

Abstract
Creatine (Cr) plays an important role in muscle energy homeostasis by its participation in the ATP–phosphocreatine phosphoryl exchange reaction mediated by creatine kinase. Given that the consequences of Cr depletion are incompletely understood, we assessed the morphological, metabolic and functional consequences of systemic depletion on skeletal muscle in a mouse model with deficiency of l-arginine:glycine amidinotransferase (AGAT−/−), which catalyses the first step of Cr biosynthesis. In vivo magnetic resonance spectroscopy showed a near-complete absence of Cr and phosphocreatine in resting hindlimb muscle of AGAT−/− mice. Compared with wild-type, the inorganic phosphate/β-ATP ratio was increased fourfold, while ATP levels were reduced by nearly half. Activities of proton-pumping respiratory chain enzymes were reduced, whereas F1F0-ATPase activity and overall mitochondrial content were increased. The Cr-deficient AGAT−/− mice had a reduced grip strength and suffered from severe muscle atrophy. Electron microscopy revealed increased amounts of intramyocellular lipid droplets and crystal formation within mitochondria of AGAT−/− muscle fibres. Ischaemia resulted in exacerbation of the decrease of pH and increased glycolytic ATP synthesis. Oral Cr administration led to rapid accumulation in skeletal muscle (faster than in brain) and reversed all the muscle abnormalities, revealing that the condition of the AGAT−/− mice can be switched between Cr deficient and normal simply by dietary manipulation. Systemic creatine depletion results in mitochondrial dysfunction and intracellular energy deficiency, as well as structural and physiological abnormalities. The consequences of AGAT deficiency are more pronounced than those of muscle-specific creatine kinase deficiency, which suggests a multifaceted involvement of creatine in muscle energy homeostasis in addition to its role in the phosphocreatine–creatine kinase system.
Key points
Creatine (Cr) plays an important role in muscle energy homeostasis as a substrate in the creatine kinase phosphoryl exchange reaction, but the consequences of creatine depletion are incompletely understood.
We assessed the morphological, metabolic and functional consequences of systemic creatine depletion on skeletal muscle in a mouse model with deficiency of an essential enzyme in the biosynthesis of creatine (AGAT−/− mice).
We show that Cr depletion leads to several metabolic abnormalities in muscle, including reduced ATP, increased inorganic phosphate levels and reduced activities of proton-pumping respiratory chain enzymes and an elevated glycolytic contribution in ischaemic circumstances.
The Cr-depleted muscle suffers from reduced grip strength, severe atrophy and abnormal mitochondrial structures, increased overall mitochondrial content and an increased number of lipid droplets.
Oral Cr administration led to rapid accumulation in skeletal muscle (faster than in brain) and reversed all the muscle abnormalities, revealing that the condition of the AGAT−/− mice can be switched between Cr deficient and normal simply by dietary manipulation.
Introduction
Phosphocreatine (PCr) is a major source of ATP replenishment in tissues with rapidly fluctuating energy demand. This supply is mediated by the creatine kinase (CK) reaction, in which creatine (Cr) and ADP are reversibly phosphorylated to PCr and ATP, respectively. The PCr–CK system, which functions as a spatial and temporal buffer of ATP levels, requires a high level of total cellular Cr; 20–40 mm in mammal skeletal muscle (Wyss & Kaddurah-Daouk, 2000). High intracellular Cr concentrations are accomplished by a combination of endogenous production and exogenous dietary intake, followed by cellular uptake of Cr from blood vessels (Fig. 1).
Figure 1. Biosynthesis of creatine.
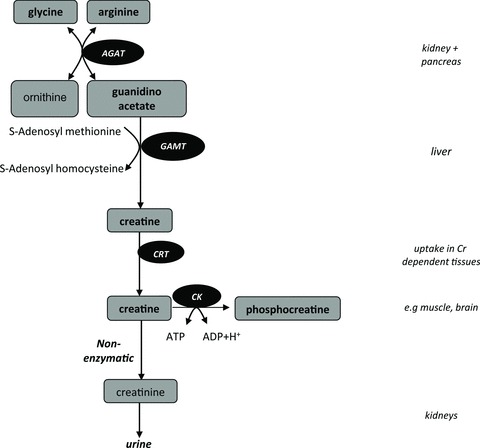
De novo synthesis of creatine mainly takes place in the kidneys, pancreas and liver. The first step of the biosynthesis of creatine (Cr) is rate limiting and is catalysed by l-arginine:glycine amidinotransferase (AGAT). The second step is catalysed by guanidinoacetate methyltransferase (GAMT). The produced Cr is transported by Cr transporters (CRT) towards tissues that have a high energy demand, such as muscle or brain, where it is phosphorylated in the creatine kinase (CK) reaction, which plays an important role in maintaining ATP levels. A proportion (∼1.5%) of the total Cr is converted non-enzymatically into creatinine (Crn), which is excreted by the kidneys.
The PCr–CK system of cardiac and skeletal muscle has been thoroughly investigated in transgenic mouse models with partial or complete deletions or overexpression of muscle-specific CK isoforms. These studies have been valuable in defining the role and importance of the PCr–CK system in skeletal muscle in different circumstances, e.g. at rest, upon stimulation or during ischaemia (for reviews see Nicolay et al. 1998; Heerschap et al. 2007; Saks et al. 2007; Salomons & Wyss, 2007; Saks, 2008). Surprisingly, skeletal muscle of double knockout mice with complete absence of both the cytosolic (M-CK) and mitochondrial (Sc-CKmit) CK isoforms still contains a substantial amount of PCr; however, this cannot easily be recruited for ATP buffering, and the consequent lack of burst activity shows the particular importance of the ATP-buffering role of the PCr–CK system during the initial phase of intense muscle contraction (de Haan et al. 1995; Steeghs et al. 1998). As these double knockout mice still contain substantial amounts of (P)Cr, they cannot be used as a model to study the ultimate consequence of Cr absence.
Previous studies have explored the administration of Cr analogues to replace creatine, in particular β-guanidinopropionic acid (β-GPA). This has several effects on muscular phenotype and biochemistry, which include reduced ATP levels, a switch from fast to slow myosin isoform expression (Moerland et al. 1989) and decreased muscle fibre diameters (Shoubridge et al. 1985). However, the supplemented β-GPA can still be phosphorylated and used as a high-energy phosphate (HEP) analogue and thus compensate at least partly for the absence of PCr in skeletal muscle.
To study the effect of pure Cr deficiency in muscle, therefore, requires disruption of Cr biosynthesis. Severely depleted Cr levels have been reported in patients with defects in the expression of one of the two enzymes of de novo Cr synthesis, namely l-arginine:glycine amidinotransferase (AGAT; EC 2.1.4.1) and guanidino acetate methyltransferase (GAMT; EC 2.1.1.2).
Although GAMT−/− mice are essentially free of (P)Cr (Schmidt et al. 2004), they accumulate guanidino acetate, one of the precursors of Cr, as well as its phosphorylated form, PGAA (Renema et al. 2003). As PGAA is able to buffer ATP in (mildly) energy-demanding conditions (Kan et al. 2004), it compensates, at least partly, for the lack of (P)Cr, which hampers the full assessment of the consequences of Cr deficiency.
The recent generation and characterization of an AGAT−/− mouse model provides new opportunities to study the consequences of Cr deficiency (Choe et al. 2012). In contrast to GAMT−/− mice, AGAT−/− mice do not accumulate any guanidino acetate (Choe et al. 2012). As no alternative HEP compounds, such as PGAA, are found in AGAT−/− mice, this mouse provides an ideal model in which to study pure Cr deficiency. Creatine depletion in AGAT−/− mice resulted in significantly reduced total body weight, reduced adiposity and improved glucose tolerance (Choe et al. 2012). The activation of AMP-activated protein kinase by intracellular energy depletion could explain the systemic metabolic phenotype (Choe et al. 2012). However, detailed tissue-specific effects of Cr depletion in skeletal muscle (the tissue containing about 90% of the total body Cr content) have not yet been investigated.
The aim of this study was to characterize energy-related metabolic, structural and functional abnormalities caused by systemic Cr deficiency in skeletal muscle of AGAT−/− mice. In addition, we explored to what extent the effects or adaptations could be reversed by Cr supplementation, and used such replenishment to compare the kinetics of Cr accumulation in skeletal muscle with that in brain.
Methods
Ethical approval and animal experiments
Generation and care of the animals as well as all experimental procedures were in accordance with institutional guidelines and national laws for the protection of experimental animals, complied with the regulations of the National Institutes of Health, and were approved by the respective local animal ethics committees (Hamburg, 110/10; Nijmegen, 2007-173). Anesthaesia has been described for the various experiments below.
l-Arginine:glycine amidinotransferase knockout mice (AGAT−/−) were generated by a gene-targeting strategy for AGAT deletion (Choe et al. 2012). Heterozygous mice were used for breeding, because homozygous knockout mice were infertile. Chow was essentially Cr free (Sniff, Soest, Germany). Creatine supplementation was achieved by addition of 1% to drinking water or 0.5% Cr to chow. Body weight was determined weekly.
Muscle histopathology
To analyse muscle morphology, wild-type (WT) mice and AGAT−/− female mice aged 16–20 weeks, either on a Cr-free diet or on a chow containing Cr (for 12 weeks), were anaesthetized (120 mg kg−1 ketamine and 16 mg kg−1 xylazine) were perfused transcardially with 2% glutaraldehyde in PBS. Tissues were postfixed in 1% OsO4, dehydrated and embedded in Epon. Light microscopic images were recorded after Toluidine Blue staining of semi-thin longitudinal sections from gastrocnemius muscle. Additionally, ultrathin sections were stained with uranyl acetate and lead citrate and examined with a Zeiss EM902 electron microscope (EM).
For morphometric analysis of skeletal muscle, fresh muscle tissue from extensor digitorum longus and gastrocnemius muscle (three mice per group) was prepared under anaesthesia (isoflurane 2.5–3% v/v in oxygen) and rapidly frozen after removal of the specimen for histological staining following standard laboratory procedures. Briefly, the specimen was put into mounting medium on a cork plate, with care being taken to yield transverse sections, and put into isopentane (cooled to −160°C). Frozen sections of 8 μm thickness were processed in a standard laboratory cryostat. Per specimen, several sections were mounted on coated slides for histological and histochemical staining procedures. For morphometry, slides routinely stained with haematoxylin and eosin were used. Myocyte diameters were determined in multiple transverse sections of muscle using the AxioVision program (Carl Zeiss Imaging Solutions GmbH, Göttingen, Germany) and calculated by averaging data from five images (Kellner et al. 2009).
Grip strength measurements
Maximal grip force of male AGAT−/− knockout mice (−/−), heterozygous (+/−) and wild-type (+/+) mice aged between 15 and 20 weeks was measured using a grip strength meter (TSE-Systems, Bad Homburg, Germany) and compared with mice supplemented with Cr for 12 weeks. Within each group, the mean grip force of each mouse was calculated from 15 appropriate trials.
In vivo magnetic resonance spectroscopy
Metabolite levels in hindlimb skeletal muscle were measured non-invasively by 31P and 1H magnetic resonance spectroscopy (MRS). For all magnetic resonance (MR) experiments, mice were anaesthetized with isoflurane (Abott, Cham, Switzerland; induction dose, 4 min at 4–3%; steady state, 1.0–1.8% isoflurane) in a gas mixture of 33/66% O2/air. The steady-state isoflurane dose was adjusted based on the breathing rate, which was monitored continuously using a pneumatic cushion respiratory monitoring system (Small Animal Instruments Inc., Stony Brook, NY, USA) and kept between 80 and 120 breaths min−1. Body temperature was controlled at 37 ± 1°C. The MR experiments were performed in a 200 mm horizontal-bore magnet (Magnex Scientific, Abingdon, UK) interfaced to an MR spectrometer (MR Solutions, Guildford, UK) operating at 300.22 MHz for 1H and at 121.53 MHz for 31P. The magnet was equipped with a gradient insert with gradient strength of 150 mT m−1, rise time of 150 μs and free bore size of 120 mm.
The 1H MR measurements of skeletal muscle of the lower hindlimb were performed using a solenoid radio frequency coil positioned at the magic angle with respect to the B0 field (in 't Zandt et al. 1999). Multislice gradient echo images (repetition time (TR) = 250 ms, echo time (TE) = 5 ms, field of view (FOV) 20 mm × 20 mm, 256 × 256 matrix) were used to guide the positioning of 16 μl voxels in the tibialis anterior/extensor digitorus longus region. Localized 1H MR spectra were recorded using stimulated echo acquisition mode (STEAM) (Bottomley, 1987), with variable pulse power and optimized relaxation delays (VAPOR) water suppression (Pfeuffer et al. 1999; Tkáčet al. 1999), TE = 15 ms, TM = 10 ms, TR = 5 s and 128 averages. All 1H spectra were analysed with AMARES (http://sermn02.uab.es/mrui/) using Gaussian line shapes. Absolute levels of total Cr (tCr) and taurine (Tau) in muscle were determined from the water signal of additional 1H spectra acquired without water suppression (eigh averages), assuming a water content of 77% (Sjøgaard & Saltin, 1982) and corrected for T2 relaxation using values determined in a previous study (Renema et al. 2003).
