

Abstract
At birth, rat adrenomedullary chromaffin cells (AMCs) respond directly to asphyxial stressors such as hypoxia and hypercapnia by triggering catecholamine secretion, which is critical for proper transition to extrauterine life. These non-neurogenic responses are suppressed postnatally in parallel with the development of splanchnic innervation, and reappear following denervation of the adult adrenal gland. To test whether neural factors released from the splanchnic nerve may regulate AMC chemosensitivity, we previously showed that nicotinic agonists in utero and in vitro suppressed hypoxia, but not hypercapnia, sensitivity. Here, we considered the potential role of opiate peptides which are also released from the splanchnic nerve and act via postsynaptic μ-, δ- and κ-opioid receptors. Treatment of neonatal rat AMC cultures for ∼1 week with μ- and/or δ- (but not κ) opioid agonists (2 μm) led to a marked suppression of both hypoxia and hypercapnia sensitivity, as measured by K+ current inhibition and membrane depolarization; co-incubation with naloxone prevented the effects of combined opioids. The suppression of hypoxia sensitivity was attributable to upregulation of KATP current density and the KATP channel subunit Kir6.2, and was reversed by the KATP channel blocker, glibenclamide. By contrast, suppression of hypercapnia sensitivity was associated with down-regulation of two key mediators of CO2 sensing, i.e. carbonic anhydrase I and II. Collectively, these studies point to a novel role for opioid receptor signalling in the developmental regulation of chromaffin cell chemosensitivity, and suggest that prenatal exposure to opioid drugs could lead to impaired arousal responses in the neonate.
Key points
Low O2 (hypoxia) and high CO2 (hypercapnia) elicit a critical, non-neurogenic catecholamine surge from neonatal rat adrenomedullary chromaffin cells (AMCs), but these chemosensing mechanisms are suppressed postnatally following splanchnic innervation.
We tested the possibility that opioids released from the splanchnic nerve during innervation contribute to the suppression of chemosensitivity using a culture model of dissociated neonatal rat AMCs exposed to opioid agonists in vitro.
Exposure of neonatal AMCs to μ- and/or δ-opioid agonists for ∼1 week led to a naloxone-sensitive blunting of both hypoxia and hypercapnia sensitivity.
The loss of hypoxia sensitivity was attributable to the increased expression of glibenclamide-sensitive KATP channels, which open during acute hypoxia favouring membrane hyperpolarization; by contrast, the loss of hypercapnic sensitivity was associated with the down-regulation of carbonic anhydrase I and II.
Thus, stimulation of postsynaptic opioid receptors on chromaffin cells following splanchnic innervation may normally contribute to the postnatal suppression of direct O2 and CO2 chemosensitivity; also, prenatal exposure to opioid drugs could lead to impaired chromaffin cell responses to asphyxial stimuli in the neonate.
Introduction
Catecholamine (CAT) secretion from adrenomedullary chromaffin cells (AMCs) is a crucial physiological response during the transition of the fetus to extrauterine life. Early in development of several mammalian species, and prior to innervation of the adrenal medulla by the splanchnic nerve, neonatal AMCs release CAT in response to asphyxial stressors including hypoxia (low 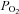 ) and hypercapnia (high
) and hypercapnia (high 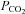 ) experienced during the birthing process (Lagercrantz & Slotkin, 1986; Thompson et al. 1997; Munoz-Cabello et al. 2005; Nurse et al. 2009; Livermore et al. 2011). CAT secretion from AMCs is required for the regulation of cardiac conduction and in the preparation of the lungs for air breathing (Lagercrantz & Bistoletti, 1977; Jones, 1980; Seidler & Slotkin, 1985; Lagercrantz & Slotkin, 1986). In general, these non-neurogenic responses to asphyxial stimuli are suppressed postnatally along a time course that parallels the development of splanchnic innervation (Seidler & Slotkin, 1985, 1986; Cheung, 1990; Thompson et al. 1997; Garcia-Fernandez et al. 2007; Nurse et al. 2009). Hence, juvenile AMCs show markedly reduced sensitivity to asphyxial stimuli (Slotkin & Seidler, 1988; Thompson et al. 1997; Munoz-Cabello et al. 2005; Rico et al. 2005; Garcia-Fernandez et al. 2007) and, moreover, adrenal denervation in the adult results in the restoration of direct hypoxia chemosensitivity (Levitsky & Lopez-Barneo, 2009).
) experienced during the birthing process (Lagercrantz & Slotkin, 1986; Thompson et al. 1997; Munoz-Cabello et al. 2005; Nurse et al. 2009; Livermore et al. 2011). CAT secretion from AMCs is required for the regulation of cardiac conduction and in the preparation of the lungs for air breathing (Lagercrantz & Bistoletti, 1977; Jones, 1980; Seidler & Slotkin, 1985; Lagercrantz & Slotkin, 1986). In general, these non-neurogenic responses to asphyxial stimuli are suppressed postnatally along a time course that parallels the development of splanchnic innervation (Seidler & Slotkin, 1985, 1986; Cheung, 1990; Thompson et al. 1997; Garcia-Fernandez et al. 2007; Nurse et al. 2009). Hence, juvenile AMCs show markedly reduced sensitivity to asphyxial stimuli (Slotkin & Seidler, 1988; Thompson et al. 1997; Munoz-Cabello et al. 2005; Rico et al. 2005; Garcia-Fernandez et al. 2007) and, moreover, adrenal denervation in the adult results in the restoration of direct hypoxia chemosensitivity (Levitsky & Lopez-Barneo, 2009).
Taken together, the above data are consistent with the hypothesis that factors released from splanchnic nerve terminals following innervation may contribute to the suppression of asphyxial sensitivity in developing AMCs, via their action at postsynaptic receptors. In one recent test of this hypothesis, we focused on the well-known preganglionic cholinergic innervation of adrenal chromaffin cells. Indeed, we found that chronic exposure to nicotinic acetylcholine (ACh) receptor agonists (i.e. nicotine) in utero and in vitro resulted in a selective blunting of hypoxia sensitivity in neonatal AMCs (Buttigieg et al. 2007, 2008). However, the sensitivity to isohydric hypercapnia (10% CO2; pH = 7.4) remained intact in these preparations (Buttigieg et al. 2007), suggesting that other neural factors released from the splanchnic nerve may also be involved. In addition to ACh, splanchnic nerve terminals release a number of neurotransmitters or neuromodulators including opiate peptides, pituitary adenylate cyclase activating peptide (PACAP), and histamine that act on postsynaptic receptors expressed on AMCs (Kobayashi et al. 1985; Holgert et al. 1998; Kuri et al. 2009). Therefore, by chronically activating postsynaptic receptors any one or more of these agents could also be involved, and particularly in the developmental regulation of hypercapnia sensitivity in AMC.
In the present study, we examined the potential role of opioid receptor signalling in the developmental regulation of chemosensitivity in AMCs, which are known to express μ-, δ- and κ-opioid receptors (Kimura et al. 1988; Wittert et al. 1996; Keating et al. 2004). Indeed, the involvement of opiate peptides in suppressing the non-neurogenic response of adrenal medulla to hypoxia has been previously considered, although these experiments involved acute exposures to opioid agonists. For example, Keating et al. (2004) reported that hypoxia-induced catecholamine secretion from ovine AMCs was inhibited in the presence of μ- and κ-opioid agonists and this was attributable to opioid-mediated enhancement of a K+ conductance (SK) and/or inhibition of voltage-gated Ca2+ channels. By contrast, similar studies in the rat failed to show any effect of opioid agonists on hypoxia-induced catecholamine secretion from neonatal AMCs (Rico et al. 2005). To investigate the effects of chronic exposures we used an in vitro model of dissociated neonatal rat AMCs exposed for ∼1 week to selective opioid agonists, either singly or in combination, and in the presence or absence of the general opioid receptor blocker naloxone. To characterize AMC sensitivity to hypoxia and isohydric hypercapnia, we used perforated-patch whole-cell recording to monitor whole-cell K+ currents and membrane potential (Thompson et al. 1997; Buttigieg et al. 2007, 2008). Interestingly, we found that exposure to μ- and δ- opioid agonists led to a suppression of both hypercapnia and hypoxia sensitivity in AMCs, and these effects were prevented during co-incubation of naloxone. To probe at potential underlying molecular mechanisms, we further investigated components in the signal transduction pathway previously known to regulate hypoxia and hypercapnia sensitivity in these cells, i.e. the expression of ATP-sensitive K+ channels (Buttigieg et al. 2008) and carbonic anhydrase (CA) isoforms I and II (Munoz-Cabello et al. 2005), respectively.
Methods
Ethical approval
All procedures for animal handling and tissue dissections were carried out according to the guidelines of the Canadian Council on Animal Care (CCAC) and institutional guidelines. The authors have read, and the experiments comply with, the policies and regulations of The Journal of Physiology given by Drummond (2009).
