoxic incubator for 45 min followed by reoxygenation (R) for the indicated time periods and active Rac1 was pulled down by a pulldown assay. Whole-cell lysate was used as a loading control. B, densitometric analysis of the Western blots presented in A; n= 3; *P < 0.05 vs. N. C, Epac activation activates Rac1. PAEC monolayers were exposed to 8-CPT-O′-Me-cAMP (8-CPT: 200 μm) or vehicle (control) and active Rac1 was analysed by a pulldown assay. D, effect of Rac1 activation on reoxygenation-induced hyperpermeability. PAEC monolayers were exposed to oxyrase (1 IU ml−1) for 30 min followed by reoxygenation in the presence or absence of 8-CPT (100 and 200 μm), ML-7 (10 μm) plus 8-CPT (200 μm), and Y27632 (1 μm) plus 8-CPT (200 μm). The maximum inhibitory effect of 8-CPT was achieved with 200 μm after 60 min while time to half-maximum response was 30 min. Mean ± SEM of three experiments of independent cell preparations; *P < 0.05 vs. normoxia, #P < 0.05 vs. E, VE-cadherin and actin staining. PAEC monolayers were subjected to normoxia or hypoxia in an hypoxic incubator for 45 min followed by reoxygenation (R) in the presence or absence of 8-CPT (200 μm), ML-7 (10 μm) plus 8-CPT (200 μm), and Y27632 (1 μm) plus 8-CPT (200 μm).
Next, we analysed whether inhibition of RhoA/Rock signalling or MLCK in addition to Rac1 activation induces an additional protective effect. As shown in Fig. 5D, inhibition of Rock with lower concentration of Y27632 (1 μm) and inhibition of contractile activation by ML-7 had an additive effect on 8-CPT-mediated barrier protection and VE-cadherin localisation (Fig. 5D, right).
Activation of Rac1 but not inhibition of RhoA or Ca2+ protects against ischaemia/reperfusion-induced increase in myocardial water content
In isolated perfused rat hearts, ischaemia/reperfusion caused a significant increase in myocardial water content compared to normoxic controls, which was reduced by perfusing hearts with either the MLCK inhibitor ML-7 or the Rac1 activator 8-CPT-cAMP. Perfusion with either Rock inhibitor, Y27632, or the Ca2+-chelator, BAPTA-AM, had no protective effect on ischaemia/reperfusion-induced increase in myocardial water content (Fig. 6).
Figure 6. Myocardial water content after ischaemia/reperfusion of saline-perfused rat heart.
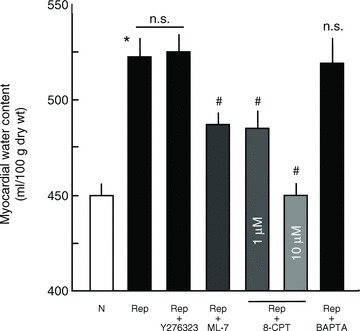
Hearts were subjected to normoxia (N) or no-flow ischaemia/reperfusion (Rep) in the presence or absence of pharmacological agents as noted and myocardial water content was measured as described in the Methods; n= 4. *P ≤ 0.05 vs. N; #P ≤ 0.05 vs. Rep; n.s., not significantly different.
Discussion
Hypoxia–reoxygenation disrupts the endothelial barrier and induces oedema formation, which jeopardises the functional recovery of vital organs such as the heart. Hypoxia–reoxygenation leads to activation of a repertoire of signalling cascades in endothelial cells, which may disrupt or protect barrier function of vascular endothelium. In the present study we show that a generalised inhibition of Ca2+ or RhoA/Rock signalling during reperfusion do not exert any protective effect on reperfusion-induced hyperpermeability both in cell culture models and in isolated perfused rat hearts. However, inhibition of contractile activation by inhibiting Ca2+–calmodulin-dependent MLCK partially rescues both cultured endothelial cells and isolated perfused rat hearts against reperfusion-induced hyperpermeability, compared with complete rescue via activation of Rac1 using a cAMP analogue.