The 31P MR spectra in muscle were obtained in AGAT−/− (n = 8) and WT mice (n = 6) by non-localized pulse-acquire experiments (40 μs pulse, α ∼90 deg, TR = 7 s, 256 averages) using a three-turn solenoid radio frequency coil with a diameter of 8 mm (in 't Zandt et al. 2003). Signal integrals were determined by fitting resonances of PCr, inorganic phosphate (Pi), phosphomonoesters and ATP. The phosphomonoesters and Pi line widths were fixed to 0.7 times the line width of β-ATP (experimentally observed in the spectra with the highest signal-to-noise ratio). Metabolite levels were corrected for T1 relaxation and expressed as a ratio with respect to the signal intensity for β-ATP (Kan et al. 2004).
We assessed possible differences in absolute ATP tissue concentrations in an additional group of mice (four WT and five AGAT−/− mice) by acquiring one-dimensional Image selected in vivo spectroscopy (ISIS) localized 31P MR spectra (5 mm thick cross-sectional slice of the hindlimb). The localisation of the corresponding muscle volume was obtained from multiple spin echo images (TR/TE = 1500/12 ms, 10 slices, 1 mm slice thickness, FOV = 12 mm × 12 mm, matrix = 128 × 128, two averages) by manual delineation of the cross-sectional muscle area in the ISIS-localized slices. In this way, we could compare the ATP signal intensity per unit muscle volume between WT and AGAT−/− mice.
Tissue pH was determined in both tissues from the chemical shift difference (S) between Pi and α-ATP; the frequency of the latter does not shift in the physiological pH range (Moon & Richards, 1973).
In brain, 1H MR specra were also recorded using an elliptical 1H surface coil (15 × 11 mm) from a 2.2 mm × 2 mm × 2 mm voxel located partly in thalamus and hippocampus (STEAM, TE = 15 ms, mixing time (TM) = 10 ms, TR = 5 s, 256 averages; Kan et al. 2007). Multislice gradient echo images were acquired in transverse (TR = 4000 ms, TE = 4 ms, 10 slices of 1 mm) and horizontal directions (TR = 4000 ms, TE = 10 ms, 4 slices of 1 mm), avoiding inclusion of ventricles in the volume of interest. The 1H MR brain spectra were analysed with LCModel software to obtain tCr concentrations (Provencher, 1993http://2-provencer.com/pages/lcmodl.shtml). Absolute quantification was performed using the water signal from the unsuppressed spectra (Kan et al. 2007), assuming a mean tissue water content of 78% (in 't Zandt et al. 2004).
Creatine accumulation and washout in muscle and brain
To study Cr accumulation in AGAT−/− muscle and brain tissue and its effect on other metabolite levels, we performed sequential MRS measurements during 35 days of ad libitum Cr supplementation via drinking water (5.32 g (500 ml)−1). Additional glucose (4.32 g (500 ml)−1) was added to compensate for the bitter taste of Cr (Ipsiroglu et al. 2001; Kan et al. 2007). The 1H spectra and 31P spectra were recorded in hindleg muscle at 0, 1, 2, 11, 16, 21, 24 and 34 days of supplementation in eight AGAT−/− mice. After supplementation, the mice were put on a Cr-free diet, to assess the rate of Cr breakdown by acquiring MR spectra at 5, 15, 35, 55 and 85 days of Cr restriction. To minimize effects of anaesthetics, the mice were divided into two groups (n = 4 per time point), which allowed for a recovery period of at least 10 days between two sequential MR measurements. Creatine accumulation in brain was determined in 11 AGAT−/− mice, divided into three groups. Each group underwent MR experiments three or four times, resulting in spectra on days 0, 1, 3, 7, 11, 15, 25, 30, 36 and 45 of Cr supplementation Breakdown rate constants of Cr were determined using the following equation representing the kinetics of non-enzymatic degradation:
| (1) |
where t represents the number of days on a Cr-free diet, [tCr] is the total Cr concentration on day t, [tCr]0 is the concentration of tCr on the last day of the Cr supplementation period and k is the Cr breakdown rate constant.
Monitoring changes in muscle volume by MR imaging
Relative muscle volumes were determined by drawing the regions of interest (ROIs) around the hindleg muscle in five subsequent cross-sectional gradient echo images (TR = 250 ms, TE = 5 ms, FOV 20 mm × 20 mm, 256 × 256 matrix) positioned 3 mm distal from the knee joint of the hindleg. The surface areas of the five ROIs were multiplied by the 1 mm slice thickness to yield muscle volume over a 5 mm length of the hindleg. Muscle growth upon Cr supplementation was expressed relative to the volume on day 0.
Muscle MRS during ischaemic occlusion
To assess the role and capacity of the the PCr–CK system in skeletal muscle, we monitored by 31P MRS the dynamic changes in ATP, PCr, Pi and pH upon ischaemic occlusion of the hindlimb in AGAT−/− and WT mice. Reversible obstruction of blood flow through hindlimb skeletal muscle was accomplished in the MR magnet by clamping the hindlimb above the knee with a diaphragm plate (in 't Zandt et al. 1999). The 31P spectra were acquired with a time resolution of 1.46 min (TR = 1400 ms, 76 averages) for 7 min prior to the ischaemic period, and during 25 min of ischaemia and 16 min of recovery (Kan et al. 2004). The ischaemia experiments in AGAT−/− were performed after a long period of Cr deprivation (i.e. day 0) and at 2 and 21 days of Cr supplementation (n = 5 per group). All metabolite signals acquired before, during or after the ischaemic occlusion were normalized to the mean signal intensity of β-ATP before ischaemia was applied (β-ATP0).
During ischaemia, ATP is supplied for basal (i.e. non-contractile) ATPase activity by the following two means: by breakdown of PCr and by glycolytic ATP synthesis (in the closed system of the ischaemic muscle, oxidative ATP synthesis can be ignored, and [ATP] does not change significantly). The component supplied by PCr breakdown is simply estimated as the rate of decline of [PCr], calculated here by linear regression analysis of the last spectrum before ischaemic occlusion and the first four spectra (7 min) of the ischaemic period (Blei et al. 1993; Marcinek et al. 2004). The glycolytic contribution to the resting ATPase flux is determined from changes in pH occurring in the same period, taking into account the H+ stoichiometry of glycolytic ATP synthesis and net PCr breakdown, and the cytosolic buffer capacity (Kemp et al. 2001; Marcinek et al. 2010). Total ATP production rate, the sum of PCr breakdown and glycolytic ATP synthesis, is therefore given by:
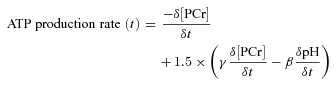 |
where −γ = −27.239 + 13.593 × pH – 2.1440 × (pH)2 + 0.10887 × (pH)3 is the (positive) amount of H+‘consumed’ as a consequence of PCr breakdown (Kushmerick, 1997) and β is the sum of the cytosolic non-Pi buffering capacity, assumed to be 16 mmol l−1 (pH unit)−1 (Marcinek et al. 2010), and the calculated buffering contribution of Pi (Roos & Boron, 1981) as follows:
where
Biochemical determination of muscle metabolites
For absolute quantification of metabolites, hindlimb muscles (hamstrings) were snap-frozen in liquid nitrogen and stored at −80°C. After freeze drying, 10–15 mg dry muscle tissue was extracted with 1 m perchloric acid. After centrifugation, the supernatant was neutralized with 2 m KHCO3. These extracts were used for spectrophotometric determination of Cr, PCr, ATP, ADP, AMP and lactate (Harris et al. 1974). The biochemically determined metabolite concentrations for each muscle sample were expressed as micromoles per gram dry weight.
Mitochondrial activity in vitro
Hamstring muscles from adult AGAT−/− and WT littermates were removed under isoflurane anaesthesia (5% for >4 min, in a gas mixture of 25/75% O2/air) and the animals were decapitated by cervical dislocation instantly afterwards. The muscle tissue was immediately stored in liquid nitrogen. Mitochondria were isolated from 70–120 mg samples of wet muscle tissue for spectrophotometric determination of mitochondrial enzyme activities of NADH dehydrogenase (complex I, EC 1.6.5.3), succinate dehydrogenase (complex II, EC 1.3.5.1), ubiquinol-cytochrome c reductase (complex III, EC 1.10.2.2), cytochrome c oxidase (complex IV, EC 1.9.3.1), succinate dehydrogenase (SCC, EC 1.3.5.1) and F1F0-ATPase (complex V, EC 3.6.3.14). All the values were expressed as milliunits per unit citrate synthase activity (CS, EC 2.3.3.1). Details of the methods have been described previously (Cooperstein & Lazarow, 1951; Mourmans et al. 1997; Janssen et al. 2007; Jonckheere et al. 2008). Citrate synthase activity was used as a marker for mitochondrial content. The ratio of complex V activity to total ATPase activity was determined in the absence and presence of oligomycin (8 mg ml−1), the specific inhibitor of the F0-part of complex V (Jonckheere et al. 2008).
Statistical analysis
Data are given as means ± SD unless stated otherwise. The following statistical tests were applied: unpaired Student's t test for comparisons between WT and AGAT−/− mice, one-way ANOVA for comparison of WT, AGAT−/− and Cr-supplemented AGAT−/− mice, and two-way ANOVA for multiparametric measurements with Bonferroni's post hoc test using GraphPad Prism (version 4; GraphPad Inc., La Jolla, CA, USA).
Results
Morphology
Light and electron microscopy of hindlimb muscle demonstrated two important morphological abnormalities in muscle fibres of AGAT−/− mice (Fig. 2): first, a large increase in lipid droplets (Fig. 2D, E and G), mainly around the mitochondria (Fig. 2G); and second, electron-dense layers with the typical appearance of crystal structures, located within numerous mitochondria (Fig. 2H). After 12 weeks of Cr supplementation (Fig. 2F), lipid droplets decreased to numbers comparable to those seen in WT mice (Fig. 2A–C), and the intramitochondrial crystal structures were no longer observed. Histopathology also showed a significantly smaller average myocyte diameter in AGAT−/− mice compared with that of WT control animals (Fig. 3A), whereas the AGAT−/− mice that were supplemented with Cr (AGAT−/−+ Cr) had normal-sized myocytes. Hence, the morphological abnormalities seen in the AGAT−/− mice on a Cr-free diet were completely abolished by oral Cr treatment.
Figure 2. Photomicrographs of skeletal muscle sections.
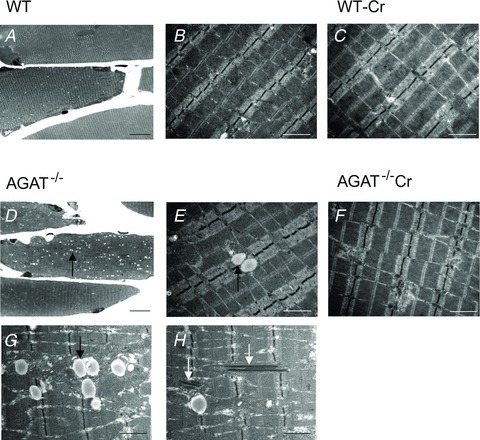
A and D, light microscopic images show Toluidine Blue-stained semi-thin longitudinal sections from hindlimb muscle of wild-type (WT; A) and AGAT−/− mice (D) on a normal Cr-free diet. D, AGAT−/− muscle shows an increased number of lipid droplets (small white dots) when compared with the muscles of the WT control animals. E, G and H, electron microscopic images of hindlimb skeletal muscle of AGAT−/− muscle demonstrate that the lipid droplets were mainly present in close proximity to the mitochondria (G). H, in AGAT−/− muscle multiple mitochondria contain electron-dense bodies between the mitochondrial cristae membranes. B, electron microscopic images of WT mice on a creatine-free diet show normal skeletal muscle for comparison. F, after 12 weeks of Cr supplementation, the number of lipid droplets in the electron microscopic images of the AGAT−/− mice decreases to normal amounts, and the abnormalities in the mitochondria are no longer observed. Creatine supplementation did not reveal any changes in the WT mice. Magnifications: A and D, ×440; and B, C and E–H, ×12,000. Scale bars: (A, D) = 20μm, (B, C, E–H) = 1μm.
Figure 3. Reduced muscle volume, myocyte diameter and grip strength in AGAT−/− mice.
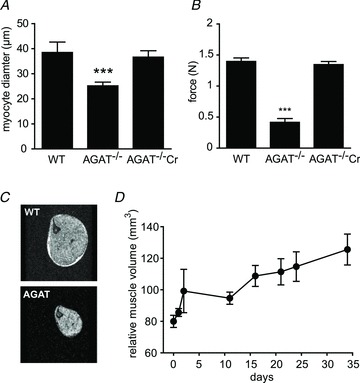
A, myocyte diameters determined from neuropathological analysis. B, muscle force determined by grip strength tests in WT (+/+), and AGAT-deficient knockout (−/−) mice on a Cr-free diet and after 12 weeks of Cr supplementation. C, cross-sectional gradient echo images of hindlimb (TR/TE = 250/5 ms, FOV = 20 mm × 20 mm, 256 × 256 matrix). D, increasing muscle volume of AGAT−/− mice upon Cr supplementation determined from cross-sectional MR images. Values are means ± SEM, n = 5–10 per group. Significant differences compared with all other groups: ***P < 0.001 (Student's unpaired t test).
Grip strength
Grip strength of AGAT−/− mice was significantly reduced, by >70%, compared with WT mice (Fig. 3B), while heterozygous littermates did not differ from WT. Creatine supplementation improved grip strength in all groups; however, the gain was statistically significant only in the AGAT−/− mice, which showed a complete recovery of strength to WT levels. The AGAT−/− mice were hypotonic (hanging down when lifted by their tail), but muscle tone and habitus normalized upon Cr supplementation.