Cell culture
Soon after birth, neonatal rat pups (postnatal day 0 (P0); Wistar, Charles River, Quebec, Canada) were first rendered unconscious by a blow to the back of the head and then killed immediately by decapitation. Adrenal glands were excised bilaterally and most of the surrounding cortex was trimmed away and discarded. Cultures of primary AMCs were prepared using a combination of enzymatic and mechanical dissociation of the medullary-rich tissue as previously described (Thompson et al. 1997; Buttigieg et al. 2008). Dissociated cells were plated on 35 mm culture dishes coated with Matrigel (Collaborative Research, Bedford, MA, USA) and grown in Dulbecco's modified Eagle medium-F12 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum and various additives as described previously (Thompson et al. 1997). Cultures were incubated in a humidified atmosphere of 95% air and 5% CO2 at 37°C for ∼1 week in the presence or absence of μ-, δ- and/or κ-opioid agonists (2 μm; see below); the general opioid receptor blocker naloxone (2 μm) was present in some experiments. The medium was changed every 2 days and fresh drugs were added at that time in the case of opioid-treated cultures.
Electrophysiology
The nystatin perforated-patch clamp technique was used for electrophysiological studies on whole-cell currents and membrane potentials in isolated AMCs as previously described (Thompson & Nurse, 1998; Thompson et al. 2002, 2007; Buttigieg et al. 2007, 2008). The pipette solution consisted of (mm): potassium gluconate, 115; KCl, 25; NaCl, 5; CaCl2, 1; Hepes, 10 at pH 7.2, and nystatin (300–450 μg ml−1). Before each experiment solutions containing nystatin were made fresh and used within 2 h after preparation; after perforation the series resistance was typically ∼25 MΩ. Experiments were carried out at 37°C in HCO3--buffered extracellular solution containing (mm): NaCl, 115; KCl, 5; CaCl2, 2; MgCl2, 2; glucose, 10; and NaHCO3, 24, bubbled with 5% CO2 at pH 7.4. Hypoxic solutions (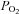 ∼ 15 mmHg) were generated by bubbling N2 gas; in the case of hypercapnic solutions (10% CO2) the bicarbonate concentration was increased to 48 mm (equimolar substitution with NaCl) so as to maintain the pH at 7.4. Tetrodotoxin (TTX; 0.5 μm) was present in some experiments. The extracellular solution was applied to the cells by gravity flow or via a rapid perfusion system (Thompson et al. 1997; Zhang & Nurse, 2004). In some experiments, cells were held at a holding potential of −60 mV and currents recorded during voltage steps to +70 mV; in other experiments, the voltage was ramped from −120 to +80 mV over a period of 500 ms at a frequency of 0.1 Hz, followed by voltage steps to +30 and +60 mV from a holding potential of −60 mV. Membrane potentials were monitored in current clamp mode (I= 0). Results are presented as mean ± SEM. Current density (pA/pF), obtained by dividing the whole-cell current (pA) by cell capacitance (pF), and membrane potentials (mV) were compared using paired or independent (Mann–Whitney) Student's t tests with significance set at P < 0.05 (Microcal Origin version 7.5).
∼ 15 mmHg) were generated by bubbling N2 gas; in the case of hypercapnic solutions (10% CO2) the bicarbonate concentration was increased to 48 mm (equimolar substitution with NaCl) so as to maintain the pH at 7.4. Tetrodotoxin (TTX; 0.5 μm) was present in some experiments. The extracellular solution was applied to the cells by gravity flow or via a rapid perfusion system (Thompson et al. 1997; Zhang & Nurse, 2004). In some experiments, cells were held at a holding potential of −60 mV and currents recorded during voltage steps to +70 mV; in other experiments, the voltage was ramped from −120 to +80 mV over a period of 500 ms at a frequency of 0.1 Hz, followed by voltage steps to +30 and +60 mV from a holding potential of −60 mV. Membrane potentials were monitored in current clamp mode (I= 0). Results are presented as mean ± SEM. Current density (pA/pF), obtained by dividing the whole-cell current (pA) by cell capacitance (pF), and membrane potentials (mV) were compared using paired or independent (Mann–Whitney) Student's t tests with significance set at P < 0.05 (Microcal Origin version 7.5).
Western immunoblot analysis
Cells were homogenized and lysed in Buffer A solution containing: 10 mm Hepes pH 7.6, 10 mm KCl, 0.1 mm EDTA pH 8, 0.1 mm EGTA pH 8, 1 mm dithiothreitol and protein inhibitors (Complete Mini, Roche, Laval, QC, Canada), 1 mm phenylmethanesulfonylfluoride, 5 mg ml−1 aprotinin and 5 mg ml−1 leupeptin. Protein samples were boiled at 95–100°C for 5 min. Ten micrograms of protein samples, measured using the Bradford assay (1:5 dilution reagent and 1 mg ml−1 bovine serum albumin), were loaded and resolved on 10% SDS-PAGE gel and transferred onto polyvinylidene difluoride membranes. Membranes were then washed and incubated with either primary rabbit polyclonal antibody against Kir6.2 (1:1000 dilution; Alomone Labs Ltd, Jerusalem, Israel), rabbit polyclonal anti-human carbonic anhydrase I antibody (1:1000 dilution; Abcam Inc., Cambridge, MA, USA), sheep polyclonal anti-human carbonic anhydrase II antibody (1:1000 dilution; AbD Serotec, Kidlington, UK), or primary rabbit monoclonal (β-actin antibody (1:10,000 dilution; Millipore, Billerica, MA, USA)) (loading control) at 4°C overnight. Membranes were then washed in phosphate-buffered saline (PBS) and incubated in a goat anti-rabbit horseradish peroxidase (HRP)-linked secondary antibody (1:10,000 dilution; Jackson Labs, Bar Harbor, ME, USA) for 1 h at room temperature. Immunoreactions were visualized using ECL and exposed to XAR-film.
Immunofluorescence
Cultures of neonatal rat AMCs were grown on glass cover slips attached to the underside of central wells of modified Nunc 35 mm dishes (Buttigieg et al. 2008). Medium was aspirated and cells were gently rinsed with pre-warmed PBS at pH 7.2, and quickly fixed in 2 ml of ice cold 4% paraformaldehyde in PBS for 1 h at 4°C. Cells were then washed three times with PBS for 3 min and incubated with 100 μl of primary antibodies (rabbit polyclonal anti-μ-opioid receptor; rabbit polyclonal anti-δ-opioid receptor; Alomone) diluted in 1% BSA/PBS overnight. Samples were washed the next day with PBS three times (10 min each) and incubated with FITC-conjugated secondary antibody (1:50; Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA) for 1 h at room temperature in the dark. Cells were then washed three times with PBS (10 min each) and covered with Vectashield to avoid photobleaching. Pre-adsorption controls were performed by incubating primary antibody overnight at 4°C in the presence of 3× excess antigen (1 μg of antibody incubated with 3 μg of antigen). Cells were visualized and imaged using a Zeiss inverted microscope (IM 35).
Drugs
All drugs, including TTX, were purchased from Sigma-Aldrich (St Louis, MO, USA). Fresh opioid drugs were applied in medium every 2 days at a concentration of 2 μm. The following opioid drugs were used for chronic treatments: μ-opioid agonist DALDA ([d-Arg2, Ly4]-dermorphin-(1–4)-amide); δ-opioid agonist DPDPE ([d-Pen2, 5, P-Cl-Phe4]-enkephalin); κ-opioid agonist U-62066 ((±)-(5α,7α,8β)-3,4-dichlo-ro-N-methyl-N-[7-(1-pyrrolidinyl)-1-oxaspiro[4.5]dec-8-yl] benzeneacetamidemesylate salt). Naloxone hydrochloride dehydrate (2 μm) was used as a general opioid antagonist.
Statistical analysis
Data are expressed as means ± SEM and statistical analyses were carried out using a non-parametric test (Mann–Whitney U test) and ANOVA as appropriate. Differences were considered significant at P < 0.05.
Results
For studies on opioid-treated cells, we incubated AMC cultures with the selective μ-, δ- and κ- opioid receptor agonists, i.e. DALDA, DPDPE and U-62066, respectively, separately or together at a concentration of 2 μm for ∼1 week. This concentration is similar to that used in previous studies (Keating et al. 2004) and is likely to be in the physiological range as the opiate concentration near single chromaffin cells has been estimated to be in the micromolar range (Castanas et al. 1985a,b). Also, a concentration of 2 μm is more than 100 times greater that the binding affinity for these agonists at their respective receptors (Cheng et al. 1992; Schlosser et al. 1995; Keating et al. 2004). Under phase contrast microscopy, the appearance of neonatal rat AMC cultures grown for ∼1 week in the presence of opioid receptor agonists was indistinguishable from that of control sister cultures (not shown). Similarly, in electrophysiological studies the mean resting potential of control untreated cells was −60.7 ± 1.2 mV (n= 12), a value not significantly different from that of opioid-treated cells (−64.4 ± 3.5 mV; n= 10; P < 0.05), after 1 week in culture.