The integrity of the endothelial barrier is highly dependent on the actin myosin-based contractile machinery and actin cytoskeleton-mediated endothelial AJs (consisting of VE-cadherin and associated catenins) and linked to the actin cytoskeleton (Lampugnani et al. 1995; Dejana et al. 2008). Changes in actin cytoskeleton dynamics therefore affect the stability of these AJs (Waschke et al. 2005). Previously, using coronary microvascular endothelial cells we have shown that reperfusion causes a robust rise in intracellular levels of Ca2+ accompanied by endothelial contractile activation and enhanced inter-endothelial cell gap formation (Gündüz et al. 2006). This reoxygenation-induced rise in intracellular Ca2+ is mediated both via release from intracellular stores and an increased influx from extracellular space via Ca2+ channels (Peters & Piper, 2007). Although an uncontrolled rise in intracellular levels of Ca2+ activates barrier disruptive signalling via activation of Ca2+–calmodulin-dependent endothelial MLCK leading to contraction and activation of RhoA/Rock signalling (Singh et al. 2007), inhibition of MLCP and increased stress fibre formation, a minimal basal level of Ca2+ is required to preserve endothelial AJ stability. Removal of either extra- or intracellular Ca2+ with EGTA or BAPTA-AM, respectively, leads to disruption of endothelial adherens and/or tight junctions and leads to endothelial hyperpermeability (Shasby & Shasby, 1986; Alexander et al. 1993).
A number of interventions have been described to antagonise this uncontrolled rise in intracellular Ca2+ in order to protect the endothelium against hypoxia and/or reoxygenation-induced hyperpermeability. One of the earliest described interventions was the use of L-type Ca2+ channel blockers (Oshiro et al. 1995). Although a few reports describe a protective effect to some extent, a general problem with the Ca2+ channel blockers is that they themselves induce vascular leakage (Taherzadeh & Warren, 1997; Taherzadeh et al. 1998) and peripheral oedema is one of the major side effects of Ca2+ channel blockers (Sica, 2003), the frequency of which may be as high as 70% depending upon the potency and dose of the agent used. Likewise, it has been reported that antagonising Ca2+ rise using BAPTA-AM, Ca2+ channel blockers or transient receptor potential channel (TRPC) inhibitors under aglycaemic conditions induces basal endothelial hyperpermeability and has no protective effect against hypoxia-induced endothelial hyperpermeability in brain endothelial monolayers (Brown et al. 2004). Accordingly, the present data show that either inhibition of Ca2+ release from the endoplasmic reticulum or complete removal of intracellular Ca2+ using BAPTA-AM leads to actin derangement and disappearance of VE-cadherin from cell–cell adhesions and that both interventions have no protective effect on reoxygenation-induced hyperpermeability; rather, they worsen the reoxygenation effect on the endothelial barrier in vitro and have no protective effect in isolated perfused rat hearts ex vivo. However, the exact mechanism by which Ca2+ channel blockers induce endothelial barrier disruption is not well understood. Sphingosine 1-phosphate (S1P) mediates barrier stabilisation via Ca2+-mediated Rac1 activation and inhibition of Ca2+ rise abrogated S1P-mediated barrier stabilisation (Mehta et al. 2005). Likewise, thrombin-induced RhoA activation in endothelial cells occurs via a PLC-mediated rise in intracellular Ca2+ (Singh et al. 2007). Although we did not measure RhoA and Rac1 activity directly during the inhibition of Ca2+ signalling, we can speculate from the more severe derangement of the actin cytoskeleton by Ca2+ inhibition that the activity of these GTPase might be further deregulated.
Ca2+ can activate the endothelial contractile machinery via activation of Ca2+-calmodulin-dependent MLCK. Activation of the contractile machinery during hypoxia–reoxygenation is depicted by increased phosphorylation of MLC, which could be completely abrogated by ML-7, a specific inhibitor of MLCK. Accordingly, a part of the barrier disrupting the effect of reoxygenation is mediated via Ca2+-mediated endothelial contraction.