Body weight and muscle atrophy
Body weight was significantly reduced in Cr-depleted AGAT−/− mice (19.7 ± 2.2 g, n = 8) compared with WT animals (37.8 ± 7.1 g, n = 5) and was accompanied by severe muscle atrophy. Cross-sectional gradient-echo MR images of hindlimb muscles demonstrate a reduction of relative muscle volume to 34 ± 9% (n = 5) compared with WT (100 ± 21%, n = 4; Fig. 3C). The decreased muscle volumes are consistent with the decreased myocyte diameters described above (see ‘Morphology’). The atrophy was apparently not confined to the leg muscles; thoracolumbar kyphosis in AGAT−/− mice suggests additional dysfunction of the postural and paraspinal muscles. Creatine supplementation for 3 months increased the body weight of AGAT−/− mice up to 56%, while age-matched WT mice showed an increase of only 19.3 ± 4.7% over that period, which is not significantly different from WT animals on a normal diet (15.3 ± 2.2%). In contrast, age-matched AGAT−/− mice showed no increase in body weight during this period. The body weight of heterozygous mice did not differ from that of WT animals regardless of diet. Besides the normalization of body weight and composition (i.e. water, fat and lean mass) during Cr supplementation (Choe et al. 2012), skeletal muscle volume also recovered, as demonstrated by the increase in cross-sectional muscle areas and amelioration of kyphosis (Fig. 3D).
Metabolite levels
In vivo 1H MR spectra of skeletal muscle of AGAT−/− mice demonstrated virtually complete absence of the Cr signals at 3.0 and 3.9 p.p.m., while taurine levels were not significantly different from those of WT mice (Fig. 4A). As expected, AGAT−/− muscles did not show PCr signals at 0 p.p.m. in 31P MR spectra either (Fig. 4B). Importantly, these spectra did not reveal any other phosphorylated guanidine compound that could replace PCr in its ATP-buffering role, such as phosphorylated forms of guanidinoacetate or arginine. Upon Cr supplementation, normal PCr and tCr signals were observed (see Table 1). Intracellular pH, as calculated from the chemical shift difference between α-ATP and Pi, was not statistically different between the two groups. Interestingly, the lack of Cr in skeletal muscle of the mutants was accompanied by significant elevation of the Pi/β-ATP ratio, approximately fourfold compared with WT levels (Table 1), as a consequence of either low ATP concentration or high Pi concentration, or both. To resolve this, we determined the ratio of β-ATP signal intensity to muscle volume over five slices, which was significantly reduced in the AGAT−/− mice (63 ± 8% of WT levels). Biochemical analysis confirmed decreased ATP concentrations by 53% in the AGAT−/− muscle (Table 1). The decreased ATP levels imply that absolute Pi concentrations are increased by approximately twofold in AGAT−/− muscle compared with WT. Biochemical determinations of tissue metabolite concentrations confirmed the nearly complete absence of PCr in AGAT−/− hamstring muscle, demonstrating a ∼87% depletion in Cr content compared with WT. Very similar PCr/ATP ratios in WT muscle were calculated from biochemical determinations (3.0 ± 0.4) and in vivo MR spectra (3.2 ± 0.1). Biochemically determined concentrations of ADP, AMP and lactate in muscle were not significantly different between WT and AGAT−/− mice, whereas ATP/ADP and ATP/AMP ratios were both much lower in the AGAT−/− muscle (Table 1).
Figure 4. Magnetic resonance spectra of muscle obtained from WT mice (top) and AGAT−/− mice on a Cr-free diet.
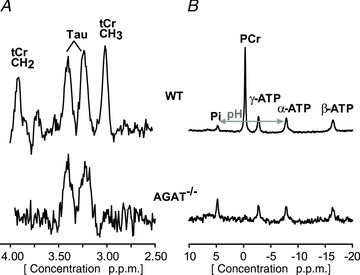
A 1H MR spectrum (A) obtained from a 16 μl voxel and a non-localized 31P MR spectrum (B) obtained in hindlimb tissue of WT (upper spectra) and AGAT−/− mice (lower spectra) on a Cr-free diet. The unlocalized 31P AGAT−/− spectrum was multiplied by four to increase visibility, which was a direct consequence of the severe reduction in muscle volume. Note the absence of total creatine (tCr) and phosphocreatine (PCr) and the relatively large inorganic phosphate (Pi) signal in the AGAT−/− muscle.
Table 1.
Metabolite levels and pH in wild-type and AGAT−/− mice
| Parameter | Wild-type (n = 5–6) | AGAT−/− (n = 8) | AGAT−/−+ Cr (n = 4) | Units |
|---|---|---|---|---|
| In vivo magnetic resonance spectroscopy | ||||
| [tCr] | 28.9 ± 2.9 | 4.1 ± 1.5† | 22.7 ± 2.0† | mm |
| PCr/β-ATP | 3.2 ± 0.1 | 0.3 ± 0.1† | 3.0 ± 0.3 | — |
| Pi/β-ATP | 0.5 ± 0.04 | 2.0 ± 0.3† | 0.6 ± 0.2 | — |
| PCr/Pi | 6.1 ± 0.6 | 0.1 ± 0.1† | 5.0 ± 1.5 | — |
| pH | 7.20 ± 0.06 | 7.25 ± 0.04 | 7.24 ± 0.04 | — |
| ATP/volume | 1.00 ± 0.06 | 0.63 ± 0.08*** | — | a.u. |
| Biochemical | ||||
| Cr | 40.0 ± 12.4 | 5.1 ± 3.7††† | — | μmol (g dry mass)−1 |
| PCr | 75.2 ± 15.7 | 1.1 ± 3.2††† | — | μmol (g dry mass)−1 |
| ATP | 25.1 ± 3.1 | 13.5 ± 3.9††† | — | μmol (g dry mass)−1 |
| ADP | 4.5 ± 1.5 | 4.1 ± 1.3 | — | μmol (g dry mass)−1 |
| AMP | 0.1 ± 0.1 | 0.4 ± 0.1 | — | μmol (g dry mass)−1 |
| Lactate | 10.6 ± 5.2 | 11.5 ± 6.2 | — | μmol (g dry mass)−1 |
| PCr/ATP | 3.0 ± 0.4 | 0.1 ± 0.2*** | — | — |
| ADP/ATP | 0.18 ± 0.03 | 0.31 ± 0.05*** | — | — |
| ATP/AMP | 256 ± 116 | 34 ± 10*** | — | — |
The mice used here were on creatine (Cr)-free diet for at least 5 months. The AGAT−/−+ Cr mice were supplemented for 35 days. Values determined by in vivo magnetic resonance spectroscopy were corrected for relaxation differences (T1, T2). Total creatine concentration ([tCr]) is expressed in millimoles per litre of tissue using a factor of 0.77 for the intracellular water fraction. Absolute quantification of phosphate metabolites in skeletal muscle was determined in vitro on freeze-clamped hindlimb muscle extracts and determined by spectrophotometry. Data are expressed as means ± SD. ***P < 0.001 compared with WT littermates, by unpaired Student's t test. †P < 0.05, †††P < 0.001 when compared with wild-type littermates, by two-way ANOVA with Bonferroni post hoc test.
Mitochondrial enzyme activities
The increased Pi and decreased ATP concentrations suggested adaptive changes in oxidative phosphorylation in AGAT−/− muscle. We therefore measured respiratory chain activities in isolated mitochondria of muscle tissue. Enzyme activities of complex II and complex V, total ATPase and citrate synthase were significantly elevated per gram of wet muscle tissue (Table 2). Given that CS is commonly used as a marker for mitochondrial mass, the elevation of its activity in AGAT−/− muscle (170% of WT levels) indicates an increased mitochondrial content. The ratio of F-type ATPase (complex V) to total ATPase activity was comparable between the two groups, hence not only is F-type ATPase increased by ∼90%, but total ATPase activity per mitochondrial content (mg wet weight) is equally increased. Using CS activity as a marker for mitochondrial content, activities of complexes III and IV per mitochondrial content in AGAT−/− muscle were significantly decreased by 36 and 27%, respectively (Table 2). A similar decrease was seen in complex I (38%), although it did not reach statistical significance. This implies that the H+-pumping respiratory enzymes are downregulated in mitochondria of AGAT−/− muscle, while other respiratory enzyme activities were not significantly different from WT muscle.
Table 2.
Respiratory enzyme activities
| Enzyme activity (mU (g wet weight)−1) | Enzyme activity (mU (U CS)−1) | |||
|---|---|---|---|---|
| Enzyme | Wild-type | AGAT−/− | Wild-type | AGAT−/− |
| Complex I | 4.4 ± 1.1 | 4.4 ± 1.6 | 567 ± 107 | 352 ± 145 |
| Complex II | 2.8 ± 0.9 | 6.5 ± 2.8* | 372 ± 124 | 481 ± 129 |
| Complex III | 12.5 ± 2.6 | 13.4 ± 3.7 | 1622 ± 208 | 1031 ± 273*** |
| SCC | 1.6 ± 0.4 | 3.2 ± 1.1 | 208 ± 39 | 240 ± 52 |
| Complex IV | 14.5 ± 3.7 | 17.7 ± 5.1 | 1866 ± 350 | 1355 ± 295*** |
| Complex V | 4.5 ± 1.0 | 10.4 ± 3.2*** | 589 ± 109 | 815 ± 250 |
| Total ATPase | 5.0 ± 1.4 | 10.8 ± 1.6*** | — | — |
| CS | 7.8 ± 1.6 | 13.3 ± 3.5*** | — | — |
Values are means ± SD; n = 9 per group. All muscle samples were taken from mice on a creatine-free diet. *P < 0.05, ***P < 0.001, when compared with wild-type, by two-way ANOVA. Abbreviations: complex I, NADH dehydrogenase; complex II, succinate dehydrogenase; complex III, ubiquinol-cytochrome c reductase; complex IV, cytochrome c oxidase; complex V, F1F0-ATPase; CS, citrate synthase; and SCC, succinate dehydrogenase.
Accumulation of creatine in muscle and brain
Creatine-supplemented AGAT−/− mice demonstrated a rapid accumulation of Cr in skeletal muscle (Fig. 5A). The tCr signal intensities of 1H MR spectra from tibialis anterior/extensor digitorum longus muscle reached normal WT levels within 1 day of supplementation, whereas the accumulation in brain was much slower (Fig. 5B). In the hypothalamic/hippocampal region of AGAT−/− mice, tCr levels gradually increased to WT levels over about 20 days. Given that we previously studied Cr supplementation in GAMT−/− mice in the same experimental set-up and with the same Cr dosing protocol (Kan et al. 2007), we can directly compare treatment responses between these two models of Cr deficiency. Two differences are apparent in Cr accumulation (Fig. 5A and B). Creatine accumulation in AGAT−/− muscle occurs faster than in GAMT−/− animals, but levels off after 2 days. In contrast, skeletal muscle tCr levels in GAMT−/− mice increased up to about 4 weeks, eventually reaching higher levels than in AGAT−/− mice. However, there is no apparent difference in the kinetics of cerebral Cr accumulation between the two models.
Figure 5. Metabolic changes in AGAT−/− and GAMT−/− mice during Cr supplementation.
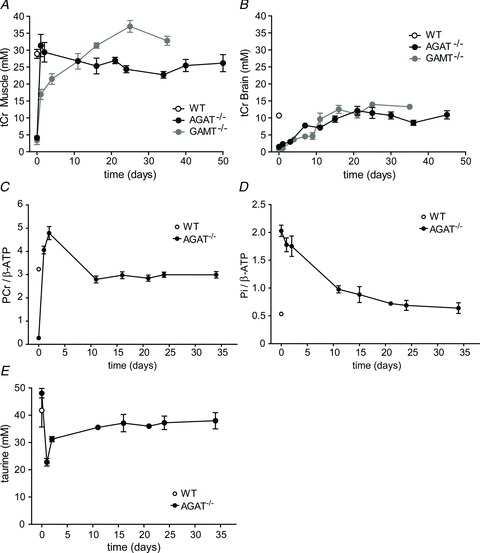
A and B, changes in total creatine (tCr) concentrations were obtained from 1H spectra of muscle from a 16 μl voxel (STEAM) in the lower limb tibialis anterior/extensor digitorum longus (A) and an 8.8 μl voxel in the hypothalamic/hippocampal region of the brain (B). Total Cr levels in AGAT−/− mouse muscle (filled circles) were compared with tCr levels obtained in triceps surae in GAMT−/− mice (grey circles; the GAMT−/− data were used with permission from Kan et al. 2007). C–E, changes in PCr/β-ATP (C) and Pi/β-ATP signal ratios (D) and taurine concentration (E) in hindleg muscle of AGAT−/− mice during Cr administration. The ratios were determined from unlocalized 31P MR spectra. Data are means ± SEM, n ≥ 3 per time point.
Most Cr taken up by muscle is immediately phosphorylated, as shown by the near-instantaneous increase of PCr/β-ATP ratios upon Cr supplementation (Fig. 5C). An initial overshoot in PCr/β-ATP during the first 2 days normalized after 11 days of supplementation. In contrast to this very rapid replenishment of (P)Cr, the high initial Pi levels in skeletal muscle of AGAT−/− mice normalized only slowly upon Cr administration, taking more than 15 days to reach normal WT levels (Fig. 5D). One other change in metabolite content was observed; after 1 day of Cr supplementation, the taurine (Tau) levels in muscle showed a transient decrease (Fig. 5E).