Electrophysiological studies of the effects of chronic opioid exposure on hypoxic sensitivity in neonatal rat AMCs
In late fetal and neonatal rat AMCs, hypoxia inhibits a variety of O2-sensitive K+ channels (SK, BK and KV channels), leading to or facilitating membrane depolarization, voltage-gated Ca2+ entry and CAT release (Mochizuki-Oda et al. 1997; Thompson et al. 1997; Thompson & Nurse, 1998; Bournaud et al. 2007; Garcia-Fernandez et al. 2007; Nurse et al. 2009). In the first experimental series we used perforated-patch, whole-cell recording to investigate the effects of μ-, δ- and κ-opioid agonists (DALDA, DPDPE and U-62066; 2 μm), present individually or in combination, on hypoxic sensitivity of AMCs cultured for 7 days. In control cultures, approximately 81% (n= 9/11) of AMCs showed the expected reversible inhibition of outward K+ current at positive potentials under voltage clamp (Fig. 1A; upper traces and I–V plot), and membrane depolarization under current clamp (Fig. 1A; lower trace), during acute hypoxia (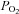 ∼15 mmHg); the mean depolarization during hypoxia was 4.5 ± 0.80 mV (n= 10). In contrast, AMCs from sister cultures chronically exposed for 7 days to combined μ-, δ- and κ-opioid agonists (2 μm) showed a marked suppression of hypoxic sensitivity, reflected by the relative lack of effect of hypoxia on outward K+ current during depolarizing voltage steps (Fig. 1B; upper traces, and I–V plot). In these experiments only ∼18% (n= 2/11) of opioid-treated cells tested appeared to show a detectable hypoxia-evoked inhibition of K+ current at positive potentials. Also, as exemplified in Fig. 1B (lower trace), in current clamp studies hypoxia had no detectable effect on membrane potential of opioid-treated cells (n= 12). The blunting effects of combined agonist exposure were mediated via the opioid receptor signalling pathway because they could be prevented by co-incubation with the general opioid receptor antagonist naloxone (2 μm) over the entire treatment period (Fig. 1C); naloxone had no significant effect on whole-cell currents or membrane potential when present alone (n= 12).
∼15 mmHg); the mean depolarization during hypoxia was 4.5 ± 0.80 mV (n= 10). In contrast, AMCs from sister cultures chronically exposed for 7 days to combined μ-, δ- and κ-opioid agonists (2 μm) showed a marked suppression of hypoxic sensitivity, reflected by the relative lack of effect of hypoxia on outward K+ current during depolarizing voltage steps (Fig. 1B; upper traces, and I–V plot). In these experiments only ∼18% (n= 2/11) of opioid-treated cells tested appeared to show a detectable hypoxia-evoked inhibition of K+ current at positive potentials. Also, as exemplified in Fig. 1B (lower trace), in current clamp studies hypoxia had no detectable effect on membrane potential of opioid-treated cells (n= 12). The blunting effects of combined agonist exposure were mediated via the opioid receptor signalling pathway because they could be prevented by co-incubation with the general opioid receptor antagonist naloxone (2 μm) over the entire treatment period (Fig. 1C); naloxone had no significant effect on whole-cell currents or membrane potential when present alone (n= 12).
Figure 1. Effects of chronic exposure of neonatal rat AMCs to opioid agonists for ∼1 week in vitro on hypoxia sensitivity.
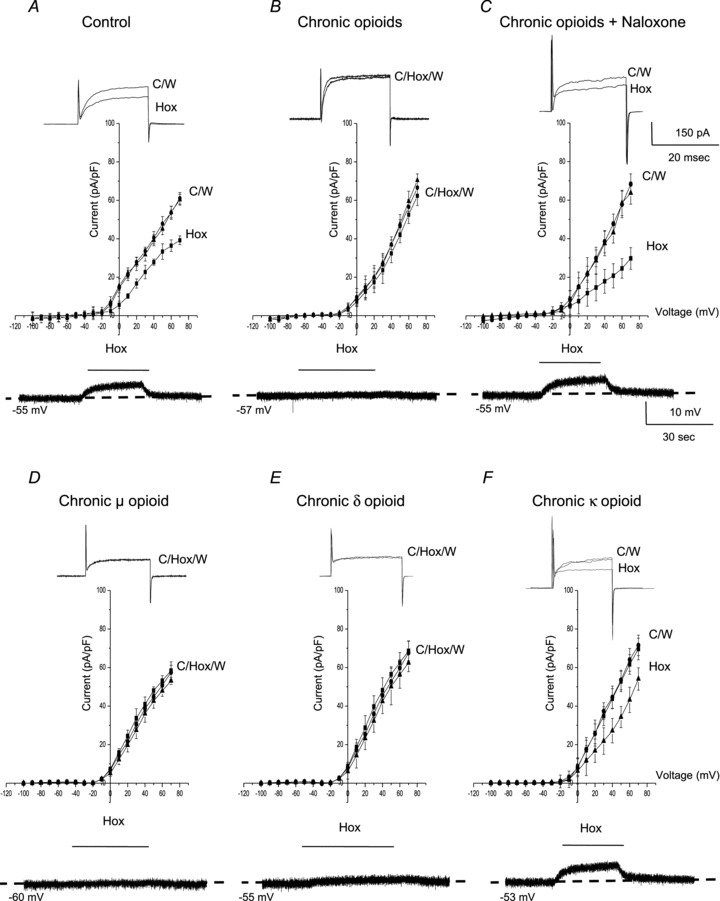
A, in control (untreated) AMCs cultured for ∼1 week, hypoxia (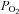 ∼ 15 mmHg) typically causes inhibition of outward K+ current (top trace) and membrane depolarization (bottom trace); current density (pA/pF) versus voltage (I–V) plot (middle) shows significant inhibition of outward current (P < 0.05) at potentials positive to +10 mV. These responses are markedly reduced or abolished in neonatal AMCs chronically exposed for ∼1 week to either a combination of μ-, δ- and κ-opioid agonists (2 μm) (B), or to either μ- or δ-opioid agonists (2 μm) alone (D and E, respectively). These blunting effects of chronic opioids on the hypoxia-induced responses are prevented during continuous co-incubation with the general opioid receptor antagonist naloxone (2 μm; C). Also, chronic exposure to the κ-opioid agonist (2 μm) alone was ineffective (F). Data are presented as mean ± SEM (n= 11). TTX was present in the extracellular solution. C, control; Hox, hypoxia; W, wash.
∼ 15 mmHg) typically causes inhibition of outward K+ current (top trace) and membrane depolarization (bottom trace); current density (pA/pF) versus voltage (I–V) plot (middle) shows significant inhibition of outward current (P < 0.05) at potentials positive to +10 mV. These responses are markedly reduced or abolished in neonatal AMCs chronically exposed for ∼1 week to either a combination of μ-, δ- and κ-opioid agonists (2 μm) (B), or to either μ- or δ-opioid agonists (2 μm) alone (D and E, respectively). These blunting effects of chronic opioids on the hypoxia-induced responses are prevented during continuous co-incubation with the general opioid receptor antagonist naloxone (2 μm; C). Also, chronic exposure to the κ-opioid agonist (2 μm) alone was ineffective (F). Data are presented as mean ± SEM (n= 11). TTX was present in the extracellular solution. C, control; Hox, hypoxia; W, wash.
We next investigated whether the blunting of hypoxic sensitivity in opioid-treated AMCs was linked to specific subtypes of opioid receptors. Following chronic exposure to either μ- (DALDA; 2 μm) or δ- (DPDPE; 2 μm) opioid agonist alone, hypoxic sensitivity in neonatal AMCs was almost completely abolished, as indicated by the relative lack of effect of acute hypoxia on whole-cell K+ currents and membrane potential (Fig. 1D and E). These blunting effects, however, were not observed when neonatal AMCs were chronically exposed to the κ-opioid agonist (U-62066; 2 μm) alone (Fig. 1F). These findings suggest that chronic exposure to μ- and/or δ-, but not κ-, opioid receptor agonists renders neonatal AMCs relatively insensitive to acute hypoxia.
Role of ATP-sensitive K+ (KATP) channels in mediating the blunting effects of chronic opioids on hypoxic sensitivity in neonatal AMCs
The blunting effect of chronic opioids on hypoxic sensitivity could occur at the level of the O2 sensor or at downstream steps in the signal transduction pathway. We previously showed that hypoxic sensitivity in neonatal AMCs is also blunted following exposure to chronic nicotine, and this was attributable to up-regulation of KATP channel expression (Buttigieg et al. 2007, 2008). These KATP channels open during hypoxia favouring membrane hyperpolarization which counteracts the depolarizing effects of hypoxia on membrane potential, arising from inhibition of other K+ (e.g. SK, BK and KV) channels (Buttigieg et al. 2008; Nurse et al. 2009). To test whether increased KATP channel activity plays a similar role in opioid-treated cells, we added the KATP channel blocker glibenclamide (Glib; 50 μm) to the recording solution. Consistent with our previous studies (Thompson & Nurse, 1998; Buttigieg et al. 2008), the hypoxic suppression of outward K+ current in control (untreated) neonatal AMCs after 7 days in vitro was augmented in the presence of glibenclamide, as exemplified in Fig. 2Aa (upper traces). A current density versus voltage plot for a group of six cells showing the enhancing effects of glibenclamide on the hypoxic inhibition of outward K+ current at positive potentials is shown in Fig. 2Aa (lower). Interestingly, in the case of opioid-treated cells cultured for 7 days, addition of glibenclamide to the recording solution resulted in the unmasking of hypoxic sensitivity in cells that initially failed to show hypoxic inhibition of outward K+ current (e.g. Fig. 2Ab, upper traces). Pooled data from a group of seven opioid-treated cells, exposed sequentially to hypoxia, glibenclamide, hypoxia plus glibenclamide and wash are shown in the lower I–V plot (Fig. 2Ab; lower); note the significant enhancing effect of glibenclamide on the hypoxic inhibition of outward current at more positive potentials. These data are consistent with the hypothesis that the O2-sensor itself is functionally intact in opioid-treated AMCs and that the blunting effect of chronic opioids on hypoxia sensitivity is attributable to increased KATP channel activity.