It is now well established that the Rho family of GTPases (RhoA, Rac1 and cdc42) are important regulators of endothelial actin cytoskeleton dynamics and AJs and that they play a crucial role in the maintenance of the endothelial barrier (Wojciak-Stothard et al. 2001; Wojciak-Stothard & Ridley, 2002; Vandenbroucke et al. 2008). RhoA mediates actin stress fibre formation via activation of Rock (Hall, 1998). Rock mediates phosphorylation-dependent inactivation of actin severing protein cofilin via LIM kinase (Maekawa et al. 1999) and thus protects actin from depolymerisation. An inhibition of RhoA/Rock activity would therefore lead to enhanced dephosphorylation and subsequently activation of cofilin leading to actin depolymerisation. On the other hand, Rac1 mediates actin polymerisation at the cell periphery (Hall, 1998) and thus supports the establishment of cell–cell adhesions. Under basal conditions, inhibition of RhoA/Rock signalling will lead to depolymerisation of actin stress fibres, providing more building blocks (G-actin) to the Rac1-mediated actin polymerisation machinery and thus strengthening cell–cell adhesions (Supplementary Fig. S2A). Therefore, inhibition of Rock using Y27632 protects against thrombin-induced endothelial hyperpermeability (Supplementary Fig. S2B). However, if both Rock and Rac1 are inhibited, a complete depolymerisation of actin and a collapse of cytoskeleton occurs leading to a loss of cell–cell adhesion (Supplementary Fig. S2C) and barrier failure. The present data show that reoxygenation causes a strong activation of RhoA, which was accompanied by enhanced phosphorylation of MYPT1 at Thr850 (Rock site) and cofilin at Ser3, and enhanced actin stress fibre formation, indicating a strong inhibition of the actin depolymerisation machinery. Based on this observation, it was hypothesised that inhibition of RhoA/Rock signalling would protect against reoxygenation-induced hyperpermeability. Surprisingly, inhibition of RhoA/Rock signalling worsened reoxygenation-induced hyperpermeability and complete derangement of the actin cytoskeleton occurred. This prompted us to analyse Rac1 activity during reoxygenation. As shown in Fig. 4A, during reoxygenation Rac1 activity was strongly inhibited. This observation explained why inhibition of RhoA/Rock signalling worsened reoxygenation-induced barrier failure. As Rac1 activity was suppressed during reoxygenation, the actin cytoskeleton at the cell periphery was lost, leading to destabilisation of AJs, and the additional inhibition of RhoA/Rock signalling resulted in complete inhibition of the actin polymerising machinery and a collapse of actin cytoskeleton, thereby worsening the condition. Therefore, in the next step we used the strategy to activate Rac1 without interfering with RhoA activity. This was achieved by using a cAMP analogue, 8-CPT, which specifically activates Epac/Rap1 and downstream Rac1 signalling. We have previously demonstrated that this cAMP analogue neither activates protein kinase A signalling nor interferes with RhoA/Rock signalling (Aslam et al. 2010). 8-CPT abrogated reoxygenation-induced loss of peripheral actin and VE-cadherin from cell–cell junctions and antagonised endothelial hyperpermeability. Moreover, when 8-CPT was combined with an MLCK inhibitor or a Rock inhibitor (low concentration only), an additive effect was observed.
In conclusion, the present data show that manipulating Ca2+ signalling and RhoA activity to preserve endothelial barrier function is context dependent. Inhibition of Ca2+ signalling might be beneficial in chronic hypoxic or inflammatory conditions where expression of certain Ca2+ channels is up-regulated, but not in acute hypoxia–reoxygenation. Similarly, RhoA inhibition could be beneficial in situations where Rac1 activity is preserved or minimally affected but can worsen the barrier function in situations where Rac1 activity is already compromised.
Acknowledgments
The study was supported by an Excellence Cluster Cardiopulmonary System (ECCPS) postdoc grant and a University of Giessen Anschubfinanzierung grant to M.A. The technical support of S. Schäfer, D. Reitz, H. Thomas, H. Holzträger and A. Reis is gratefully acknowledged. The authors have no conflicts of interest to disclose.
Glossary
- AJ
adherens junctions
- 8-CPT
8-CPT-O′-Me-cAMP
- Epac
exchange protein directly activated by cAMP
- HUVEC
human umbilical vein endothelial cell
- IP3
inositol trisphosphate
- MLC
myosin light chain
- MLCK
myosin light chain kinase
- MLCP
myosin light chain phosphatase
- PAEC
porcine aortic endothelial cells
- Rock
rho kinase
Author contributions
M.A. designed the study, performed experiments, analysed data and wrote the paper; K-D. S., A.R., and S.N. performed experiments and analysed data; and S.R., H.M.P., T.N., R.S. and D.G. analysed data and drafted and critically reviewed the manuscript. All authors have read and approved the final version of the manuscript. All experiments were performed at the institutes of Physiology and Anatomy and Cell Biology, Justus Liebig University, Giessen, and the Department of Cardiology and Angiology, University Hospital, Giessen, Germany.