Creatine breakdown in muscle and brain
During Cr restriction, both tCr and PCr levels exponentially decreased towards zero (Fig. 6A). The breakdown rates of tCr and PCr in hindlimb muscle were 1.0 ± 1.2 and 1.5 ± 0.2% day−1, respectively. Interestingly, during Cr restriction the Pi/β-ATP ratios did not change within the first 2 months and started to increase at PCr/β-ATP ratios of ∼1 (Fig. 6A). The breakdown rate tCr in brain (Fig. 6B) was 1.8 ± 0.9% per day.
Figure 6. Changes in PCr and Pi levels in hindleg muscle and tCr levels in brain during Cr restriction.
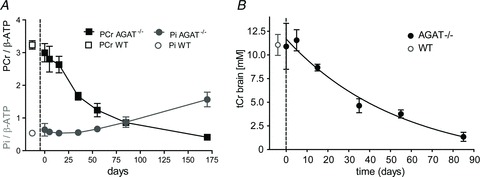
A, PCr/β-ATP follows an exponential decay in AGAT−/− mice upon Cr restriction, whereas Pi/β-ATP ratios gradually increase when Cr intake is restricted. The Pi levels start to increase when PCr/β-ATP ratios decrease below ∼1.5. Metabolite levels in muscle were determined from unlocalized 31P MR spectra. B, tCr concentrations in the thalamic/hippocampal region (8.8 μl volume) were determined by 1H MRS after 35 days of supplementation using water as a reference signal. Day 0 indicates the last day of the Cr administration. The WT reference values are given to the left of the dashed line. Data are means ± SD, n = 3–4 per time point.
Figure 7 illustrates in a time-independent form the different kinetics of PCr/β-ATP (x-axis) and Pi/β-ATP (y-axis) in AGAT−/− muscle during the Cr accumulation and washout periods (Fig. 7, combined data of Fig. 5C and D and 6A). Strikingly, total phosphate content (or its close surrogate, Pi + PCr) is not maintained at a constant level. During Cr supplementation, replenishment of (P)Cr occurred instantly, while the high initial Pi levels in skeletal muscle of AGAT−/− mice normalized only slowly; therefore, it was not directly depleted as a mass-balance consequence of phosphorylation of the Cr taken up.
Figure 7. Changes in PCr and Pi levels in hindleg muscle of AGAT−/− mice during Cr administration and restriction.
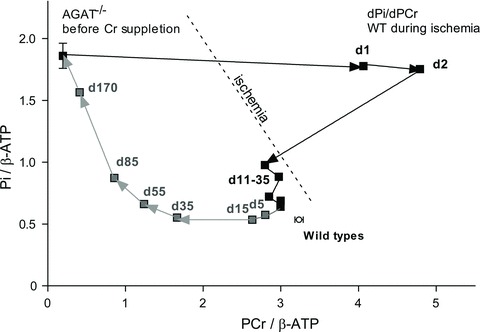
Ratios of PCr/β-ATP and Pi/β-ATP during Cr supplementation (filled squares) and Cr restriction (grey squares). Note that the fast accumulation of PCr and slow adaptation of Pi levels during Cr treatment results in an immediate increase of total phosphate content in muscle. During Cr restriction, Pi levels start to increase when PCr/β-ATP ratios decrease below ∼1.5. Metabolite levels in muscle were determined from unlocalized 31P MR spectra. Data are means, n = 3–4 per time point. Note that the overshoot in PCr/β-ATP during the first 2 days of Cr supplementation can be explained by adaptations in gene expression, resulting in decreases in [ATP] that are likely still to be present after only 2 days of Cr supplementation.
Ischaemia experiments
The PCr–CK system functions as a rapid energy buffer in situations of high energy demand. Therefore, we investigated the effect of Cr deficiency in response to a metabolic challenge by performing ischaemia–reperfusion experiments on AGAT−/− and WT hindlimb muscles. Analysis of 31P MRS spectra during 25 min of ischaemic occlusion demonstrated the expected responses to ischaemia in WT muscle, i.e. a decrease of PCr, an equal increase of Pi and, slightly delayed, a drop in pH (Fig. 8), which reflects the respective roles of the PCr–CK system and glycolysis in maintaining ATP levels in the absence of oxygen. We determined the flux through ATPase indirectly from decreasing PCr levels (WT, 0.78 ± 0.86 mm min−1) during the first 7 min of ischaemia (Fig. 8D). In WT mice, initial glycolytic ATP synthesis rates, calculated from the pH change over the first 7 min of ischaemia, were small compared with ATP generation via the CK–PCr system (Fig. 8E).
Figure 8. Metabolic responses to ischaemia and calculated ATPase flux.

A–C, changes of PCr (A) and Pi signal intensities (B), as well as pH (C), in skeletal muscle before and during an ischaemic period. Signal intensities and pH levels were determined from 31P MR spectra in WT mice (black), AGAT−/− mice on a Cr-free diet (day 0; blue), and after 2 (red) and 21 days (grey) of Cr administration. The PCr and Pi signal intensities were normalized to the β-ATP signal intensity before ischaemia (β-ATP0). Data are means ± SEM, n = 5–7 per group. D–F, rates of ATP generation by net breakdown of PCr (D), by glycolysis (E), and their sum (total ATPase rate; F), determined from the decreases in PCr and pH during the first 7 min of the ischaemic period, assuming normal ATP levels (7.8 mm) in WT and AGAT−/− mice on day 21 and reduced levels (60%) in AGAT−/− mice on day 0 and day 2. Data are means ± SD, n = 5–7 per group. Significant differences were determined by ANOVA. # Value obtained from the summed spectrum of the entire AGAT−/− group, because of low individual signal intensities.
In the AGAT−/− mice (day 0) intracellular pH immediately started to decrease on occlusion, reaching a significantly lower pH at the end of the ischaemic period (6.52 ± 0.16) compared with WT mice (6.96 ± 0.04). The initially elevated Pi levels demonstrated only a small increase upon ischaemia. The ATPase rates could not be determined for individual AGAT−/− mice, owing to the very low PCr levels in the AGAT−/− mice (i.e. PCr/β-ATP ∼0.5). However, summation of all PCr time courses of AGAT−/− in this group (n = 7) enabled estimation of the initial slope of the average time course, which was subsequently multiplied by an ATP concentration of 4.9 mm (60% of WT ATP levels) to obtain an estimate of average ATP synthesis rate from PCr depletion; this was decreased at 0.12 mm min−1, compared with 0.78 ± 0.14 mm min−1 for WT mice. Initial glycolytic ATP synthesis completely compensated for the near absence of the PCr–CK buffer, as is shown by a total ATP synthesis (sum of initial ATP production by PCr depletion and glycolysis; Fig. 8F) that was not different from WT levels. The ATP levels of these mice did not change during the ischaemic period (data not shown).
The AGAT−/− mice supplemented with Cr for 2 days demonstrated a similar decreased muscle pH at the end of the ischaemic period to that of the WT mice. Changes in Pi/β-ATP ratios after 25 min of ischaemia with respect to baseline (ΔPi/β-ATP) were significantly larger in AGAT−/− (3.9 ± 1.5) than in WT muscle (2.1 ± 0.7, P = 0.04, unpaired Student's t test), matching the larger drop in PCr (ΔPCr/β-ATP = 2.4 ± 0.5; see also Fig. 8). The ATPase flux as determined from the initial PCr depletion in the AGAT−/− mice after 2 days of Cr supplementation, and assuming ATP levels still to be 4.9 mm (60% of WT levels), was larger than that of WT mice, although not significantly so. Initial ATP synthesis from glycolytic and PCr depletion, however, contributed almost equally to total initial ATP synthesis in the AGAT−/− mice after 2 days of Cr supplementation (Fig. 8D and E). At 21 days, all changes (pH, PCr depletion and Pi increases) during ischaemic occlusion were similar to those of WT. Total initial ATP synthesis was mainly dependent on PCr depletion, as the glycolytic contribution decreased towards normal WT levels.
Discussion
In this study, we demonstrate that hindlimb skeletal muscle of AGAT−/− mice kept on a Cr-free diet is depleted of Cr and PCr and results in reduced ATP and elevated Pi levels. The absence of Cr was associated with muscular atrophy, reduced strength, dysfunction of mitochondrial respiratory chain enzymes, morphological alterations and an increased dependence on glycolytic ATP production in energy-demanding situations. Those effects were all reversible by oral Cr supplementation. Furthermore, Cr accumulation and degradation showed tissue-specific kinetics in skeletal muscle and brain.
Changes in phosphate metabolites due to Cr deficiency
Levels Pi are twofold increased in skeletal muscle of AGAT−/− mice with Cr deficiency, which cannot be explained by systemic hyperphosphataemia, because the plasma phosphate concentration in AGAT−/− mice was not elevated (data not shown). In order to understand the conditions for hyperphosphataemia to occur, we must consider some relevant physiological principles. Any stable cytosolic [Pi] is in steady state with transmembrane Pi transport (such that passive Pi efflux equals secondary active Pi influx) and also with intracellular metabolic fluxes (such that the rates of Pi-consuming and Pi-generating processes are equal). Both aspects are part of the metabolic ‘setting’ of the cell, which in muscle defines the resting baseline from which perturbations (e.g. exercise, ischaemia) take off. Direct ex vivo measurements of sarcolemmal Pi transport would be needed to explain how the transmembrane steady state is maintained at this high cytosolic [Pi], but one might speculate that an increase in Pi uptake is a consequence of the absence of the CK–PCr system. An increased [Pi] might have several consequences; for instance, it may promote a cellular milieu that can maintain sustained mechanical output at the expense of maximal power of the fibres (Kushmerick et al. 1992). Although [ADP] is classically assumed to have the key feedback role in mitochondrial ATP production, recent studies point to the importance of [Pi] as a feedback signal, especially at low rates of ATP turnover as in low and moderate exercise (Scheibye-Knudsen & Quistorff, 2009; Jeneson et al. 2011). From this perspective, an increased [Pi] represents an increase in feedback signal, in response either to an increased ATP demand or to decreased capacity of oxidative synthesis (glycolytic ATP synthesis is negligible in resting, normally perfused muscle). In this context, it would be interesting to confirm a potentially increased ability for endurance exercise in AGAT−/− mice, similar to CK-deficient muscle that demonstrates a resistance to fatigue during low-intensity exercise (Dahlstedt et al. 2000). Increased Pi is also associated with inhibited force production (e.g. Pathare et al. 2005), although changes in resting [Pi] make proportionately less difference to exercise [Pi] the higher the intensity of exercise.
The mechanism of the decreased [ATP] must involve relative activation of the pathways of adenine nucleotide synthesis and breakdown, which again can be thought of as aspects of the metabolic setting of the cell.
The ischaemia experiments, used as a stress test for integrated ATP supply, provide a non-invasive technique to determine ATP generation from the initial PCr depletion once oxidative ATP synthesis is halted by ischaemia. Unfortunately, the lack of PCr hampered this assessment in truly Cr-depleted muscle. However, the rate of glycolysis to lactate seems to compensate for this lack in order to maintain a similar total initial ATP synthesis rate (i.e. the resting ATP demand) compared with that of WT or Cr-supplemented AGAT−/− mice. The situation is strikingly different on the second day of Cr supplementation, when the rate is substantially increased due to a normal contribution from the PCr–CK buffering system in addition to a still-elevated glycolytic contribution.
From another point of view, the combination of elevated Pi and decreased ATP levels compared with WT is similar to that observed in cat, rat and mouse soleus muscles, which have a high fraction of slow-twitch oxidative type I fibres (Meyer et al. 1985; Kushmerick et al. 1992). Independent of fibre type, the elevated activity of citrate synthase and F-type ATPase and the increased availability of intramyocellular lipid droplets point towards an upregulation of oxidative ATP production in mitochondria of resting AGAT−/− muscle. While the proton-pumping respiratory enzymes are downregulated, the majority of the metabolic alterations suggest an increased oxidative metabolism in AGAT−/− muscle. This may be related to the enhanced energy expenditure, as indicated by the increased food intake in combination with a low body fat content (Choe et al. 2012).
Creatine deficiency and a non-functional PCr–CK system
Skeletal muscle normally contains substantial concentrations of Cr (∼30 mm), which comprises > 90% of the total body Cr content. At normal resting conditions, about 85% of the total Cr is present in the phosphorylated form. The depletion of Cr in the AGAT−/− mice disables the PCr–CK system for all tissues, including skeletal muscle. Double CK knockout mice (CK=/= mice) with deletions of muscle-specific CK-isoforms, i.e. cytosolic M-CK and mitochondrial ScCKmit (Steeghs et al. 1998; in 't Zandt et al. 2003), also have to cope without this system for ATP buffering. It is of interest to compare them with Cr-depleted AGAT−/− mice.