Figure 2. Effects of glibenclamide on outward K+ current and membrane potential during hypoxia in control vs. opioid-treated neonatal AMCs.
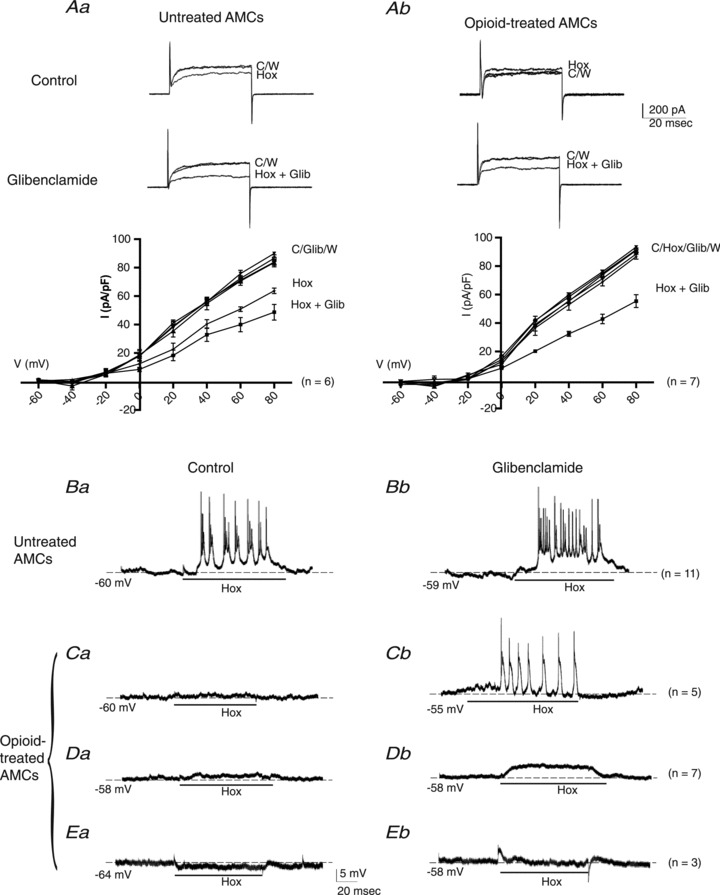
In control (untreated) AMCs, glibenclamide (50 μm) enhances the hypoxia-induced inhibition of outward K+ current as shown in upper sample traces (steps to + 30 mV) and lower current density vs. voltage (I–V) plot (Aa); significant difference (P < 0.05) between current density in hypoxia (Hox) and hypoxia plus glibenclamide (Hox + Glib) at potentials ≥+30 mV. By contrast, opioid-treated cells failed to respond to hypoxia alone, but when combined with glibenclamide, there was a pronounced inhibition of outward current at positive potentials (> +20 mV), as shown in upper traces and lower I–V plot (Ab). Data are presented as mean ± SEM. (P < 0.05). Under current clamp, the hypoxia-induced membrane depolarization and increased excitability (Ba) was potentiated when glibenclamide was applied to the same control cell (Bb) (n= 11). By contrast, membrane depolarization was absent or weak when hypoxia was applied to opioid-treated cells (Ca, Da), and even a weak hyperpolarization occurred in a few cases (Ea). However, when these same cells were exposed to hypoxia in the presence of glibenclamide, there was a marked positive or depolarizing shift in membrane potential as shown in Cb, Db and Eb, respectively; note the cell Ca that was initially quiescent during hypoxia actually fired action potentials when hypoxia was combined with glibenclamide (Cb). The ‘n’ values for each type of response are shown on the right. C, control; Hox, hypoxia; W, wash; Glib, glibenclamide.
To address whether the increased KATP channel activity after chronic opioids is effective in blunting the hypoxia-induced receptor potential in neonatal AMCs, we tested the effects of glibenclamide under current clamp conditions. As exemplified in Fig. 2Ba and Bb, respectively, control (untreated) cells that initially responded to hypoxia with bursts of action potentials appeared to show increased hypoxia-induced spike activity superimposed on a more depolarized membrane potential after glibenclamide (cf. Fig. 2Ba and Bb; n= 10) (see also Buttigieg et al. 2008). On the other hand, many opioid-treated cells that initially failed to show hypoxia-induced membrane depolarization responded with positive shifts in membrane potential when glibenclamide was present in the bathing solution. Among these opioid-treated cells we identified several categories: (i) cells which showed no obvious response to hypoxia initially but responded to hypoxia plus glibenclamide with a burst of action potentials (Fig. 2Ca and Cb; n= 5); (ii) cells which showed no obvious response to hypoxia initially but responded to hypoxia plus glibenclamide with a subthreshold membrane depolarization (Fig. 2Da and Db; mean depolarization = 5.8 ± 1.06 mV, n= 7); and (iii) cells which even showed a slight hyperpolarization to hypoxia initially but responded to hypoxia plus glibenclamide with a positive shift in membrane potential (Fig. 2Ea and Eb; n= 3). These categories probably reflect cell groups with different expression levels of KATP channels relative to the other O2-sensitive K+ channels that are inhibited by hypoxia.
Chronic opioids upregulate expression of Kir6.2 subunit of the KATP channel
One possible explanation for the enhancement of hypoxic inhibition of K+ current and the decreased hypoxia-sensitivity in opioid-treated AMCs exposed to glibenclamide (see Fig. 2) is an increased expression of functional KATP channels. To obtain a quantitative estimate of the increased contribution of KATP channel currents in control vs. opioid-treated cells we calculated the glibenclamide-sensitive difference current density under hypoxia, using data similar to those in Fig. 2Aa and Ab. The resulting estimate of IKATP current density (pA/pF) was plotted against voltage (mV) over the voltage range −120 to +80 mV as shown in Fig. 3A (n= 10). As expected, the IKATP currents reversed near the K+ equilibrium potential (∼−84 mV) and was significantly larger in opioid-treated cells; this was especially noticeable at positive membrane potentials as the driving force on K+ increased. For a voltage step to +30 mV (from a holding potential of −60 mV) the glibenclamide-sensitive difference current density increased from 11.5 ± 1.85 pA pF-1 (n= 11) in control to 21.6 ± 3.03 pA pF-1 in opioid-treated cells (n= 15; P < 0.05; Fig. 3B). To address whether this increase was attributable to increased expression of KATP channels at the protein level, we used Western blot analysis on extracts from 7-day-old control and opioid-treated AMC cultures. As indicated in Fig. 3C, the expression level of the channel pore-forming subunit Kir6.2 (relative to β-actin) increased ∼7-fold following opioid exposure. These data suggest that chronic opioids cause an increased expression of KATP channel proteins in neonatal AMCs during 1 week of exposure.
Figure 3. Upregulation of functional KATP channels and Kir6.2 subunit in opioid-treated neonatal rat AMCs.
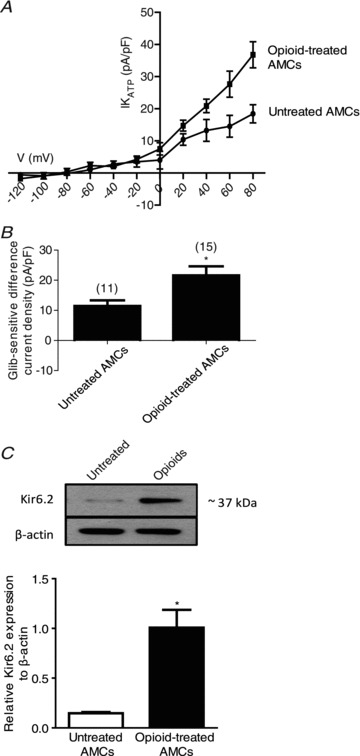
The glibenclamide-sensitive difference current density (IKATP (pA/pF)) in untreated versus opioid-treated AMCs is plotted against voltage in the I–V plot (A), and during steps to +30 mV (B). Note the significant increase in KATP current density in opioid-treated relative to control untreated AMCs (P < 0.05 in B). C, Western blot analysis of KATP channel subunit, Kir6.2, in untreated AMCs and in AMCs cultured with combined μ-, δ- and κ-opioid agonists (2 μm) for 7 days. Note increased expression of Kir6.2 during chronic opioid exposure; β-actin was used as an internal control. Values are presented as mean ± SEM of three independent experiments (*P < 0.05).