Supplementary material
Supplementary Fig. S1
Supplementary Fig. S2
References
- Alexander JS, Blaschuk OW, Haselton FR. An N-cadherin-like protein contributes to solute barrier maintenance in cultured endothelium. J Cell Physiol. 1993;156:610–618. doi: 10.1002/jcp.1041560321. [DOI] [PubMed] [Google Scholar]
- Aslam M, Gündüz D, Schuler D, Li L, Sharifpanah F, Sedding D, Piper HM, Noll T. Intermedin induces loss of coronary microvascular endothelial barrier via derangement of actin cytoskeleton: role of RhoA and Rac1. Cardiovasc Res. 2011;92:276–286. doi: 10.1093/cvr/cvr213. [DOI] [PubMed] [Google Scholar]
- Aslam M, Härtel FV, Arshad M, Gündüz D, Abdallah Y, Sauer H, Piper HM, Noll T. cAMP/PKA antagonizes thrombin-induced inactivation of endothelial myosin light chain phosphatase: role of CPI-17. Cardiovasc Res. 2010;87:375–384. doi: 10.1093/cvr/cvq065. [DOI] [PubMed] [Google Scholar]
- Baumer Y, Drenckhahn D, Waschke J. cAMP induced Rac 1-mediated cytoskeletal reorganization in microvascular endothelium. Histochem Cell Biol. 2008;129:765–778. doi: 10.1007/s00418-008-0422-y. [DOI] [PubMed] [Google Scholar]
- Brown RC, Mark KS, Egleton RD, Davis TP. Protection against hypoxia-induced blood–brain barrier disruption: changes in intracellular calcium. Am J Physiol Cell Physiol. 2004;286:C1045–C1052. doi: 10.1152/ajpcell.00360.2003. [DOI] [PubMed] [Google Scholar]
- Dejana E, Orsenigo F, Lampugnani MG. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J Cell Sci. 2008;121:2115–2122. doi: 10.1242/jcs.017897. [DOI] [PubMed] [Google Scholar]
- Gündüz D, Kasseckert SA, Härtel FV, Aslam M, Abdallah Y, Schäfer M, Piper HM, Noll T, Schäfer C. Accumulation of extracellular ATP protects against acute reperfusion injury in rat heart endothelial cells. Cardiovasc Res. 2006;71:764–773. doi: 10.1016/j.cardiores.2006.06.011. [DOI] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- Ho KC, Leach JK, Eley K, Mikkelsen RB, Lin PS. A simple method of producing low oxygen conditions with oxyrase for cultured cells exposed to radiation and tirapazamine. Am J Clin Oncol. 2003;26:e86–e91. doi: 10.1097/01.COC.0000077937.91824.44. [DOI] [PubMed] [Google Scholar]
- Huang Q, Xu W, Ustinova E, Wu M, Childs E, Hunter F, Yuan S. Myosin light chain kinase-dependent microvascular hyperpermeability in thermal injury. Shock. 2003;20:363–368. doi: 10.1097/01.shk.0000079425.0000.db. [DOI] [PubMed] [Google Scholar]
- Ishizaki T, Uehata M, Tamechika I, Keel J, Nonomura K, Maekawa M, Narumiya S. Pharmacological properties of Y-27632, a specific inhibitor of rho-associated kinases. Mol Pharmacol. 2000;57:976–983. [PubMed] [Google Scholar]
- Jung F, Haendeler J, Hoffmann J, Reissner A, Dernbach E, Zeiher AM, Dimmeler S. Hypoxic induction of the hypoxia-inducible factor is mediated via the adaptor protein Shc in endothelial cells. Circ Res. 2002;91:38–45. doi: 10.1161/01.res.0000024412.24491.ca. [DOI] [PubMed] [Google Scholar]
- Lampugnani MG, Corada M, Caveda L, Breviario F, Ayalon O, Geiger B, Dejana E. The molecular organization of endothelial cell to cell junctions: differential association of plakoglobin,b-catenin, and a-catenin with vascular endothelial cadherin (VE-cadherin) J Cell Biol. 1995;129:203–217. doi: 10.1083/jcb.129.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa M, Ishizaki T, Boku S, Watanabe N, Fujita A, Iwamatsu A, Obinata T, Ohashi K, Mizuno K, Narumiya S. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science. 1999;285:895–898. doi: 10.1126/science.285.5429.895. [DOI] [PubMed] [Google Scholar]
- Mehta D, Konstantoulaki M, Ahmmed GU, Malik AB. Sphingosine 1-phosphate-induced mobilization of intracellular Ca2+ mediates rac activation and adherens junction assembly in endothelial cells. J Biol Chem. 2005;280:17320–17328. doi: 10.1074/jbc.M411674200. [DOI] [PubMed] [Google Scholar]
- Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86:279–367. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- Noll T, Wozniak G, McCarson K, Hajimohammad A, Metzner HJ, Inserte J, Kummer W, Hehrlein FW, Piper HM. Effect of factor XIII on endothelial barrier function. J Exp Med. 1999;189:1373–1382. doi: 10.1084/jem.189.9.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshiro H, Kobayashi I, Kim D, Takenaka H, Hobson RW, Duran WN. L-type calcium channel blockers modulate the microvascular hyperpermeability induced by platelet-activating factor in vivo. J Vasc Surg. 1995;22:732–739. doi: 10.1016/s0741-5214(95)70064-1. [DOI] [PubMed] [Google Scholar]
- Peters SC, Piper HM. Reoxygenation-induced Ca2+ rise is mediated via Ca2+ influx and Ca2+ release from the endoplasmic reticulum in cardiac endothelial cells. Cardiovasc Res. 2007;73:164–171. doi: 10.1016/j.cardiores.2006.09.015. [DOI] [PubMed] [Google Scholar]
- Pfeil U, Aslam M, Paddenberg R, Quanz K, Chang CL, Park JI, Gries B, Rafiq A, Faulhammer P, Goldenberg A, Papadakis T, Noll T, Hsu SY, Weissmann N, Kummer W. Intermedin/adrenomedullin-2 is a hypoxia-induced endothelial peptide that stabilizes pulmonary microvascular permeability. Am J Physiol Lung Cell Mol Physiol. 2009;297:L837–L845. doi: 10.1152/ajplung.90608.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin E, Guzy RD, Loor G, Iwase H, Waypa GB, Marks JD, Hoek TL, Schumacker PT. Oxidant stress during simulated ischemia primes cardiomyocytes for cell death during reperfusion. J Biol Chem. 2007;282:19133–19143. doi: 10.1074/jbc.M701917200. [DOI] [PubMed] [Google Scholar]
- Sandoval R, Malik AB, Minshall RD, Kouklis P, Ellis CA, Tiruppathi C. Ca2+ signalling and PKCa activate increased endothelial permeability by disassembly of VE–cadherin junctions. J Physiol. 2001;533:433–445. doi: 10.1111/j.1469-7793.2001.0433a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shasby DM, Shasby SS. Effects of calcium on transendothelial albumin transfer and electrical resistance. J Appl Physiol. 1986;60:71–79. doi: 10.1152/jappl.1986.60.1.71. [DOI] [PubMed] [Google Scholar]
- Sica DA. Calcium channel blocker-related periperal edema: can it be resolved. J Clin Hypertens (Greenwich) 2003;5:291–294. doi: 10.1111/j.1524-6175.2003.02402.x. 297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh I, Knezevic N, Ahmmed GU, Kini V, Malik AB, Mehta D. Galphaq-TRPC6-mediated Ca2+ entry induces RhoA activation and resultant endothelial cell shape change in response to thrombin. J Biol Chem. 2007;282:7833–7843. doi: 10.1074/jbc.M608288200. [DOI] [PubMed] [Google Scholar]
- Stocker R, Keaney JF., Jr Role of oxidative modifications in atherosclerosis. Physiol Rev. 2004;84:1381–1478. doi: 10.1152/physrev.00047.2003. [DOI] [PubMed] [Google Scholar]
- Stocker R, Keaney JF., Jr New insights on oxidative stress in the artery wall. J Thromb Haemost. 2005;3:1825–1834. doi: 10.1111/j.1538-7836.2005.01370.x. [DOI] [PubMed] [Google Scholar]
- Taherzadeh M, Das AK, Warren JB. Nifedipine increases microvascular permeability via a direct local effect on postcapillary venules. Am J Physiol Heart Circ Physiol. 1998;275:H1388–H1394. doi: 10.1152/ajpheart.1998.275.4.H1388. [DOI] [PubMed] [Google Scholar]
- Taherzadeh M, Warren JB. Comparison of diltiazem and verapamil on rat microvascular permeability. Microvasc Res. 1997;54:206–213. doi: 10.1006/mvre.1997.2040. [DOI] [PubMed] [Google Scholar]
- Vandenbroucke E, Mehta D, Minshall R, Malik AB. Regulation of endothelial junctional permeability. Ann N Y Acad Sci. 2008;1123:134–145. doi: 10.1196/annals.1420.016. [DOI] [PubMed] [Google Scholar]
- Waschke J, Curry FE, Adamson RH, Drenckhahn D. Regulation of actin dynamics is critical for endothelial barrier functions. Am J Physiol Heart Circ Physiol. 2005;288:H1296–H1305. doi: 10.1152/ajpheart.00687.2004. [DOI] [PubMed] [Google Scholar]
- Wojciak-Stothard B, Potempa S, Eichholtz T, Ridley AJ. Rho and Rac but not Cdc42 regulate endothelial cell permeability. J Cell Sci. 2001;114:1343–1355. doi: 10.1242/jcs.114.7.1343. [DOI] [PubMed] [Google Scholar]
- Wojciak-Stothard B, Ridley AJ. Rho GTPases and the regulation of endothelial permeability. Vascul Pharmacol. 2002;39:187–199. doi: 10.1016/s1537-1891(03)00008-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.