In contrast to the Cr-depleted AGAT−/− mice, in which Cr and PCr are absent, CK=/= muscles have near-normal concentrations of tCr and a substantial amount of PCr (in 't Zandt et al. 2003). Further experiments suggested that the presence of PCr in CK=/= muscle could be due to minute amounts of B-CK (cytosolic CK isoform), deposited in myotubes during the fusion of myoblasts, which start their differentiation with substantial B-CK levels. The presence of some B-CK makes sense, because CK=/= mice are subject to active muscle degeneration–regeneration processes (Momken et al. 2005). As the PCr–CK system seems to be essential for myoblast differentiation and fusion (O’Connor et al. 2008), the potential for muscle regeneration in Cr-depleted AGAT−/− mice is likely to be severely deficient. Although CK=/= mice show reduced mass of some muscles (Momken et al. 2005), muscle atrophy is much more severe in Cr-depleted AGAT−/− mice. In CK=/= mice, muscle atrophy may be caused by limited voluntary exercise (Momken et al. 2005), but as locomotor activity in Cr-depleted AGAT−/− does not appear to be different from that of WT mice (see Choe et al. 2012), it is more plausible that impaired muscle regeneration contributes to muscle atrophy, possibly due to other effects of severe Cr deficiency. It is worth noting here that in addition to a defect in metabolism the atrophy may be the main contributor to the decreased grip strength of the AGAT−/− mice, because the muscle cross-sectional area correlates with contractile power (Maughan et al. 1983; del Porto et al. 2010; Akima et al. 2012).
In some other aspects, Cr-depleted AGAT−/− skeletal muscle resembles that of CK=/= mice. Both demonstrate a larger number of intramyocellular lipid droplets near mitochondria, as well as elevated GLUT4 activation, presumably reflecting similar metabolic adaptations (Steeghs et al. 1998). In CK=/= muscle, mitochondrial density is elevated, but no electron-dense mitochondrial intermembrane inclusions were reported (Steeghs et al. 1997a, 1998). The latter may be associated with mitochondrial ScCKmit. While, strikingly, ATP levels are decreased and Pi is increased in Cr-depleted AGAT−/− muscle, ATP levels in CK=/= muscle were comparable to normal WT muscle, and only mild elevations in Pi/β-ATP ratios were observed in basal conditions (Steeghs et al. 1997a,b). However, in CK=/= mice the Pi/ATP ratios can be increased by an ischaemic period, without recovery for at least a day thereafter (in 't Zandt et al. 1999). Moreover, both CK=/= and AGAT−/− muscle demonstrate abnormal functionality; depleted AGAT−/− mice suffer from a reduced grip strength, while the absence of the PCr–CK system results in a lack of burst activity upon electric stimulation (de Haan et al. 1995; Steeghs et al. 1998).
Taken together, in comparison to muscle of CK=/= mice, Cr-depleted AGAT−/− skeletal muscle shows a more severe phenotype at a metabolic and morphological level. This can be attributed to additional effects of inoperable PCr–CK systems of non-muscle cells present in (immature) skeletal muscle tissue and/or effects of the pure absence of Cr.
Energy metabolism in the absence of Cr, compared with other models of Cr depletion
Absence or depletion of Cr in skeletal muscle and other body tissues has been studied in humans and mice with a defect in the genome encoding for GAMT, which catalyses the final step in Cr synthesis. The GAMT−/− muscle demonstrated accumulation of GAA and PGAA. The latter can serve as an alternative substrate for the CK reaction (Stöckler et al. 1997; Kan et al. 2004); in contrast, muscle of AGAT−/− mice has no alternative HEP compound that can be used as an ATP buffer. In GAMT−/− mice, the grip strength was reduced by only 16% compared with WT littermates (Schmidt et al. 2004), the specific force of the medial gastrocnemius muscle was decreased by 34% (Kan et al. 2005), and Pi/β-ATP in GAMT−/− muscle was increased by only ∼60% (Kan et al. 2004). These aspects indicate a milder phenotype compared with the AGAT−/− mice in the present study.
Upregulation of the activities of respiratory enzymes has also been observed in GAMT−/− brain and muscle (CS and complex V; Schmidt et al. 2004) and in GAMT−/− fibroblasts incubated in Cr-free medium (complexes I, III and V; Das et al. 2000). In the latter, the enhanced enzyme activities were still present after 1 week, but were restored to normal after 2 weeks when the incubation medium was supplemented with Cr.
Creatine can also be depleted by administration of Cr analogues, in particular β-GPA. This has several effects on muscular phenotype and biochemistry (Moerland et al. 1989), some of them similar to those we have observed in AGAT−/− muscle. Systemic GPA supplementation competitively inhibits Cr accumulation by muscle fibres, where cytosolic GPA is reversibly phosphorylated to P-GPA by CK (Boehm et al. 1996). Thus, GPA is acting as a HEP trap and reduces the Cr concentration (Clark et al. 1994). The P-GPA can act as an alternative HEP in vivo, exactly like P-GAA in GAMT−/− mice, although in vitro MM-CK is much less active on these substrates compared with Cr (about 1500-fold lower for P-GPA and 100-fold lower for P-GAA; Roberts & Walker, 1983).
Interestingly, β-GPA-fed rats showed a 50% decrease in ATP concentration in both slow- and fast-twitch skeletal muscle, which is in agreement with the decrease that we observed in Cr-depleted AGAT−/− muscle. Upon β-GPA accumulation, glycolytic muscles demonstrate enhanced respiratory chain enzyme activities (complex IV and CS) and higher mitochondrial density (Shoubridge et al. 1985; Moerland et al. 1989), as well as a switch from fast to slow myosin isoform expression and a 3.5-fold faster post-stimulation PCr recovery (Ren et al. 1993). Slow-twitch muscle undergoes milder changes on β-GPA feeding, although post-stimulus oxidative recovery was still twofold faster (Moerland et al. 1989).
The increased expression of GLUT4 transporters and a shift towards a more oxidative metabolism in skeletal muscle of β-GPA-fed rats (Shoubridge et al. 1985; Ren et al. 1993) suggest that absence of Cr leads to increased glucose uptake and catabolism in muscle, which is in agreement with the increased glucose tolerance and insulin sensitivity that we have observed in the AGAT−/− mice (Choe et al. 2012). Our data suggested that (P)Cr depletion and decreased ATP levels result in chronic activation of AMP-activated protein kinase (AMPK) and insulin-independent glucose uptake in skeletal muscle. It is worth noting that AMPK promotes mitochondrial biogenesis in muscle of WT mice and AMPK knockouts, both treated with β-GPA (Zong et al. 2002). Activation of AMPK counteracts energy deficiency by increasing glucose transport via enhanced GLUT4 translocation (Holmes et al. 1999; Hayashi et al. 2000; Bergeron et al. 2001), stimulating β-oxidation by inhibition of acetyl CoA carboxylase (Kudo et al. 1995; Dyck et al. 1999) and increasing mitochondrial enzyme activities (Winder et al. 2000). Those adaptations indicate an upregulation of oxidative metabolism and fit well, in particular with the large elevation in AMP/ATP ratio, which determines the phosphorylation (i.e. activation) of AMPK. The upregulation of AMPK activity is worth further study to elucidate the mechanism linking Cr depletion with increased GLUT4 expression.
The severe atrophy in the AGAT−/− mice occurs to a certain extent also in β-GPA-fed rats, as shown by a decrease in the diameter of fast-twitch fibres (Shoubridge et al. 1985). Moreover, electron-dense mitochondrial inner membrane inclusion bodies have been reported in muscle of β-GPA-fed rats, presenting initially only in slow-twitch muscle, but during longer β-GPA supplementation also in fast-twitch muscle and heart (O’Gorman et al. 1997). These structures were associated with high levels of ScCKmit that co-crystallise within the mitochondrial cristae with unknown structures, possibly in the absence of a proper substrate (Stadhouders et al. 1994; O’Gorman et al. 1996, 1997).
Taken together, these studies indicate that enhanced aerobic metabolism occurs in muscle depleted of Cr, whether by β-GPA administration or by GAMT or AGAT deficiency. The present results suggest that the fully Cr-depleted muscles in AGAT−/− mice are most severely affected, perhaps because PGAA in GAMT−/− mice, β-GPA in rodents fed with this Cr analogue, or residual PCr still can serve as an HEP buffer, whereas the PCr–CK system in Cr-depleted AGAT−/− muscle is completely non-functional (Wyss & Kaddurah-Daouk, 2000). Although valuable information on consequences of Cr deficiency have been gained from studies performed on Cr-analogue-fed models and GAMT−/− mice, the AGAT−/− mice in this study are a model for pure Cr deficiency. Recently, a complete Cr deficiency has also been demonstrated in a mouse model for creatine transporter (CrT) deficiency, but to our knowledge the consequences for muscle metabolism and morphology have not yet been investigated (Skelton et al. 2011).
Ischaemia and upregulated glycolytic ATP production
To characterize the metabolic effects of pure Cr deficiency further, we subjected Cr-depleted AGAT−/− muscle to a period of ischaemia. The AGAT−/− mice showed an enhanced drop in pH in occluded muscle, which is at least partly due to the absence of the pH-buffering capacity of the CK reaction. In agreement with this, a similar end-pH during the same ischaemic protocol was observed in the double CK knockout, CK=/= (in 't Zandt et al. 1999). Interestingly, in those mice the ATP declined by 30% during the ischaemic period, with a proportional elevation in Pi and phosphomonoesters. Although ATP levels were lower in resting AGAT−/− muscle, they did not decline further during ischaemic occlusion. This might be explained at least partly by ATP production from residual PCr (∼3 mm) in the muscles of the knockout mice that were used for the ischaemia study. Nevertheless, in the absence of almost all of the ATP-buffering and pH-buffering effects of the PCr–CK system, the muscle was able to generate sufficient ATP from glycolysis to maintain the increased ATP demand without significant loss of ATP.
Together with the upregulated ATPase activity in resting muscle after 2 days of Cr supplementation, the slow changes in Pi levels upon supplementation and breakdown suggest that there are slow adaptations in ATPase activity at the transcriptional level. Unfortunately, the low signal-to-noise ratio of the 31P MR spectra of the small muscle volumes in AGAT−/− mice hampered accurate determination of postischaemic recovery in AGAT−/− mice after 2 days of Cr supplementation.
Creatine supplementation
In patients with AGAT or with GAMT deficiency, oral administration of Cr increases its levels in the central nervous system and improves neurological condition (Stöckler et al. 1994; Bianchi et al. 2000). However, treatment responses in other tissues have not been thoroughly investigated in patients, except for two CrT-deficient patients who did show Cr in muscle in contrast to a complete deficiency in brain (deGrauw et al. 2003; Pyne-Geithman et al. 2004). In the present study, we explored the accumulation of Cr by muscle of AGAT−/− mice and demonstrated that muscle was replenished within 1 day of creatine supplementation. The overshoot in PCr/β-ATP ratios during the first 2 days of Cr supplementation can be explained by the lower ATP concentrations in the AGAT−/− muscle that are presumably caused by transcriptional adaptations and take several days to increase from 13.5 μmol (g dry weight)−1 to normal WT levels (25.1 μmol (g dry weight)−1).
As muscle volume drastically increased upon Cr treatment, depleted AGAT−/− muscle tissue clearly benefits from Cr treatment, not only from a metabolic point of view. The gain in muscle mass accounted for at least a part of the increase in body weight in response to Cr treatment, and is likely to involve increases in the volume of individual muscle fibres. A relation between muscle volume and Cr concentration was not only shown in β-GPA-fed rats, in which Cr depletion was accompanied by decreases in the diameter of glycolytic muscle fibres; Cr supplementation in healthy subjects has also frequently been associated with an increase in cellular volume, which might be due to an osmotic effect of creatine (Low et al. 1996; Juhn & Tarnopolsky, 1998; van Loon et al. 2003). Interestingly, we observed a transient change in the level of another potential osmolyte, taurine, in response to the rapid and large increase in tCr levels, which might suggest a balance between taurine and Cr in osmolytic homeostasis.
A comparison between the rates of Cr accumulation in AGAT−/− and GAMT−/− mice revealed that Cr in muscle of AGAT−/− mice immediately reached normal levels, whereas Cr concentrations in GAMT−/− mice increased substantially, but less pronounced during the first day of supplementation (Kan et al. 2007), which was then followed by a slower increase to a supranormal steady-state level. This small difference in the accumulation curves between the two groups could be related to the fact that the 1H MRS voxels in AGAT−/− and GAMT−/− mice were placed in different hindlimb muscles. Thus, we cannot rule out a potential effect of fibre-type composition on the accumulation curves. The delayed uptake in the GAMT−/− mice was explained by competition between the newly available Cr and the accumulated plasma GAA (Boehm et al. 1996). However, it is not known whether AGAT−/− mammals have a more profound upregulation of Cr transporters.
In contrast to the complete tCr replenishment in muscle within 1 day of supplementation, the brain demonstrated a much more gradual increase of Cr, taking about 20 days to reach normal WT values. This pattern has also been observed in a GAMT-deficient patient (Ensenauer et al. 2004) and in GAMT−/− mice (Kan et al. 2007). Previous in vivo labelling studies in rats and mice showed that the brain is able to take up Cr from the blood, even against a large concentration gradient (Ohtsuki et al. 2002; Perasso et al. 2003), via the Na+- and Cl−-dependent Cr transporter protein, SLC6A8 (Guimbal & Kilimann, 1993; Sora et al. 1994). This transporter is expressed in the microcapillary endothelial cells that form the blood–brain barrier (Braissant et al. 2001; Ohtsuki et al. 2002; Nakashima et al. 2004; Acosta et al. 2005), but not in the astrocytes that surround the capillaries, which might be the main reason for the relatively inefficient cerebral uptake of Cr (Braissant et al. 2001; Ohtsuki et al. 2002; Tachikawa et al. 2004; Braissant, 2012).