Chronic opioid exposure blunts CO2 chemosensitivity in neonatal AMCs
Sensitivity of neonatal rat AMCs to hypercapnia (10% CO2) also decreases postnatally in parallel with splanchnic innervation, and is mediated by inhibition of outward K+ current, membrane depolarization, voltage-gated Ca2+ entry and CAT secretion (Munoz-Cabello et al. 2005; Buttigieg et al. 2007, 2008). To address whether hypercapnic sensitivity is also regulated by chronic opioid exposure we monitored membrane currents and membrane potential in AMCs exposed to high CO2 (10% CO2) under isohydric conditions (pH = 7.4). In control conditions, isohydric hypercapnia caused inhibition of outward K+ current under voltage clamp (Fig. 4A; upper trace), and induced a depolarizing receptor potential under current clamp (Fig. 4A; lower trace) in several neonatal AMCs cultured for 7 days. For this experimental series, pooled data from a group of cells (n= 11) examined under voltage clamp are summarized in the I–V plot in Fig. 4A, where isohydric hypercapnia caused a significant suppression of outward K+ current at positive potentials. By contrast, exposure to combined μ-, δ- and κ-opioid agonists (DALDA, DPDPE and U-62066; 2 μm) for 7 days resulted in a marked reduction or blunting of CO2 sensitivity as illustrated in the exemplar traces in Fig. 4B (upper and lower trace), and in the I–V plot of Fig. 4B (middle). Moreover, continuous co-incubation of these combined agonists with the general opioid receptor blocker naloxone (2 μm) resulted in the retention of CO2 sensitivity (Fig. 4C). As was the case for opioid-mediated blunting of hypoxic sensitivity, the blunting of CO2 sensitivity involved mainly μ- and δ-opioid receptors. For example, as illustrated in Fig. 4D–F, incubation for 7 days with either the μ- or the δ-opioid receptor agonist alone resulted in a marked suppression of CO2 sensitivity (Fig. 4D and E), whereas incubation with the κ-opioid agonist alone was ineffective (Fig. 4F).
Figure 4. Effects of chronic opioid exposure on CO2 sensitivity in neonatal rat AMCs.
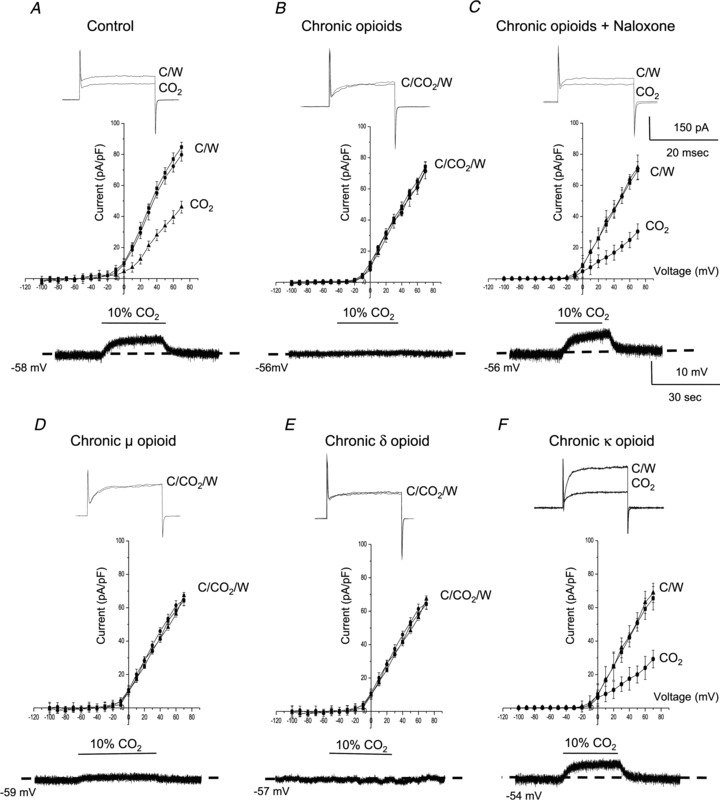
In control neonatal AMCs cultured for ∼1 week, isohydric hypercapnia (10% CO2; pH 7.4) typically causes inhibition of outward K+ current (top trace) and membrane depolarization (bottom trace); current density (pA/pF) versus voltage (I–V) plot (middle) shows significant inhibition of outward current (P < 0.05) at positive potentials (A). These responses are markedly reduced or abolished in neonatal AMCs chronically exposed for ∼1 week to either a combination of μ-, δ- and κ-opioid agonists (2 μm) (B), or to either μ- or δ-opioid agonists (2 μm) alone (D and E, respectively). These blunting effects of chronic opioids on the CO2-induced responses are prevented during continuous co-incubation with the general opioid receptor antagonist naloxone (2 μm) (C). Also, chronic exposure to the κ-opioid agonist (2 μm) alone was ineffective (F). Data are presented as mean ± SEM (n= 11). TTX was present in the extracellular solution. C, control; W, wash.
Whereas inhibition of outward K+ current is thought to contribute to CO2 sensitivity of neonatal AMCs via prolongation of the action potential duration, the origin of the receptor potential or initial depolarization at rest is attributable to activation of a cation conductance (Munoz-Cabello et al. 2005). To examine whether this component of the CO2 sensing pathway was also impaired in opioid-treated cells we first monitored the receptor potential in control versus opioid-treated cells exposed briefly to isohydric hypercapnia (10% CO2; pH 7.4). As exemplified in Fig. 4A and B (lower traces) and Fig. 5Ba and Bb, whereas hypercapnia readily induced a subthreshold membrane depolarization (mean = 5.5 ± 0.25 mV; n= 4) or action potential firing (n= 12) in the majority (∼80%) of control cells, opioid-treated cells were largely (Fig. 4B, lower trace; Fig. 5Ca), although not exclusively (Fig. 5Cb), unresponsive (>80% non-responsive cells). Quantification of the relative proportions of responsive versus non-responsive cells in the two conditions is summarized in Fig. 5D for this experimental series. The blunting effect of opioids on hypercapnia sensitivity was prevented during continuous co-incubation with 2 μm naloxone (Fig. 4C; lower trace; n= 12) and, as in the case of hypoxia (see above), involved mainly μ- and δ- (but not κ-) opioid receptors (Fig. 4D–F).
Figure 5. Comparative estimates of the blunting effects of chronic opioids on CO2-mediated responses in neonatal AMCs.
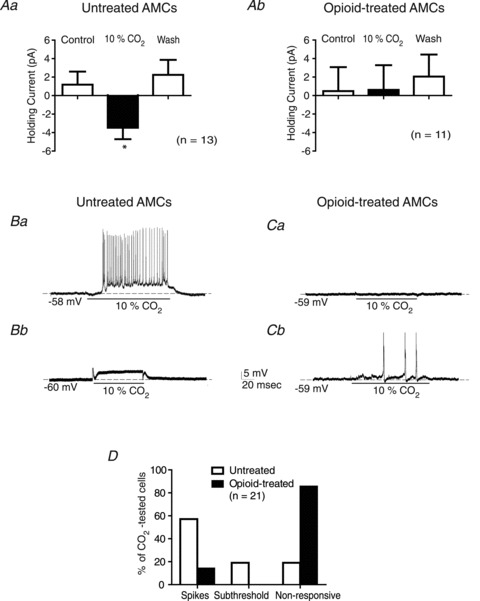
Comparison of the CO2-induced changes in holding current at −60 mV in control (Aa) versus opioid-treated (Ab) cells; note the significant (*P < 0.05) inward shift in holding current normally seen in control cells during high CO2 is abolished in opioid-treated cells. The variability in CO2-induced changes in membrane potential is shown for control cells (Ba, Bb) versus opioid-treated cells (Ca, Cb); data of response frequency for each condition are summarized in D. Note that CO2-induced action potential firing or spikes (Ba) or subthreshold depolarizations (Bb) occur frequently in untreated (control) cells (D), but is rare in opioid-treated cells (Cb, D). Also, the majority of opioid-treated cells fail to show either CO2-induced membrane depolarization or spikes, i.e. are non-responsive (Ca, D), in contrast to untreated cells (D).
We also compared changes in holding current in control versus opioid-treated cells during a switch from normocapnic (5% CO2) to hypercapnic (10% CO2) solutions at constant pH. As indicated in Fig. 5Aa, and consistent with a previous report (Munoz-Cabello et al. 2005), under voltage clamp there was a significant inward shift in the holding current (at −60 mV) when control AMCs were exposed to hypercapnia, and the effect was readily reversible. By contrast, under similar conditions hypercapnia failed to induce a significant shift in the holding current in opioid-treated cells, consistent with the current clamp data showing suppression or loss of hypercapnia-induced membrane depolarization at the resting potential (Fig. 4B and E, lower traces; Fig. 5Ca).