Creatine administration studies in healthy humans, rats and mice have shown no change (Horn et al. 1998; Wilkinson et al. 2006) or a limited elevation of Cr in the brain (Dechent et al. 1999; Ipsiroglu et al. 2001; Lyoo et al. 2003; Hersch et al. 2006) with respect to basal concentrations. Our results show saturation of the cerebral Cr accumulation, which levels off at the normal concentrations of WT control animals. This is most likely to be due to downregulation of transporter proteins at high Cr concentrations (Guerrero-Ontiveros & Wallimann, 1998; ten Hove et al. 2008).
The virtually complete replenishment of cerebral Cr in AGAT−/− mice seems to be in accordance with previous reports on AGAT-deficient patients, in whom oral Cr intake increased Cr levels in the brain and improved neurological condition (Bianchi et al. 2000, 2007; Battini et al. 2002). However, a comparative study in a few patients with either AGAT or GAMT deficiency suggested a less effective treatment response in GAMT−/− patients (Bianchi et al. 2007; Schulze & Battini, 2007), which has been ascribed to a potential competition between the elevated GAA levels in blood and the administered Cr (Boehm et al. 1996) and/or a neurotoxic effect of GAA accumulation. In contrast, we did not observe a difference in Cr accumulation between the two mouse models. Furthermore, GAA is not taken up into brains of AGAT–GAMT double knockout mice on a 0.5% GAA-containing diet (data not shown), indicating that GAA cannot pass the blood–brain barrier and thus might not compete with Cr at the Cr transporter.
Creatine breakdown
The irreversible conversion of Cr and PCr into Crn is one of the few non-enzymatic reactions in vivo. Membrane permeability for Crn results in a constant diffusion out of the Cr-containing tissues into the blood, from where it is excreted in urine (Wyss & Kaddurah-Daouk, 2000). Whole-body Cr turnover can be determined by 15N-labelling studies. We have reported a 13C-labelling study of local turnover in human muscle where Cr content was only mildly increased by 13C-labelled supplementation (Kan et al. 2006). The present study enabled the determination of tissue-specific turnover/degradation of Cr in a fully depleted to normal range. The results are in accordance with the reported total body Cr turnover of ∼1.7% (Walker, 1979; Wyss & Kaddurah-Daouk, 2000) and the local turnover we reported (Kan et al. 2006), as expected for a non-enzymatic process with no involvement of rate-limiting transporters.
The depletion of Cr from normal to a complete absence takes several months, as shown by the breakdown rates. This explains why the MRS data still show some residual tCr and PCr, because these mice were measured after 5–6 months of Cr-free diet. The accumulation of Cr, in contrast, is much faster and shows that AGAT−/− mice have the useful property of being switchable between states of complete tissue Cr depletion and normal Cr levels simply by withholding Cr or feeding them with Cr. This avoids complications due to any pre- and postnatal adaptation that occurs in mice lacking the various isoforms of brain- or muscle-specific CK or the Cr transporter.
In rapid perturbations, such as ischaemia or exercise, changes in the concentration of PCr and Pi are opposite, so that total Pi + PCr content is maintained; there are no changes in net Pi or Cr flux across the sarcolemma on this time scale. (This linear inverse relation between Pi and PCr can be seen in the dotted line in Fig. 8 representing changes during ischaemia in WT mice). However, changes in resting concentration during long-term perturbations depend on different physiological processes, largely those of trans-sarcolemmal transport. Total phosphate content (or its close surrogate, Pi + PCr) is not maintained during supplementation or depletion; during Cr supplementation replenishment of (P)Cr occurred instantly, while the high initial Pi levels in skeletal muscle of AGAT−/− mice normalized only slowly (Fig. 7). Therefore, Pi was not directly depleted as a mass-balance consequence of phosphorylation of the Cr taken up. The slow kinetics of the normalization of Pi suggest transcriptional processes, and the general principles of the regulation of cell [Pi] imply that these are at work in trans-sarcolemmal Pi transport, perhaps as a part of the reversal of a general process of adaptation towards a predominantly oxidative metabolic phenotype in Cr-deficient muscle. A similar phenomenon was observed in GAMT−/− mice during Cr supplementation, because (PCr + PGAA)/ATP ratios after 2 days of Cr administration were increased when compared with normal PCr/ATP ratios, while Pi/β-ATP was also still beyond normal WT levels (Kan et al. 2004).
Conclusion
In conclusion, the mouse model with AGAT deficiency, which can be switched between Cr-depleted and Cr-repleted states, provides a unique opportunity to study muscle metabolism and morphology in the absence of Cr without compensation by accumulated guanidino compounds. Compared with CK-deficient muscle, which does contain Cr although it cannot be used in the CK system, the more severe phenotype of AGAT−/− mice on a Cr-free diet provides evidence for an important role of Cr in skeletal muscle integrity, apart from being a substrate in the CK reaction. The effects of Cr depletion on muscle show some similarities with the phenotype of Cr-depleted GAMT−/− mice and β-GPA-fed rodents. The phenotype in Cr-depleted AGAT−/− skeletal muscle appears more pronounced, which is not surprising given that it is the only model in which Cr is completely depleted and no alternative HEP buffer is available. As in the other models of PCr–CK system deficiencies, depleted AGAT−/− muscle adapts towards a more oxidative metabolism. Transport of Cr into muscle tissue is very efficient; therefore, Cr deficiency in skeletal muscle is easily rescued by dietary Cr intake.
Acknowledgments
We would like to thank The Netherlands organisation for scientific research (NWO) (investment grant 834.04.007), the Prinses Beatrixfonds (WAR06-0217) and the Deutsche Forschungsgemeinschaft (DFG; CH872/1-1, IS63/3-1 and IS63/3-2) for financial support. In particular, we thank M. Romeijn, J. Senden, J. van Asten, L. Wintjes and B. Stoltenborg for excellent technical assistance, M. Schweizer, C. Raithore and S. Fehr for help with histology and J. Jeneson for helpful discussions.
Glossary
- AGAT
l-arginine:glycine amidinotransferase
- AMPK
adenosine monophosphate-activated protein kinase
- β-GPA
β-guanidinopropionic acid
- CK
creatine kinase
- Cr
creatine
- Crn
creatinine
- CrT
creatine transporter
- CS
citrate synthase
- EM
electron microscopy
- GAA
guanidine acetate
- GAMT
guanidine acetate methyltransferase
- GLUT4
glucose transporter 4
- HEP
high-energy phosphate
- MR
magnetic resonance
- MRS
magnetic resonance spectroscopy
- ROI
region of interest
- PCr
phosphocreatine
- (P)Cr
either phosphocreatine or creatine
- PGAA
phosphorylated guanidino acetate
- Pi
inorganic phosphate
- Tau
taurine
- tCr
total creatine (Cr + PCr)
- WT
wild-type
- TR
repetition time
- TE
echo time
- FOV
field of view
- TM
mixing time
- STEAM
stimulated echo acquisition mode
- ISIS
image selected in vivo spectroscopy
Author contributions
All authors approved the final version for publication. Conception and design of the experiments: C.I.N., C.U.C., A.V., H.E.K. and A.H. Collection and analysis of data: C.I.N., C.U.C., J.M. and R.J.T.R. Interpretation of data: C.I.N., C.U.C., H.E.K., B.W., D.I., G.J.K. and A.H. Drafting and revising article: C.I.N., C.U.C., H.E.K., B.W., L.J.C.L., R.J.T.R., D.I., G.J.K. and A.H. The MR experiments and determinations of mitochondrial activities were performed in Nijmegen, Grip strength tests and hisopathology was performed in Hamburg, and biochemical metabolite concentrations were determined in Maastricht.
Author's present address
C. I. Nabuurs: Department of Radiology, Maastricht University Medical Center, PO Box 5800, 6202 AZ Maastricht, The Netherlands.
References
- Acosta ML, Kalloniatis M, Christie DL. Creatine transporter localization in developing and adult retina: importance of creatine to retinal function. Am J Physiol Cell Physiol. 2005;289:C1015–C1023. doi: 10.1152/ajpcell.00137.2005. [DOI] [PubMed] [Google Scholar]
- Akima H, Lott D, Senesac C, Deol J, Germain S, Arpan I, Bendixen R, Lee Sweeney H, Walter G, Vandenborne K. Relationships of thigh muscle contractile and non-contractile tissue with function, strength, and age in boys with Duchenne muscular dystrophy. Neuromuscul Disord. 2012;22:16–25. doi: 10.1016/j.nmd.2011.06.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battini R, Leuzzi V, Carducci C, Tosetti M, Bianchi MC, Item CB, Stöckler-Ipsiroglu S, Cioni G. Creatine depletion in a new case with AGAT deficiency: clinical and genetic study in a large pedigree. Mol Genet Metab. 2002;77:326–331. doi: 10.1016/s1096-7192(02)00175-0. [DOI] [PubMed] [Google Scholar]
- Bergeron R, Previs SF, Cline GW, Perret P, Russell RR, 3rd, Young LH, Shulman GI. Effect of 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside infusion on in vivo glucose and lipid metabolism in lean and obese Zucker rats. Diabetes. 2001;50:1076–1082. doi: 10.2337/diabetes.50.5.1076. [DOI] [PubMed] [Google Scholar]
- Bianchi MC, Tosetti M, Battini R, Leuzzi V, Alessandri' MG, Carducci C, Antonozzi I, Cioni G. Treatment monitoring of brain creatine deficiency syndromes: a 1H- and 31P-MR spectroscopy study. AJNR Am J Neuroradiol. 2007;28:548–554. [PMC free article] [PubMed] [Google Scholar]
- Bianchi MC, Tosetti M, Fornai F, Alessandri MG, Cipriani P, De Vito G, Canapicchi R. Reversible brain creatine deficiency in two sisters with normal blood creatine level. Ann Neurol. 2000;47:511–513. [PubMed] [Google Scholar]
- Blei ML, Conley KE, Kushmerick MJ. Separate measures of ATP utilization and recovery in human skeletal muscle. J Physiol. 1993;465:203–222. doi: 10.1113/jphysiol.1993.sp019673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm EA, Radda GK, Tomlin H, Clark JF. The utilisation of creatine and its analogues by cytosolic and mitochondrial creatine kinase. Biochim Biophys Acta. 1996;1274:119–128. doi: 10.1016/0005-2728(96)00018-7. [DOI] [PubMed] [Google Scholar]
- Bottomley PA. Spatial localization in NMR spectroscopy in vivo. Ann N Y Acad Sci. 1987;508:333–348. doi: 10.1111/j.1749-6632.1987.tb32915.x. [DOI] [PubMed] [Google Scholar]
- Braissant O. Creatine and guanidinoacetate transport at blood-brain and blood-cerebrospinal fluid barriers. J Inherit Metab Dis. 2012;35:655–664. doi: 10.1007/s10545-011-9433-2. [DOI] [PubMed] [Google Scholar]
- Braissant O, Henry H, Loup M, Eilers B, Bachmann C. Endogenous synthesis and transport of creatine in the rat brain: an in situ hybridization study. Brain Res Mol Brain Res. 2001;86:193–201. doi: 10.1016/s0169-328x(00)00269-2. [DOI] [PubMed] [Google Scholar]
- Choe CU, Nabuurs C, Stockebrand M, Neu A, Nunes P, Morellini F, Sauter K, Schillemeit S, Hermans-Borgmeyer I, Marescau B, Heerschap A, Isbrandt D. L-Arginine:glycine amidinotransferase (AGAT) deficiency protects from metabolic syndrome. Hum Mol Genet. 2012 doi: 10.1093/hmg/dds407. DOI. [DOI] [PubMed] [Google Scholar]
- Clark JF, Khuchua Z, Kuznetsov AV, Vassil’eva E, Boehm E, Radda GK, Saks V. Actions of the creatine analogue beta-guanidinopropionic acid on rat heart mitochondria. Biochem J. 1994;300:211–216. doi: 10.1042/bj3000211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooperstein SJ, Lazarow A. A microspectrophotometric method for the determination of cytochrome oxidase. J Biol Chem. 1951;189:665–670. [PubMed] [Google Scholar]
- Dahlstedt AJ, Katz A, Wieringa B, Westerblad H. Is creatine kinase responsible for fatigue? Studies of isolated skeletal muscle deficient in creatine kinase. FASEB J. 2000;14:982–990. doi: 10.1096/fasebj.14.7.982. [DOI] [PubMed] [Google Scholar]
- Das AM, Ullrich K, Isbrandt D. Upregulation of respiratory chain enzymes in guanidinoacetate methyltransferase deficiency. J Inherit Metab Dis. 2000;23:375–377. doi: 10.1023/a:1005643617251. [DOI] [PubMed] [Google Scholar]
- Dechent P, Pouwels PJ, Wilken B, Hanefeld F, Frahm J. Increase of total creatine in human brain after oral supplementation of creatine-monohydrate. Am J Physiol. 1999;277:R698–R704. doi: 10.1152/ajpregu.1999.277.3.R698. [DOI] [PubMed] [Google Scholar]
- deGrauw TJ, Cecil KM, Byars AW, Salomons GS, Ball WS, Jakobs C. The clinical syndrome of creatine transporter deficiency. Mol Cell Biochem. 2003;244:45–48. [PubMed] [Google Scholar]
- de Haan A, Koudijs JC, Wevers RA, Wieringa B. The effects of MM-creatine kinase deficiency on sustained force production of mouse fast skeletal muscle. Exp Physiol. 1995;80:491–494. doi: 10.1113/expphysiol.1995.sp003863. [DOI] [PubMed] [Google Scholar]
- del Porto LA, Nicholson GA, Ketheswaren P. Correlation between muscle atrophy on MRI and manual strength testing in hereditary neuropathies. J Clin Neurosci. 2010;17:874–878. doi: 10.1016/j.jocn.2009.11.006. [DOI] [PubMed] [Google Scholar]
- Dyck JR, Kudo N, Barr AJ, Davies SP, Hardie DG, Lopaschuk GD. Phosphorylation control of cardiac acetyl-CoA carboxylase by cAMP-dependent protein kinase and 5′-AMP activated protein kinase. Eur J Biochem. 1999;262:184–190. doi: 10.1046/j.1432-1327.1999.00371.x. [DOI] [PubMed] [Google Scholar]
- Ensenauer R, Thiel T, Schwab KO, Tacke U, Stöckler-Ipsiroglu S, Schulze A, Hennig J, Lehnert W. Guanidinoacetate methyltransferase deficiency: differences of creatine uptake in human brain and muscle. Mol Genet Metab. 2004;82:208–213. doi: 10.1016/j.ymgme.2004.04.005. [DOI] [PubMed] [Google Scholar]
- Guerrero-Ontiveros ML, Wallimann T. Creatine supplementation in health and disease. Effects of chronic creatine ingestion in vivo: down-regulation of the expression of creatine transporter isoforms in skeletal muscle. Mol Cell Biochem. 1998;184:427–437. [PubMed] [Google Scholar]
- Guimbal C, Kilimann MW. A Na+-dependent creatine transporter in rabbit brain, muscle, heart, and kidney. cDNA cloning and functional expression. J Biol Chem. 1993;268:8418–8421. [PubMed] [Google Scholar]
- Harris RC, Hultman E, Nordesjö LO. Glycogen, glycolytic intermediates and high-energy phosphates determined in biopsy samples of musculus quadriceps femoris of man at rest. Methods and variance of values. Scand J Clin Lab Invest. 1974;33:109–120. [PubMed] [Google Scholar]
- Hayashi T, Hirshman MF, Fujii N, Habinowski SA, Witters LA, Goodyear LJ. Metabolic stress and altered glucose transport: activation of AMP-activated protein kinase as a unifying coupling mechanism. Diabetes. 2000;49:527–531. doi: 10.2337/diabetes.49.4.527. [DOI] [PubMed] [Google Scholar]
- Heerschap A, Kan HE, Nabuurs CI, Renema WK, Isbrandt D, Wieringa B. In vivo magnetic resonance spectroscopy of transgenic mice with altered expression of guanidinoacetate methyltransferase and creatine kinase isoenzymes. Subcell Biochem. 2007;46:119–148. doi: 10.1007/978-1-4020-6486-9_7. [DOI] [PubMed] [Google Scholar]
- Hersch SM, Gevorkian S, Marder K, Moskowitz C, Feigin A, Cox M, Como P, Zimmerman C, Lin M, Zhang L, Ulug AM, Beal MF, Matson W, Bogdanov M, Ebbel E, Zaleta A, Kaneko Y, Jenkins B, Hevelone N, Zhang H, Yu H, Schoenfeld D, Ferrante R, Rosas HD. Creatine in Huntington disease is safe, tolerable, bioavailable in brain and reduces serum 8OH2′dG. Neurology. 2006;66:250–252. doi: 10.1212/01.wnl.0000194318.74946.b6. [DOI] [PubMed] [Google Scholar]
- Holmes BF, Kurth-Kraczek EJ, Winder WW. Chronic activation of 5′-AMP-activated protein kinase increases GLUT-4, hexokinase, and glycogen in muscle. J Appl Physiol. 1999;87:1990–1995. doi: 10.1152/jappl.1999.87.5.1990. [DOI] [PubMed] [Google Scholar]
- Horn M, Frantz S, Remkes H, Laser A, Urban B, Mettenleiter A, Schnackerz K, Neubauer S. Effects of chronic dietary creatine feeding on cardiac energy metabolism and on creatine content in heart, skeletal muscle, brain, liver and kidney. J Mol Cell Cardiol. 1998;30:277–284. doi: 10.1006/jmcc.1997.0590. [DOI] [PubMed] [Google Scholar]
- in 't Zandt HJ, de Groof AJ, Renema WK, Oerlemans FT, Klomp DW, Wieringa B, Heerschap A. Presence of (phospho)creatine in developing and adult skeletal muscle of mice without mitochondrial and cytosolic muscle creatine kinase isoforms. J Physiol. 2003;548:847–858. doi: 10.1113/jphysiol.2002.034538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- in 't Zandt HJ, Oerlemans F, Wieringa B, Heerschap A. Effects of ischemia on skeletal muscle energy metabolism in mice lacking creatine kinase monitored by in vivo31P nuclear magnetic resonance spectroscopy. NMR Biomed. 1999;12:327–334. doi: 10.1002/(sici)1099-1492(199910)12:6<327::aid-nbm570>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- in 't Zandt HJ, Renema WK, Streijger F, Jost C, Klomp DW, Oerlemans F, Van der Zee CE, Wieringa B, Heerschap A. Cerebral creatine kinase deficiency influences metabolite levels and morphology in the mouse brain: a quantitative in vivo1H and 31P magnetic resonance study. J Neurochem. 2004;90:1321–1330. doi: 10.1111/j.1471-4159.2004.02599.x. [DOI] [PubMed] [Google Scholar]
- Ipsiroglu OS, Stromberger C, Ilas J, Höger H, Muhl A, Stöckler-Ipsiroglu S. Changes of tissue creatine concentrations upon oral supplementation of creatine-monohydrate in various animal species. Life Sci. 2001;69:1805–1815. doi: 10.1016/s0024-3205(01)01268-1. [DOI] [PubMed] [Google Scholar]
- Janssen AJ, Trijbels FJ, Sengers RC, Smeitink JA, van den Heuvel LP, Wintjes LT, Stoltenborg-Hogenkamp BJ, Rodenburg RJ. Spectrophotometric assay for complex I of the respiratory chain in tissue samples and cultured fibroblasts. Clin Chem. 2007;53:729–734. doi: 10.1373/clinchem.2006.078873. [DOI] [PubMed] [Google Scholar]
- Jeneson JA, ter Veld F, Schmitz JP, Meyer RA, Hilbers PA, Nicolay K. Similar mitochondrial activation kinetics in wild-type and creatine kinase-deficient fast-twitch muscle indicate significant Pi control of respiration. Am J Physiol Regul Integr Comp Physiol. 2011;300:R1316–R1325. doi: 10.1152/ajpregu.00204.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonckheere AI, Hogeveen M, Nijtmans LG, van den Brand MA, Janssen AJ, Diepstra JH, van den Brandt FC, van den Heuvel LP, Hol FA, Hofste TG, Kapusta L, Dillmann U, Shamdeen MG, Smeitink JA, Rodenburg RJ. A novel mitochondrial ATP8 gene mutation in a patient with apical hypertrophic cardiomyopathy and neuropathy. J Med Genet. 2008;45:129–133. doi: 10.1136/jmg.2007.052084. [DOI] [PubMed] [Google Scholar]
- Juhn MS, Tarnopolsky M. Potential side effects of oral creatine supplementation: a critical review. Clin J Sport Med. 1998;8:298–304. doi: 10.1097/00042752-199810000-00007. [DOI] [PubMed] [Google Scholar]
- Kan HE, Buse-Pot TE, Peco R, Isbrandt D, Heerschap A, de Haan A. Lower force and impaired performance during high-intensity electrical stimulation in skeletal muscle of GAMT-deficient knockout mice. Am J Physiol Cell Physiol. 2005;289:C113–C119. doi: 10.1152/ajpcell.00040.2005. [DOI] [PubMed] [Google Scholar]
- Kan HE, Meeuwissen E, van Asten JJ, Veltien A, Isbrandt D, Heerschap A. Creatine uptake in brain and skeletal muscle of mice lacking guanidinoacetate methyltransferase assessed by magnetic resonance spectroscopy. J Appl Physiol. 2007;102:2121–2127. doi: 10.1152/japplphysiol.01327.2006. [DOI] [PubMed] [Google Scholar]
- Kan HE, Renema WK, Isbrandt D, Heerschap A. Phosphorylated guanidinoacetate partly compensates for the lack of phosphocreatine in skeletal muscle of mice lacking guanidinoacetate methyltransferase. J Physiol. 2004;560:219–229. doi: 10.1113/jphysiol.2004.067926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan HE, van der Graaf M, Klomp DW, Vlak MH, Padberg GW, Heerschap A. Intake of 13C-4 creatine enables simultaneous assessment of creatine and phosphocreatine pools in human skeletal muscle by 13C MR spectroscopy. Magn Reson Med. 2006;56:953–957. doi: 10.1002/mrm.21068. [DOI] [PubMed] [Google Scholar]
- Kellner A, Matschke J, Bernreuther C, Moch H, Ferrer I, Glatzel M. Autoantibodies against β-amyloid are common in Alzheimer's disease and help control plaque burden. Ann Neurol. 2009;65:24–31. doi: 10.1002/ana.21475. [DOI] [PubMed] [Google Scholar]
- Kemp GJ, Roussel M, Bendahan D, Le Fur Y, Cozzone PJ. Interrelations of ATP synthesis and proton handling in ischaemically exercising human forearm muscle studied by 31P magnetic resonance spectroscopy. J Physiol. 2001;535:901–928. doi: 10.1111/j.1469-7793.2001.00901.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo N, Barr AJ, Barr RL, Desai S, Lopaschuk GD. High rates of fatty acid oxidation during reperfusion of ischemic hearts are associated with a decrease in malonyl-CoA levels due to an increase in 5′-AMP-activated protein kinase inhibition of acetyl-CoA carboxylase. J Biol Chem. 1995;270:17513–17520. doi: 10.1074/jbc.270.29.17513. [DOI] [PubMed] [Google Scholar]
- Kushmerick MJ. Multiple equilibria of cations with metabolites in muscle bioenergetics. Am J Physiol. 1997;272:C1739–C1747. doi: 10.1152/ajpcell.1997.272.5.C1739. [DOI] [PubMed] [Google Scholar]
- Kushmerick MJ, Moerland TS, Wiseman RW. Mammalian skeletal muscle fibers distinguished by contents of phosphocreatine, ATP, and Pi. Proc Natl Acad Sci U S A. 1992;89:7521–7525. doi: 10.1073/pnas.89.16.7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low SY, Rennie MJ, Taylor PM. Modulation of glycogen synthesis in rat skeletal muscle by changes in cell volume. J Physiol. 1996;495:299–303. doi: 10.1113/jphysiol.1996.sp021594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyoo IK, Kong SW, Sung SM, Hirashima F, Parow A, Hennen J, Cohen BM, Renshaw PF. Multinuclear magnetic resonance spectroscopy of high-energy phosphate metabolites in human brain following oral supplementation of creatine-monohydrate. Psychiatry Res. 2003;123:87–100. doi: 10.1016/s0925-4927(03)00046-5. [DOI] [PubMed] [Google Scholar]
- Marcinek DJ, Kushmerick MJ, Conley KE. Lactic acidosis in vivo: testing the link between lactate generation and H+ accumulation in ischemic mouse muscle. J Appl Physiol. 2010;108:1479–1486. doi: 10.1152/japplphysiol.01189.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcinek DJ, Schenkman KA, Ciesielski WA, Conley KE. Mitochondrial coupling in vivo in mouse skeletal muscle. Am J Physiol Cell Physiol. 2004;286:C457–C463. doi: 10.1152/ajpcell.00237.2003. [DOI] [PubMed] [Google Scholar]
- Maughan RJ, Watson JS, Weir J. Strength and cross-sectional area of human skeletal muscle. J Physiol. 1983;338:37–49. doi: 10.1113/jphysiol.1983.sp014658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer RA, Brown TR, Kushmerick MJ. Phosphorus nuclear magnetic resonance of fast- and slow-twitch muscle. Am J Physiol. 1985;248:C279–C287. doi: 10.1152/ajpcell.1985.248.3.C279. [DOI] [PubMed] [Google Scholar]
- Moerland TS, Wolf NG, Kushmerick MJ. Administration of a creatine analogue induces isomyosin transitions in muscle. Am J Physiol. 1989;257:C810–C816. doi: 10.1152/ajpcell.1989.257.4.C810. [DOI] [PubMed] [Google Scholar]
- Momken I, Lechene P, Koulmann N, Fortin D, Mateo P, Doan BT, Hoerter J, Bigard X, Veksler V, Ventura-Clapier R. Impaired voluntary running capacity of creatine kinase-deficient mice. J Physiol. 2005;565:951–964. doi: 10.1113/jphysiol.2005.086397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon RB, Richards JH. Determination of intracellular pH by 31P magnetic resonance. J Biol Chem. 1973;248:7276–7278. [PubMed] [Google Scholar]