Chronic opioid exposure reduces expression of CA isoforms I and II in neonatal AMCs
Tests for the presence of seven different CA isoforms using RT-PCR and in situ hybridization revealed that only CAI and CAII mRNA were expressed in the neonatal rat adrenal gland and, interestingly, expression of both isoforms was virtually absent (>1000-fold lower) in the adult gland (Munoz-Cabello et al. 2005). Moreover, in electrophysiological studies the hypercapnic response of neonatal AMCs was markedly inhibited by the CA inhibitor methazolamide, leading these authors to conclude that the CO2 sensitivity of these cells was at least partially dependent on high expression levels of the CO2-hydrating enzymes CAI and CAII (Munoz-Cabello et al. 2005). These considerations led us to examine whether the opioid-induced suppression of CO2 sensitivity in cultured neonatal AMCs was associated with reduced expression of CAI and CAII at the protein level. Indeed, using Western blot analysis with β-actin as a control, we found that expression of both CAI and CAII proteins was significantly reduced in opioid-treated neonatal AMCs compared to control cells after 7 days in vitro. Exemplar gels of CAI and CAII expression patterns for control versus opioid-treated cells and summary data from triplicate experiments are illustrated in Fig. 6A and B.
Figure 6. Effects of chronic opioid exposure on CA enzyme expression in neonatal rat AMCs.
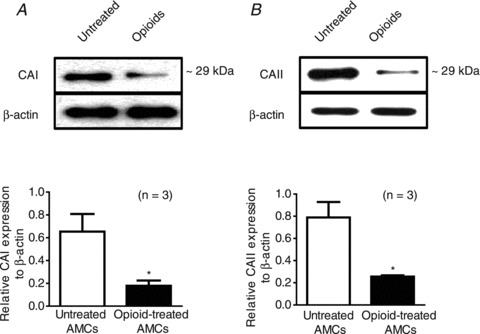
A, Western blot analysis showing expression of CA isoforms CAI and CAII in 7-day-old cultures of control untreated AMCs versus AMCs treated with combined μ-, δ- and κ-opioid agonists (2 μm); β-actin was used as an internal control. Note the downregulation in CAI and CAII expression in neonatal AMCs following chronic opioid exposure. Values are represented as mean ± SEM of three independent experiments (*P < 0.05).
Immunocytochemical detection of μ- and δ-opioid receptors on cultured neonatal rat chromaffin cells
Based on gene expression studies using RT-PCR analysis, the three major opioid receptors, μ, δ and κ, have been identified in adult rat adrenal gland (Wittert et al. 1996). To corroborate the above electrophysiological findings demonstrating key roles for μ- and δ-opioid receptor signalling in regulation of O2 and CO2 sensing in neonatal AMCs we used fluorescence immunocytochemistry. As shown in Fig. 7, both μ- and δ-opioid receptor immunostaining was localized selectively to chromaffin cell clusters in dissociated neonatal AMC cultures. These data suggest that the blunting effects of chronic opioid exposure on chemosensing were mediated via direct interactions with μ- or δ-opioid receptors on neonatal AMCs.
Figure 7. Immunofluorescence staining of neonatal rat AMC cultures for μ- and δ-opioid receptor expression.
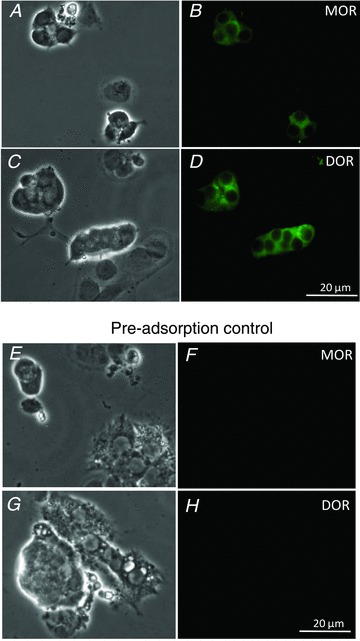
Corresponding phase and fluorescence (FITC) images of cultures showing positive immunostaining of AMCs for μ-opioid receptor (MOR) (A and B, respectively), and for δ-opioid receptor (DOR) (C and D, respectively). Pre-absorption controls with excess antigen (see Methods) confirming staining specificity for each antibody are shown in E–H. The data are representative of three independent experiments for each antibody staining.
Discussion
In this study we demonstrate that chronic exposure of neonatal rat AMCs to μ- and/or δ-opioid receptor agonists for ∼1 week in vitro leads to a naloxone-sensitive suppression of direct hypoxic and hypercapnic chemosensitivity. These results are of interest because they suggest a general mechanism that could account for the developmental loss or suppression of direct O2 and CO2 sensitivity in AMCs that normally occurs during preganglionic splanchnic innervation (Slotkin & Seidler, 1988; Thompson et al. 1997; Munoz-Cabello et al. 2005). In particular, because the amount of opiate peptides (i.e. enkephalin) increases in the developing rat preganglionic splanchnic nerve in parallel with the suppression of direct hypoxic chemosensitivity (Holgert et al. 1998; Nurse et al. 2009), the possibility is raised that activation of postsynaptic receptors on AMCs by released opiate peptides is involved. In this regard, there are conflicting reports on the suppressive effects of opioid agonists on hypoxic sensitivity of ovine and rat AMCs when these agonists are applied acutely (Keating et al. 2004; Rico et al. 2005). While the difference between these two studies may be species related, it should be noted that in the present study opioid agonist exposure lasted for ∼1 week in vitro and, importantly, opioid agonists were not present in the bathing solution when chemosensitivity was tested. Chronic exposure of neonatal AMCs to opioids is more likely to mimic the developmental effects of splanchnic innervation given that the latter matures gradually over the first 1–2 postnatal weeks in the rat (Seidler & Slotkin, 1985). Moreover, our finding that both hypoxic and hypercapnic sensitivity are suppressed in neonatal AMCs after chronic opioid exposure in vitro closely mimics the in vivo developmental pattern seen in these cells, when examined acutely before and after splanchnic innervation (Slotkin & Seidler, 1988; Thompson et al. 1997; Munoz-Cabello et al. 2005). It is noteworthy that the splanchnic nerve also supplies an important nicotinic cholinergic innervation to the adrenal gland; however, in our previous studies chronic exposure of neonatal AMCs to nicotine in utero and in vitro led to a selective blunting of hypoxic, but not hypercapnic, sensitivity (Buttigieg et al. 2007, 2008). It therefore appears that the opioid receptor signalling pathway contributes to the developmental regulation of both hypoxic and hypercapnic sensitivity in AMCs, whereas the nicotinic receptor pathway contributes only to the regulation of hypoxic sensitivity.
Mechanisms underlying opioid-mediated suppression of hypoxic chemosensitivity
Hypoxic sensitivity of neonatal AMCs involves regulation of various subtypes of K+ channels, leading to membrane depolarization, voltage-gated Ca2+ entry and CAT secretion (reviewed by Nurse et al. 2009). Both small (SK) and large (BK) conductance K+, as well as voltage-dependent K+ (Kv), channels are inhibited by hypoxia leading to or facilitating membrane depolarization. However, at the resting membrane potential, inhibition of apamin-sensitive SK channels appears to be the primary event leading to membrane depolarization and CAT secretion (Lee et al. 2000; Keating et al. 2004; Bournaud et al. 2007). This depolarizing action, however, is blunted by the simultaneous activation of ATP-sensitive K+ (KATP) channels, which favours membrane hyperpolarization during hypoxia (Thompson & Nurse, 1998; Bournaud et al. 2007). After chronic exposure to μ- and/or δ- (but not κ-) opioid agonists, at concentrations (2 μm) greatly exceeding their Kd values but comparable to estimated opiate extracellular concentrations near AMCs in vivo (Castanas et al. 1985a,b; Keating et al. 2004), the normal hypoxia-induced inhibition of outward K+ currents at positive potentials was markedly reduced or eliminated. Moreover, the hypoxia-induced membrane depolarization or receptor potential, due mainly to inhibition of SK channels at the resting potential (Lee et al. 2000; Keating et al. 2005; Bournaud et al. 2007), was also significantly suppressed. Dramatically, however, hypoxia-sensitivity as determined by both assays could be restored in these opioid-treated cells if the KATP channel blocker glibenclamide was present in the recording extracellular solution. In fact, some opioid-treated cells that were initially silent when exposed to acute hypoxia generated a burst of action potentials when the same stimulus was applied to the same cell in the presence of glibenclamide. These data suggest that upregulation of KATP channel function, which would favour membrane hyperpolarization during acute hypoxia, is a major contributor to the blunting of hypoxic sensitivity in these cells. Supportive evidence for this mechanism was obtained from electrophysiological estimates of KATP current density, determined from the glibenclamide-sensitive difference current during hypoxia, and by Western blot analysis of the expression of the KATP channel subunit, Kir6.2, in control versus opioid-treated cells. Taken together, these data are reminiscent of a similar role played by KATP channel upregulation in the blunting of hypoxia sensitivity in perinatal AMCs following exposure to chronic nicotine in utero and in vitro (Buttigieg et al. 2008; Salman et al. 2012). However, further experiments are required to elucidate the intracellular signalling pathway that leads to KATP channel upregulation during opioid exposure. It should also be noted that we cannot presently exclude the possibility that other potential targets in the O2 chemotransduction pathway may also be modified by chronic opioid exposure. These include subtle changes in expression levels of the other O2-sensitive K+ channels (i.e. SK, BK and Kv), or of T-type Ca2+ channels which have been reported to play a key role in the hypoxic sensitivity of neonatal AMCs (Levitsky & Lopez-Barneo, 2009).