- Mourmans J, Wendel U, Bentlage HA, Trijbels JM, Smeitink JA, de Coo IF, Gabreels FJ, Sengers RC, Ruitenbeek W. Clinical heterogeneity in respiratory chain complex III deficiency in childhood. J Neurol Sci. 1997;149:111–117. doi: 10.1016/s0022-510x(97)05379-3. [DOI] [PubMed] [Google Scholar]
- Nakashima T, Tomi M, Katayama K, Tachikawa M, Watanabe M, Terasaki T, Hosoya K. Blood-to-retina transport of creatine via creatine transporter (CRT) at the rat inner blood–retinal barrier. J Neurochem. 2004;89:1454–1461. doi: 10.1111/j.1471-4159.2004.02437.x. [DOI] [PubMed] [Google Scholar]
- Nicolay K, van Dorsten FA, Reese T, Kruiskamp MJ, Gellerich JF, van Echteld CJ. In situ measurements of creatine kinase flux by NMR. The lessons from bioengineered mice. Mol Cell Biochem. 1998;184:195–208. [PubMed] [Google Scholar]
- O’Connor RS, Steeds CM, Wiseman RW, Pavlath GK. Phosphocreatine as an energy source for actin cytoskeletal rearrangements during myoblast fusion. J Physiol. 2008;586:2841–2853. doi: 10.1113/jphysiol.2008.151027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Gorman E, Beutner G, Wallimann T, Brdiczka D. Differential effects of creatine depletion on the regulation of enzyme activities and on creatine-stimulated mitochondrial respiration in skeletal muscle, heart, and brain. Biochim Biophys Acta. 1996;1276:161–170. doi: 10.1016/0005-2728(96)00074-6. [DOI] [PubMed] [Google Scholar]
- O’Gorman E, Fuchs KH, Tittmann P, Gross H, Wallimann T. Crystalline mitochondrial inclusion bodies isolated from creatine depleted rat soleus muscle. J Cell Sci. 1997;110:1403–1411. doi: 10.1242/jcs.110.12.1403. [DOI] [PubMed] [Google Scholar]
- Ohtsuki S, Tachikawa M, Takanaga H, Shimizu H, Watanabe M, Hosoya K, Terasaki T. The blood-brain barrier creatine transporter is a major pathway for supplying creatine to the brain. J Cereb Blood Flow Metab. 2002;22:1327–1335. doi: 10.1097/01.WCB.0000033966.83623.7D. [DOI] [PubMed] [Google Scholar]
- Pathare N, Walter GA, Stevens JE, Yang Z, Okerke E, Gibbs JD, Esterhai JL, Scarborough MT, Gibbs CP, Sweeney HL, Vandenborne K. Changes in inorganic phosphate and force production in human skeletal muscle after cast immobilization. J Appl Physiol. 2005;98:307–314. doi: 10.1152/japplphysiol.00612.2004. [DOI] [PubMed] [Google Scholar]
- Perasso L, Cupello A, Lunardi GL, Principato C, Gandolfo C, Balestrino M. Kinetics of creatine in blood and brain after intraperitoneal injection in the rat. Brain Res. 2003;974:37–42. doi: 10.1016/s0006-8993(03)02547-2. [DOI] [PubMed] [Google Scholar]
- Pfeuffer J, Tkáč I, Provencher SW, Gruetter R. Toward an in vivo neurochemical profile: quantification of 18 metabolites in short-echo-time 1H NMR spectra of the rat brain. J Magn Reson. 1999;141:104–120. doi: 10.1006/jmre.1999.1895. [DOI] [PubMed] [Google Scholar]
- Provencher SW. Estimation of metabolite concentrations from localized in vivo proton NMR spectra. Magn Reson Med. 1993;30:672–679. doi: 10.1002/mrm.1910300604. [DOI] [PubMed] [Google Scholar]
- Pyne-Geithman GJ, deGrauw TJ, Cecil KM, Chuck G, Lyons MA, Ishida Y, Clark JF. Presence of normal creatine in the muscle of a patient with a mutation in the creatine transporter: a case study. Mol Cell Biochem. 2004;262:35–39. doi: 10.1023/b:mcbi.0000038213.15646.4a. [DOI] [PubMed] [Google Scholar]
- Ren JM, Semenkovich CF, Holloszy JO. Adaptation of muscle to creatine depletion: effect on GLUT-4 glucose transporter expression. Am J Physiol. 1993;264:C146–C150. doi: 10.1152/ajpcell.1993.264.1.C146. [DOI] [PubMed] [Google Scholar]
- Renema WK, Schmidt A, van Asten JJ, Oerlemans F, Ullrich K, Wieringa B, Isbrandt D, Heerschap A. MR spectroscopy of muscle and brain in guanidinoacetate methyltransferase (GAMT)-deficient mice: validation of an animal model to study creatine deficiency. Magn Reson Med. 2003;50:936–943. doi: 10.1002/mrm.10627. [DOI] [PubMed] [Google Scholar]
- Roberts JJ, Walker JB. Synthesis and accumulation of an extremely stable high-energy phosphate compound by muscle, heart, and brain of animals fed the creatine analog, 1-carboxyethyl-2-iminoimidazolidine (homocyclocreatine) Arch Biochem Biophys. 1983;220:563–571. doi: 10.1016/0003-9861(83)90448-4. [DOI] [PubMed] [Google Scholar]
- Roos A, Boron WF. Intracellular pH. Physiol Rev. 1981;61:296–434. doi: 10.1152/physrev.1981.61.2.296. [DOI] [PubMed] [Google Scholar]
- Saks V. The phosphocreatine–creatine kinase system helps to shape muscle cells and keep them healthy and alive. J Physiol. 2008;586:2817–2818. doi: 10.1113/jphysiol.2008.155358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saks V, Kaambre T, Guzun R, Anmann T, Sikk P, Schlattner U, Wallimann T, Aliev M, Vendelin M. The creatine kinase phosphotransfer network: thermodynamic and kinetic considerations, the impact of the mitochondrial outer membrane and modelling approaches. Subcell Biochem. 2007;46:27–65. doi: 10.1007/978-1-4020-6486-9_3. [DOI] [PubMed] [Google Scholar]
- Salomons G, Wyss M. Creatine and creatine kinase in health and disease—a bright future ahead? Subcell Biochem. 2007;46:309–334. doi: 10.1007/978-1-4020-6486-9_16. [DOI] [PubMed] [Google Scholar]
- Scheibye-Knudsen M, Quistorff B. Regulation of mitochondrial respiration by inorganic phosphate; comparing permeabilized muscle fibers and isolated mitochondria prepared from type-1 and type-2 rat skeletal muscle. Eur J Appl Physiol. 2009;105:279–287. doi: 10.1007/s00421-008-0901-9. [DOI] [PubMed] [Google Scholar]
- Schmidt A, Marescau B, Boehm EA, Renema WK, Peco R, Das A, Steinfeld R, Chan S, Wallis J, Davidoff M, Ullrich K, Waldschutz R, Heerschap A, De Deyn PP, Neubauer S, Isbrandt D. Severely altered guanidino compound levels, disturbed body weight homeostasis and impaired fertility in a mouse model of guanidinoacetate N-methyltransferase (GAMT) deficiency. Hum Mol Genet. 2004;13:905–921. doi: 10.1093/hmg/ddh112. [DOI] [PubMed] [Google Scholar]
- Schulze A, Battini R. Pre-symptomatic treatment of creatine biosynthesis defects. Subcell Biochem. 2007;46:167–181. doi: 10.1007/978-1-4020-6486-9_9. [DOI] [PubMed] [Google Scholar]
- Shoubridge EA, Challiss RA, Hayes DJ, Radda GK. Biochemical adaptation in the skeletal muscle of rats depleted of creatine with the substrate analogue beta-guanidinopropionic acid. Biochem J. 1985;232:125–131. doi: 10.1042/bj2320125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjøgaard G, Saltin B. Extra- and intracellular water spaces in muscles of man at rest and with dynamic exercise. Am J Physiol. 1982;243:R271–R280. doi: 10.1152/ajpregu.1982.243.3.R271. [DOI] [PubMed] [Google Scholar]
- Skelton MR, Schaefer TL, Graham DL, Degrauw TJ, Clark JF, Williams MT, Vorhees CV. Creatine transporter (CrT; Slc6a8) knockout mice as a model of human CrT deficiency. PLoS One. 2011;6:e16187. doi: 10.1371/journal.pone.0016187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sora I, Richman J, Santoro G, Wei H, Wang Y, Vanderah T, Horvath R, Nguyen M, Waite S, Roeske WR, Yamamura HI. The cloning and expression of a human creatine transporter. Biochem Biophys Res Commun. 1994;204:419–427. doi: 10.1006/bbrc.1994.2475. [DOI] [PubMed] [Google Scholar]
- Stadhouders AM, Jap PH, Winkler HP, Eppenberger HM, Wallimann T. Mitochondrial creatine kinase: a major constituent of pathological inclusions seen in mitochondrial myopathies. Proc Natl Acad Sci U S A. 1994;91:5089–5093. doi: 10.1073/pnas.91.11.5089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steeghs K, Benders A, Oerlemans F, de Haan A, Heerschap A, Ruitenbeek W, Jost C, van Deursen J, Perryman B, Pette D, Brückwilder M, Koudijs J, Jap P, Veerkamp J, Wieringa B. Altered Ca2+ responses in muscles with combined mitochondrial and cytosolic creatine kinase deficiencies. Cell. 1997a;89:93–103. doi: 10.1016/s0092-8674(00)80186-5. [DOI] [PubMed] [Google Scholar]
- Steeghs K, Heerschap A, de Haan A, Ruitenbeek W, Oerlemans F, van Deursen J, Perryman B, Pette D, Brückwilder M, Koudijs J, Jap P, Wieringa B. Use of gene targeting for compromising energy homeostasis in neuro-muscular tissues: the role of sarcomeric mitochondrial creatine kinase. J Neurosci Methods. 1997b;71:29–41. doi: 10.1016/s0165-0270(96)00124-0. [DOI] [PubMed] [Google Scholar]
- Steeghs K, Oerlemans F, de Haan A, Heerschap A, Verdoodt L, de Bie M, Ruitenbeek W, Benders A, Jost C, van Deursen J, Tullson P, Terjung R, Jap P, Jacob W, Pette D, Wieringa B. Cytoarchitectural and metabolic adaptations in muscles with mitochondrial and cytosolic creatine kinase deficiencies. Mol Cell Biochem. 1998;184:183–194. [PubMed] [Google Scholar]
- Stöckler S, Holzbach U, Hanefeld F, Marquardt I, Helms G, Requart M, Hänicke W, Frahm J. Creatine deficiency in the brain: a new, treatable inborn error of metabolism. Pediatr Res. 1994;36:409–413. doi: 10.1203/00006450-199409000-00023. [DOI] [PubMed] [Google Scholar]
- Stöckler S, Marescau B, De Deyn PP, Trijbels JM, Hanefeld F. Guanidino compounds in guanidinoacetate methyltransferase deficiency, a new inborn error of creatine synthesis. Metabolism. 1997;46:1189–1193. doi: 10.1016/s0026-0495(97)90215-8. [DOI] [PubMed] [Google Scholar]
- Tachikawa M, Fukaya M, Terasaki T, Ohtsuki S, Watanabe M. Distinct cellular expressions of creatine synthetic enzyme GAMT and creatine kinases uCK-Mi and CK-B suggest a novel neuron–glial relationship for brain energy homeostasis. Eur J Neurosci. 2004;20:144–160. doi: 10.1111/j.1460-9568.2004.03478.x. [DOI] [PubMed] [Google Scholar]
- ten Hove M, Makinen K, Sebag-Montefiore L, Hunyor I, Fischer A, Wallis J, Isbrandt D, Lygate C, Neubauer S. Creatine uptake in mouse hearts with genetically altered creatine levels. J Mol Cell Cardiol. 2008;45:453–459. doi: 10.1016/j.yjmcc.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkáč I, Starčuk Z, Choi IY, Gruetter R. In vivo 1H NMR spectroscopy of rat brain at 1 ms echo time. Magn Reson Med. 1999;41:649–656. doi: 10.1002/(sici)1522-2594(199904)41:4<649::aid-mrm2>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- van Loon LJ, Oosterlaar AM, Hartgens F, Hesselink MK, Snow RJ, Wagenmakers AJ. Effects of creatine loading and prolonged creatine supplementation on body composition, fuel selection, sprint and endurance performance in humans. Clin Sci (Lond) 2003;104:153–162. doi: 10.1042/CS20020159. [DOI] [PubMed] [Google Scholar]
- Walker JB. Creatine: biosynthesis, regulation, and function. Adv Enzymol Relat Areas Mol Biol. 1979;50:177–242. doi: 10.1002/9780470122952.ch4. [DOI] [PubMed] [Google Scholar]
- Wilkinson ID, Mitchel N, Breivik S, Greenwood P, Griffiths PD, Winter EM, Van Beek EJ. Effects of creatine supplementation on cerebral white matter in competitive sportsmen. Clin J Sport Med. 2006;16:63–67. doi: 10.1097/01.jsm.0000176372.67398.c8. [DOI] [PubMed] [Google Scholar]
- Winder WW, Holmes BF, Rubink DS, Jensen EB, Chen M, Holloszy JO. Activation of AMP-activated protein kinase increases mitochondrial enzymes in skeletal muscle. J Appl Physiol. 2000;88:2219–2226. doi: 10.1152/jappl.2000.88.6.2219. [DOI] [PubMed] [Google Scholar]
- Wyss M, Kaddurah-Daouk R. Creatine and creatinine metabolism. Physiol Rev. 2000;80:1107–1213. doi: 10.1152/physrev.2000.80.3.1107. [DOI] [PubMed] [Google Scholar]
- Zong H, Ren JM, Young LH, Pypaert M, Mu J, Birnbaum MJ, Shulman GI. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc Natl Acad Sci U S A. 2002;99:15983–15987. doi: 10.1073/pnas.252625599. [DOI] [PMC free article] [PubMed] [Google Scholar]