Mechanisms underlying opioid-mediated suppression of CO2 chemosensitivity
An especially novel aspect of our study was the demonstration that chronic opioids also blunted the sensitivity of neonatal AMCs to high CO2 (hypercapnia), thereby mimicking the developmental loss or reduction in hypercapnic sensitivity following splanchnic innervation (Munoz-Cabello et al. 2005). To our knowledge, this evidence is the first to suggest how innervation of AMCs could lead to loss of CO2 sensitivity. A previous study highlighted several aspects associated with hypercapnic sensitivity in neonatal rat AMCs (Munoz-Cabello et al. 2005). These include: (i) high CO2-mediated activation of a resting cationic conductance leading to membrane depolarization or the receptor potential; (ii) high CO2-mediated inhibition of voltage-dependent K+ channels, leading to prolongation of action potential duration and increased CAT secretion; and (iii) blockade of high CO2-mediated responses by the membrane-permeable CA inhibitor methazolamide (Munoz-Cabello et al. 2005). In the present study, chronic exposure to μ- and/or δ-opioid agonists markedly reduced or abolished not only the high CO2-mediated receptor potential, but also inhibition of voltage-dependent outward K+ current. Moreover, because CAI and CAII are the main CA isoforms expressed in neonatal rat AMCs, and both isoforms are downregulated during splanchnic innervation (Munoz-Cabello et al. 2005), we compared their expression patterns in control versus opioid-treated AMCs using Western blot analysis. Indeed, we found that both CAI and CAII proteins were significantly downregulated in neonatal AMCs exposed to chronic opioids in vitro. The intracellular signalling cascade leading to reduced CAI and CAII expression in opioid-treated cells remains to be elucidated. Nevertheless, these data suggest a molecular mechanism by which opioid innervation can lead to suppression of CO2 sensitivity in AMCs during postnatal development.
Clinical significance
Our main finding that chronic opioid exposure can impair the ability of neonatal AMCs to sense asphyxial stimuli such as hypoxia and hypercapnia has important clinical implications. The deleterious consequence is that the CAT surge that normally occurs in response to asphyxial stressors at birth would become seriously impaired, thereby compromising the proper transition of the neonate to extra-uterine life (Seidler & Slotkin, 1985; Slotkin & Seidler, 1988). This CAT release is critical for maintenance of cardiac conduction and for transformation of the lung into an air-breathing epithelium (Slotkin & Seidler, 1988). Although the use of opioid medication is widespread for pain management, opioid drugs (e.g. heroin) are major contributors to drug misuse and fatalities. In the adult, these fatalities often result from the adverse side effects of opioids, particularly the marked respiratory depression that becomes exacerbated during sleep (Walker & Farney, 2009). To date, brain sites involved in respiratory control (e.g. the pre-Bötzinger complex) have received the most attention for the targets of opioid-mediated respiratory depression in the adult (Morin-Surun et al. 1992; Montandon et al. 2011). In the neonate, however, hypoxic activation of catecholaminergic pathways, especially via the adrenal medulla, stimulates breathing and arousal responses that are critical for survival (Sawnani et al. 2004; Cohen et al. 2005). Thus, blunted adrenal catecholaminergic responses during perinatal asphyxia contributes to the failure of arousal and elevated perinatal mortality as occurs during sudden infant death syndrome (SIDS) (Sawnani et al. 2004; Cohen et al. 2005; Buttigieg et al. 2008). In this regard, opioid intake by mothers during pregnancy has been linked to an increased incidence of infant mortality due mainly to SIDS (Burns et al. 2010). It is possible that the mechanisms underlying opioid-mediated suppression of O2 and CO2 sensing in neonatal chromaffin cells, as uncovered in the present study, are broadly applicable to other chemosensitive sites involved in respiratory control. If so, they could help clarify the relationship between opiate misuse during pregnancy and abnormal arousal responses associated with perinatal disorders that result in infant morbidity, e.g. SIDS.
Acknowledgments
This work was supported by a Discovery grant from the Natural Sciences and Engineering Research Council of Canada (NSERC) and an operating grant from the Canadian Institutes of Health Research (MOP-119501) to C.A.N. We thank Cathy Vollmer for expert technical assistance.
Glossary
- ACh
acetylcholine
- AMCs
adrenomedullary chromaffin cells
- CA
carbonic anhydrase
- CAT
catecholamine
- SIDS
sudden infant death syndrome
- TTX
tetrodotoxin
Author contributions
S.S. assisted with some of the electrophysiological data acquisition and analysis, performed the molecular and immunocytochemical experiments, and wrote the first draft of the manuscript. J.B. performed the initial electrophysiological experiments and analysed the data. M.Z. was involved in data acquisition for some of the electrophysiological experiments. C.A.N. was involved in initiating the study, planning and design of the experiments, and in editing the final manuscript. All authors approved the final version of the manuscript. All experiments were performed in the Department of Biology, McMaster University.
References
- Bournaud R, Hidalgo J, Yu H, Girard E, Shimahara T. Catecholamine secretion from rat foetal adrenal chromaffin cells and hypoxia sensitivity. Pflugers Arch. 2007;454:83–92. doi: 10.1007/s00424-006-0185-z. [DOI] [PubMed] [Google Scholar]
- Burns L, Conroy E, Mattick R. Infant mortality among women on a methadone program during pregnancy. Drug Alcohol Rev. 2010;29:551–556. doi: 10.1111/j.1465-3362.2010.00176.x. [DOI] [PubMed] [Google Scholar]
- Buttigieg J, Brown S, Zhang M, Lowe M, Holloway AC, Nurse CA. Chronic nicotine in utero selectively suppresses hypoxic sensitivity in neonatal rat adrenal chromaffin cells. FASEB J. 2007;22:1317–1326. doi: 10.1096/fj.07-9194com. [DOI] [PubMed] [Google Scholar]
- Buttigieg J, Brown ST, Holloway AC, Nurse CA. Chronic nicotine blunts hypoxic sensitivity in perinatal rat adrenal chromaffin cells via upregulation of KATP channels: role of α7 nicotinic AChR and hypoxia inducible factor-2α. J Neurosci. 2008;29:7137–7147. doi: 10.1523/JNEUROSCI.0544-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castanas E, Bourhim N, Giraud P, Boudouresque F, Cantau P, Oliver C. Interaction of opiates with opioid binding sites in the bovine adrenal medulla: I. Interaction with delta and mu sites. J Neurochem. 1985a;45:677–687. doi: 10.1111/j.1471-4159.1985.tb04046.x. [DOI] [PubMed] [Google Scholar]
- Castanas E, Bourhim N, Giraud P, Boudouresque F, Cantau P, Oliver C. Interaction of opiates with opioid binding sites in the bovine adrenal medulla: II. Interaction with kappa sites. J Neurochem. 1985b;45:688–699. doi: 10.1111/j.1471-4159.1985.tb04047.x. [DOI] [PubMed] [Google Scholar]
- Cheng CY, Wu SC, Hsin LW, Tam SW. Selective reversible and irreversible ligands for the kappa opioid receptor. J Med Chem. 1992;35:2243–2247. doi: 10.1021/jm00090a015. [DOI] [PubMed] [Google Scholar]
- Cheung CY. Fetal adrenal medulla catecholamine response to hypoxia – direct and neural components. Am J Physiol Regul Integr Comp Physiol. 1990;258:R1340–R1346. doi: 10.1152/ajpregu.1990.258.6.R1340. [DOI] [PubMed] [Google Scholar]
- Cohen G, Roux JC, Grailhe R, Malcolm G, Changeux JP, Lagercrantz H. Perinatal exposure to nicotine causes deficits associated with a loss of nicotinic receptor function. Proc Natl Acad Sci U S A. 2005;102:3817–3821. doi: 10.1073/pnas.0409782102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Fernandez M, Mejias R, Lopez-Barneo J. Developmental changes of chromaffin cell secretory response to hypoxia studied in thin adrenal slices. Pflugers Arch. 2007;454:93–100. doi: 10.1007/s00424-006-0186-y. [DOI] [PubMed] [Google Scholar]
- Holgert H, Dagerlind A, Hokfelt T. Immunohistochemical characterization of the peptidergic innervation of the rat adrenal gland. Horm Metab Res. 1998;30:315–322. doi: 10.1055/s-2007-978891. [DOI] [PubMed] [Google Scholar]
- Jones CT. Circulating catecholamines in the fetus, their origin, actions and significance. In: Parvez H, Parvez S, editors. Biogenic Amines in Development. Amsterdam: Elsevier/North Holland Biomedical Press; 1980. pp. 63–68. [Google Scholar]
- Keating DJ, Rychkov GY, Adams MB, Holgert H, McMillen IC, Roberts ML. Opioid receptor stimulation suppresses the adrenal medulla hypoxic response in sheep by actions on Ca2+ and K+ channels. J Physiol. 2004;555:489–502. doi: 10.1113/jphysiol.2003.056176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating DJ, Rychkov GY, Giacomin P, Roberts ML. Oxygen-sensing pathway for SK channels in the ovine adrenal medulla. Clin Exp Pharmacol Physiol. 2005;32:882–887. doi: 10.1111/j.1440-1681.2010.04279.x. [DOI] [PubMed] [Google Scholar]
- Kimura T, Katoh M, Satoh S. Inhibition by opioid agonists and enhancement by antagonists of the release of catecholamines from the dog adrenal gland in response to splanchnic nerve stimulation: evidence for the functional role of opioid receptors. J Pharmacol Exp Ther. 1988;244:1098–1102. [PubMed] [Google Scholar]
- Kobayashi S, Miyabayashi T, Uchida T, Yanaihara N. Met-enkephalin-Arg6-Gly7-Leu8 in large-cored vesicles of splanchnic nerve terminals innervating guinea pig adrenal chromaffin cells. Neurosci Lett. 1985;53:247–252. doi: 10.1016/0304-3940(85)90545-2. [DOI] [PubMed] [Google Scholar]
- Kuri BA, Chan SA, Smith CB. PACAP regulates immediate catecholamine release from adrenal chromaffin cells in an activity-dependent manner through a protein kinase C-dependent pathway. J Neurochem. 2009;110:1214–1225. doi: 10.1111/j.1471-4159.2009.06206.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagercrantz H, Bistoletti P. Catecholamine release in the newborn infant at birth. Pediatr Res. 1977;11:889–893. doi: 10.1203/00006450-197708000-00007. [DOI] [PubMed] [Google Scholar]
- Lagercrantz H, Slotkin TA. The ‘stress’ of being born. Sci Am. 1986;254:100–107. doi: 10.1038/scientificamerican0486-100. [DOI] [PubMed] [Google Scholar]
- Lee J, Lim W, Eun SY, Kim SJ, Kim J. Inhibition of apamin-sensitive K+ current by hypoxia in adult rat adrenal chromaffin cells. Pflugers Arch. 2000;439:700–704. doi: 10.1007/s004249900228. [DOI] [PubMed] [Google Scholar]
- Levitsky KL, Lopez-Barneo J. Developmental change of T-type Ca2+ channel expression and its role in rat chromaffin cell responsiveness to acute hypoxia. J Physiol. 2009;587:1917–1929. doi: 10.1113/jphysiol.2009.168989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livermore S, Piskuric NA, Buttigieg J, Zhang M, Nurse CA. Low glucose sensitivity and polymodal chemosensing in neonatal rat adrenomedullary chromaffin cells. Am J Physiol Cell Physiol. 2011;301:C1104–C1115. doi: 10.1152/ajpcell.00170.2011. [DOI] [PubMed] [Google Scholar]
- Mochizuki-Oda N, Takeuchi Y, Matsumura K, Oosawa Y, Watanabe Y. Hypoxia-induced catecholamine release and intracellular Ca2+ increase via suppression of K+ channels in cultured rat adrenal chromaffin cells. J Neurochem. 1997;69:377–387. doi: 10.1046/j.1471-4159.1997.69010377.x. [DOI] [PubMed] [Google Scholar]
- Montandon G, Qin W, Liu H, Ren J, Greer JJ, Horner RL. PreBotzinger complex neurokinin-1 receptor-expressing neurons mediate opioid-induced respiratory depression. J Neurosci. 2011;31:1292–1301. doi: 10.1523/JNEUROSCI.4611-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin-Surun MP, Boudinot E, Fournie-Zaluski MC, Champagnat J, Roques BP, Denavit-Saubie M. Control of breathing by endogenous opioid peptides: possible involvement in sudden infant death syndrome. Neurochem Int. 1992;20:103–107. doi: 10.1016/0197-0186(92)90132-b. [DOI] [PubMed] [Google Scholar]
- Munoz-Cabello AM, Toledo-Aral JJ, Lopez-Barneo J, Echevarria M. Rat adrenal chromaffin cells are neonatal CO2 sensors. J Neurosci. 2005;25:6631–6640. doi: 10.1523/JNEUROSCI.1139-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurse CA, Buttigieg J, Brown S, Holloway AC. Regulation of oxygen sensitivity in adrenal chromaffin cells. Ann N Y Acad Sci. 2009;1177:132–139. doi: 10.1111/j.1749-6632.2009.05031.x. [DOI] [PubMed] [Google Scholar]
- Rico AJ, Prieto-Lloret J, Gonzalez C, Rigual R. Hypoxia and acidosis increase the secretion of catecholamines in the neonatal rat adrenal medulla: an in vitro study. Am J Physiol Cell Physiol. 2005;289:C1417–C1425. doi: 10.1152/ajpcell.00023.2005. [DOI] [PubMed] [Google Scholar]
- Salman S, Brown ST, Nurse CA. Chronic nicotine induces hypoxia inducible factor-2α in perinatal rat adrenal chromaffin cells: role in transcriptional upregulation of KATP channel subunit Kir6.2. Am J Physiol Cell Physiol. 2012;302:C1531–C1538. doi: 10.1152/ajpcell.00052.2012. [DOI] [PubMed] [Google Scholar]
- Sawnani H, Jackson T, Murphy T, Beckerman R, Simakajornboon N. The effect of maternal smoking on respiratory and arousal patterns in preterm infants during sleep. Am J Respir Crit Care Med. 2004;169:733–738. doi: 10.1164/rccm.200305-692OC. [DOI] [PubMed] [Google Scholar]
- Schlosser B, Kudernatsch MB, Sutor B, ten Bruggencate G. Delta, mu and kappa opioid receptor agonists inhibit dopamine overflow in rat neostriatal slices. Neurosci Lett. 1995;191:126–130. doi: 10.1016/0304-3940(94)11552-3. [DOI] [PubMed] [Google Scholar]
- Seidler FJ, Slotkin TA. Adrenomedullary function in the neonatal rat: responses to acute hypoxia. J Physiol. 1985;358:1–16. doi: 10.1113/jphysiol.1985.sp015536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidler FJ, Slotkin TA. Non-neurogenic adrenal catecholamine release in the neonatal rat: exocytosis or diffusion. Brain Res. 1986;393:274–277. doi: 10.1016/0165-3806(86)90031-3. [DOI] [PubMed] [Google Scholar]
- Slotkin TA, Seidler FJ. Adrenomedullary catecholamine release in the fetus and newborn: secretory mechanisms and their role in stress and survival. J Dev Physiol. 1988;10:1–16. [PubMed] [Google Scholar]
- Thompson RJ, Buttigieg J, Zhang M, Nurse CA. A rotenone-sensitive site and H2O2 are key components of hypoxia-sensing in neonatal rat adrenomedullary chromaffin cells. Neuroscience. 2007;145:130–141. doi: 10.1016/j.neuroscience.2006.11.040. [DOI] [PubMed] [Google Scholar]
- Thompson RJ, Farragher SM, Cutz E, Nurse CA. Developmental regulation of O2-sensing in neonatal adrenal chromaffin cells from wild-type and NADPH-oxidase-deficient mice. Pflugers Arch. 2002;444:539–548. doi: 10.1007/s00424-002-0853-6. [DOI] [PubMed] [Google Scholar]
- Thompson RJ, Jackson A, Nurse CA. Developmental loss of hypoxic chemosensitivity in rat adrenomedullary chromaffin cells. J Physiol. 1997;498:503–510. doi: 10.1113/jphysiol.1997.sp021876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RJ, Nurse CA. Anoxia differentially modulates multiple K+ currents and depolarizes neonatal rat adrenal chromaffin cells. J Physiol. 1998;512:421–434. doi: 10.1111/j.1469-7793.1998.421be.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JM, Farney RJ. Are opioids associated with sleep apnea? A review of the evidence. Curr Pain Headache Rep. 2009;13:120–126. doi: 10.1007/s11916-009-0021-1. [DOI] [PubMed] [Google Scholar]
- Wittert G, Hope P, Pyle D. Tissue distribution of opioid receptor gene expression in the rat. Biochem Biophys Res Commun. 1996;218:877–881. doi: 10.1006/bbrc.1996.0156. [DOI] [PubMed] [Google Scholar]
- Zhang M, Nurse CA. CO2/pH chemosensory signaling in co-cultures of rat carotid body receptors and petrosal neurons: role of ATP and ACh. J Neurophysiol. 2004;92:3433–3445. doi: 10.1152/jn.01099.2003. [DOI] [PubMed] [Google Scholar]